Abstract
In this study, based on the OneKP database and through comparative genetic analysis, we found that HMT and HDM may originate from Chromista and are highly conserved in green plants, and that during the evolution from algae to land plants, histone methylation modifications gradually became complex and diverse, which is more conducive to the adaptation of plants to complex and variable environments. We also characterized the number of members, genetic similarity, and phylogeny of HMT and HDM families in barley using the barley pangenome and the Tibetan Lasa Goumang genome. The results showed that HMT and HDM were highly conserved in the domestication of barley, but there were some differences in the Lasa Goumang SDG subfamily. Expression analysis showed that HvHMTs and HvHDMs were highly expressed in specific tissues and had complex expression patterns under multiple stress treatments. In summary, the amplification and variation of HMT and HDM facilitate plant adaptation to complex terrestrial environments, while they are highly conserved in barley and play an important role in barley growth and development with abiotic stresses. In brief, our findings provide a novel perspective on the origin and evolutionary history of plant HvHMTs and HvHDMs, and lay a foundation for further investigation of their functions in barley.
1. Introduction
Histone methylation and demethylation modifications are the main epigenetic mechanisms, often referred to as the “second genetic code”, and play important roles in regulating transcription, genome integrity and epigenetics [1,2,3]. Histone methylation is dynamically and reversibly regulated by histone methyltransferases (HMTs) and histone demethylases (HDMs). HMT contains SDG (SET domain group) and PRMT (protein arginine methyltransferase) subfamilies. The HDM contains the HDMA (SWIRM and C-terminal domain) and JMJ (JmjC domain-containing proteins) subfamilies [3]. Histone lysine modification is catalysed by SDGs, which have been classified into seven classes in previous studies in plants such as Arabidopsis [4], Brassica napus [5] and Dendrobium catenatum [6]. Class I members, E (Z) homologues, can transfer methyl groups to H3K27; class II members, ASH1 homologues, are responsible for H3K4/H3K36 methylation; class III members, Trx homologues and related proteins are responsible for H3K4me1/3; class IV members, plant-specific proteins that contain SET and PHD domains, are responsible for monomethylation at H3K27; class V, SU (var) homologues are responsible for H3K9 methylation; and class VI and VII genes include disrupted SET domains (S-ET) or poorly functionally defined SET-related domains [6]. Arginine methylation is catalysed by a family of enzymes known as protein arginine methyltransferases (PRMTs). PRMTs are classified as type I or type II, depending on the position of the methyl group on the guanidine of the methylated arginine [7]. Seven type I arginine methyltransferases (PRMT1a, PRMT1b, PRMT3, PRMT4a, PRMT4b, PRMT6 and PRMT10) and one type II enzyme (PRMT5) have been found in Arabidopsis [2]. On the other hand, histone methylation can be removed by the action of histone demethylases (HDMs) [8]. HDMA is a flavin adenine dinucleotide (FAD)-dependent enzyme that can only remove single/double lysine residue methylation. However, JMJ subfamily proteins that use Fe (II) and -ketoglutarate (KG) as cofactors will catalyse the removal of mono/di/tri-lysine residue methylation [1]. The JMJ subfamily can be divided into five categories according to sequence similarity and catalytic specificity in plants [1]. KDM5/JARID1 group proteins have been found to catalyse the demethylation of H3K4me1/2/3, KDM4/JHDM3 family proteins have been shown to actively demethylate histone H3K9me2/3, H3K36me2/3 and KDM3A/JHDM2 can demethylate H3K9me2 and H3K9me1 but not H3K9me3, the JMJD6 group can demethylate H3R2me2 and H4R3me2, and the JmjC domain-only subfamily can remove the methylation of H3K27me3 [3].
A combination of epigenetics and environmental stress dynamics has led scientists to investigate the impact of epigenetics on evolution in plants [9]. Theoretical models and simulations predict that epigenetic variation has the potential to affect the rate and outcomes of adaptation [10,11,12]. The adjustment of gene expression is a key mechanism used by plants during growth and developmental processes. Because plants are immobile organisms, control of gene expression becomes more essential when plants are subjected to inescapable environmental stressors [13,14]. Histone methylation can promote plant adaptation to stressful environments by regulating the expression of stress-responsive genes [15,16]. ARABIDOPSIS TRITHORAX4 (ATX4) and ATX5 play essential roles in the drought stress response [17]. Atx4 and atx5 single loss-of-function mutants showed drought stress-tolerant and ABA-hypersensitive phenotypes during seed germination and seedling development [17]. Downregulation of stress-responsive genes upon dehydration treatment is accompanied by an obvious decrease in H3K4me and H3K4me3, while H3K4me2 levels are only slightly reduced, indicating that HDMs might modulate the expression of dehydration stress-responsive genes [18]. During salt stress, SKB1 mediates the salt response by altering the methylation status of H4R3sme2 of stress-responsive genes [19]. Partial histone methylation modifications have been shown to be missing in some algae, suggesting that plants may have acquired more diverse histone methylation modifications through evolution, and that this evolution may have facilitated plant adaptation to adversity [20,21]. However, studies have mainly focused on angiosperms, and nothing is known about the origin of HMT and HDM in the plant kingdom and the evolutionary relationship between species. Further studies on the evolutionary history of HMT and HDM will help to better understand their important roles in plant evolution.
Barley (Hordeum vulgare L.) is among the world’s earliest domesticated crop species, being cultivated in the Nile River Valley of Egypt at least 17,000 years ago [22]. It is the world’s fourth largest cereal crop and has many uses, such as feeding, food and beer [23]. Grain yield is the most important driver of grower profitability and is thus a key breeding target for genetic improvement. Yield is, however, a challenging breeding target as it is controlled by a large number of genes with varying effects, and strongly influenced by genotype and by environmental interactions (complex traits) [24]. Research has indicated that histone methylation can regulate gene expression, thereby modulating the growth and development of barley and its adaptation to stress. High chromatin H3K27me3 levels before vernalization inhibited the expression of the HvVRN1 gene that induced vernalization in barley (Hv-Hordeum vulgare L.), and after vernalization, chromatin H3K4me3 levels increased, while H3K27me3 levels decreased, promoting its expression [25]. Under salt stress, salt-tolerant varieties of rye were significantly enriched in H3K4me3 and H3K27me3 compared to salt-sensitive varieties, and the significant enrichment of both modifications was associated with oxidative processes [26]. However, the characterization of barley HMT and HDM families has not yet been reported. Furthermore, barley has a large and complex genome, similar to other Triticae crops such as wheat, making genome-based selection difficult. In recent years, the publication of the barley pangenome and the Tibetan naked barley (Lasa Goumang) genome has greatly facilitated the genetic investigation of barley [27,28]. Limited research has been conducted on the comprehensive characterization of gene families across various germplasms. The identification and analysis of HMT and HDM in multiple germplasms are particularly valuable for comprehending their significant roles in the evolutionary process of barley adaptation.
