
The Phosphoproteome of the Rd1 Mouse Retina, a Model of Inherited Photoreceptor Degeneration, Changes after Protein Kinase G Inhibition


Abstract
1. Introduction
2. Results
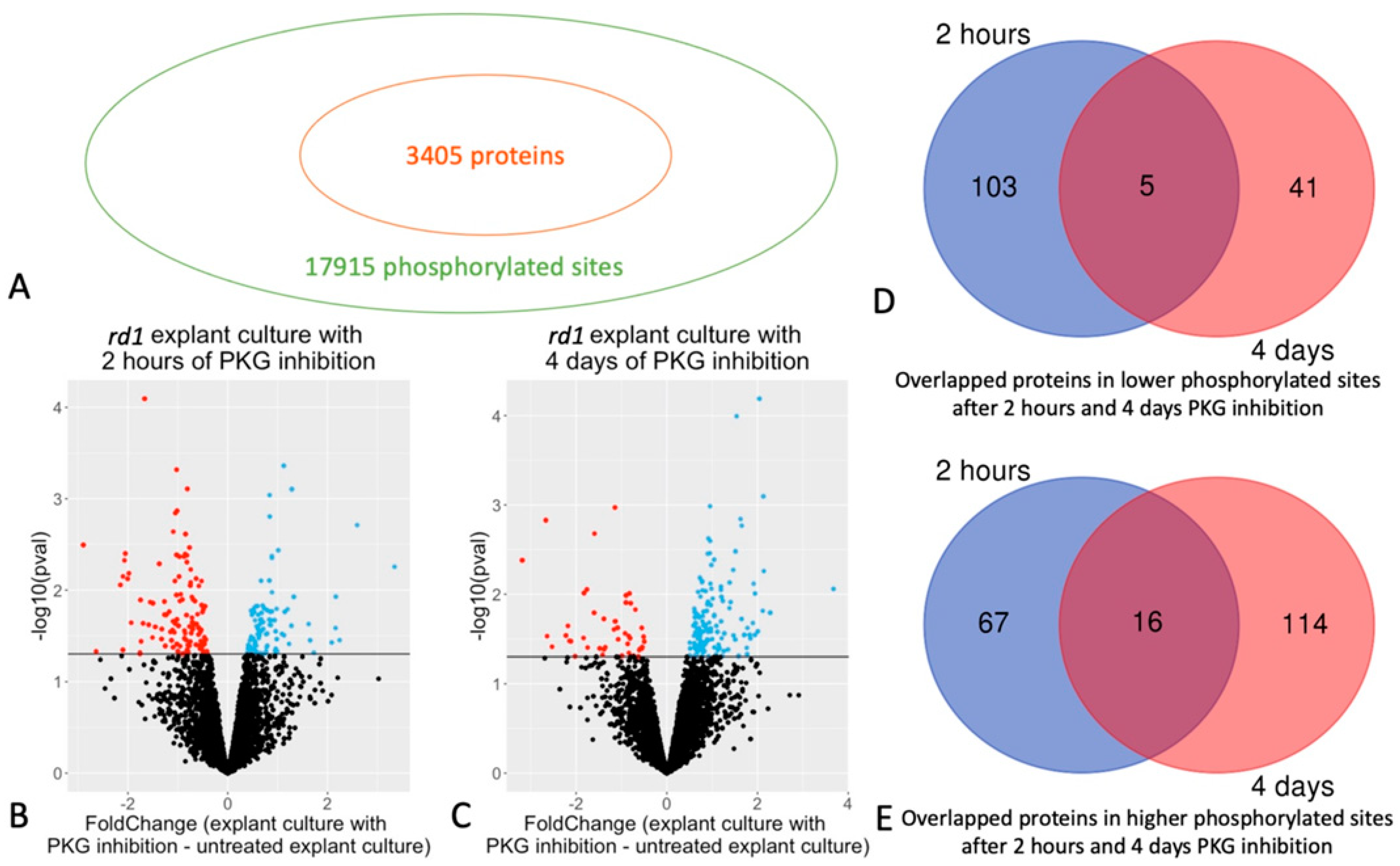
2.1. Quantitative