

Efficient Redirection of NK Cells by Genetic Modification with Chemokine Receptors CCR4 and CCR2B

Abstract
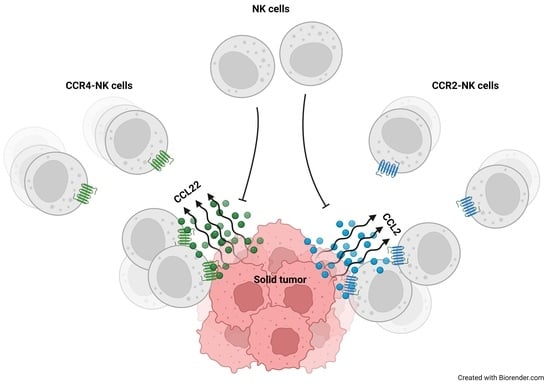
1. Introduction
2. Results
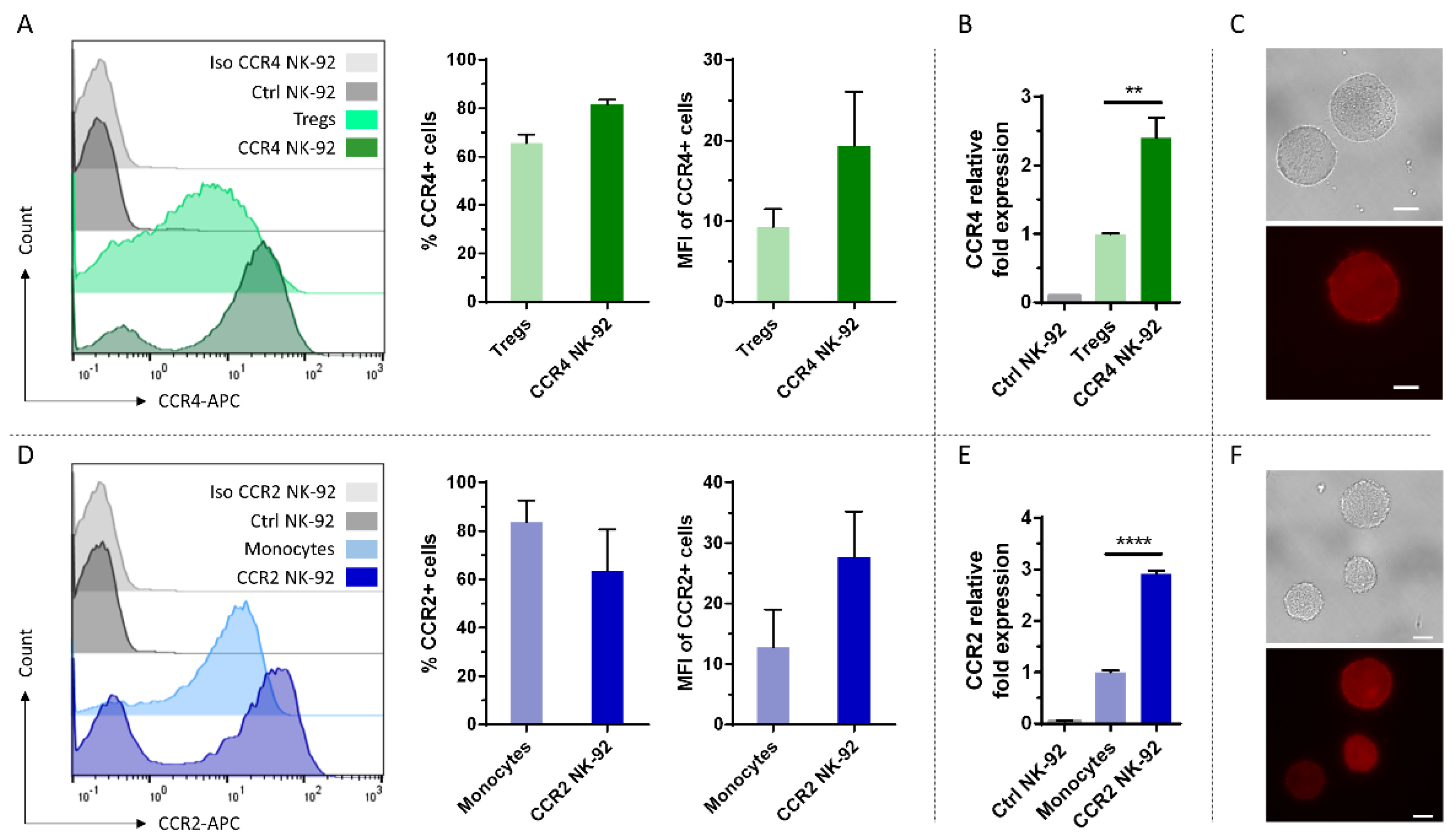
2.1. CCR4 and CCR2B Can Be Efficiently Expressed on NK-92 Cells
2.2. Genetically Engineered NK-92 Cells Efficiently Migrate towards CCL22 or CCL2
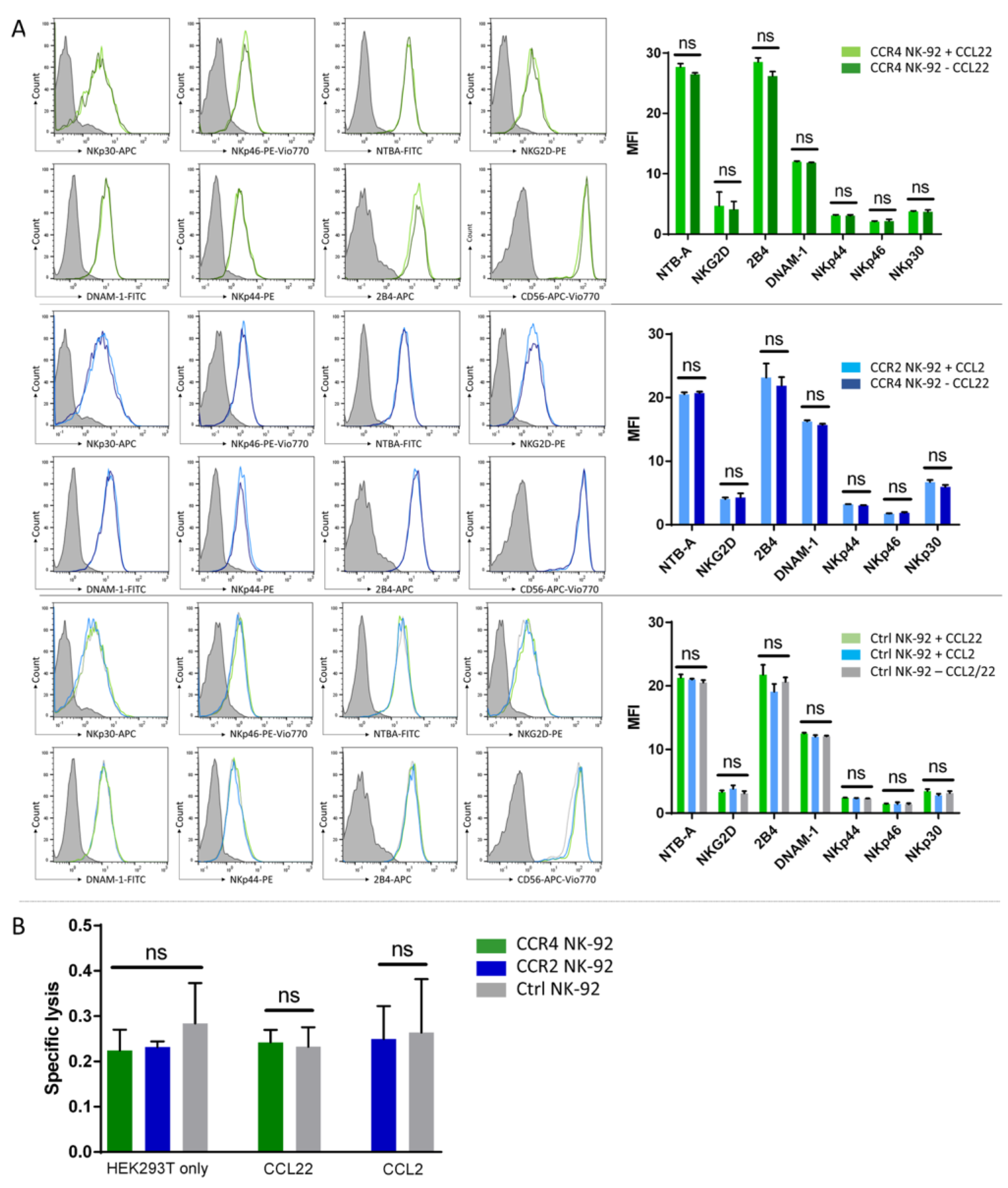
2.3. CCR4 or CCR2B Expression Does Not Impact the Cytotoxic Activity of NK-92 Cells
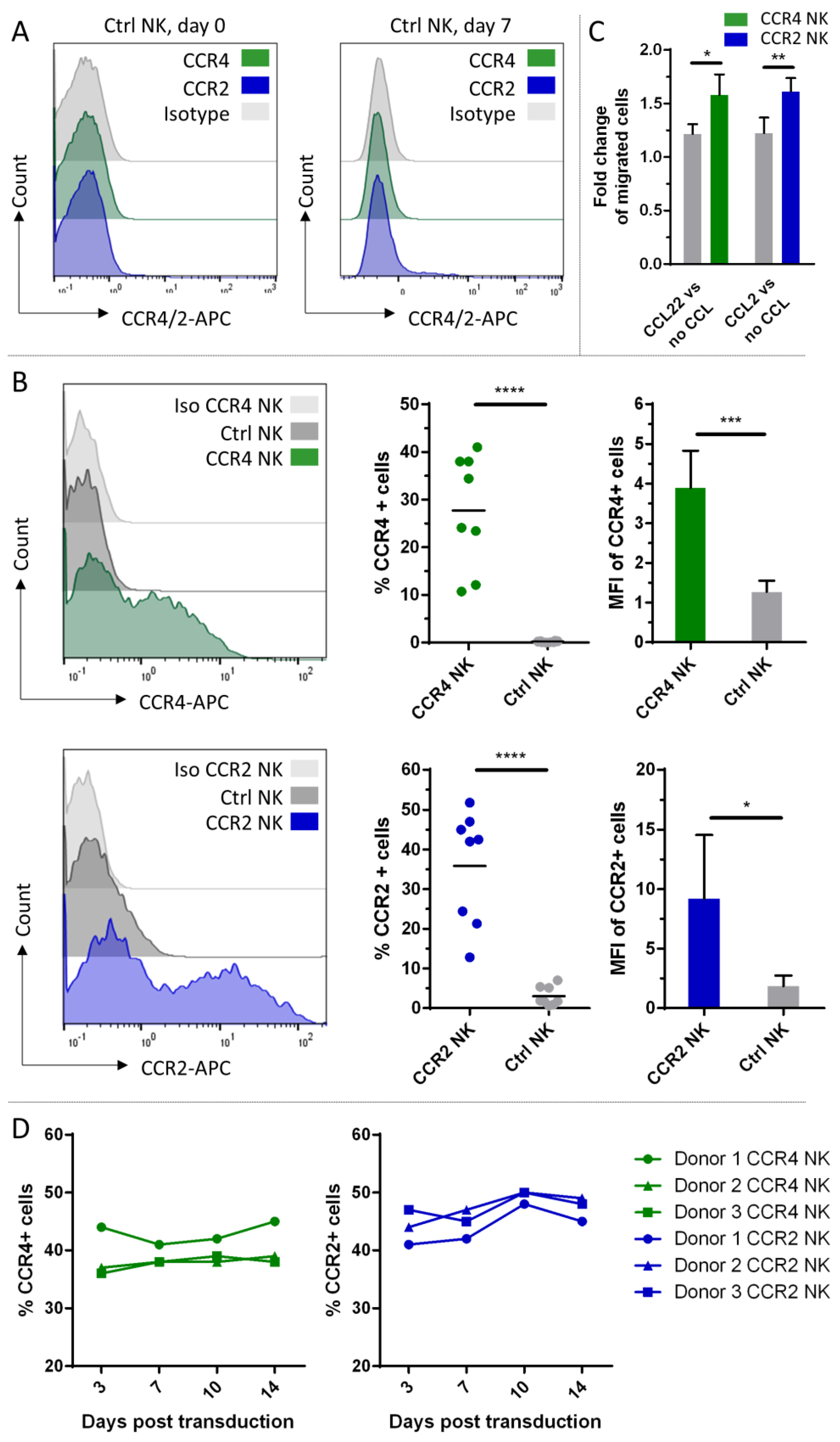
2.4. CCR4 and CCR2B Expression and Functional Testing on Peripheral Blood NK Cells
2.5. Characterization of NK Cell Populations between Transduced and Untransduced Fractions
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Primary Cells
4.2. Transgene Constructs
4.3. γ-Retroviral Vector Generation
4.4. Transduction
4.5. Flow Cytometry
4.6. qPCR
4.7. Migration Assay
4.8. Cytotoxicity Assay
4.9. Statistic Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.-Z.; Jin, W.-L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The Prognostic Value of Infiltrating Immune Cells in Cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef]
- Liu, Q.-F.; Feng, Z.-Y.; Jiang, L.-L.; Xu, T.-T.; Li, S.-M.; Liu, K.-R. Immune Cell Infiltration as Signatures for the Diagnosis and Prognosis of Malignant Gynecological Tumors. Front. Cell Dev. Biol. 2021, 9, 702451. [Google Scholar] [CrossRef]
- Sui, S.; An, X.; Xu, C.; Li, Z.; Hua, Y.; Huang, G.; Sui, S.; Long, Q.; Sui, Y.; Xiong, Y.; et al. An Immune Cell Infiltration-Based Immune Score Model Predicts Prognosis and Chemotherapy Effects in Breast Cancer. Theranostics 2020, 10, 11938–11949. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, M.; Wang, S.; Dong, J.; Wei, J. Pan-Cancer Analysis of Immune Cell Infiltration Identifies a Prognostic Immune-Cell Characteristic Score (ICCS) in Lung Adenocarcinoma. Front. Immunol. 2020, 11, 1218. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Borcherding, N.; Kolb, R.; Gullicksrud, J.; Vikas, P.; Zhu, Y.; Zhang, W. Keeping Tumors in Check: A Mechanistic Review of Clinical Response and Resistance to Immune Checkpoint Blockade in Cancer. J. Mol. Biol. 2018, 430, 2014–2029. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Huang, Q.; Deng, Y.; Guo, F.; Wang, G.; Liu, M. Crosstalk Between the Tumor Microenvironment and Cancer Cells: A Promising Predictive Biomarker for Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2021, 9, 738373. [Google Scholar] [CrossRef]