In this study, we analysed the origin of HMT and HDM in the plant kingdom and the evolutionary relationship between different species. The HMT and HDM families in barley were characterized through the barley pangenome and the Tibetan barley variety (Lasa Goumang), and their expression profiles in different organs and under various abiotic stresses were also analysed. This provides a basis for future exploration of the regulatory role of HMT and HDM in barley growth, development and stress response.
2. Results
2.1. Origin and Evolution of HMT and HDM in Green Plants
To explore the origin of HMT and HDM in the plant kingdom, we performed a comparative genetic analysis among green plants [29]. The amino acid sequences of 55 HMTs and 29 HDMs from Arabidopsis were set as seed sequences to search the OneKP Phytozome genome database. Phylogenetics showed that HMT and HDM are conserved in land plants and algae, and that both may have evolved from Chromista (such as Petalonia fascia, Scytosiphon dotyo, Colpomenia sinuosa, etc.) (Figure S1). Phylogenetic topology clearly separates algae from land plants. To further explore the evolutionary relationship between HMT and HDM between algae and land plants, a total of 3846 homologous genes were identified from 35 species, including monocots, dicots, gymnosperms, ferns, lycophytes, mosses, green algae, red algae, and brown algae (Figure 1a). Dramatic changes in the environment accompanied the evolution from algae to land plants; asteroids hit the Earth several times, and a particularly severe impact preceded the emergence of land plants, with continual increases in atmospheric oxygen, fluctuations in carbon dioxide levels, and continually rising light intensity (Figure 1a) [30]. The number of HMT and HDM family members among species was significantly higher in land plants than in algae, including the gene copy number of SDG, JMJ, PRMT (there were slightly fewer ferns and gymnosperms) and HDMA subfamilies (Figure 1a). Therefore, a comparative genetic analysis grouped subfamilies according to Arabidopsis classification based on the multiple protein sequence ratios of 35 different species, with all Arabidopsis HMT and HDM gene families as references (Figure 1b). We found that some subgroup members of the SDG and JMJ subfamilies were missing in algae. Among the SDG subfamily, class I and class IV share H3K27 methylation modifications, but their homologues were not found in brown algae. In addition, although class I and class IV share H3K27 methylation modification, class IV appeared late in the evolution of species, and its homologues were not found throughout algae until land plants. Similarly, class ii and class III share H3K4 methylation modification, but class III appears later than class II. This suggests that as species evolved, the same histone methylation modification changed from being regulated by a single grouping to being regulated by multiple groupings, which contributed to a more complex modification mechanism of histone methylation. Class V (modified H3K9 methylation) is the largest subfamily, including SUVH and SUVR homologues. SUVR is conserved in algae, but SUVH is only found in Chlamydomonas (green algae). In the JMJ subfamily, KDM3 (modified H3K9me1/2) was missing, and its homologues were also found only in Chlamydomonas. Hence, as algae evolved to land plants, family expansion led to the emergence of several new groupings and homologues.
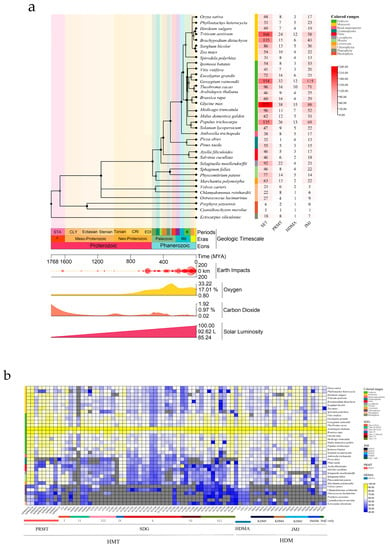
Figure 1.
(a) Predicted number of HMT and HDM in different plant and algal species. Coloured squares indicate the protein predicted number from 0 (white) to 240 (red). (b) Similarity heatmap of HMT and HDM proteins in plant and algal species. Coloured squares indicate protein sequence similarity from zero (blue) to 100% (yellow). Gray squares indicate no proteins that satisfied the selection criteria.
2.2. Number of HMTs and HDMs in Barley
A total of 1671 homologous genes were identified from 21 materials of the barley pangenome and Lasa Goumang (Table 1; Supplementary Materials: Table S1). In HvHMTs, there were a total of 7 HvPRMTs in all germplasms, with between 49 and 53 HvSDGs in wild and cultivated barley (except Lasa Goumang), while in Lasa Goumang, there were significantly fewer HvSDGs than in other germplasms (only 42). Regarding HvHDMs, there were 4 HvHDMAs in all germplasms, with between 17 and 19 HvJMJs members in all germplasms.

Table 1.
Number of HMT and HDM family members in different barley germplasms.
2.3. Variation in HMT and HDM Family Members in Barley
To investigate the variation among different germplasms, HvHMTs and HvHDMs in Morex were used as a reference to observe the sequence homology of homologous genes among different germplasms (Figure 2). The homology of most genes was above 99%, including the homology found in the HvHDMA and HvPRMT subfamilies. However, the individual gene sequences of HvJMJ and HvSDG had clear variations; for example, the homologous gene sequence identity of HORVU.MOREX.r3.1HG0093350.1 (SDG) in B1K-04-12 was 26.0%, and that of HORVU.MOREX.r3.5HG0474610.1 (SDG) in HOR_13821 was 69.1%, whereas the homologous gene sequence homology of HORVU.MOREX.r3.4HG0411310.1 (JMJ) in Lasa Goumang was 44.1%. These results indicate that homologous genes among different barley germplasms are highly homologous, but few genes have undergone gene sequence variation.

Figure 2.
Sequence homology of homologous genes among different germplasms. Coloured squares indicate protein sequence similarity from zero (blue) to 100% (red). Gray squares indicate no proteins that satisfied the selection criteria.
2.4. Phylogenetic, Conserved Domain and Motif Analysis of HMT and HDM in Barley
To investigate the structural characteristics and phylogenetics among the HvHMTs and HvHDMs, the 217 HMT and 89 HDM protein sequences from wild barley (B1K-04-12), cultivated barley (Morex), Tibetan barley (Lasa Goumang) and Arabidopsis were used (Figure 3 and Figure S2, Supplementary Materials: Table S2). The HvHMT family is composed of two subfamilies, HvSDG and HvPRMT. All HvSDGs are classified into seven groups, each of which contains group-specific conserved domains in addition to the SET domain. Class I contains CXC and SET domains; class II contains zf-CW, Pre-SET and AWS domains; class III contains zf-HC5HC2H_2, PWWP and PHD domains; class IV contains the Zf_RING and PHD domains; the class V subgroup I contains the Pre-SET and SAD_SRA domains, and subgroup II contains the Pre-SET and WIYLD domains; and classes VI and VII contain the Rubis-subs-bind and zf-MY ND domains. HvPRMTs were highly conserved, including PrmA, Methyltransf_25 and PRMT5 domains, and they were divided into type I and type II. Phylogenetic analysis revealed that all AtPRMTs have homologous genes in barley, except AtPRMT7. The HvJMJ subfamily in HvHDM was divided into five groups, and KDM4 and KDM5 could be further divided into two subgroups. KDM3 features the zf-4CXXC_R1 domain and the JmjC domain. The HvJMJs of KDM4 all contain JmjC, JmjN and zf-C2H2 domains, and the HvJMJs of the KDM4 class falls into two main subclasses. Subgroup I is characterized by the zf-H2C2_2 domain, while subgroup II contains the zf-C5HC2 domain. The HvJMJs of the KDM5 class all contain JmjN, zf-C5HC2 and Jmjc domains. Additionally, subgroup I contains FYRN and FYRC domains, while subgroup II contains 1 ARID, 2 PHD and 2 PLU-1 domains. Jmjc-only contains only one Cupin_8 domain; JMJD6 contains cupin_Rmlc-like, F-box-like and APH domains. All genes of HvHDMA were highly conserved, containing only one N-terminal Amino_oxidase and one C-terminal SWIRM domain.
