
The Role of NF-κB in Intracranial Aneurysm Pathogenesis: A Systematic Review




Abstract
:1. Introduction
2. Methods
2.1. Systematic Literature Search
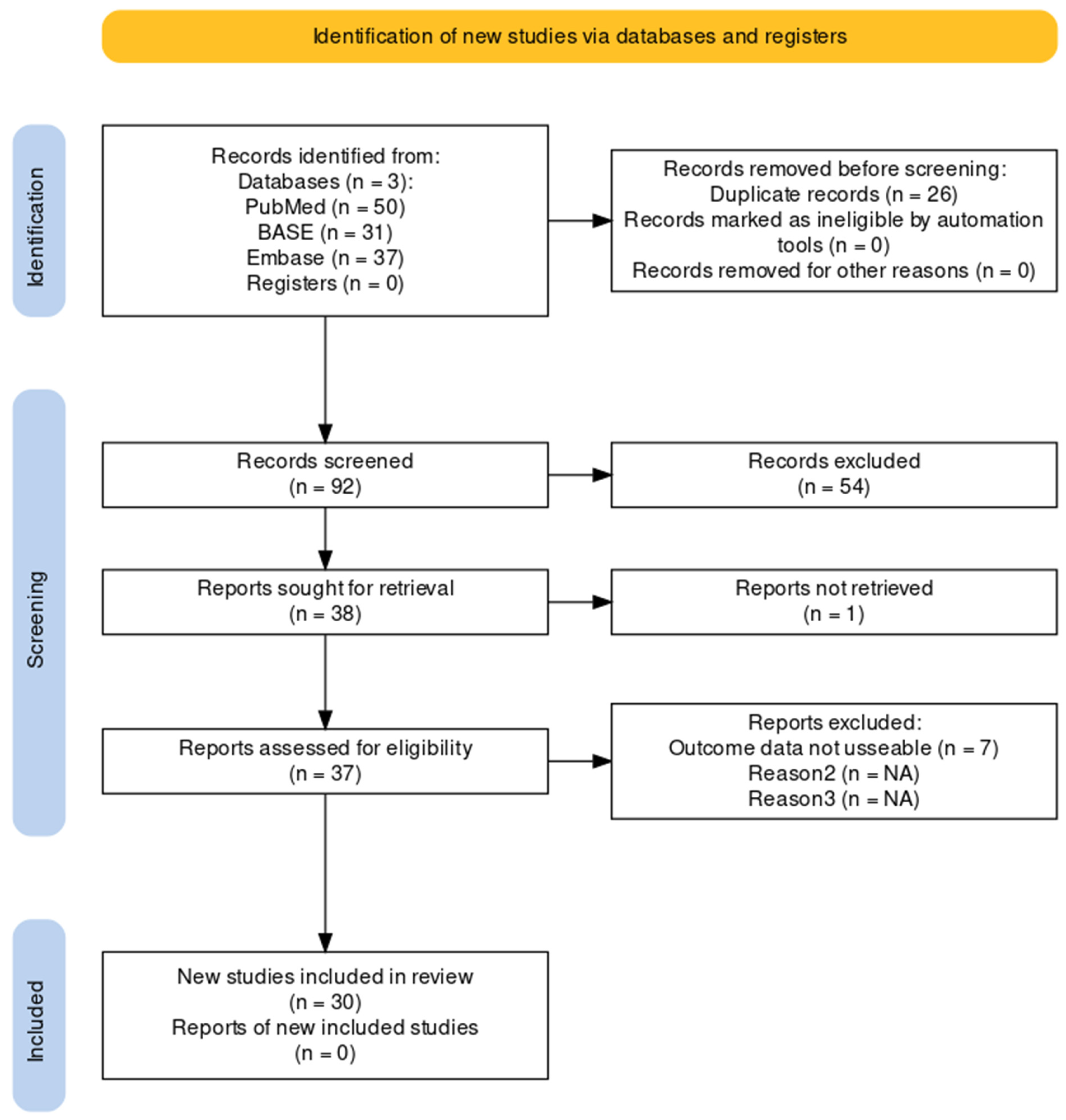
2.2. Literature Screening Criteria
3. Results
3.1. Clinical Studies
3.2. Animal Experimental Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Etminan, N.; Rinkel, G.J. Unruptured intracranial aneurysms: Development, rupture and preventive management. Nat. Rev. Neurol. 2016, 12, 699–713. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, K.-Y.; Ha, S.W.; Suh, S.H. Prevalence of Unruptured Intracranial Aneurysms: A Single Center Experience Using 3T Brain MR Angiography. Neurointervention 2021, 16, 117–121. [Google Scholar] [CrossRef]
- Etminan, N.; Chang, H.-S.; Hackenberg, K.; de Rooij, N.K.; Vergouwen, M.D.I.; Rinkel, G.J.E.; Algra, A. Worldwide Incidence of Aneurysmal Subarachnoid Hemorrhage According to Region, Time Period, Blood Pressure, and Smoking Prevalence in the Population. JAMA Neurol. 2019, 76, 588–597. [Google Scholar] [CrossRef]
- Hackenberg, K.A.; Hänggi, D.; Etminan, N. Unruptured Intracranial Aneurysms. Stroke 2018, 49, 2268–2275. [Google Scholar] [CrossRef]
- Rehman, S.; Phan, H.T.; Reeves, M.J.; Thrift, A.G.; Cadilhac, D.A.; Sturm, J.; Breslin, M.; Callisaya, M.L.; Vemmos, K.; Parmar, P.; et al. Case-Fatality and Functional Outcome after Subarachnoid Hemorrhage (SAH) in INternational STRoke oUtComes sTudy (INSTRUCT). J. Stroke Cerebrovasc. Dis. 2021, 31, 106201. [Google Scholar] [CrossRef]
- Muhammad, S.; Chaudhry, S.R.; Dobreva, G.; Lawton, M.T.; Niemelä, M.; Hänggi, D. Vascular Macrophages as Therapeutic Targets to Treat Intracranial Aneurysms. Front. Immunol. 2021, 12, 630381. [Google Scholar] [CrossRef]
- Pawlowska, E.; Szczepanska, J.; Wisniewski, K.; Tokarz, P.; Jaskólski, D.J.; Blasiak, J. NF-κB-Mediated Inflammation in the Pathogenesis of Intracranial Aneurysm and Subarachnoid Hemorrhage. Does Autophagy Play a Role? Int. J. Mol. Sci. 2018, 19, 1245. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Shimamura, M.; Nakagami, H.; Wakayama, K.; Moriwaki, T.; Ishibashi, R.; Nozaki, K.; Morishita, R.; Hashimoto, N.; et al. NF-κB Is a Key Mediator of Cerebral Aneurysm Formation. Circulation 2007, 116, 2830–2840. [Google Scholar] [CrossRef]
- Bond, M.; Chase, A.J.; Baker, A.H.; Newby, A.C. Inhibition of transcription factor NF-κB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc. Res. 2001, 50, 556–565. [Google Scholar] [CrossRef]
- Monaco, C.; Andreakos, E.; Kiriakidis, S.; Mauri, C.; Bicknell, C.; Foxwell, B.; Cheshire, N.; Paleolog, E.; Feldmann, M. Canonical pathway of nuclear factor κB activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 5634–5639. [Google Scholar] [CrossRef]
- Lai, X.-L.; Deng, Z.-F.; Zhu, X.-G.; Chen, Z.-H. Apc gene suppresses intracranial aneurysm formation and rupture through inhibiting the NF-κB signaling pathway mediated inflammatory response. Biosci. Rep. 2019, 39, BSR20181909. [Google Scholar] [CrossRef]
- Zhou, H.; Khan, D.; Gerdes, N.; Hagenbeck, C.; Rana, M.; Cornelius, J.F.; Muhammad, S. Colchicine Protects against Ethanol-Induced Senescence and Senescence-Associated Secretory Phenotype in Endothelial Cells. Antioxidants 2023, 12, 960. [Google Scholar] [CrossRef]
- Li, X.; Khan, D.; Rana, M.; Hänggi, D.; Muhammad, S. Doxycycline Attenuated Ethanol-Induced Inflammaging in Endothelial Cells: Implications in Alcohol-Mediated Vascular Diseases. Antioxidants 2022, 11, 2413. [Google Scholar] [CrossRef]
- Starke, R.M.; Thompson, J.W.; Ali, M.S.; Pascale, C.L.; Lege, A.M.; Ding, D.; Chalouhi, N.; Hasan, D.M.; Jabbour, P.; Owens, G.K.; et al. Cigarette Smoke Initiates Oxidative Stress-Induced Cellular Phenotypic Modulation Leading to Cerebral Aneurysm Pathogenesis. Arter. Thromb. Vasc. Biol. 2018, 38, 610–621. [Google Scholar] [CrossRef]