- Maruyama, T.; Kono, K.; Izawa, S.; Mizukami, Y.; Kawaguchi, Y.; Mimura, K.; Watanabe, M.; Fujii, H. CCL17 and CCL22 Chemokines within Tumor Microenvironment Are Related to Infiltration of Regulatory T Cells in Esophageal Squamous Cell Carcinoma. Dis. Esophagus 2010, 23, 422–429. [Google Scholar] [CrossRef]
- Zalfa, C.; Paust, S. Natural Killer Cell Interactions with Myeloid Derived Suppressor Cells in the Tumor Microenvironment and Implications for Cancer Immunotherapy. Front. Immunol. 2021, 12, 633205. [Google Scholar] [CrossRef]
- Röhrle, N.; Knott, M.M.L.; Anz, D. CCL22 Signaling in the Tumor Environment. Adv. Exp. Med. Biol. 2020, 1231, 79–96. [Google Scholar]
- Ye, T.; Zhang, X.; Dong, Y.; Liu, J.; Zhang, W.; Wu, F.; Bo, H.; Shao, H.; Zhang, R.; Shen, H. Chemokine CCL17 Affects Local Immune Infiltration Characteristics and Early Prognosis Value of Lung Adenocarcinoma. Front. Cell Dev. Biol. 2022, 10, 816927. [Google Scholar] [CrossRef]
- Jin, J.; Lin, J.; Xu, A.; Lou, J.; Qian, C.; Li, X.; Wang, Y.; Yu, W.; Tao, H. CCL2: An Important Mediator Between Tumor Cells and Host Cells in Tumor Microenvironment. Front. Oncol. 2021, 11, 722916. [Google Scholar] [CrossRef]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key Chemokines Direct Migration of Immune Cells in Solid Tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef]
- Marshall, L.A.; Marubayashi, S.; Jorapur, A.; Jacobson, S.; Zibinsky, M.; Robles, O.; Hu, D.X.; Jackson, J.J.; Pookot, D.; Sanchez, J.; et al. Tumors Establish Resistance to Immunotherapy by Regulating T Recruitment via CCR4. J. Immunother. Cancer 2020, 8, e000764. [Google Scholar] [CrossRef]
- Yoshie, O. CCR4 as a Therapeutic Target for Cancer Immunotherapy. Cancers 2021, 13, 5542. [Google Scholar] [CrossRef]
- Berlato, C.; Khan, M.N.; Schioppa, T.; Thompson, R.; Maniati, E.; Montfort, A.; Jangani, M.; Canosa, M.; Kulbe, H.; Hagemann, U.B.; et al. A CCR4 Antagonist Reverses the Tumor-Promoting Microenvironment of Renal Cancer. J. Clin. Investig. 2017, 127, 801–813. [Google Scholar] [CrossRef]
- Moore, D.C.; Elmes, J.B.; Shibu, P.A.; Larck, C.; Park, S.I. Mogamulizumab: An Anti-CC Chemokine Receptor 4 Antibody for T-Cell Lymphomas. Ann. Pharmacother. 2020, 54, 371–379. [Google Scholar] [CrossRef]
