
Ribbon Synapses and Retinal Disease: Review


Abstract
1. Introduction
2. Nonsyndromic Congenital Progressive Retinal Disorders
2.1. Human Retinal Gene 4 (HRG4/Unc119/Munc119)
2.2. Regulating Synaptic Membrane Exocytosis Protein 1/Rab3 Interacting Molecule 1 (Rim1)
2.3. Tubby-like Protein-1 (Tulp1)
3. Nonsyndromic Congenital Nonprogressive Retinal Disorders
3.1. Calcium Channels
3.1.1. α1. F Subunit (CACNA1F)
3.1.2. α2δ4. Subunit (CACNA2D4)
3.2. CaBP4
4. Syndromic Congenital Nonprogressive Retinal Disorders
Regulating Synaptic Membrane Exocytosis 2/Rab3 Interacting Molecule 2 (Rim2)
5. Inherited Syndromic Diseases with Retinal Dysfunction: Muscular Dystrophy
Pikachurin
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sterling, P.; Matthews, G. Structure and function of ribbon synapses. Trends Neurosci. 2005, 28, 20–29. [Google Scholar] [CrossRef]
- Dieck, S.; Altrock, W.D.; Kessels, M.M.; Qualmann, B.; Regus, H.; Brauner, D.; Fejtova, A.; Bracko, O.; Gundelfinger, E.D.; Brandsta, J.H. Molecular dissection of the photoreceptor ribbon synapse: Physical interaction of Bassoon and RIBEYE is essential for the assembly of the ribbon complex. J. Cell Biol. 2005, 168, 825–836. [Google Scholar] [CrossRef]
- Prescott, E.D.; Zenisek, D. Recent progress towards understanding the synaptic ribbon. Curr. Opin. Neurobiol. 2005, 15, 431–436. [Google Scholar] [CrossRef]
- Schmitz, F.; Konigstorfer, A.; Sudhof, T. RIBEYE, a component of synaptic ribbons: A protein’s journey through evolution provides insight into synaptic ribbon function. Neuron 2000, 28, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Kantardzhieva, A.; Peppi, M.; Lane, W.S.; Sewell, W.F. Protein Composition of Immunoprecipitated Synaptic Ribbons. J. Proteome Res. 2011, 11, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Zenisek, D.; Horst, N.K.; Merrifield, C.; Sterling, P.; Matthews, G. Visualizing synaptic ribbons in the living cell. J. Neurosci. 2004, 24, 9752–9759. [Google Scholar] [CrossRef]
- Von Kriegstein, K.; Schmitz, F.; Link, E.; Südhof, T.C. Distribution of synaptic vesicle proteins in the mammalian retina identifies obligatory and facultative components of ribbon synapses. Eur. J. Neurosci. 1999, 11, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, B.; Südhof, T. Distribution of synaptic markers in the retina: Implications for synaptic vesicle traffic in ribbon synapses. J. Physiol. 1994, 88, 249–257. [Google Scholar] [CrossRef]
- Brandstätter, J.H.; Wässle, H.; Betz, H.; Morgans, C.W. The Plasma Membrane Protein SNAP-25, but not Syntaxin, is Present at Photoreceptor and Bipolar Cell Synapses in the Rat Retina. Eur. J. Neurosci. 1996, 8, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Zenisek, D.; Steyer, J.A.; Almers, W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature 2000, 406, 849–854. [Google Scholar] [CrossRef]
- Heidelberger, R.; Thoreson, W.B.; Witkovsky, P. Synaptic transmission at retinal ribbon synapses. Prog. Retin. Eye Res. 2005, 24, 682–720. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-H.; Li, G.-L.; von Gersdorff, H.; Peiró, C.; Vallejo, S.; Gembardt, F.; Palacios, E.; Novella, S.; Azcutia, V.; Rodríguez-Mañas, L.; et al. Single Ca2+ channels and exocytosis at sensory synapses. J. Physiol. 2013, 591, 3167–3178. [Google Scholar] [CrossRef] [PubMed]
- Mesnard, C.S.; Barta, C.L.; Sladek, A.L.; Zenisek, D.; Thoreson, W.B. Eliminating Synaptic Ribbons from Rods and Cones Halves the Releasable Vesicle Pool and Slows Down Replenishment. Int. J. Mol. Sci. 2022, 23, 6429. [Google Scholar] [CrossRef] [PubMed]
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Daiger, S.P. Inherited retinal degeneration: Exceptional genetic and clinical heterogeneity. Mol. Med. Today 1996, 2, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P. Cone rod dystrophies. Orphanet. J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Marazova, K.; Audo, I. Clinical Characteristics and Current Therapies for Inherited Retinal Degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef]
- Hagstrom, S.A.; Duyao, M.; North, M.A.; Li, T. Retinal degeneration in tulp1-/- mice: Vesicular accumulation in the interphotoreceptor matrix. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2795–2802. [Google Scholar]