- Zhou, H.; Khan, D.; Hussain, S.M.; Gerdes, N.; Hagenbeck, C.; Rana, M.; Cornelius, J.F.; Muhammad, S. Colchicine inhibited oxidative stress-induced endothelial cell senescence via blocking NF-κB and MAPKs: Implications in vascular diseases. bioRxiv 2023. [Google Scholar] [CrossRef]
- Haddaway, N.R.; Page, M.J.; Pritchard, C.C.; McGuinness, L.A. PRISMA2020: An R package and Shiny app for producing PRISMA 2020-compliant flow diagrams, with interactivity for optimised digital transparency and Open Synthesis. Campbell Syst. Rev. 2022, 18, e1230. [Google Scholar] [CrossRef]
- Sima, X.; Xu, J.; Li, J.; You, C. Association Between NFKB1 −94 Insertion/Deletion ATTG Polymorphism and Risk of Intracranial Aneurysm. Genet. Test. Mol. Biomarkers 2013, 17, 620–624. [Google Scholar] [CrossRef]
- Kamińska, J.; Tylicka, M.; Dymicka-Piekarska, V.; Mariak, Z.; Matowicka-Karna, J.; Koper-Lenkiewicz, O.M. Canonical NF-κB signaling pathway and GRO-α/CXCR2 axis are activated in unruptured intracranial aneurysm patients. Sci. Rep. 2022, 12, 21375. [Google Scholar] [CrossRef]
- Cheng, W.-T.; Wang, N. Correlation between MMP-2 and NF-κ B expression of intracranial aneurysm. Asian Pac. J. Trop. Med. 2013, 6, 570–573. [Google Scholar] [CrossRef]
- Sun, X.; Zheng, X.; Zhang, X.; Zhang, Y.; Luo, G. Exosomal microRNA-23b-3p from bone marrow mesenchymal stem cells maintains T helper/Treg balance by downregulating the PI3k/Akt/NF-κB signaling pathway in intracranial aneurysm. Brain Res. Bull. 2020, 165, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wan, Y.; Feng, J.; Li, M.; Jiang, Z. Involvement of TLR2/4-MyD88-NF-κB signaling pathway in the pathogenesis of intracranial aneurysm. Mol. Med. Rep. 2021, 23, 230. [Google Scholar] [CrossRef]
- Fan, X.; Zhao, H.; Yu, G.; Zhong, X.; Yao, H.; Yang, Q. Role of inflammatory responses in the pathogenesis of human cerebral aneurysm. Genet. Mol. Res. 2015, 14, 9062–9070. [Google Scholar] [CrossRef]
- Wei, L.; Yang, C.; Li, K.Q.; Zhong, C.L.; Sun, Z.Y. 3-Aminobenzamide protects against cerebral artery injury and inflammation in rats with intracranial aneurysms. Die Pharm.-Int. J. Pharm. Sci. 2019, 74, 142–146. [Google Scholar] [CrossRef]
- Mandelbaum, M.; Kolega, J.; Dolan, J.M.; Siddiqui, A.H.; Meng, H. A Critical Role for Proinflammatory Behavior of Smooth Muscle Cells in Hemodynamic Initiation of Intracranial Aneurysm. PLoS ONE 2013, 8, e74357. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, D.; Tian, Y.; Wei, H.; Zhou, Z.; Liu, L.; Wang, D.; Dong, J.-F.; Jiang, R.; Zhang, J. Aspirin Inhibits Degenerative Changes of Aneurysmal Wall in a Rat Model. Neurochem. Res. 2015, 40, 1537–1545. [Google Scholar] [CrossRef]
- Hayashi, K.; Kataoka, H.; Minami, M.; Ikedo, T.; Miyata, T.; Shimizu, K.; Nagata, M.; Yang, T.; Yamamoto, Y.; Yokode, M.; et al. Association of zinc administration with growth suppression of intracranial aneurysms via induction of A20. J. Neurosurg. 2020; ahead-of-print. [Google Scholar] [CrossRef]
- Liu, Y.F.; Zhang, Y.; Dai, D.; Xu, Z. Expression of NF-κB, MCP-1 and MMP-9 in a Cerebral Aneurysm Rabbit Model. Can. J. Neurol. Sci./J. Can. des Sci. Neurol. 2014, 41, 200–205. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Egashira, K.; Hashimoto, N. Impact of Monocyte Chemoattractant Protein-1 Deficiency on Cerebral Aneurysm Formation. Stroke 2009, 40, 942–951. [Google Scholar] [CrossRef]
- Li, S.; Tian, Y.; Huang, X.; Zhang, Y.; Wang, D.; Wei, H.; Dong, J.; Jiang, R.; Zhang, J. Intravenous transfusion of endothelial colony-forming cells attenuates vascular degeneration after cerebral aneurysm induction. Brain Res. 2014, 1593, 65–75. [Google Scholar] [CrossRef]
- Jiang, Z.; Huang, J.; You, L.; Zhang, J.; Li, B. Pharmacological inhibition of STAT3 by BP-1-102 inhibits intracranial aneurysm formation and rupture in mice through modulating inflammatory response. Pharmacol. Res. Perspect. 2021, 9, e00704. [Google Scholar] [CrossRef]