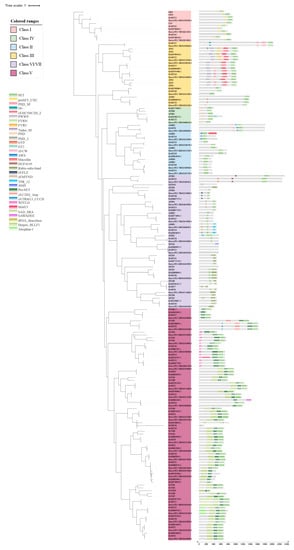
Figure 3.
Phylogenetic and conserved domain analysis of SDG.
The above results indicate that HMT and HDM are highly conserved in barley. However, domain observation among wild barley, cultivated barley, and Lasa Goumang revealed that domestication also led to the emergence of some new structural domains and the loss of some structures of a few genes in the HvSDG and HvJMJ subfamilies, with more significant losses in the SDG subclade of Lasa Goumang. For example, Morex and Lasa Goumang contain the SPICE domain (JMJ subfamily), which is missing in wild barley B1K-04-12; Morex lacks the zf-TRM13_CCCH and zf-C2H2_3rep domains contained in Lasa Goumang and wild barley (SDG subfamily); and Lasa Goumang contains germplasm-specific Med15, tRNA_deacylase and SAWADEE domains (SDG subfamily). In addition, we explored the source of deletion of SDG family members in Lasa Goumang and found that the missing genes in Lasa Goumang were mainly from class II, and AWS and zf-CW domains unique to class II were missing (Supplementary Materials: Table S3, Figure 3).
To better understand the conservation and diversification of HvHMTs and HvHDMs, the putative motifs of all HvHMT and HvHDM proteins were predicted by MEME motif analysis (Figure S3). HvHMT and HvHDM proteins in the same group had similar motifs, which aligned with the results of the phylogenetic analysis. Additionally, there were a few differences in motifs between the HvSDG and HvJMJ subfamilies among germplasms, and they were mainly concentrated in HvSDG subfamily genes of Lasa Goumang. In general, family members within the same group had similar functions.
2.5. Chromosomal Location Analysis of HvHMTs and HvHDMs
We performed a chromosomal localization analysis of histone methylation gene families in three different barley cultivars (Morex, B1K-04-12, OUN333) (Figure S4). The results showed highly similar distribution of homologous genes across the different cultivars. All genes were located on barley chromosomes 1 to 7. Chromosomes 3 and 5 contained the largest number of genes, while chromosome 4 had the fewest. SET subfamily genes were distributed on all chromosomes, PRMT subfamily genes were located on chromosomes 1, 2, 5, 6, and 7, JMJ subfamily genes were located on all chromosomes except chromosome 2, and HDMA subfamily genes were located on chromosomes 2, 6, and 7. The gene distribution of HMT and HDM subfamilies among different germplasms was highly consistent, which indicated that HMT and HDM subfamilies were highly conserved in barley. In the barley genome, genes with different histone methylation modifications are distributed on different chromosomes, which may reflect their functional differences in different biological processes.
2.6. Analysis of Cis-Acting Elements of HvHMTs and HvHDMs
Plant growth and development are regulated by different cis-regulatory elements in genes. Various cis-acting elements, containing hormone-sensitive, light-responsive, stress-responsive, tissue-specific and other elements were mined and analyzed from the 2000-bp upstream regulatory regions of HvHMTs and HvHDMs genes using the PlantCARE web site (Figure 4). The most abundant cis-acting element was the light-responsive element; tissue-specific elements, including in the endosperm, meristems, roots, seeds and palisade mesophyll cells, were found in the putative promoters of HvHMT and HvHDM genes. Hormone-responsive elements, in response to auxin, ABA, GA, MeJA and SA, were also widely observed; multiple abiotic and biotic stress-related elements, including in response to drought, anaerobic, hypothermia, injury, defense and stress, were largely enriched. These results suggest that HvHMTs and HvHDMs might play important roles in plant growth and development, hormone signaling, and resistance to biotic and abiotic stresses.

Figure 4.
Analysis of promoter cis-acting elements of HvHMTs and HvHDMs. Gray squares indicates that the number of cis-acting elements in the gene is zero.
2.7. HvHMTs and HvHDMs Expression Patterns in Different Organs
Analysis of gene expression patterns can provide directions and strategies for prediction of gene function [31]. The expression profiles of HMTs and HDMs at 16 developmental stages of barley were investigated using RNA-seq data (Figure 5). All HvHMTs and HvHDMs were expressed in at least one tissue or stage. The same genes, such as HvJMJ6, HvSET15 and HvSET38, were highly expressed in almost all tested tissues across diverse developmental stages; hence, they played an integral role in organ formation and development. Most genes were highly expressed in the developing inflorescence (INF1 and INF2), and histone methylation modification genes might play an important role in the development of barley inflorescence. In addition, expression profiling revealed that some genes were tissue- and stage specific. HvSET48, HvSET6, HvSET36, and HvSET40 were only highly expressed in senescent leaves and etiolated seedlings, suggesting that they might have functions in regulating cell senescence and death. HvSET2, HvSET46, HvJMJ10, and HvSET26 are highly expressed only in the early stage of grain development (CAR5), while HvSET3, HvSET19, and HvSET31 are highly expressed in both stages of grain development (CAR5 and CAR15), indicating a specific role in grain development. HvJMJ13, HvSET3, HvSET30 and HvPRMT1, HvPRMT5 and HvPRMT10 were also specifically expressed in the epidermis and roots of surrounding seedlings.
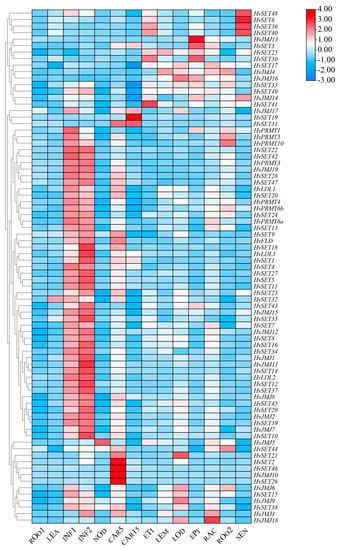
Figure 5.