- Löhner, M.; Babai, N.; Müller, T.; Gierke, K.; Atorf, J.; Joachimsthaler, A.; Peukert, A.; Martens, H.; Feigenspan, A.; Kremers, J.; et al. Analysis of RIM Expression and Function at Mouse Photoreceptor Ribbon Synapses. J. Neurosci. 2017, 37, 7848–7863. [Google Scholar] [CrossRef]
- Liu, X.; Kerov, V.; Haeseleer, F.; Majumder, A.; Artemyev, N.; A Baker, S.; Lee, A. Dysregulation of Cav1.4 channels disrupts the maturation of photoreceptor synaptic ribbons in congenital stationary night blindness type 2. Channels 2013, 7, 514–523. [Google Scholar] [CrossRef]
- De Sevilla Muller, L.P.; Liu, J.; Solomon, A.; Rodriguez, A.; Brecha, N.C. Expression of voltage-gated calcium channel alpha(2)delta(4) subunits in the mouse and rat retina. J. Comp. Neurol. 2013, 521, 2486–2501. [Google Scholar] [CrossRef]
- Maeda, T.; Lem, J.; Palczewski, K.; Haeseleer, F. A Critical Role of CaBP4 in the Cone Synapse. Investig. Opthalmology Vis. Sci. 2005, 46, 4320–4327. [Google Scholar] [CrossRef] [PubMed]
- Bucher, F.; Friedlander, M.S.; Aguilar, E.; Kurihara, T.; Krohne, T.U.; Usui, Y.; Friedlander, M. The long dystrophin gene product Dp427 modulates retinal function and vascular morphology in response to age and retinal ischemia. Neurochem. Int. 2019, 129, 104489. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Omori, Y.; Katoh, K.; Kondo, M.; Kanagawa, M.; Miyata, K.; Funabiki, K.; Koyasu, T.; Kajimura, N.; Miyoshi, T.; et al. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat. Neurosci. 2008, 11, 923–931. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet. J. Rare Dis. 2006, 1, 40. [Google Scholar]
- Higashide, T.; Murakami, A.; McLaren, M.J.; Inana, G. Cloning of the cDNA for a novel photoreceptor protein HRG4. J. Biol. Chem. 1996, 271, 1797–1804. [Google Scholar] [CrossRef]
- Murakami, A.; Yajima, T.; Inana, G. Isolation of human retinal genes: Recoverin cDNA and gene. Biochem. Biophys. Res. Commun. 1992, 187, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Higashide, T.; McLaren, M.J.; Inana, G. Localization of HRG4, a photoreceptor protein homologous to Unc-119, in ribbon synapse. Investig. Opthalmology Vis. Sci. 1998, 39, 690–698. [Google Scholar]
- Maduro, M.; Pilgrim, D. Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics 1995, 141, 977–988. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kubota, S.; Mori, N.; McLaren, M.J.; Inana, G. Photoreceptor synaptic protein HRG4 (UNC119) interacts with ARL2 via a putative conserved domain. FEBS Lett. 2002, 534, 26–32. [Google Scholar] [CrossRef]
- Kubota, S.; Kobayashi, A.; Mori, N.; Higashide, T.; McLaren, M.J.; Inana, G. Changes in retinal synaptic proteins in the transgenic model expressing a mutant HRG4 (UNC119). Investig. Opthalmology Vis. Sci. 2002, 43, 308–313. [Google Scholar]
- Alpadi, K.; Magupalli, V.G.; Käppel, S.; Köblitz, L.; Schwarz, K.; Seigel, G.M.; Sung, C.-H.; Schmitz, F. RIBEYE Recruits Munc119, a Mammalian Ortholog of the Caenorhabditis elegans Protein unc119, to Synaptic Ribbons of Photoreceptor Synapses. J. Biol. Chem. 2008, 283, 26461–26467. [Google Scholar] [CrossRef]
- Haeseleer, F. Interaction and Colocalization of CaBP4 and Unc119 (MRG4) in Photoreceptors. Investig. Opthalmology Vis. Sci. 2008, 49, 2366–2375. [Google Scholar] [CrossRef] [PubMed]
- Higashide, T.; Inana, G. Characterization of the Gene for HRG4 (UNC119), a Novel Photoreceptor Synaptic Protein Homologous to Unc-119. Genomics 1999, 57, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Higashide, T.; Hamasaki, D.; Kubota, S.; Sakuma, H.; An, W.; Fujimaki, T.; McLaren, M.J.; Weleber, R.G.; Inana, G. HRG4 (UNC119) mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Investig. Opthalmology Vis. Sci. 2000, 41, 3268–3277. [Google Scholar]
- Miller, R.F.; E Dowling, J. Intracellular responses of the Müller (glial) cells of mudpuppy retina: Their relation to b-wave of the electroretinogram. J. Neurophysiol. 1970, 33, 323–341. [Google Scholar] [CrossRef]
- Ishiba, Y.; Higashide, T.; Mori, N.; Kobayashi, A.; Kubota, S.; McLaren, M.J.; Satoh, H.; Wong, F.; Inana, G. Targeted inactivation of synaptic HRG4 (UNC119) causes dysfunction in the distal photoreceptor and slow retinal degeneration, revealing a new function. Exp. Eye Res. 2007, 84, 473–485. [Google Scholar] [CrossRef]