- Xiao, G.; Zhang, M.; Peng, X.; Jiang, G. Protocatechuic acid attenuates cerebral aneurysm formation and progression by inhibiting TNF-alpha/Nrf-2/NF-κB-mediated inflammatory mechanisms in experimental rats. Open Life Sci. 2021, 16, 128–141. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Nishimura, M.; Ishibashi, R.; Morishita, R.; Miyamoto, S. Regression of Intracranial Aneurysms by Simultaneous Inhibition of Nuclear Factor-κB and Ets With Chimeric Decoy Oligodeoxynucleotide Treatment. Neurosurgery 2012, 70, 1534–1543. [Google Scholar] [CrossRef]
- Jin, T.; An, Q.; Qin, X.; Hu, Y.; Hu, J.; Zhou, B.; Leng, B. Resveratrol inhibits cerebral aneurysms in mice via downregulating the NF-κB pathway. Acta Biochim. Pol. 2022, 69, 613–618. [Google Scholar] [CrossRef]
- Aoki, T.; Nishimura, M.; Ishibashi, R.; Kataoka, H.; Takagi, Y.; Hashimoto, N.; Liu, L.; Zhang, Q.; Xiong, X.-Y.; Gong, Q.-W.; et al. Toll-like receptor 4 expression during cerebral aneurysm formation. J. Neurosurg. 2010, 113, 851–858. [Google Scholar] [CrossRef]
- Ishibashi, R.; Aoki, T.; Nishimura, M.; Hashimoto, N.; Miyamoto, S. Contribution of Mast Cells to Cerebral Aneurysm Formation. Curr. Neurovascular Res. 2010, 7, 113–124. [Google Scholar] [CrossRef]
- Aoki, T.; Fukuda, M.; Nishimura, M.; Nozaki, K.; Narumiya, S. Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol. Commun. 2014, 2, 34. [Google Scholar] [CrossRef]
- Ikedo, T.; Minami, M.; Kataoka, H.; Hayashi, K.; Nagata, M.; Fujikawa, R.; Higuchi, S.; Yasui, M.; Aoki, T.; Fukuda, M.; et al. Dipeptidyl Peptidase-4 Inhibitor Anagliptin Prevents Intracranial Aneurysm Growth by Suppressing Macrophage Infiltration and Activation. J. Am. Heart Assoc. 2017, 6, e004777. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Hashimoto, N. Nifedipine Inhibits the Progression of An Experimentally Induced Cerebral Aneurysm in Rats with Associated Down-Regulation of NF-Kappa B Transcriptional Activity. Curr. Neurovascular Res. 2008, 5, 37–45. [Google Scholar] [CrossRef]
- Aoki, T.; Nishimura, M.; Matsuoka, T.; Yamamoto, K.; Furuyashiki, T.; Kataoka, H.; Kitaoka, S.; Ishibashi, R.; Ishibazawa, A.; Miyamoto, S.; et al. PGE2-EP2signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-κB. Br. J. Pharmacol. 2011, 163, 1237–1249. [Google Scholar] [CrossRef]
- Aoki, T.; Frȍsen, J.; Fukuda, M.; Bando, K.; Shioi, G.; Tsuji, K.; Ollikainen, E.; Nozaki, K.; Laakkonen, J.; Narumiya, S. Prostaglandin E2–EP2–NF-κB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci. Signal. 2017, 10, eaah6037. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Morishita, R.; Hashimoto, N. Reduced Collagen Biosynthesis Is the Hallmark of Cerebral Aneurysm. Arter. Thromb. Vasc. Biol. 2009, 29, 1080–1086. [Google Scholar] [CrossRef]
- Ma, J.; Hou, D.; Wei, Z.; Zhu, J.; Lu, H.; Li, Z.; Wang, X.; Li, Y.; Qiao, G.; Liu, N. Tanshinone IIA attenuates cerebral aneurysm formation by inhibiting the NF-κB-mediated inflammatory response. Mol. Med. Rep. 2019, 20, 1621–1628. [Google Scholar] [CrossRef]
- Wang, D.; Lai, D.; Peng, C. TWIST1 silencing attenuates intracranial aneurysms by inhibiting NF-κB signaling. Trop. J. Pharm. Res. 2022, 21, 927–932. [Google Scholar] [CrossRef]
- Moriwaki, T.; Takagi, Y.; Sadamasa, N.; Aoki, T.; Nozaki, K.; Hashimoto, N. Impaired Progression of Cerebral Aneurysms in Interleukin-1β–Deficient Mice. Stroke 2006, 37, 900–905. [Google Scholar] [CrossRef]
- Starke, R.M.; Chalouhi, N.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, L.F.; Rosenwasser, R.H.; Wada, K.; Shimada, K.; Hasan, D.M.; Greig, N.H.; et al. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J. Neuroinflamm. 2014, 11, 77. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Morimoto, M.; Nozaki, K.; Hashimoto, N. Macrophage-Derived Matrix Metalloproteinase-2 and -9 Promote the Progression of Cerebral Aneurysms in Rats. Stroke 2007, 38, 162–169. [Google Scholar] [CrossRef]