Phosphoproteomics of Retinal Explants with PKG Inhibition
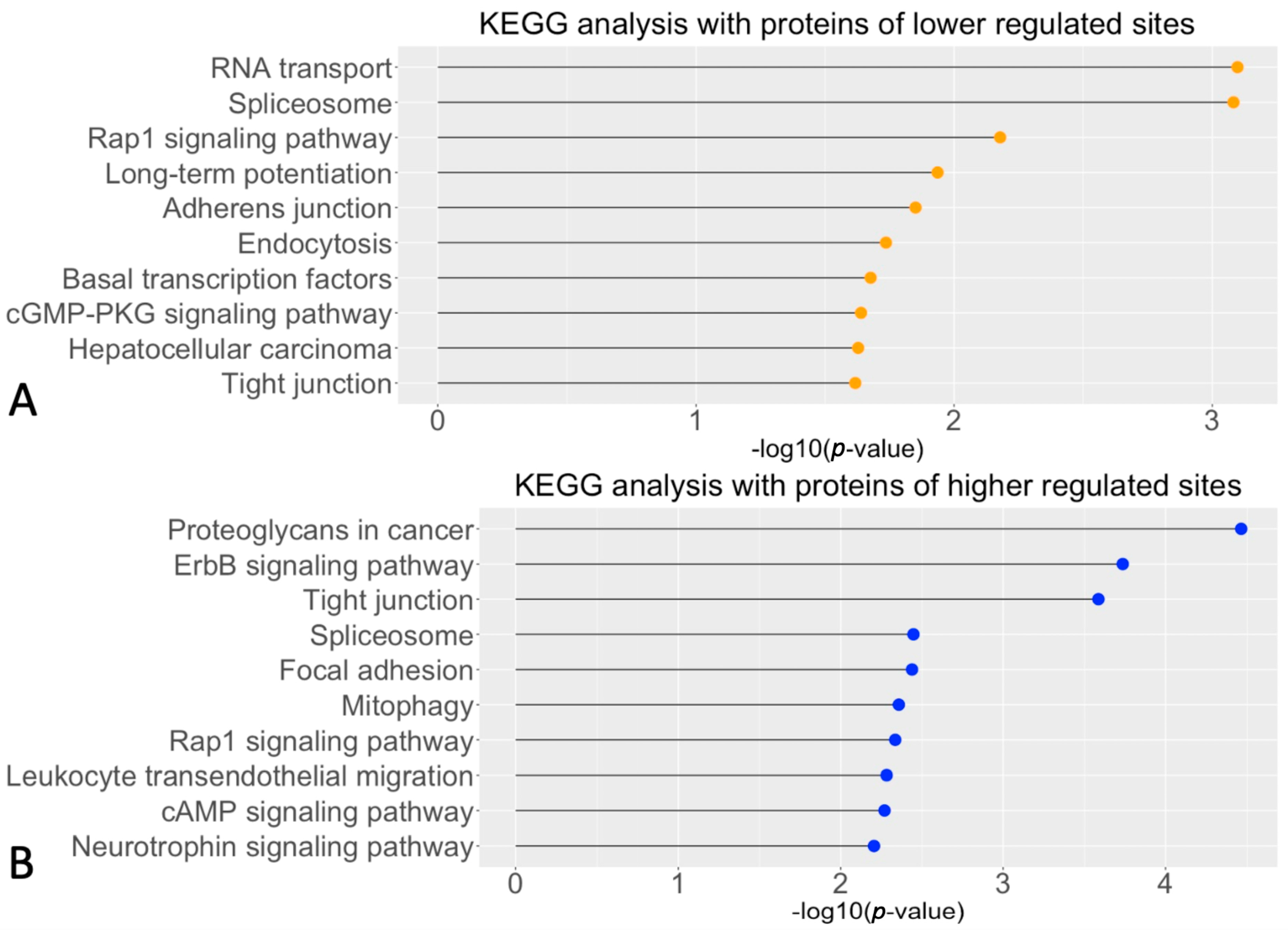
2.2. Potential cGMP-PKG