- Mori, N.; Ishiba, Y.; Kubota, S.; Higashide, T.; McLaren, M.J.; Inana, G.; Kobayashi, A. Truncation Mutation in HRG4 (UNC119) Leads to Mitochondrial ANT-1–Mediated Photoreceptor Synaptic and Retinal Degeneration by Apoptosis. Investig. Opthalmology Vis. Sci. 2006, 47, 1281–1292. [Google Scholar] [CrossRef][Green Version]
- Johnson, S.; Halford, S.; Morris, A.G.; Patel, R.J.; E Wilkie, S.; Hardcastle, A.; Moore, A.T.; Zhang, K.; Hunt, D.M. Genomic organisation and alternative splicing of human RIM1, a gene implicated in autosomal dominant cone-rod dystrophy (CORD7). Genomics 2003, 81, 304–314. [Google Scholar] [CrossRef]
- Kelsell, R.E.; Gregory-Evans, K.; Gregory-Evans, C.Y.; Holder, G.E.; Jay, M.R.; Weber, B.H.; Moore, A.T.; Bird, A.C.; Hunt, D.M. Localization of a Gene (CORD7) for a Dominant Cone-Rod Dystrophy to Chromosome 6q. Am. J. Hum. Genet. 1998, 63, 274–279. [Google Scholar] [CrossRef]
- Sisodiya, S.M.; Thompson, P.J.; Need, A.; E Harris, S.; E Weale, M.; E Wilkie, S.; Michaelides, M.; Free, S.L.; Walley, N.; Gumbs, C.; et al. Genetic enhancement of cognition in a kindred with cone-rod dystrophy due to RIMS1 mutation. J. Med. Genet. 2007, 44, 373–380. [Google Scholar] [CrossRef]
- Michaelides, M.; E Holder, G.; Hunt, D.M.; Fitzke, F.W.; Bird, A.C.; Moore, A.T. A detailed study of the phenotype of an autosomal dominant cone-rod dystrophy (CORD7) associated with mutation in the gene for RIM1. Br. J. Ophthalmol. 2005, 89, 198–206. [Google Scholar] [CrossRef]
- E Holder, G. Pattern Electroretinography (PERG) and an Integrated Approach to Visual Pathway Diagnosis. Prog. Retin. Eye Res. 2001, 20, 531–561. [Google Scholar] [CrossRef]
- Wang, Y.; Okamoto, M.; Schmitz, F.; Hofmann, K.; Südhof, T.C. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature 1997, 388, 593–598. [Google Scholar] [CrossRef]
- Dai, H.; Tomchick, D.R.; García, J.; Südhof, T.C.; Machius, M.; Rizo, J. Crystal structure of the RIM2 C2A-domain at 1.4 A resolution. Biochemistry 2005, 44, 13533–13542. [Google Scholar] [CrossRef]
- Coppola, T.; Magnin-Luthi, S.; Perret-Menoud, V.; Gattesco, S.; Schiavo, G.; Regazzi, R. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J. Biol. Chem. 2001, 276, 32756–32762. [Google Scholar] [CrossRef]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Schlüter, O.M.; Schmitz, F.; Jahn, R.; Rosenmund, C.; Südhof, T.C. A Complete Genetic Analysis of Neuronal Rab3 Function. J. Neurosci. 2004, 24, 6629–6637. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.C.M.; Holopainen, J.M.; Molday, L.L.; Foster, L.J.; Molday, R.S. Proteomics of Photoreceptor Outer Segments Identifies a Subset of SNARE and Rab Proteins Implicated in Membrane Vesicle Trafficking and Fusion. Mol. Cell. Proteom. 2008, 7, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Schoch, S.; Mittelstaedt, T.; Kaeser, P.S.; Padgett, D.; Feldmann, N.; Chevaleyre, V.; Castillo, P.; E Hammer, R.; Han, W.; Schmitz, F.; et al. Redundant functions of RIM1α and RIM2α in Ca2+-triggered neurotransmitter release. EMBO J. 2006, 25, 5852–5863. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, S.A.; North, M.A.; Nishina, P.M.; Berson, E.L.; Dryja, T.P. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with Retinitis pigmentosa. Nat. Genet. 1998, 18, 174–176. [Google Scholar] [CrossRef]
- Coleman, D.L.; Eicher, E.M. Fat (fat) and Tubby (tub): Two Autosomal Recessive Mutations Causing Obesity Syndromes in the Mouse. J. Hered. 1990, 81, 424–427. [Google Scholar] [CrossRef]
- Ohlemiller, K.K.; Hughes, R.M.; Mosinger-Ogilvie, J.; Speck, J.D.; Grosof, D.H.; Silverman, M.S. Cochlear and retinal degeneration in the tubby mouse. Neuroreport 1995, 6, 845–849. [Google Scholar] [CrossRef]
- Kleyn, P.W.; Fan, W.; Kovats, S.G.; Lee, J.J.; Pulido, J.C.; Wu, Y.; Berkemeier, L.R.; Misumi, D.J.; Holmgren, L.; Charlat, O.; et al. Identification and Characterization of the Mouse Obesity Gene tubby: A Member of a Novel Gene Family. Cell 1996, 85, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Noben-Trauth, K.; Naggert, J.K.; North, M.A.; Nishina, P.M. A candidate gene for the mouse mutation tubby. Nature 1996, 380, 534–538. [Google Scholar] [CrossRef] [PubMed]