- Perera, L.P.; Zhang, M.; Nakagawa, M.; Petrus, M.N.; Maeda, M.; Kadin, M.E.; Waldmann, T.A.; Perera, P.-Y. Chimeric Antigen Receptor Modified T Cells That Target Chemokine Receptor CCR4 as a Therapeutic Modality for T-Cell Malignancies. Am. J. Hematol. 2017, 92, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb Selectively Depletes Effector-Type FoxP3+CD4+ Regulatory T Cells, Evoking Antitumor Immune Responses in Humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Miller, J.S. Exploring the NK Cell Platform for Cancer Immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK Cells: Surface Receptors, Inhibitory Checkpoints, and Translational Applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Koehl, U.; Kalberer, C.; Spanholtz, J.; Lee, D.A.; Miller, J.S.; Cooley, S.; Lowdell, M.; Uharek, L.; Klingemann, H.; Curti, A.; et al. Advances in Clinical NK Cell Studies: Donor Selection, Manufacturing and Quality Control. Oncoimmunology 2016, 5, e1115178. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Ruppel, K.E.; Fricke, S.; Köhl, U.; Schmiedel, D. Taking Lessons from CAR-T Cells and Going Beyond: Tailoring Design and Signaling for CAR-NK Cells in Cancer Therapy. Front. Immunol. 2022, 13, 822298. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK Cell-Based Cancer Immunotherapy: From Basic Biology to Clinical Development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Marofi, F.; Saleh, M.M.; Rahman, H.S.; Suksatan, W.; Al-Gazally, M.E.; Abdelbasset, W.K.; Thangavelu, L.; Yumashev, A.V.; Hassanzadeh, A.; Yazdanifar, M.; et al. CAR-Engineered NK Cells; a Promising Therapeutic Option for Treatment of Hematological Malignancies. Stem Cell Res. Ther. 2021, 12, 374. [Google Scholar] [CrossRef]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U.; et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front. Immunol. 2019, 10, 3123. [Google Scholar] [CrossRef]
- Sivori, S.; Meazza, R.; Quintarelli, C.; Carlomagno, S.; Della Chiesa, M.; Falco, M.; Moretta, L.; Locatelli, F.; Pende, D. NK Cell-Based Immunotherapy for Hematological Malignancies. J. Clin. Med. 2019, 8, 1702. [Google Scholar] [CrossRef]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-Modified Memory-like NK Cells Exhibit Potent Responses to NK-Resistant Lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. [Google Scholar] [CrossRef]
- Khorasani, A.B.S.; Yousefi, A.-M.; Bashash, D. CAR NK Cell Therapy in Hematologic Malignancies and Solid Tumors; Obstacles and Strategies to Overcome the Challenges. Int. Immunopharmacol. 2022, 110, 109041. [Google Scholar] [CrossRef]
- Portillo, A.L.; Hogg, R.; Poznanski, S.M.; Rojas, E.A.; Cashell, N.J.; Hammill, J.A.; Chew, M.V.; Shenouda, M.M.; Ritchie, T.M.; Cao, Q.T.; et al. Expanded Human NK Cells Armed with CAR Uncouple Potent Anti-Tumor Activity from off-Tumor Toxicity against Solid Tumors. iScience 2021, 24, 102619. [Google Scholar] [CrossRef]