- Morimoto, M.; Kume, N.; Miyamoto, S.; Mizoguchi, A.; Nozaki, K.; Sadamasa, N.; Kita, T.; Hashimoto, N. The Roles of MMPs for Cerebral Aneurysm Formation; Springer: Tokyo, Japan, 2002; pp. 223–233. [Google Scholar] [CrossRef]

{kind=link}
| No. | NF-κB Status | Inflammatory Markers and MMPs | Ref. |
|---|---|---|---|
| 1 | Lower nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκB-α) and higher NF-κB P65 protein expression in IA tissue | Higher serum levels of MCP-1, TNF-α, IL-1β, and IL-6 in IA patients than in control, the serum levels of MCP-1, TNF-α, IL-1β, and IL-6 were higher in patients with ruptured CAs than in patients with unruptured IAs, lower APC protein expression in IA tissue | [11] |
| 2 | Lower median cerebrospinal fluid (CSF) and serum NF-κB p65 concentrations | Higher median CSF GRO alpha chemokine/C-X-C motif ligand 1 (CXCL1) (GRO-α) and CSF C-X-C Motif Chemokine Receptor 2 (CXCR2) concentration | [18] |
| 3 | Higher NF-κB mRNA and protein expression in IAs than in control, NF-κB protein expression was detected in the intima, media, and extima | Higher MMP-2 mRNA and protein expression in IAs than in control, MMP-2 protein expression was detected in the intima, media, and extima | [19] |
| 4 | High positive protein expression of NF-κB P65 in IA walls detected by immunohistochemical staining | Higher levels of IL-17, interferon-gamma (IFN-γ), IL-17a, MCP-1, TNF-α, IL-6, and lower levels of IL-10 in peripheral blood, high positive protein expression of phosphoinositide-3-kinase (PI3K) and protein kinase B (Akt) detected by immunohistochemical staining, high mRNA expression of T helper 17 cells (Th17) transcription-factor-related orphan receptor C (RORC) and low mRNA expression of regulatory T-cell (Treg) transcription factor forkhead box p3 (Foxp3) | [20] |
| 5 | In serum higher NF-κB 65 concentration | In serum higher mRNA expression and concentration of toll-like receptor (TLR) 2, TLR 4, and myeloid differentiation primary response 88 (MyD88) | [21] |
| 6 | Increased immunohistostaining for NF-κB P65 in the aneurysmal wall | [8] | |
| 7 | Increased protein levels and staining intensity for NF-κB P65 in IA tissue | Higher protein levels and increased staining intensity for ICAM-1 and higher MCP-1 mRNA expression in IA tissue | [22] |
| Animals | NF-κB Activation | Inflammatory Markers and MMPs | IA Features | Ref. | |
|---|---|---|---|---|---|
| 1 | Rats | Higher NF-κB P65 mRNA expression, increased protein levels of NF-κB p-p65 | Higher mRNA levels of TLR4, Poly (ADP-ribose) polymerase-1 (PARP-1), TNF-α, inducible nitric oxide synthase (iNOS), MMP-2, and MMP-9, increased protein expression of TLR4 and PARP-1 | Stratification in the cerebral artery wall, decreased SMCs, inward depressing exited in the vascular wall, infiltration, and accumulation of macrophages, neutrophils, and T cells | [23] |
| 2 | Rabbits | Increased NF-κB-p65 staining intensity | MMP-2 and MMP-9 in SMCs, increased staining intensity of MMP-2, MMP-9, and MCP-1, lower smooth muscle actin (SMA) and calponin | IEL loss, media thinning, and bulge formation within one week, larger zones of media thinning and bulging 6 months later | [24] |
| 3 | Rats | Higher mRNA and protein expression of NF-κB P65 and lower mRNA and protein expression of IκBα, increased NF-κB p65 phosphorylation | Higher mRNA protein expression of MMP-2, MMP-9, TNF-α, IL-1β, Il-6, and lower mRNA and protein expression of APC | Damaged endothelium, degenerated VSMCs, lower number of VSMCs and its layers, thinner artery wall, fractured elastic fiber, and inflammatory cell infiltration | [11] |