- North, M.A.; Naggert, J.K.; Yan, Y.; Noben-Trauth, K.; Nishina, P.M. Molecular characterization of TUB, TULP1, and TULP 2, members of the novel tubby gene family and their possible relation to ocular diseases. Proc. Natl. Acad. Sci. USA 1997, 94, 3128–3133. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Boggon, T.J.; Baird, C.L.; Gomez, C.A.; Zhao, J.; Shan, W.S.; Myszka, D.G.; Shapiro, L. G-Protein Signaling Through Tubby Proteins. Science 2001, 292, 2041–2050. [Google Scholar] [CrossRef]
- Xi, Q.; Pauer, G.J.T.; Ball, S.L.; Rayborn, M.; Hollyfield, J.G.; Peachey, N.S.; Crabb, J.W.; Hagstrom, S.A. Interaction between the Photoreceptor-Specific Tubby-like Protein 1 and the Neuronal-Specific GTPase Dynamin-1. Investig. Opthalmology Vis. Sci. 2007, 48, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.A.; Shugart, Y.; Banerjee, P.; Gilllam, T.; Lewis, C.A.; Jacobson, S.; Ott, J. Identification of a locus, distinct from RDS-peripherin, for autosomal recessive retinitis pigmentosa on chromosome 6p. Hum. Mol. Genet. 1994, 3, 1401–1403. [Google Scholar] [CrossRef] [PubMed]
- Shugart, Y.Y.; Banerjee, P.; A Knowles, J.; A Lewis, C.; Jacobson, S.G.; Matise, T.C.; Penchaszadeh, G.; Gilliam, T.C.; Ott, J. Fine genetic mapping of a gene for autosomal recessive retinitis pigmentosa on chromosome 6p21. Am. J. Hum. Genet. 1995, 57, 499–502. [Google Scholar]
- Gu, S.; Lennon, A.; Li, Y.; Lorenz, B.; Fossarello, M.; North, M.; Gal, A.; Wright, A. Tubby-like protein-1 mutations in autosomal recessive retinitis pigmentosa. Lancet 1998, 351, 1103–1104. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Batlle, I.R.; Batlle, K.G.; Banerjee, P.; Cideciyan, A.V.; Huang, J.; Alemán, T.S.; Huang, Y.; Ott, J.; Gilliam, T.C.; et al. Tubby-like protein 1 homozygous splice-site mutation causes early-onset severe retinal degeneration. Investig. Opthalmology Vis. Sci. 1999, 40, 2106–2144. [Google Scholar]
- Mataftsi, A.; Schorderet, D.F.; Chachoua, L.; Boussalah, M.; Nouri, M.T.; Barthelmes, D.; Borruat, F.-X.; Munier, F.L. Novel TULP1 Mutation Causing Leber Congenital Amaurosis or Early Onset Retinal Degeneration. Investig. Opthalmology Vis. Sci. 2007, 48, 5160–5167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grossman, G.H.; Pauer, G.J.; Narendra, U.; Hagstrom, S.A. Tubby-Like Protein 1 (Tulp1) Is Required for Normal Photoreceptor Synaptic Development. In Retinal Degenerative Diseases: Laboratory and Therapeutic Investigations; Anderson, R., Hollyfield, J., LaVail, M., Eds.; Springer: New York, NY, USA, 2010; pp. 89–96. [Google Scholar]
- Wahl, S.; Magupalli, V.G.; Dembla, M.; Katiyar, R.; Schwarz, K.; Köblitz, L.; Alpadi, K.; Krause, E.; Rettig, J.; Sung, C.-H.; et al. The Disease Protein Tulp1 Is Essential for Periactive Zone Endocytosis in Photoreceptor Ribbon Synapses. J. Neurosci. 2016, 36, 2473–2493. [Google Scholar] [CrossRef]
- Ebke, L.; Sinha, S.; Pauer, G.; Hagstrom, S. Photoreceptor Compartment-Specific TULP1 Interactomes. Int. J. Mol. Sci. 2021, 22, 8066. [Google Scholar] [CrossRef]
- Dolphin, A.C.; Lee, A. Presynaptic calcium channels: Specialized control of synaptic neurotransmitter release. Nat. Rev. Neurosci. 2020, 21, 213–229. [Google Scholar] [CrossRef]
- Dolphin, A.C. Voltage-gated calcium channels: Their discovery, function and importance as drug targets. Brain Neurosci. Adv. 2018, 2, 2398212818794805. [Google Scholar] [CrossRef]
- Maddox, J.W.; Lee, A. Calcium Channels in Retinal Function and Disease. Annu. Rev. Vis. Sci. 2022, 8, 53–77. [Google Scholar] [CrossRef]
- Audo, I.; Kohl, S.; Leroy, B.P.; Munier, F.L.; Guillonneau, X.; Mohand-Saïd, S.; Bujakowska, K.; Nandrot, E.; Lorenz, B.; Preising, M.; et al. TRPM1 Is Mutated in Patients with Autosomal-Recessive Complete Congenital Stationary Night Blindness. Am. J. Hum. Genet. 2009, 85, 720–729. [Google Scholar] [CrossRef]
- Bech-Hansen, N.; Naylor, M.J.; Maybaum, T.A.; Sparkes, R.L.; Koop, B.; Birch, D.; Bergen, A.A.; Prinsen, C.F.; Polomeno, R.C.; Gal, A.; et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000, 26, 319–323. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Berson, E.L.; Fishman, G.A.; Sandberg, M.A.; Alexander, K.R.; Derlacki, D.J.; Rajagopalan, A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA 2005, 102, 4884–4889. [Google Scholar] [CrossRef]