The spatiotemporal expression profile of HvHMTs and HvHDMs in different tissues or stages of barley. CAR15: bracts removed grains at 15 DPA; CAR5: bracts removed grains at 5 DPA; EPI: epidermis at 4 weeks old; ETI: etiolated from 10-day-old seedling; INF1: young inflorescences at 5 mm; INF2: young inflorescences at 1–1.5 cm; LEA: shoot at 10 cm from the seedlings; LEM: lemma at 6 weeks after anthesis; LOD: lodicule at 6 weeks after anthesis; NOD: developing tillers at six-leaf stage; RAC: rachis at 5 weeks after anthesis; ROO2: root from 4-week-old seedlings; ROO1: roots from the seedlings at 10 cm shoot stage; SEN: senescing leaf.
2.8. HvHMTs and HvHDMs Expression Profiling under Abiotic Stresses
To confirm whether HvHMTs and HvHDMs are involved in the stress response, comprehensive expression patterns were analyzed under different abiotic stresses, including low temperature, drought, salt, and waterlogging (Figure 6a). A total of 36 HvHMTs (30 HvSDGs and 6 HvPRMTs) and 18 HvHDMs (14 HvJMJs and 4 HvHDMAs) were differentially expressed under different stresses (Supplementary Materials: Table S4). Different response patterns were noted in a single subclade, and some were specifically stress-dependent. For example, of a total of seven members of the PRMT subfamily, six were significantly upregulated under low-temperature stress, whereas no members of the PRMT subfamily were differentially expressed under flooding stress. A total of four HDMA subfamily members were differentially expressed under low temperature, salt and flooding stresses, while no HDMA subfamily members were differentially expressed under drought stress. Among the differentially expressed genes, the expression patterns of certain genes differed under different stress treatments. For instance, HvJMJ4 expression was downregulated under drought stress but upregulated under low-temperature stress, and HvSET20 expression was upregulated under low temperature stress but downregulated under salt stress. In addition, HvHMTs and HvHDMs were differentially expressed in different germplasms. Whereas HvPRMT3, HvPRMT10, HvJMJ4, and HvSET22 were differentially expressed only in sensitive varieties under drought stress, under salt stress, HvPRMT3, HvLDL1, HvJMJ1, and HvSET9 were differentially expressed only in tolerant varieties. These results suggest that HvHMTs and HvHDMs have complex expression patterns and are variety-dependent.
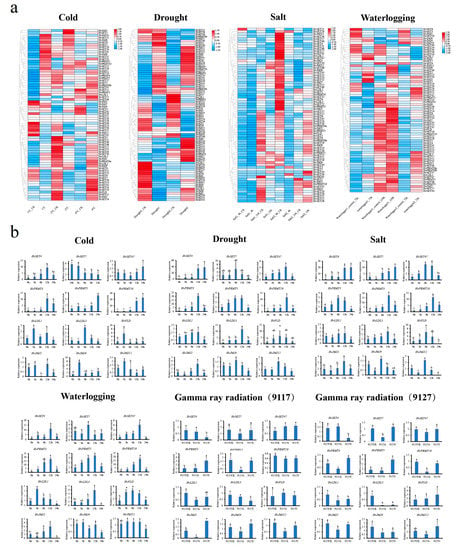
Figure 6.
(a) RNA-seq: Cold, the expression patterns of HvHMTs and HvHDMs at 1 °C, −4 °C, and −8 °C. Drought, the expression patterns of HvHMTs and HvHDMs in sensitive varieties (Drought 1) and drought-tolerant varieties (Drought 2) under drought stress. Salt, the expression patterns of HvHMTs and HvHDMs in salt-sensitive varieties (Salt 1) and salt-tolerant varieties (Salt 2) under salt treatment for 4 h and 16 h. Waterlogging, the expression patterns of HvHMTs and HvHDMs of waterlogging tolerant varieties (Waterlogging 1) and medium waterlogging tolerant varieties (Waterlogging 2) under flooding. (b) RT-PCR: Expression of barley at 0, 3, 6, 12, and 24 h under low temperature, drought, salt, and flooding stress, and the expression of sensitive varieties (9127) and tolerant varieties (9117) under 50 Gy and 200 Gy intensity of gamma ray irradiation. Error bars represent ± SD from three biological repeats. Lowercase letters above bars indicate a significant difference (p < 0.05, LSD) among the treatments.
To eliminate intervarietal differences and to further validate the above results, we subjected the barley varieties of Efupi N0.1 to low temperature, drought, salt and flooding stress treatments. Furthermore, in addition to these stresses under natural conditions, we further explored the expression patterns of HvHMTs and HvHDMs under ionizing radiation (gamma) using different sensitive varieties (tolerant variety, 9117; sensitive variety, 9127). We selected 12 genes differentially expressed in the barley transcriptome under low temperature, drought, salt, and flooding for qRT–PCR, and the primers are listed in Table S5 (Figure 6b). For example, HvLDL3 and HvPRMT3 showed an expression pattern that peaked at 12 h under all four stresses except radiation. The upregulation of HvPRMT3 was also similar, ranging from 17-fold to 20-fold under salt, drought, and flooding stresses. However, most of them showed specific expression patterns under different stress treatments; for example, HvJMJ4 showed significant inhibition after 3 h of drought but was significantly induced at 3 h under low temperature, and HvSET4 expression gradually increased at 0–12 h and significantly decreased after 24 h under low temperature and salt stress, while its expression continued to increase under drought and flooding stress and remained at a high level after 24 h. In addition, the expression of 13 genes under radiation was correlated with both material sensitivity and radiation dose, and HvPRMT5 expression was significantly suppressed in the tolerant material (9117) at a dose of 50 Gy, while there was no difference in expression in the sensitive material (9127). In tolerant material (9117), HvSET4 gene expression did not change significantly at 50 Gy, while expression was significantly repressed at 200 Gy. The above results indicate that some HvHMTs and HvHDMs had similar expression patterns, and that most genes had complex expression pattern variations. Therefore, differential expression of HvHMTs and HvHDMs under ionizing radiation further highlights that histone methylation modification is an epigenetic modification that widely responds to multiple stresses.