| 4 | Rats | Higher mRNA expression of NF-κB in IA walls | Higher mRNA expression of MMP-2, MMP-9, MCP-1, and VCAM-1 in IA walls | Increased macrophage infiltration in IA walls | [25] |
| 5 | Rats | Higher levels of phosphorylated NF-κB P65 and IκBα, lower levels of IκBα protein | Decreased tumor necrosis alpha-induced protein 3 (A20) protein expression | Disrupted IEL | [26] |
| 6 | Mice | mRNA expression of NF-κB increased in unruptured IAs and even more in ruptured IAs | Myocardin, smooth muscle alpha-actin (SM-α-actin), smooth muscle myosin heavy chain (SM-MHC), and SM-22α mRNA levels decreased, while MCP-1, MMP-3, MMP-9, TNF-α, IL-1β, iNOS, VCAM, and Krüppel-like factor 4 (KLF4) increased in unruptured IAs and even more in ruptured IAs | Layers of discontinuous endothelial cells and scattered VSMCs, disorganized elastic lamina, macrophage infiltration, and NADPH oxidase-1 (NOX1) immunoreactivity was significantly higher in unruptured IAs, and highest in ruptured IAs, colocalizing with both SMCs and macrophages | [14] |
| 7 | Rabbits | Increased the protein expression of phosphorylated inhibitory-κB kinase alpha (p-IKKα) and t-IKKα and positive expression rate of NF-κB P65 | Decreased eNOS mRNA expression and increased iNOS mRNA expression. Staining intensity and mRNA expression of MMP-2 and MMP-9 increased. The expression of Th17-related factors RORYT, IL-17, IL-22, IL-23, and RORC were increased, and the expression of Treg-related factors IL-10, TGF-β, and Foxp3 was decreased, increased protein expression of t-PI3K, p-PI3K, t-AKT, p-AKT. | Increased length of IEL loss and media thinning, reduced SMCs, broken elastic fibers, staining intensity, and mRNA expression of α-SMA was decreased. The number of Th17 cells was increased and the number of Treg cells was decreased in IA walls. | [20] |
| 8 | Rabbits | The mRNA and protein expression of NF-κB peaked one week after IA induction. | The mRNA and protein expression of MCP-1 peaked one week after IA induction. MMP-9 protein expression increased gradually. | Fractured elastic fiber, lower number of SMCs, damaged endothelial cells | [27] |
| 9 | Rats | NF-κB p65 expression colocalized with MCP-1 | MCP-1 expressed in intima, media, and adventitia, localized to IA walls, increase in MCP-1 protein expression with IA progression | Macrophage accumulation in IA walls increased with IA progression. | [28] |
| 10 | Rats | Increased mRNA expression of NF-κB | Increased mRNA expression of MMP-2, MMP-9, VCAM-1, MCP-1 and decreased mRNA expression of eNOS and (issue inhibitor matrix metalloproteinase 1 (TIMP-1) | Increased macrophage infiltration and increased SMC apoptosis, decreased mRNA expression of B-cell lymphoma 2 (Bcl-2), and increased mRNA expression of iNOS | [29] |
| 11 | Rats | Increased staining intensity for NF-κB P65, NF-κB was activated in both endothelial cells and macrophages | MCP-1 and VCAM-1 costained with NF-κB P65 | [8] | |
| 12 | Mice | Increased protein expression of janus kinase 2 (JAK2), signal transducer and activator of transcription 3 (STAT3), and NF-κB P65, increased phosphorylation of STAT3 and NF-κB P65 | Increased relative mRNA expression and release of TNF-α, IL-1β, IL-6, MCP-1, and IFN-γ and reduced IL-10 Reduced mRNA expression of MHC, SMA, and SM22 and increased mRNA expression of MMP-2 and MMP-9 | [30] | |
| 13 | Rats | increased mRNA expression of NF-κB | Increased concentration of IL-1β, IL-2, IL-6, IL-8, IL-17, and TNF-α, and increased MMP-2 and MMP-9 levels in IA walls, increased IFN-γ and SM22, increased NAD(P)H quinone dehydrogenase 1 (NQO-1) levels Decreased cytoplasmic nuclear factor erythroid-2-related factor (Nrf)-2 and increased nuclear Nrf-2 | Increased macrophage infiltration and increased reactive oxygen species (ROS) | [31] |