- Bashiri Dezfouli, A.; Yazdi, M.; Pockley, A.G.; Khosravi, M.; Kobold, S.; Wagner, E.; Multhoff, G. NK Cells Armed with Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development. Cells 2021, 10, 3390. [Google Scholar] [CrossRef]
- Nayyar, G.; Chu, Y.; Cairo, M.S. Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Front. Oncol. 2019, 9, 51. [Google Scholar] [CrossRef]
- Melero, I.; Rouzaut, A.; Motz, G.T.; Coukos, G. T-Cell and NK-Cell Infiltration into Solid Tumors: A Key Limiting Factor for Efficacious Cancer Immunotherapy. Cancer Discov. 2014, 4, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.; Lundqvist, A. Targeting the Tumor Microenvironment to Improve Natural Killer Cell-Based Immunotherapies: On Being in the Right Place at the Right Time, with Resilience. Hum. Vaccines Immunother. 2016, 12, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Ran, G.H.; Lin, Y.Q.; Tian, L.; Zhang, T.; Yan, D.M.; Yu, J.H.; Deng, Y.C. Natural Killer Cell Homing and Trafficking in Tissues and Tumors: From Biology to Application. Signal Transduct. Target. Ther. 2022, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Reger, R.; Segerberg, F.; Lambert, M.; Leijonhufvud, C.; Baumer, Y.; Carlsten, M.; Childs, R. Enhanced Bone Marrow Homing of Natural Killer Cells Following mRNA Transfection with Gain-of-Function Variant CXCR4. Front. Immunol. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Jamali, A.; Hadjati, J.; Madjd, Z.; Mirzaei, H.R.; Thalheimer, F.B.; Agarwal, S.; Bonig, H.; Ullrich, E.; Hartmann, J. Highly Efficient Generation of Transgenically Augmented CAR NK Cells Overexpressing CXCR4. Front. Immunol. 2020, 11, 2028. [Google Scholar] [CrossRef]
- Pesce, S.; Moretta, L.; Moretta, A.; Marcenaro, E. Human NK Cell Subsets Redistribution in Pathological Conditions: A Role for CCR7 Receptor. Front. Immunol. 2016, 7, 414. [Google Scholar] [CrossRef]
- Schomer, N.T.; Jiang, Z.K.; Lloyd, M.I.; Klingemann, H.; Boissel, L. CCR7 Expression in CD19 Chimeric Antigen Receptor-Engineered Natural Killer Cells Improves Migration toward CCL19-Expressing Lymphoma Cells and Increases Tumor Control in Mice with Human Lymphoma. Cytotherapy 2022, 24, 827–834. [Google Scholar] [CrossRef]
- Khan, I.A.; Thomas, S.Y.; Moretto, M.M.; Lee, F.S.; Islam, S.A.; Combe, C.; Schwartzman, J.D.; Luster, A.D. CCR5 Is Essential for NK Cell Trafficking and Host Survival Following Toxoplasma Gondii Infection. PLoS Pathog. 2006, 2, e49. [Google Scholar] [CrossRef]