- Audo, I.; Bujakowska, K.; Orhan, E.; Poloschek, C.M.; Defoort-Dhellemmes, S.; Drumare, I.; Kohl, S.; Luu, T.D.; Lecompte, O.; Zrenner, E.; et al. Whole-Exome Sequencing Identifies Mutations in GPR179 Leading to Autosomal-Recessive Complete Congenital Stationary Night Blindness. Am. J. Hum. Genet. 2012, 90, 321–330. [Google Scholar] [CrossRef]
- Neuillé, M.; Cao, Y.; Caplette, R.; Guerrero-Given, D.; Thomas, C.; Kamasawa, N.; Sahel, J.-A.; Hamel, C.P.; Audo, I.; Picaud, S.; et al. LRIT3 Differentially Affects Connectivity and Synaptic Transmission of Cones to ON- and OFF-Bipolar Cells. Investig. Opthalmology Vis. Sci. 2017, 58, 1768–1778. [Google Scholar] [CrossRef]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Pearce, W.G.; Koop, B.; Fishman, G.A.; Mets, M.; Musarella, M.A.; Boycott, K.M. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 264–267. [Google Scholar] [CrossRef]
- Bijveld, M.M.; Florijn, R.J.; Bergen, A.A.; Born, L.I.V.D.; Kamermans, M.; Prick, L.; Riemslag, F.C.; van Schooneveld, M.J.; Kappers, A.M.; van Genderen, M.M. Genotype and Phenotype of 101 Dutch Patients with Congenital Stationary Night Blindness. Ophthalmology 2013, 120, 2072–2081. [Google Scholar] [CrossRef]
- Boycott, K.M.; Pearce, W.G.; Bech-Hansen, N.T. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can. J. Ophthalmol. 2000, 35, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.; Maybaum, T.; Naylor, M.; Weleber, R.; Robitaille, J.; Miyake, Y.; Bergen, A.; Pierpont, M.; Pearce, W.; Bech-Hansen, N. A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants. Hum. Genet. 2001, 108, 91–97. [Google Scholar] [CrossRef]
- Burtscher, V.; Schicker, K.; Novikova, E.; Pöhn, B.; Stockner, T.; Kugler, C.; Singh, A.; Zeitz, C.; Lancelot, M.-E.; Audo, I.; et al. Spectrum of Cav1.4 dysfunction in congenital stationary night blindness type 2. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Koschak, A.; Fernandez-Quintero, M.L.; Heigl, T.; Ruzza, M.; Seitter, H.; Zanetti, L. Cav1.4 dysfunction and congenital stationary night blindness type 2. Pflügers Archiv Eur. J. Physiol. 2021, 473, 1437–1454. [Google Scholar] [CrossRef] [PubMed]
- Hemara-Wahanui, A.; Berjukow, S.; Hope, C.I.; Dearden, P.K.; Wu, S.-B.; Wilson-Wheeler, J.; Sharp, D.M.; Lundon-Treweek, P.; Clover, G.M.; Hoda, J.-C.; et al. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proc. Natl. Acad. Sci. USA 2005, 102, 7553–7558. [Google Scholar] [CrossRef]
- Hoda, J.C.; Zaghetto, F.; Koschak, A.; Striessnig, J. Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Ca(v)1.4 L-type Ca2+ channels. J. Neurosci. 2005, 25, 252–259. [Google Scholar] [CrossRef]
- I Hope, C.; Sharp, D.M.; Hemara-Wahanui, A.; Dbo, J.I.S.; Lundon, P.; A Mitchell, E.; A Maw, M.; Clover, G.M. Clinical manifestations of a unique X-linked retinal disorder in a large New Zealand family with a novel mutation in CACNA1F, the gene responsible for CSNB2. Clin. Exp. Ophthalmol. 2005, 33, 129–136. [Google Scholar] [CrossRef]
- Regus-Leidig, H.; Atorf, J.; Feigenspan, A.; Kremers, J.; Maw, M.A.; Brandstätter, J.H. Photoreceptor Degeneration in Two Mouse Models for Congenital Stationary Night Blindness Type 2. PLoS ONE 2014, 9, e86769. [Google Scholar] [CrossRef]
- Ba-Abbad, R.; Arno, G.; Carss, K.; Stirrups, K.; Penkett, C.J.; Moore, A.T.; Michaelides, M.; Raymond, F.L.; Webster, A.R.; Holder, G.E. Mutations in CACNA2D4 Cause Distinctive Retinal Dysfunction in Humans. Ophthalmology 2016, 123, 668–671.e2. [Google Scholar] [CrossRef] [PubMed]
- Wycisk, K.A.; Zeitz, C.; Feil, S.; Wittmer, M.; Forster, U.; Neidhardt, J.; Wissinger, B.; Zrenner, E.; Wilke, R.; Kohl, S.; et al. Mutation in the Auxiliary Calcium-Channel Subunit CACNA2D4 Causes Autosomal Recessive Cone Dystrophy. Am. J. Hum. Genet. 2006, 79, 973–977. [Google Scholar] [CrossRef]
- Huang, X.-F.; Huang, F.; Wu, K.-C.; Wu, J.; Chen, J.; Pang, C.-P.; Lu, F.; Qu, J.; Jin, Z.-B. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Anesth. Analg. 2015, 17, 271–278. [Google Scholar] [CrossRef]
- Wycisk, K.A.; Budde, B.; Feil, S.; Skosyrski, S.; Buzzi, F.; Neidhardt, J.; Glaus, E.; Nu¨rnberg, P.; Ruether, K.; Berger, W. Structural and Functional Abnormalities of Retinal Ribbon Synapses due to Cacna2d4 Mutation. Investig. Opthalmology Vis. Sci. 2006, 47, 3523–3530. [Google Scholar] [CrossRef] [PubMed]