3. Discussion
Proof of similarity between eukaryotic and archaeal chromatin suggests that histone and chromatin structures evolved before archaea and eukaryotes diverged [32,33]. This also suggests that the initial function of nucleosomes and chromatin formation might have been the regulation of gene expression rather than the packaging of DNA. Histone methylation and acetylation are considered to be the most important and prevalent epigenetic markers associated with gene regulation [34]. The regulatory effects of histone methylation modifications can facilitate rapid adaptation of organisms to extreme fluctuations in environmental conditions [35]. In this study, we found that the evolution from algae to land plants was accompanied by great changes in the environment. At the same time, the colonization of terrestrial habitats by plants is accompanied by exposure to a number of abiotic stress factors, such as high irradiance, lack of mineral nutrients (bare rocks and no soils), varying temperature, and dehydration [36]. In this study, the number of HMTs (PRMTs and SETs) and HDMs (JMJs and HDMAs) was found to increase significantly during the aquatic-terrestrial transition from algae to land plants, while their sequence homology also changed significantly. In addition, all subfamilies and subgroups of HMT and HDM are commonly found in land plants, and only the KDM3 subgroup is missing in Selaginella moellendorffii. In algae, a large number of homologous genes of the SDG and JMJ subfamilies were missing. H3K27me3 was absent in brown algae, while H3K9me2/3 was detected only at very low levels, which is consistent with the results of the present study [37]. The homologue of E(z) (SDG class I) that controls H3K27 methylation was not detected in brown algae during this study. Furthermore, the SUVH (SDG) homologue was absent in brown algae, suggesting that low levels of H3K9me2/3 may be regulated by the SUVR homologue in class V (SDG). In whole algae, KDM3 (JMJ) and SUVH homologues are only present in Chlamydomonas reinhardtii, and the SUVH homologue SET3p has been reported, which functions in vitro as a specific H3K9 mono-methyltransferase [38]. Some subsequent subgroups, such as class III and class IV of the SDG subfamily, commodify H3K4 and H3K9 methylation with the previously existing class II and class I, which makes histone methylation modifications more complex and diverse. In summary, during the evolution of the species, HMT and HDM showed an amplified state, and along with the emergence of some new subgroups in the HMT and HDM subfamilies, histone methylation gradually became complex and diverse. The emergence of new resistance genes often takes hundreds of thousands of years, and in the face of abrupt and dramatic environmental changes, histone methylation modifications, which have become more complex and diverse with evolution, may be more rapid and effective in regulating the expression of existing resistance genes than in generating new resistance genes. Complex and diverse histone methylation modifications can promote better adaptation of plants to complex terrestrial environments and help them cope with sudden environmental stresses.
The domestication process of barley and its wide adaptability have long been the focus of researchers around the world. In the past decade, individual reference genomes have not been fully representative of species diversity due to the high degree of genomic variation. Sequencing of the barley pangenome and Lasa Goumang provides an excellent opportunity for this purpose [39]. On this basis, we identified HMT and HDM using the barley pangenome and the Lasa Goumang genome. Plant domestication is an evolutionary process whereby humans have used wild species to develop new and altered forms of plants with morphological or physiological traits that meet human needs [40]. The domestication process has resulted in reduced diversity at both the genome and local level. For example, more than half of genetic variation has been lost in cultivated soybean, 2–4% of maize genes have experienced artificial selection, and cultivated rice has also suffered genetic erosion [41,42,43,44]. In this study, we found that only a small number of genes in the SDG and JMJ subfamilies of wild barley and cultivated barley (except Lasa Goumang) have low homology, with few differences in the conserved domains. However, the number of family members is nearly identical, and most genes in farmed and wild germplasm are nearly identical. This suggests that the HMT and HDM families have an important role and remain highly conserved in the domestication process of barley. The PRMT, HDMA and JMJ subfamilies in Lasa Goumang were not significantly different from those in wild barley and cultivated barley (except Lasa Goumang). However, there were significantly fewer SDG subfamily members in Lasa Goumang, and they showed more variation than other germplasms, accompanied by the addition and loss of structural domains. Members of the PRMT, HDMA and JMJ subfamilies in Lasa Goumang were not significantly different from those in wild barley and cultivated barley, but the number of members of the SDG subfamilies in Lasa Goumang was significantly lower. At the same time, there is obvious addition and loss of domains, and the class II grouping of the SDG subfamily is missing. However, this phenomenon was not found for other barley varieties in the pangenome. Therefore, we speculate that this may be due to the loss of some SDG subfamily genes in Lasa Goumang during the artificial domestication process, or major mutations in some SDG subfamily genes, which made it impossible to identify them by Blastp.
Histone methylation can occur at various sites in histone proteins, primarily on lysine and arginine residues, and it can be governed by multiple positive and negative regulators, even at a single site, to either activate or repress transcription [45]. Cis-acting elements in promoter regions are always associated with their transcriptional control and function, and in this study, functional elements associated with tissue-specific stress responses were identified in the predicted promoters of HvHMTs and HvHDMs. The important role of histone methylation in regulating flowering, grain development, leaf senescence and root growth has been revealed in Arabidopsis, rice and Eucalyptus grandis, and the specific high expression of HvHMTs and HvHDMs in barley flowers, grains, senescing leaves and roots indicates their important role in barley growth and development [46,47,48,49]. Abiotic stresses, including heat, drought, cold, flooding, and salinity, negatively impact crop productivity. Histone methylation has been shown to represent key modulators in plant stress responses [50]. At low temperatures, COLD-REGULATED15A (COR15A) and GALACTINOL SYNTHASE3 (GOLS3) are activated by a decrease in the H3K27me3 modification level in the gene region, thereby helping plants to defend against coldness [51]. Under drought stress, drought response factors and genes in the ABA biosynthesis pathway are activated by H3K4me3 modification [52]. Salt stress generally results in the deposition of active histone markers such as H3K9K14Ac and H3K4me3, and reduces the deposition of H3K9me2 and H3K27me3 inhibitory histone markers on salt-tolerant genes [13]. Dynamic and reversible histone modifications occur in rice seedlings under submergence stress, and ADH1 and PDC1, which are submergence-inducible genes, respond to stress as a result of increased H3K4 methylation and H3 acetylation [53]. In this study, experimental data from RNA-seq and qRT–PCR showed that HvHMTs and HvHDMs exhibited significant transcriptional responses with complex expression patterns under abiotic stresses. Generally, histone H3K9 and H3K27 methylation is associated with transcriptional gene silencing, whereas H3K4 and H3K36 methylation is associated with gene activation [13]. This suggests that HvHMTs and HvHDMs may modify the histone methylation of key responsive genes under different stresses to improve plant stress resistance by regulating their transcriptional levels. The transcriptional responses of HvHMT and HvHDM to low temperature, drought, salt, waterlogging and ionizing radiation also indicate that HvHMT and HvHDM respond extensively to various stresses.
4. Materials and Methods
4.1. Evolutionary Analysis
The HMT and HDM proteins of Arabidopsis were downloaded from TAIR (https://www.arabidopsis.org/index.jsp (accessed on 15 April 2022)), which were used as query sequences, and candidate protein sequences orthologous to HMT and HDM proteins were searched using the One Thousand Plant Transcriptome (OneKP) database (https://db.cngb.org/blast/blast/blastp/ (accessed on 20 April 2022)) [54]. Multiple sequence alignments were conducted using MAFFT software V7 with the default parameters (https://mafft.cbrc.jp/alignment/server/ (accessed on 8 May 2022)). Phylogenetic trees were constructed using IQ-TREE v2.0.6, and support for each node was assessed by performing a bootstrap analysis with 1000 replicates [55]. The results were displayed using iTOL 6.0 visualization (https://itol.embl.de/ (accessed on 21 May 2022)) [56].