- Carlin, L.E.; Hemann, E.A.; Zacharias, Z.R.; Heusel, J.W.; Legge, K.L. Natural Killer Cell Recruitment to the Lung During Influenza A Virus Infection Is Dependent on CXCR3, CCR5, and Virus Exposure Dose. Front. Immunol. 2018, 9, 781. [Google Scholar] [CrossRef]
- Li, F.; Sheng, Y.; Hou, W.; Sampath, P.; Byrd, D.; Thorne, S.; Zhang, Y. CCL5-Armed Oncolytic Virus Augments CCR5-Engineered NK Cell Infiltration and Antitumor Efficiency. J. Immunother. Cancer 2020, 8, e000131. [Google Scholar] [CrossRef]
- Kremer, V.; Ligtenberg, M.A.; Zendehdel, R.; Seitz, C.; Duivenvoorden, A.; Wennerberg, E.; Colón, E.; Scherman-Plogell, A.-H.; Lundqvist, A. Correction to: Genetic Engineering of Human NK Cells to Express CXCR2 Improves Migration to Renal Cell Carcinoma. J. Immunother. Cancer 2017, 5, 88. [Google Scholar] [CrossRef]
- Tanaka, S.; Green, S.R.; Quehenberger, O. Differential Expression of the Isoforms for the Monocyte Chemoattractant Protein-1 Receptor, CCR2, in Monocytes. Biochem. Biophys. Res. Commun. 2002, 290, 73–80. [Google Scholar] [CrossRef]
- Park, H.-K.; Na, Y.H.; Nguyen, H.T.; Nguyen, L.P.; Hurh, S.; Seong, J.Y.; Lee, C.S.; Ham, B.-J.; Hwang, J.-I. Analysis of CCR2 Splice Variant Expression Patterns and Functional Properties. Cell Biosci. 2022, 12, 59. [Google Scholar] [CrossRef]
- Boyden, S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962, 115, 453–466. [Google Scholar] [CrossRef]
- Shin, M.H.; Kim, J.; Lim, S.A.; Kim, J.; Kim, S.-J.; Lee, K.-M. NK Cell-Based Immunotherapies in Cancer. Immune Netw. 2020, 20, e14. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord Blood NK Cells Engineered to Express IL-15 and a CD19-Targeted CAR Show Long-Term Persistence and Potent Antitumor Activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur, M.; Lu, W.; Krause, D.; Van Etten, R.; Wels, W.; Klingemann, H. Retargeting NK-92 Cells by Means of CD19- and CD20-Specific Chimeric Antigen Receptors Compares Favorably with Antibody-Dependent Cellular Cytotoxicity. OncoImmunology 2013, 2, e26527. [Google Scholar] [CrossRef]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Lin, K.-W.; Jares, A.; Firor, A.E.; Shuai, X.; Salman, H.; Golightly, M.; et al. Preclinical Targeting of Aggressive T-Cell Malignancies Using Anti-CD5 Chimeric Antigen Receptor. Leukemia 2017, 31, 2151–2160. [Google Scholar] [CrossRef]
- You, F.; Wang, Y.; Jiang, L.; Zhu, X.; Chen, D.; Yuan, L.; An, G.; Meng, H.; Yang, L. A Novel CD7 Chimeric Antigen Receptor-Modified NK-92MI Cell Line Targeting T-Cell Acute Lymphoblastic Leukemia. Am. J. Cancer Res. 2019, 9, 64–78. [Google Scholar]