- Haeseleer, F.; Sokal, I.; Verlinde, C.L.M.J.; Erdjument-Bromage, H.; Tempst, P.; Pronin, A.N.; Benovic, J.L.; Fariss, R.N.; Palczewski, K. Five Members of a Novel Ca2+-binding Protein (CABP) Subfamily with Similarity to Calmodulin. J. Biol. Chem. 2000, 275, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Kloeckener-Gruissem, B.; Forster, U.; Kohl, S.; Magyar, I.; Wissinger, B.; Matyas, G.; Borruat, F.-X.; Schorderet, D.F.; Zrenner, E.; et al. Mutations in CABP4, the Gene Encoding the Ca2+-Binding Protein 4, Cause Autosomal Recessive Night Blindness. Am. J. Hum. Genet. 2006, 79, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; van Genderen, M.M.; Collin, R.W.J.; Roosing, S.; de Brouwer, A.P.M.; Riemslag, F.C.C.; Venselaar, H.; Thiadens, A.A.H.J.; Hoyng, C.B.; Rohrschneider, K.; et al. A Novel Homozygous Nonsense Mutation in CABP4 Causes Congenital Cone–Rod Synaptic Disorder. Investig. Opthalmology Vis. Sci. 2009, 50, 2344–2350. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Al-Owain, M.; Alqahtani, F.; Hazzaa, S.; Alkuraya, F.S. A null mutation in CABP4 causes Leber’s congenital amaurosis-like phenotype. Mol. Vis. 2010, 16, 207–212. [Google Scholar] [PubMed]
- Khan, A.; Alrashed, M.; Alkuraya, F.S. Clinical characterisation of the CABP4-related retinal phenotype. Br. J. Ophthalmol. 2012, 97, 262–265. [Google Scholar] [CrossRef]
- Khan, A.O. CABP4 Mutations Do Not Cause Congenital Stationary Night Blindness. Ophthalmology 2014, 121, e15. [Google Scholar] [CrossRef] [PubMed]
- Haeseleer, F.; Imanishi, Y.; Maeda, T.; E Possin, D.; Maeda, A.; Lee, A.; Rieke, F.; Palczewski, K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat. Neurosci. 2004, 7, 1079–1087. [Google Scholar] [CrossRef]
- Mechaussier, S.; Almoallem, B.; Zeitz, C.; Van Schil, K.; Jeddawi, L.; Van Dorpe, J.; Rey, A.D.; Condroyer, C.; Pelle, O.; Polak, M.; et al. Loss of Function of RIMS2 Causes a Syndromic Congenital Cone-Rod Synaptic Disease with Neurodevelopmental and Pancreatic Involvement. Am. J. Hum. Genet. 2020, 106, 859–871. [Google Scholar] [CrossRef]
- Kaeser, P.S.; Deng, L.; Wang, Y.; Dulubova, I.; Liu, X.; Rizo, J.; Südhof, T.C. RIM Proteins Tether Ca2+ Channels to Presynaptic Active Zones via a Direct PDZ-Domain Interaction. Cell 2011, 144, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Liu, X.; Südhof, T.C.; Acuna, C. Efficient stimulus-secretion coupling at ribbon synapses requires RIM-binding protein tethering of L-type Ca 2+ channels. Proc. Natl. Acad. Sci. USA 2017, 114, E8081–E8090. [Google Scholar] [CrossRef]
- Weleber, R.G.; Pillers, D.-A.M.; Powell, B.R.; Hanna, C.E.; Magenis, R.E.; Buist, N.R.M. A land Island Eye Disease (Forsius-Eriksson Syndrome) Associated with Contiguous Deletion Syndrome at Xp21. Arch. Ophthalmol. 1989, 107, 1170–1179. [Google Scholar] [CrossRef]
- A Pillers, D.; A Towbin, J.; Chamberlain, J.S.; Wu, D.; Ranier, J.; Powell, B.R.; McCabe, E.R. Deletion mapping of Aland Island eye disease to Xp21 between DXS67 (B24) and Duchenne muscular dystrophy. Am. J. Hum. Genet. 1990, 47, 795–801. [Google Scholar]
- Pillers, D.A.; Weleber, R.G.; Powell, B.R.; Hanna, C.E.; Magenis, R.E.; Buist, N.R. Aland Island eye disease (Forsius-Eriksson ocular albinism) and an Xp21 deletion in a patient with Duchenne muscular dystrophy, glycerol kinase deficiency, and congenital adrenal hypoplasia. Am. J. Med. Genet. 1990, 36, 23–28. [Google Scholar] [CrossRef]
- Sigesmund, D.A.; Weleber, R.G.; Pillers, D.A.; Westall, C.A.; Panton, C.M.; Powell, B.R.; Héon, E.; Murphey, W.H.; Musarella, M.A.; Ray, P.N. Characterization of the ocullar phenotype of Duchenne and Becker muscular dystrophy. Ophthalmology 1994, 101, 856–865. [Google Scholar]
- Jensen, H.; Warburg, M.; Sjo, O.; Schwartz, M. Duchenne muscular dystrophy: Negative electroretinograms and normal dark adaptation. Reappraisal of assignment of X linked incomplete congenital stationary night blindness. J. Med. Genet. 1995, 32, 348–351. [Google Scholar] [CrossRef]
- Fitzgerald, K.M.; Cibis, G.W.; A Giambrone, S.; Harris, D.J. Retinal signal transmission in Duchenne muscular dystrophy: Evidence for dysfunction in the photoreceptor/depolarizing bipolar cell pathway. J. Clin. Investig. 1994, 93, 2425–2430. [Google Scholar] [CrossRef]