| 14 | Rats | Increased DNA binding activities of NF-κB | Increased DNA binding activities of protein C-ets-1 (Ets-1) | Disrupted IEL and media thinning | [32] |
| 15 | Mice | Increased mRNA and protein levels of NF-κB | Increased mRNA and protein expression of MMP-2 and MMP-9 | Decreased thickness of the arterial wall Increased macrophage infiltration | [33] |
| 16 | Rats | Increased protein expression and phosphorylation of NF-κB P65 | Expression of TLR10 mRNA gradually increased with cerebral aneurysm progression. mRNA, protein expression, and staining intensity of TLR-4 increased in IA walls after one month and decreased after three months. Expression of TLR4 coincided well with NF-κB P65 activation. | [34] |
| No. | Animals | Treatments | Genetic Manipulation | NF-κB Status and Other Pathways | Inflammatory Markers and MMPs | IA Features | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Rats | 3-amino benzamide (3-AB) | Reduced NF-κB P65 mRNA expression and decreased protein levels of NF-κB p-p65 | Suppressed mRNA expression of TLR-4, PARP-1, TNF-α, iNOS, MMP-9, and MMP-2 and reduced protein expression of TLR-4, PARP-1 | IA formation 66.7% in control vs. 54.8% in 3-AB, decreased cerebral artery wall damage, weaker inward depressing, reduced accumulation of macrophages, neutrophils, and T cells | [23] | |
| 2 | Rat | APC-siRNA | Enhanced mRNA and protein expression of NF-κB P65 and inhibited mRNA and protein expression of IκBα, increased NF-κB p65 phosphorylation | Enhanced mRNA and protein expression of MMP-2, MMP-9, TNF-α, IL-1β, Il-6, and inhibited mRNA and protein expression of APC | Endothelium disappeared, worsened VSMC degeneration, lowered the number of VSMCs and its layers, with the thinnest artery wall, severely fractured elastic fiber, and increased inflammatory cell infiltration | [11] | |
| 3 | Rats | Aspirin | Lowered mRNA expression of NF-κB in IA walls | Lowered the mRNA expression of MMP-2, MMP-9, MCP-1, and VCAM-1 in IA walls | Smaller aneurysm size with reduced macrophage infiltration, IEL score, and media thinning | [25] | |
| 4 | Rats | ZnSO4 | Reduced phosphorylation of NF-κB p65 and IκBα in IA walls | Increased A20 expression in IA walls | Prevented IA growth, smaller IA size, increased wall thickness ratio, suppressed macrophage infiltration | [26] | |
| 5a | Mice | Apocynin | Decreased mRNA expression of NF-κB | Decreased mRNA expression of MCP-1, MMP-3, MMP-9, TNF-α, IL-1β, iNOS, VCAM, and KLF4 Increased myocardin, SM-α-actin, SM-MHC, and SM-22α mRNA levels | IA formation 84% in control vs. 32% in apocynin, aneurysm rupture 60% in control vs. 12% in apocynin | [14] | |
| 5b | Mice | p47phox−/− | Decreased mRNA expression of NF-κB | Decreased mRNA expression of MCP-1, MMP-3, MMP-9, TNF-α, IL-1β, iNOS, VCAM, and KLF4 Increased myocardin, SM-α-actin, SM-MHC, and SM-22α mRNA levels | IA formation 84% in control vs. 16.7% in p47phox−/−, IA rupture 60% in control vs. 8.3% in p47phox−/− | [14] | |
| 6 | Rat | Tranilast | Inhibited the protein expression of NF-κB p65 in IA walls | Reduced mRNA expression and inhibited protein expression of IL-1 beta, MCP-1, MMP-2, and MMP-9 in IA walls | Suppressed the size of the induced IAs and thinning of the media, prevented the disruption of the IEL and the degeneration of the media, lowered the number of infiltrated macrophages | [35] | |
| 7 | Mice | N/A | Tumor necrosis factor receptor superfamily (TNFR)−/− | Suppressed NF-κB activation in IA lesions | Suppressed mRNA and protein expression of MCP-1 and cyclooxygenase-2 (COX-2) in IA lesions | IA formation 68% in control vs. 12% in TNFR−/−. Inhibited macrophage infiltration in IA lesions | [36] |
| 8 | Rats | Anagliptin | Reduced NF-κB P65 phosphorylation in macrophages in IA walls | Reduced MCP-1 protein expression in IA walls and lowered MCP-1 mRNA expression in cerebral arteries | Suppressed IA growth, decreased size of IEL disruption and IAs. Increased wall thickness ratio, decreased lumen area of aneurysm, less macrophage infiltration into the IA walls | [37] | |