- Guha, P.; Heatherton, K.R.; O’Connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef]
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2020, 11, 622509. [Google Scholar] [CrossRef] [PubMed]
- Ukidve, A.; Cu, K.; Kumbhojkar, N.; Lahann, J.; Mitragotri, S. Overcoming Biological Barriers to Improve Solid Tumor Immunotherapy. Drug Deliv. Transl. Res. 2021, 11, 2276–2301. [Google Scholar] [CrossRef] [PubMed]
- Berahovich, R.D.; Lai, N.L.; Wei, Z.; Lanier, L.L.; Schall, T.J. Evidence for NK Cell Subsets Based on Chemokine Receptor Expression. J. Immunol. 2006, 177, 7833–7840. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Qin, S.; Unutmaz, D.; Soler, D.; Murphy, K.E.; Hodge, M.R.; Wu, L.; Butcher, E.C. Unique Subpopulations of CD56+ NK and NK-T Peripheral Blood Lymphocytes Identified by Chemokine Receptor Expression Repertoire. J. Immunol. 2001, 166, 6477–6482. [Google Scholar] [CrossRef] [PubMed]
- Inngjerdingen, M.; Damaj, B.; Maghazachi, A.A. Expression and Regulation of Chemokine Receptors in Human Natural Killer Cells. Blood 2001, 97, 367–375. [Google Scholar] [CrossRef]
- Matsuo, K.; Yoshie, O.; Nakayama, T. Multifaceted Roles of Chemokines and Chemokine Receptors in Tumor Immunity. Cancers 2021, 13, 6132. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of Tumour-Infiltrating Macrophages via CCL2/CCR2 Signalling as a Therapeutic Strategy against Hepatocellular Carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef]
- Goyne, H.E.; Stone, P.J.B.; Burnett, A.F.; Cannon, M.J. Ovarian Tumor Ascites CD14 Cells Suppress Dendritic Cell-activated CD4 T-Cell Responses Through IL-10 Secretion and Indoleamine 2,3-Dioxygenase. J. Immunother. 2014, 37, 163–169. [Google Scholar] [CrossRef]
- Lubowicka, E.; Przylipiak, A.; Zajkowska, M.; Piskór, B.M.; Malinowski, P.; Fiedorowicz, W.; Ławicki, S. Plasma Chemokine CCL2 and Its Receptor CCR2 Concentrations as Diagnostic Biomarkers for Breast Cancer Patients. BioMed Res. Int. 2018, 2018, 2124390. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Yang, X.; Yang, J.; Lu, P.; Zhao, L.; Li, B.; Pan, H.; Jiang, Z.; Shen, X.; et al. Chemokine Receptor CCR2b Enhanced Anti-Tumor Function of Chimeric Antigen Receptor T Cells Targeting Mesothelin in a Non-Small-Cell Lung Carcinoma Model. Front. Immunol. 2021, 12, 407. [Google Scholar] [CrossRef]