- De Becker, I.; Riddell, D.C.; Dooley, J.M.; Tremblay, F. Correlation between electroretinogram findings and molecular analysis in the Duchenne muscular dystrophy phenotype. Br. J. Ophthalmol. 1994, 78, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.M.; Cibis, G.W.; Gettel, A.H.; Rinaldi, R.; Harris, D.J.; A White, R. ERG phenotype of a dystrophin mutation in heterozygous female carriers of Duchenne muscular dystrophy. J. Med Genet. 1999, 36, 316–322. [Google Scholar] [PubMed]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- Moser, H. Duchenne muscular dystrophy: Pathogenetic aspects and genetic prevention. Hum. Genet. 1984, 66, 17–40. [Google Scholar] [CrossRef]
- Pilgram, G.S.K.; Potikanond, S.; Baines, R.A.; Fradkin, L.G.; Noordermeer, J.N. The Roles of the Dystrophin-Associated Glycoprotein Complex at the Synapse. Mol. Neurobiol. 2009, 41, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Gumerson, J.D.; Michele, D.E. The Dystrophin-Glycoprotein Complex in the Prevention of Muscle Damage. J. Biomed. Biotechnol. 2011, 2011, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Omairi, S.; Hau, K.-L.; Collins-Hooper, H.; Scott, C.; Vaiyapuri, S.; Torelli, S.; Montanaro, F.; Matsakas, A.; Patel, K. Regulation of the dystrophin-associated glycoprotein complex composition by the metabolic properties of muscle fibres. Sci. Rep. 2019, 9, 2770. [Google Scholar] [CrossRef] [PubMed]
- Bogdanik, L.; Framery, B.; Frölich, A.; Franco, B.; Mornet, D.; Bockaert, J.; Sigrist, S.J.; Grau, Y.; Parmentier, M.-L. Muscle Dystroglycan Organizes the Postsynapse and Regulates Presynaptic Neurotransmitter Release at the Drosophila Neuromuscular Junction. PLoS ONE 2008, 3, e2084. [Google Scholar] [CrossRef]
- Yoshida, A.; Kobayashi, K.; Manya, H.; Taniguchi, K.; Kano, H.; Mizuno, M.; Inazu, T.; Mitsuhashi, H.; Takahashi, S.; Takeuchi, M.; et al. Muscular Dystrophy and Neuronal Migration Disorder Caused by Mutations in a Glycosyltransferase, POMGnT1. Dev. Cell 2001, 1, 717–724. [Google Scholar] [CrossRef]
- Taniguchi, K.; Kobayashi, K.; Saito, K.; Yamanouchi, H.; Ohnuma, A.; Hayashi, Y.K.; Manya, H.; Jin, D.K.; Lee, M.; Parano, E.; et al. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum. Mol. Genet. 2003, 12, 527–534. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ogawa, M.; Tagawa, K.; Noguchi, S.; Ishihara, T.; Nonaka, I.; Arahata, K. Selective deficiency of -dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001, 57, 115–121. [Google Scholar] [CrossRef]
- Falsaperla, R.; Praticò, A.D.; Ruggieri, M.; Parano, E.; Rizzo, R.; Corsello, G.; Vitaliti, G.; Pavone, P. Congenital muscular dystrophy: From muscle to brain. Ital. J. Pediatr. 2016, 42, 78. [Google Scholar] [CrossRef]
- Nishida, A.; Furukawa, A.; Koike, C.; Tano, Y.; Aizawa, S.; Matsuo, I.; Furukawa, T. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat. Neurosci. 2003, 6, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, M.; Omori, Y.; Sato, S.; Kobayashi, K.; Miyagoe-Suzuki, Y.; Takeda, S.; Endo, T.; Furukawa, T.; Toda, T. Post-translational Maturation of Dystroglycan Is Necessary for Pikachurin Binding and Ribbon Synaptic Localization. J. Biol. Chem. 2010, 285, 31208–31216. [Google Scholar] [CrossRef] [PubMed]
- Grewal, P.K.; Holzfeind, P.J.; Bittner, R.E.; Hewitt, J.E. Mutant glycosyltransferase and altered glycosylation of α-dystroglycan in the myodystrophy mouse. Nat. Genet. 2001, 28, 151–154. [Google Scholar] [CrossRef]
- Omori, Y.; Araki, F.; Chaya, T.; Kajimura, N.; Irie, S.; Terada, K.; Muranishi, Y.; Tsujii, T.; Ueno, S.; Koyasu, T.; et al. Presynaptic Dystroglycan–Pikachurin Complex Regulates the Proper Synaptic Connection between Retinal Photoreceptor and Bipolar Cells. J. Neurosci. 2012, 32, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Stewart, W.J.; Akanyeti, O.; Frederick, C.; Zhu, J.; Santos-Sacchi, J.; Sheets, L.; Liao, J.C.; Zenisek, D. Synaptic Ribbons Require Ribeye for Electron Density, Proper Synaptic Localization, and Recruitment of Calcium Channels. Cell Rep. 2016, 15, 2784–2795. [Google Scholar] [CrossRef]
- Maxeiner, S.; Luo, F.; Tan, A.; Schmitz, F.; Südhof, T.C. How to make a synaptic ribbon: RIBEYE deletion abolishes ribbons in retinal synapses and disrupts neurotransmitter release. EMBO J. 2016, 35, 1098–1114. [Google Scholar] [CrossRef]