Genomes of 35 plant species from aquatic to land evolution were downloaded from the NCBI (https://www.ncbi.nlm.nih.gov (accessed on 3 June 2022)), Ensembl plant (https://plants.ensembl.org/index.html (accessed on 4 June 2022)), and Phytozome (https://phytozome-next.jgi.doe.gov/pz/portal.html (accessed on 5 June 2022)) databases. HMTs and HDMs of Arabidopsis were used as query sequences to predict the number of HMT and HDM proteins in different plant and algal species. Two methods were applied to identify the members of the HMT and HDM gene families in barley. In the first approach, the HMM files of HMT and HDM (HMT: SDG-PF00856, PRMT-PF05185; HDM: HDMA-PF04433, Amino_oxidase-PF05193, Jmjc-PF02373) were retrieved from the Pfam database (version 32.0; https://pfam.xfam.org/ (accessed on 23 July 2022)) and used as a query to search the barley proteins using HMMER software (version 3.3.2; https://hmmer.org/ (accessed on 28 July 2022)) with the default parameters. In the second approach, the Arabidopsis and rice HMT and HDM protein sequences were used to search against the barley protein sequences using Blast v2.9.0 software at an Evalue < 1 × 10−5 and coverage > 50% [57]. The HMT and HDM protein evolution time tree of the current phylogeny of plant species was constructed using the online tool TIMETREE (https://www.timetree.org/ (accessed on 15 August 2022)). Protein homology values with the highest similarity to Arabidopsis HMT and HDM proteins in 35 plant species were used to draw a protein similarity heatmap [57].
4.2. Biogenic Analysis of HMT and HDM in Barley
To identify HMT and HDM genes in H. vulgare, the barley pangenome and Lasa Goumang genome were downloaded from IPK (https://galaxy-web.ipk-gatersleben.de/libraries (accessed on 25 August 2022)) and NCBI (https://www.ncbi.nlm.nih.gov (accessed on 26 August 2022)). Blastp and HMMER search methods were used to identify HMT and HDM family homologous genes in barley. Overlapping genes identified by both approaches were considered the final members of the HMT and HDM gene families in barley.
To identify conserved protein domains, the Conserved Domains Database (CDD) of NCBI (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi (accessed on 2 September 2022)) and PfamScan (https://www.ebi.ac.uk/Tools/pfa/pfamscan/ (accessed on 5 September 2022)) with a threshold of Evalue < 0.01 was employed. Conserved motif identification within the HMT and HDM gene sequences was performed using the online tool MEME v5.4.1 (https://meme-suite.org/meme/tools/meme (accessed on 13 September 2022)) with a maximum number of 10. To identify promoter regulatory elements, the PlantCARE (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/ (accessed on 15 September 2022)) online tool was used to annotate the 2 kb genomic region upstream of the initiation codon of the HMT and HDM genes.
The gene expression levels of 14 different growth stages and different tissue samples were obtained through the IPK Barley BLAST Server (https://galaxy-web.ipk-gatersleben.de/libraries (accessed on 23 September 2022)). RNA-seq data of abiotic stresses (salt, waterlogging, drought and cold) were downloaded from the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra (accessed on 29 September 2022)) database (PRJNA578897, GSE144077, PRJEB40905 and PRJNA322603) to investigate the expression profiles of HMT and HDM genes in barley.
4.3. Plant Material, Stress Treatment, RNA Extraction, and qRT–PCR Analysis
Barley seeds (Efupi N0.1) were sterilized with 70% alcohol for one minute, rinsed three times with deionized water, sterilized with 2.5% sodium hypochlorite for 20 min, thoroughly rinsed with distilled water, and soaked for 30 min for sprouting. The soaked seeds were placed flat on a Petri dish with wet filter paper, at low density. Seeds germinated at 27 °C in the dark for two days. Then, plants were grown for 7 days under 16 h light: 8 h dark at 27 °C. For cold, drought and salt treatments, plants were incubated at 4 °C, with 20% PEG2000 and 200 mM NaCl. For the waterlogging treatment, the plants were moved to a growth box and covered with tap water to 1–2 cm above the soil surface level [58]. Seedlings at 0 h were considered the control, and seedling leaves from all treatments and the control were carefully harvested at 0, 3, 6, 12 and 24 h, immediately frozen in liquid nitrogen, and stored at −80 °C for subsequent analysis. For radiation treatment, the dry seeds of 9127 (radiation-sensitive) and 9117 (radiation-tolerant) were exposed to 7Li-ion beam gamma rays at doses of 50 Gy and 200 Gy, respectively.
Total RNA was isolated from barley leaves treated under various stress conditions using TRIzol reagent (TaKaRa, Gunma, Japan). A FastKing RT Kit (with gDNase) (TIANGEN, Beijing, China) was used to remove genomic DNA contamination and perform cDNA synthesis following the manufacturer’s instructions. Quantitative real-time PCR (qRT–PCR) was performed using SuperReal PreMix Color (SYBR Green) (TIANGEN, Beijing, China). The following PCR parameter conditions were set: denaturation at 95 °C for 2 min, 40 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 40 s, and extension at 72 °C for 15 s. The relative expression level was calculated by the 2−ΔΔCT method.
5. Conclusions
In the plant kingdom, HMT and HDM probably originated from Chromista and underwent significant amplification and mutation during evolution from algae to land plants, promoting a complex diversification of histone methylation modifications, which in turn facilitated plant adaptation to variable terrestrial environments. Meanwhile, identification and analysis of different barley germplasms revealed that although HvHMTs and HvHDMs were more conserved among different barley germplasms, some variation existed, with the most significant variation in Lasa Goumang. Both development and stress response showed distinct expression profiles throughout different tissues and under different abiotic stresses.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ijms24098043/s1.