| 9 | Rabbits | Bone marrow mesenchymal stem cells (BMSCs) exosomes | Reduced protein expression of p-IKK-a, t-IKK-a and decreased positive expression rate of NF-κB P65 | Increased eNOS mRNA expression and decreased iNOS mRNA expression Reduced mRNA expression and staining intensity of MMP-2 and MMP-9 Decreased the expression of Th17-related factors RORYT, IL-17, IL-22, IL-23, and RORC Increased the expression of Treg-related factors IL-10, TGF-β, and Foxp3, reduced protein expression of t-PI3K, p-PI3K, t-AKT, p-AKT | Reduced length of IEL loss and media thinning, staining intensity, and mRNA expression of α-SMA were increased Decreased T17 cells and increased T-regs in IA walls | [20] | |
| 10 | Mice | N/A | MCP-1−/− | Did not affect the number of cells expressing NF-κB P65 in IA walls | Reduced staining intensity and mRNA expression of MMP-2, MMP-9, and iNOS in IA walls | Reduced aneurysmal changes, decreased IEL disruption, reduced macrophage infiltration | [28] |
| 11 | Rats | Endothelial colony-forming cells (ECFCs) transfusion | Decreased mRNA expression of NF-κB | Decreased mRNA expression of MMP-2, MMP-9, VCAM-1, and MCP-1, increased mRNA expression of eNOS, TIMP-1, and Bcl-2, and decreased mRNA expression of iNOS | Decreased aneurysm size and SMC apoptosis Increased media thickness and inhibited macrophage infiltration | [29] | |
| 12a | Mice | N/A | p50−/− | Inhibited elevation in MCP-1, VCAM-1, MMP-2, MMP-9, IL-1β, and iNOS mRNA expression | Aneurysm formation 70% in p50+/+ vs. 10% in p50−/−. Reduced IEL disruption, smaller aneurysm size, reduced macrophage accumulation | [8] | |
| 12b | Rat | NF-κB decoy ODN | Lower mRNA expression of MCP-1, VCAM-1 MMP-2, MMP-9, IL-1, and iNOS. Reduced staining intensity for MCP-1 and VCAM-1 | Aneurysmal changes 100% in control vs. 40% in NF-κB decoy ODN, Reduced IEL disruption, smaller aneurysm size, reduced macrophage infiltration | [8] | ||
| 13 | Rats | Nifedipine | Decreased staining intensity for NF-κB P65 in CA walls. Reduced DNA binding of NF-κB P65 in IA walls | Reduced staining intensity and mRNA expression of MCP-1 and MMP2 | Reduced aneurysm size No effect on IEL Increased thickness of media Decreased macrophage infiltration | [38] | |
| 14a | Mice | Prostaglandin E receptor 2 (Ptger2)−/− | Suppressed NF-κB activation and staining intensity for phosphorylated NF-κB P65 | Suppressed COX-2 expression in CA walls Suppressed protein expression of iNOS and MMP2 Reduced staining intensity for MCP-1, IL-1beta, iNOS, and MMP2 | IA formation is almost absent, reduced IEL disruption, reduced macrophage infiltration in IA walls | [39] | |
| 14b | Rats | celecoxib | Suppressed NF-κB activation | Suppressed EP2 expression in CA walls. Reduced MMP-2, MCP-1 and IL-1β expression | Reduced IA formation, decreased IA size, reduced macrophage infiltration in IA walls | [39] | |
| 15 | Mice | BP-1-102 | Reduced the protein expression of JAK2, NF-κB P65, and STAT3 Decreased the phosphorylation of NF-κB P65 and STAT3 | Increased mRNA expression of SMCs markers MHC, SMA, and SM22 Reduced mRNA expression of MMP-2 and MMP-9 and reduced the mRNA expression and release of TNF-α, IL-1β, IL-6, MCP-1, and IFN-γ Increased the mRNA expression and release of IL-10 | IA rupture 81% in control vs. 37% in BP-1-102 | [30] | |
| 16a | Mice | Ptger2f/ fLyz2Cre | Suppressed NF-κB activation | Less IA formation and suppressed macrophage recruitment in IA walls | [40] | ||
| 16b | Mice | Ptger2f/ fCdh5Cre | Did not suppress NF-κB activation | Did not affect IA formation and macrophage infiltration | [40] | ||