- Nouvian, R.; Beutner, D.; Parsons, T.; Moser, T. Structure and Function of the Hair Cell Ribbon Synapse. J. Membr. Biol. 2006, 209, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Snellman, J.; Mehta, B.; Babai, N.; Bartoletti, T.M.; Akmentin, W.; Francis, A.; Matthews, G.; Thoreson, W.; Zenisek, D. Acute destruction of the synaptic ribbon reveals a role for the ribbon in vesicle priming. Nat. Neurosci. 2011, 14, 1135–1141. [Google Scholar] [CrossRef]
- Uthaiah, R.C.; Hudspeth, A. Molecular anatomy of the hair cell’s ribbon synapse. J. Neurosci. 2010, 30, 12387–12399. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.N.; Strauss, R.W.; Singh, M.S.; Dalkara, D.; Roska, B.; Picaud, S.; Sahel, J.-A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016, 8, 3686. [Google Scholar] [CrossRef]
- Kindt, K.S.; Sheets, L. Transmission Disrupted: Modeling Auditory Synaptopathy in Zebrafish. Front. Cell Dev. Biol. 2018, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Leroy, B.P.; Black, G.; Ong, T.; Yoon, D.; Trzupek, K. Genetic testing and diagnosis of inherited retinal diseases. Orphanet J. Rare Dis. 2021, 16, 514. [Google Scholar] [CrossRef]
- Chen, T.-C.; Huang, D.-S.; Lin, C.-W.; Yang, C.-H.; Yang, C.-M.; Wang, V.Y.; Lin, J.-W.; Luo, A.C.; Hu, F.-R.; Chen, P.-L. Genetic characteristics and epidemiology of inherited retinal degeneration in Taiwan. NPJ Genom. Med. 2021, 6, 16. [Google Scholar] [CrossRef]
- Hejtmancik, J.F.; Daiger, S.P. Understanding the genetic architecture of human retinal degenerations. Proc. Natl. Acad. Sci. USA 2020, 117, 3904–3906. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
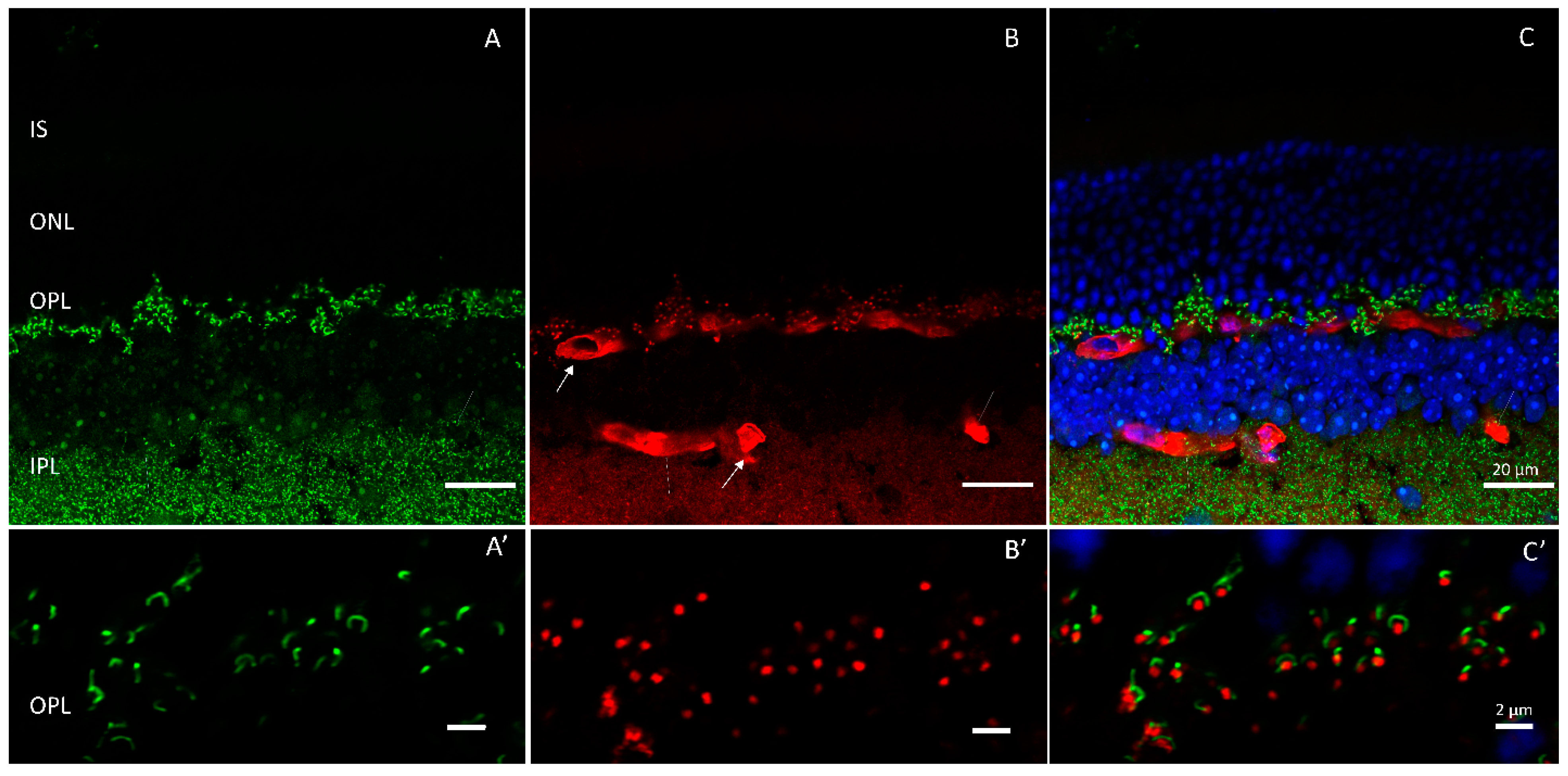{kind=link}
| Protein | Gene | Disease | Phenotype | Antibody | Source |
|---|---|---|---|---|---|
| HRG4/Unc119 | UNC119 | Cone Rod Dystrophy | Poor night vision, defective color vision, photophobia, reduced visual acuity, myopia, macular atrophy, and pericentral ring scotomas. Late onset retinal degeneration. | 13065–1-AP | Proteintech |
| Tulp1 | TULP1 | Retinitis Pigmentosa | Nystagmus (involuntary eye movement), hemeralopia (day blindness), mild myopia, low visual acuity, and nyctalopia (night blindness). Loss of mid-peripheral visual field followed by peripheral field loss and central vision with disease progression. | Custom [18] | Gift from Dr. Stephanie Hagstrom |
| Rim1 | RIMS1 | Cone Rod Dystrophy | Reduced visual clarity, especially in bright light. Disorganization of the retinal pigment epithelium (RPE). Macular disorganization and impairment. | 140–023 [19] | Synaptic systems not represented |
| Cav1.4 α1.F | CACNA1F | Congenital Stationary Night Blindness | Mild to moderate nearsightedness, farsightedness, and nystagmus. Night blindness may or may not be experienced. | Custom [20] | Gift from Dr. Amy Lee |
| Cav1.4 α2δ4 | CACNA2D | Cone Dystrophy | Mildly reduced visual acuity with onset in mid-adulthood. Early-onset photophobia. Foveal pigmentary deposits. | HPA031952 [21] | (Sigma) Not represented |
| CaBP4 | cabp4 | Cone Rod Synaptic Disorder | Nystagmus, stable low vision, photophobia, and a normal or near-normal fundus appearance. | Custom [22] | unavailable |
| Rim2 | RIMS2 | Syndromic Cone Rod Synaptic Disorder | Poor visual acuity. Developmental disabilities. Poor glucose regulation. Early onset retinal degeneration. | 140–103 [19] | Synaptic Systems |
| Dystroglycan | DAG1 | Muscular Dystrophy | Electronegative ERG not associated with ocular pathology. | 11017–1-AP | Proteintech |
| Dystrophin | DMD | Muscular Dystrophy | AB15277–1006 [23] | Abcam | |
| Pikachurin | EGFLAM | Muscular Dystrophy | 011–22631 [24] | Wako Chemicals unavailable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frederick, C.E.; Zenisek, D. Ribbon Synapses and Retinal Disease: Review. Int. J. Mol. Sci. 2023, 24, 5090. https://doi.org/10.3390/ijms24065090
Frederick CE, Zenisek D. Ribbon Synapses and Retinal Disease: Review. International Journal of Molecular Sciences. 2023; 24(6):5090. https://doi.org/10.3390/ijms24065090
Chicago/Turabian StyleFrederick, Courtney E., and David Zenisek. 2023. "Ribbon Synapses and Retinal Disease: Review" International Journal of Molecular Sciences 24, no. 6: 5090. https://doi.org/10.3390/ijms24065090
APA StyleFrederick, C. E., & Zenisek, D. (2023). Ribbon Synapses and Retinal Disease: Review. International Journal of Molecular Sciences, 24(6), 5090. https://doi.org/10.3390/ijms24065090