- Kloess, S.; Kretschmer, A.; Stahl, L.; Fricke, S.; Koehl, U. CAR-Expressing Natural Killer Cells for Cancer Retargeting. Transfus. Med. Hemotherapy 2019, 46, 4. [Google Scholar] [CrossRef]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK Cell CD16 Surface Expression and Function Is Regulated by a Disintegrin and Metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef]
- Kared, H.; Martelli, S.; Ng, T.P.; Pender, S.L.F.; Larbi, A. CD57 in Human Natural Killer Cells and T-Lymphocytes. Cancer Immunol. Immunother. 2016, 65, 441–452. [Google Scholar] [CrossRef]
- Nielsen, C.M.; White, M.J.; Goodier, M.R.; Riley, E.M. Functional Significance of CD57 Expression on Human NK Cells and Relevance to Disease. Front. Immunol. 2013, 4, 422. [Google Scholar] [CrossRef]
- Lopez-Vergès, S.; Milush, J.M.; Pandey, S.; York, V.A.; Arakawa-Hoyt, J.; Pircher, H.; Norris, P.J.; Nixon, D.F.; Lanier, L.L. CD57 Defines a Functionally Distinct Population of Mature NK Cells in the Human CD56dimCD16+ NK-Cell Subset. Blood 2010, 116, 3865–3874. [Google Scholar] [CrossRef]
- Pende, D.; Falco, M.; Vitale, M.; Cantoni, C.; Vitale, C.; Munari, E.; Bertaina, A.; Moretta, F.; Del Zotto, G.; Pietra, G.; et al. Killer Ig-Like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front. Immunol. 2019, 10, 1179. [Google Scholar] [CrossRef]
- Bari, R.; Granzin, M.; Tsang, K.S.; Roy, A.; Krueger, W.; Orentas, R.; Schneider, D.; Pfeifer, R.; Moeker, N.; Verhoeyen, E.; et al. A Distinct Subset of Highly Proliferative and Lentiviral Vector (LV)-Transducible NK Cells Define a Readily Engineered Subset for Adoptive Cellular Therapy. Front. Immunol. 2019, 10, 2001. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Abazari, M.F.; Sadeghi, S.; Mahmoudi, R.; Kheirollahi, A.; Askari, H.; Wickström, S.L.; Poortahmasebi, V.; Lundqvist, A.; Kiessling, R.; et al. Genetically Modified Immune Cells Targeting Tumor Antigens. Pharmacol. Ther. 2020, 214, 107603. [Google Scholar] [CrossRef]
- Calderon, G.A.; Thai, P.; Hsu, C.W.; Grigoryan, B.; Gibson, S.M.; Dickinson, M.E.; Miller, J.S. Tubulogenesis of Co-Cultured Human iPS-Derived Endothelial Cells and Human Mesenchymal Stem Cells in Fibrin and Gelatin Methacrylate Gels. Biomater. Sci. 2017, 5, 1652–1660. [Google Scholar] [CrossRef]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.-L.; Verhoeyen, E. Baboon Envelope Pseudotyped LVs Outperform VSV-G-LVs for Gene Transfer into Early-Cytokine-Stimulated and Resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.A.; Weijtens, M.E.M.; Ronteltap, C.; Eshhar, Z.; Gratama, J.W.; Chames, P.; Bolhuis, R.L.H. Grafting Primary Human T Lymphocytes with Cancer-Specific Chimeric Single Chain and Two Chain TCR. Gene Ther. 2000, 7, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Soneoka, Y.; Cannon, P.M.; Ramsdale, E.E.; Griffiths, J.C.; Romano, G.; Kingsman, S.M.; Kingsman, A.J. A Transient Three-Plasmid Expression System for the Production of High Titer Retroviral Vectors. Nucleic Acids Res. 1995, 23, 628–633. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Conjugate | Manufacturer |
|---|---|---|
| 2B4 (REA112) | APC | Miltenyi Biotec, Bergisch Gladbach, Germany |
| CCR2 (REA264) | APC | |
| CCR4 (REA279) | APC | |
| CD14 (REA599) | APC | |
| CD16 (REA423) | PerCP-Vio 700 | |
| CD19 (REA675) | PE-Vio770 | |
| CD3 (REA613) | VioBlue | |
| CD4 (REA623) | PE | |
| CD45 (REA747) | Vio-Green | |
| CD56 (REA196) | APC-Vio 770 | |
| CD57 (REA769) | PE-Vio770 | |
| CD8 (REA734) | FITC | |
| DNAM-1 (REA1040) | FITC | |
| Isotype Control (REA293) | APC | |
| Isotype Control (REA293) | FITC | |
| Isotype Control (REA293) | PE | |
| Isotype Control (REA293) | PE-Vio770 | |
| Isotype Control (REA293) | APC-Vio770 | |
| KIR2D (REA1042) | PE | |
| NKG2A (S19004C) | Pacific Blue | Biolegend, San Diego, CA, USA |
| NKG2D (REA1228) | PE | Miltenyi Biotec, Bergisch Gladbach, Germany |
| NKp30 (REA823) | APC | |
| NKp44 (REA1163) | PE | |
| NKp46 (REA808) | PE-Vio770 | |
| NTB-A (REA339) | FITC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feigl, F.F.; Stahringer, A.; Peindl, M.; Dandekar, G.; Koehl, U.; Fricke, S.; Schmiedel, D. Efficient Redirection of NK Cells by Genetic Modification with Chemokine Receptors CCR4 and CCR2B. Int. J. Mol. Sci. 2023, 24, 3129. https://doi.org/10.3390/ijms24043129
Feigl FF, Stahringer A, Peindl M, Dandekar G, Koehl U, Fricke S, Schmiedel D. Efficient Redirection of NK Cells by Genetic Modification with Chemokine Receptors CCR4 and CCR2B. International Journal of Molecular Sciences. 2023; 24(4):3129. https://doi.org/10.3390/ijms24043129
Chicago/Turabian StyleFeigl, Frederik Fabian, Anika Stahringer, Matthias Peindl, Gudrun Dandekar, Ulrike Koehl, Stephan Fricke, and Dominik Schmiedel. 2023. "Efficient Redirection of NK Cells by Genetic Modification with Chemokine Receptors CCR4 and CCR2B" International Journal of Molecular Sciences 24, no. 4: 3129. https://doi.org/10.3390/ijms24043129
APA StyleFeigl, F. F., Stahringer, A., Peindl, M., Dandekar, G., Koehl, U., Fricke, S., & Schmiedel, D. (2023). Efficient Redirection of NK Cells by Genetic Modification with Chemokine Receptors CCR4 and CCR2B. International Journal of Molecular Sciences, 24(4), 3129. https://doi.org/10.3390/ijms24043129