| 16c | Mice | IkB mutant– Lyz2Cre | Reduced staining intensity for NF-κB p-P65 in IA walls | The mRNA expression of MCP-1 and Ptger2 was abolished. | Less IA formation and reduced macrophage infiltration | [40] | |
| 16d | Mice | IkB mutant–Cdh5Cre | Did not affect mRNA expression Ptger2 | Did not affect IA formation and macrophage infiltration | [40] | ||
| 16e | Mice | F-04418948 | Reduced phosphorylation of NF-κB P65 | Reduced staining intensity for COX-2 and MCP-1 | Decreased the size of IAs, reduced thinning of media, suppressed macrophage infiltration | [40] | |
| 17 | Rats | PCA | N/A | Reduced mRNA expression and activation of NF-κB | Reduced concentration of IL-1β, IL-2, IL-6, IL-8, IL-17, and TNF-α and reduced MMP-2 and MMP-9 levels in CA walls Decreased IFN-γ and SM22 and increased NQO-1 levels Decreased cytoplasmic Nrf-2 and increased nuclear Nrf-2 | Reduced aneurysm size, decreased tunica-media thickness, reduced macrophage infiltration, increased ROS | [31] |
| 18 | Rats | NF-κB Decoy ODN | N/A | Higher mRNA expression of procollagen 1(I), 1(III), and lysyl oxidase (LOX) in IAs | [41] | ||
| 19a | Rats | NF-κB decoy ODN | Inhibited the up-regulated mRNA expression and staining intensity of MCP-1 mRNA in CA walls Partially restored the decreased mRNA expression of procollagen α1 and increased staining intensity for procollagen α1 | Suppressed IEL disruption and prevented media thinning Did not restore medial thickness, suppressed IA enlargement, did not reduce the size of preexisting IAs, inhibited macrophage infiltration | [32] | ||
| 19b | Rats | Chimeric decoy ODN | Inhibited DNA binding activities of both NF-κB and Ets-1 | Inhibited the up-regulated expression of MCP-1 mRNA and suppressed staining intensity for MCP-1 in IA walls Restored the mRNA expression of procollagen α1 (type I and type III), increased staining intensity for procollagen α1 (type I and type III) | Suppressed IEL disruption, prevented media thinning, increased medial thickness, suppressed the IA enlargement, diminished the size of preexisting IAs, inhibited macrophage infiltration | [32] | |
| 20 | Mice | Resveratrol | N/A | Reduced mRNA and protein level of NF-κB | Reduced mRNA and protein levels of MMP-2 and MMP-9. | IA formation 66.7% in control vs. 16.6% in resveratrol Reduced IA size and thicker arterial wall Reduced macrophage infiltration | [33] |
| 21 | Rats | Tan IIA | N/A | Reduced NF-κB mRNA expression and activation in IAs | Reduced mRNA expression of MCP-1, MMP-2, and MMP-9 | Suppressed IA growth, decreased IA size, increased arterial wall thickness, less macrophage infiltration | [42] |
| 22 | Rats | N/A | shTWIST1 | Decreased phosphorylation of NF-κB P65 and IκB Increased IκB protein expression | Increased expression of Bcl-2 and decreased expression of Bax and cleaved caspase-3 Reduced serum levels of TNF-α and IL-6 | Ameliorated vessel tissue degeneration in IA walls, suppressed VSMC apoptosis | [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, D.; Cornelius, J.F.; Muhammad, S. The Role of NF-κB in Intracranial Aneurysm Pathogenesis: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 14218. https://doi.org/10.3390/ijms241814218
Khan D, Cornelius JF, Muhammad S. The Role of NF-κB in Intracranial Aneurysm Pathogenesis: A Systematic Review. International Journal of Molecular Sciences. 2023; 24(18):14218. https://doi.org/10.3390/ijms241814218
Chicago/Turabian StyleKhan, Dilaware, Jan Frederick Cornelius, and Sajjad Muhammad. 2023. "The Role of NF-κB in Intracranial Aneurysm Pathogenesis: A Systematic Review" International Journal of Molecular Sciences 24, no. 18: 14218. https://doi.org/10.3390/ijms241814218
APA StyleKhan, D., Cornelius, J. F., & Muhammad, S. (2023). The Role of NF-κB in Intracranial Aneurysm Pathogenesis: A Systematic Review. International Journal of Molecular Sciences, 24(18), 14218. https://doi.org/10.3390/ijms241814218