Author Contributions
Y.X. and L.X. conceived the project and designed the experiments; B.A., B.L. and Y.X. performed experiments; B.A., B.L., C.J. and Y.X. writing the original draft; B.A., B.L. and R.W. conducted the experiment and data visualization; H.C., L.X., Y.G. and Y.X., reviewed, edited and revised the manuscript; S.Z. and Y.H., data acquisition and sample collection. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Hubei Outstanding Youth Fund (2021CFA064), the Hubei Key Research and Development Program (2021BBA225), and the Hubei Agricultural Science and Technology Innovation Centre Innovation Team Project (2021-620-000-001-01).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, J.; Jiang, X.; Bai, H.; Liu, C. Genome-wide identification, classification and expression analysis of the JmjC domain-containing histone demethylase gene family in Jatropha curcas L. Sci. Rep. 2022, 12, 6543. [Google Scholar] [CrossRef]
- Hernando, C.E.; Sanchez, S.E.; Mancini, E.; Yanovsky, M.J. Genome wide comparative analysis of the effects of PRMT5 and PRMT4/CARM1 arginine methyltransferases on the Arabidopsis thaliana transcriptome. BMC Genom. 2015, 16, 192. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ali, S.; Zhang, X.; Zhang, Y.; Wang, M.; Zhang, Q.; Xie, L. Genome-wide identification, classification, and expression analysis of the JmjC domain-containing histone demethylase gene family in birch. BMC Genom. 2021, 22, 6543. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Yang, J.; Yi, J.; An, G. Controlling flowering time by histone methylation and acetylation in Arabidopsis and rice. J. Plant Biol. 2015, 58, 203–210. [Google Scholar] [CrossRef]
- Sehrish, S.; Sumbal, W.; Xie, M.; Zhao, C.; Zuo, R.; Gao, F.; Liu, S. Genome-Wide Identification and Characterization of SET Domain Family Genes in Brassica napus L. Int. J. Mol. Sci. 2022, 23, 1936. [Google Scholar] [CrossRef]
- Chen, D.-H.; Qiu, H.-L.; Huang, Y.; Zhang, L.; Si, J.-P. Genome-wide identification and expression profiling of SET DOMAIN GROUP family in Dendrobium catenatum. BMC Plant Biol. 2020, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Sethunath, V.; Metaferia, N.Y.; Nogueira, M.F.; Gallant, D.S.; Garner, E.R.; Lairson, L.A.; Penney, C.M.; Li, J.; Gelbard, M.K. A genome-scale CRISPR screen reveals PRMT1 as a critical regulator of androgen receptor signaling in prostate cancer. Cell Rep. 2022, 38, 110417. [Google Scholar] [CrossRef]
- Lopez, L.; Perrella, G.; Calderini, O.; Porceddu, A.; Panara, F. Genome-Wide Identification of Histone Modification Gene Families in the Model Legume Medicago truncatula and Their Expression Analysis in Nodules. Plants 2022, 11, 322. [Google Scholar] [CrossRef]
- Kronholm, I.; Bassett, A.; Baulcombe, D.; Collins, S. Epigenetic and genetic contributions to adaptation in Chlamydomonas. Mol. Biol. Evol. 2017, 34, 2285–2306. [Google Scholar] [CrossRef]
- Day, T.; Bonduriansky, R. A unified approach to the evolutionary consequences of genetic and nongenetic inheritance. Am. Nat. 2011, 178, E18–E36. [Google Scholar] [CrossRef]
- Klironomos, F.D.; Berg, J.; Collins, S. How epigenetic mutations can affect genetic evolution: Model and mechanism. BioEssays 2013, 35, 571–578. [Google Scholar] [CrossRef]
- Kronholm, I.; Collins, S. Epigenetic mutations can both help and hinder adaptive evolution. Mol. Ecol. 2016, 25, 1856–1868. [Google Scholar] [CrossRef] [PubMed]
- Miryeganeh, M. Plants’ epigenetic mechanisms and abiotic stress. Genes 2021, 12, 1106. [Google Scholar] [CrossRef]
- Yaish, M.W. Epigenetic modifications associated with abiotic and biotic stresses in plants: An implication for understanding plant evolution. Front. Plant Sci. 2017, 8, 1983. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, X.; Luo, M.; Yang, S.; Wu, K. Involvement of histone modifications in plant abiotic stress responses. J. Integr. Plant Biol. 2013, 55, 892–901. [Google Scholar] [CrossRef]
- Kong, L.; Liu, Y.; Wang, X.; Chang, C. Insight into the role of epigenetic processes in abiotic and biotic stress response in wheat and barley. Int. J. Mol. Sci. 2020, 21, 1480. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, A.; Yin, H.; Meng, Q.; Yu, X.; Huang, S.; Wang, J.; Ahmad, R.; Liu, B.; Xu, Z.Y. Trithorax-group proteins ARABIDOPSIS TRITHORAX4 (ATX4) and ATX 5 function in abscisic acid and dehydration stress responses. New Phytol. 2018, 217, 1582–1597. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.S.; Lee, D.; Choi, G.; Chung, W.I. Histone occupancy-dependent and-independent removal of H3K27 trimethylation at cold-responsive genes in Arabidopsis. Plant J. 2009, 60, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, S.; Zhang, Y.; Wang, X.; Li, D.; Li, Q.; Yue, M.; Li, Q.; Zhang, Y.-E.; Xu, Y. Arabidopsis floral initiator SKB1 confers high salt tolerance by regulating transcription and pre-mRNA splicing through altering histone H4R3 and small nuclear ribonucleoprotein LSM4 methylation. Plant Cell 2011, 23, 396–411. [Google Scholar] [CrossRef]
- Bourdareau, S.; Tirichine, L.; Lombard, B.; Loew, D.; Scornet, D.; Wu, Y.; Coelho, S.M.; Cock, J.M. Histone modifications during the life cycle of the brown alga Ectocarpus. Genome Biol. 2021, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Bacova, R.; Kolackova, M.; Klejdus, B.; Adam, V.; Huska, D. Epigenetic mechanisms leading to genetic flexibility during abiotic stress responses in microalgae: A review. Algal Res. 2020, 50, 101999. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Chen, Y.; Wang, S.; Fang, Y.; Zhang, X.; Wu, Y.; Xue, D. Genome-wide identification of the SOD gene family and expression analysis under drought and salt stress in barley. Plant Growth Regul. 2021, 94, 49–60. [Google Scholar] [CrossRef]
- Cai, K.; Zeng, F.; Wang, J.; Zhang, G. Identification and characterization of HAK/KUP/KT potassium transporter gene family in barley and their expression under abiotic stress. BMC Genom. 2021, 22, 317. [Google Scholar] [CrossRef]
- She, K.; Pan, W.; Yan, Y.; Shi, T.; Chu, Y.; Cheng, Y.; Ma, B.; Song, W. Genome-wide identification, evolution and expressional analysis of OSCA gene family in barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2022, 23, 13027. [Google Scholar] [CrossRef]
- Sharma, N.; Geuten, K.; Giri, B.S.; Varma, A. The molecular mechanism of vernalization in Arabidopsis and cereals: Role of Flowering Locus C and its homologs. Physiol. Plant. 2020, 170, 373–383. [Google Scholar] [CrossRef]
- Yuzhen, B.; Sang, Z.; Mu, W.; Yu, M.; Wang, Y.; Yuan, H.; Xu, Q. Whole-genome analysis of the trimethylation of histone H3 lysine 4 and lysine 27 in two contrasting Tibetan hulless barley genotypes under salinity stress. Acta Physiol. Plant. 2021, 43, 89. [Google Scholar] [CrossRef]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Stein, N. The barley pangenome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, T.; Ling, Z.; Wang, Y.; Li, X.; Xu, S.; Xu, Q.; Zha, S.; Qimei, W.; Basang, Y. An improved high-quality genome assembly and annotation of Tibetan hulless barley. Sci. Data 2020, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Buitrago, S.; Peng, Y.; Abou-Elwafa, S.F.; Wan, K.; Liu, Y.; Wang, R.; Yang, X.; Zhang, W. Genome-wide identification of cold-tolerance genes and functional analysis of IbbHLH116 gene in sweet potato. Gene 2022, 837, 146690. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An expanded resource for species divergence times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Ai, Q.; Pan, W.; Zeng, Y.; Li, Y.; Cui, L. CCCH Zinc finger genes in Barley: Genome-wide identification, evolution, expression and haplotype analysis. BMC Plant Biol. 2022, 22, 117. [Google Scholar]
- Ammar, R.; Torti, D.; Tsui, K.; Gebbia, M.; Durbic, T.; Bader, G.D.; Giaever, G.; Nislow, C. Chromatin is an ancient innovation conserved between Archaea and Eukarya. eLife 2012, 1, e00078. [Google Scholar] [CrossRef]
- Nalabothula, N.; Xi, L.; Bhattacharyya, S.; Widom, J.; Wang, J.-P.; Reeve, J.N.; Santangelo, T.J.; Fondufe-Mittendorf, Y.N. Archaeal nucleosome positioning in vivo and in vitro is directed by primary sequence motifs. BMC Genom. 2013, 14, 391. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, S.; Lin, S.; Guo, Y.; Deng, W.; Zhang, Y.; Xue, Y. WERAM: A database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 2016, 45, gkw1011. [Google Scholar]
- Rastogi, A.; Lin, X.; Lombard, B.; Loew, D.; Tirichine, L. Probing the evolutionary history of epigenetic mechanisms: What can we learn from marine diatoms. Aims Genet. 2015, 2, 173–191. [Google Scholar] [CrossRef]
- Becker, B.; Feng, X.; Yin, Y.; Holzinger, A. Desiccation tolerance in streptophyte algae and the algae to land plant transition: Evolution of LEA and MIP protein families within the Viridiplantae. J. Exp. Bot. 2020, 71, 3270–3278. [Google Scholar] [CrossRef]
- Vigneau, J.; Borg, M. The epigenetic origin of life history transitions in plants and algae. Plant Reprod. 2021, 34, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Casas-Mollano, J.A.; van Dijk, K.; Eisenhart, J.; Cerutti, H. SET3p monomethylates histone H3 on lysine 9 and is required for the silencing of tandemly repeated transgenes in Chlamydomonas. Nucleic Acids Res. 2007, 35, 939–950. [Google Scholar] [CrossRef]
- Morgante, M.; De Paoli, E.; Radovic, S. Transposable elements and the plant pangenomes. Curr. Opin. Plant Biol. 2007, 10, 149–155. [Google Scholar] [CrossRef]
- Zhang, H.; Mittal, N.; Leamy, L.J.; Barazani, O.; Song, B.H. Back into the wild-Apply untapped genetic diversity of wild relatives for crop improvement. Evol. Appl. 2017, 10, 5–24. [Google Scholar] [CrossRef]
- Hyten, D.L.; Song, Q.; Zhu, Y.; Choi, I.-Y.; Nelson, R.L.; Costa, J.M.; Specht, J.E.; Shoemaker, R.C.; Cregan, P.B. Impacts of genetic bottlenecks on soybean genome diversity. Proc. Natl. Acad. Sci. USA 2006, 103, 16666–16671. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.I.; Bi, I.V.; Schroeder, S.G.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S. The effects of artificial selection on the maize genome. Science 2005, 308, 1310–1314. [Google Scholar] [CrossRef]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2012, 30, 105–111. [Google Scholar] [CrossRef]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, J.; Li, T.; Yang, H.; Wang, P.; Su, L.; Zheng, Y.; Bao, C.; Zhou, C. SDG711 is involved in rice seed development through regulation of starch metabolism gene expression in coordination with other histone modifications. Rice 2021, 14, 25. [Google Scholar] [CrossRef]
- Zheng, L.; Ma, S.; Shen, D.; Fu, H.; Wang, Y.; Liu, Y.; Shah, K.; Yue, C.; Huang, J. Genome-wide identification of Gramineae histone modification genes and their potential roles in regulating wheat and maize growth and stress responses. BMC Plant Biol. 2021, 21, 543. [Google Scholar] [CrossRef]
- Ay, N.; Irmler, K.; Fischer, A.; Uhlemann, R.; Reuter, G.; Humbeck, K. Epigenetic programming via histone methylation at WRKY53 controls leaf senescence in Arabidopsis thaliana. Plant J. 2009, 58, 333–346. [Google Scholar] [CrossRef]
- Plett, K.L.; Raposo, A.E.; Bullivant, S.; Anderson, I.C.; Piller, S.C.; Plett, J.M. Root morphogenic pathways in Eucalyptus grandis are modified by the activity of protein arginine methyltransferases. BMC Plant Biol. 2017, 17, 62. [Google Scholar] [CrossRef]
- Begcy, K.; Dresselhaus, T. Epigenetic responses to abiotic stresses during reproductive development in cereals. Plant Reprod. 2018, 31, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Y.; Liang, Y.; Zhou, D.; Li, S.; Lin, S.; Dong, H.; Huang, L. The function of histone lysine methylation related SET domain group proteins in plants. Protein Sci. 2020, 29, 1120–1137. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Avramova, Z.; Fromm, M. The Arabidopsis trithorax-like factor ATX1 functions in dehydration stress responses via ABA-dependent and ABA-independent pathways. Plant J. 2011, 66, 735–744. [Google Scholar] [CrossRef]
- Qiao, W.; Fan, L. Epigenetics, a mode for plants to respond to abiotic stresses. Front. Cell Dev. Biol. 2011, 6, 477–481. [Google Scholar] [CrossRef]
- Monat, C.; Schreiber, M.; Stein, N.; Mascher, M. Prospects of pan-genomics in barley. Theor. Appl. Genet. 2019, 132, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Tong, T.; Li, Q.; Jiang, W.; Chen, G.; Xue, D.; Deng, F.; Zeng, F.; Chen, Z.-H. Molecular evolution of calcium signaling and transport in plant adaptation to abiotic stress. Int. J. Mol. Sci. 2021, 22, 12308. [Google Scholar] [CrossRef]
- Jiang, W.; Tong, T.; Chen, X.; Deng, F.; Zeng, F.; Pan, R.; Zhang, W.; Chen, G.; Chen, Z.-H. Molecular response and evolution of plant anion transport systems to abiotic stress. Plant Mol. Biol. 2021, 110, 397–412. [Google Scholar] [CrossRef]
- Xu, Z.; Shen, Q.; Zhang, G. The mechanisms for the difference in waterlogging tolerance among sea barley, wheat and barley. Plant Growth Regul. 2022, 96, 431–441. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).