Dependent Biological Pathways
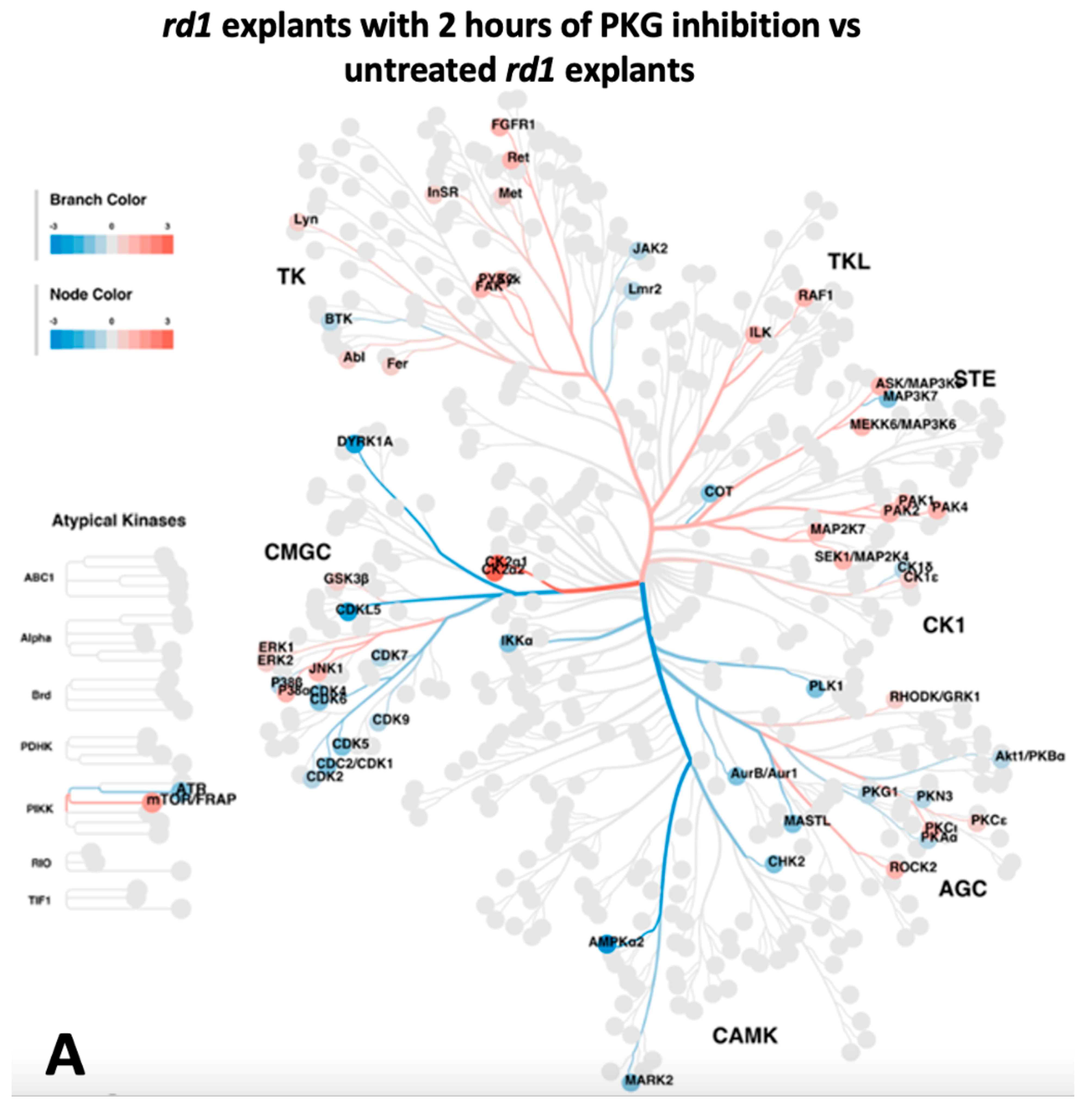
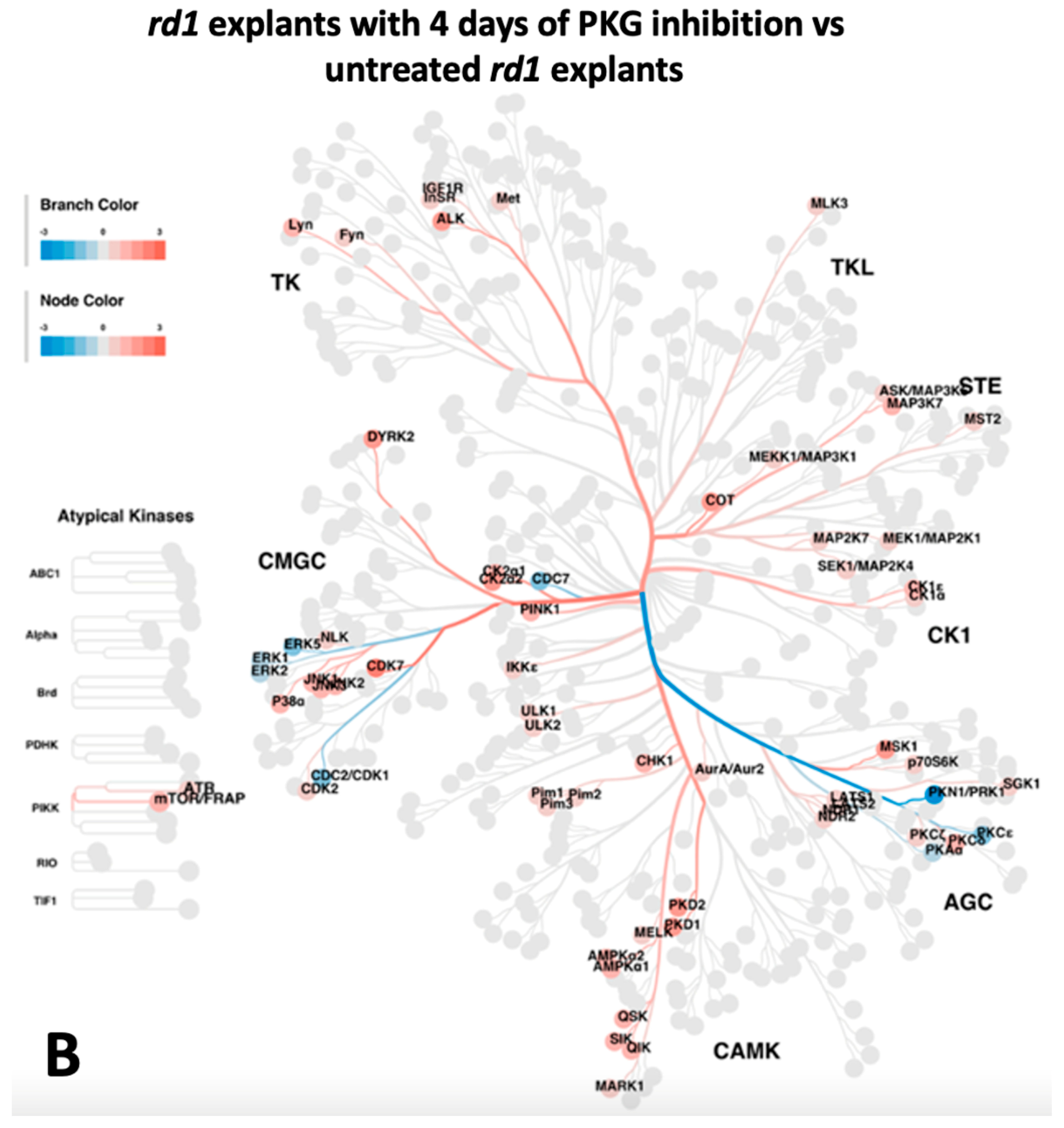
2.3. Upstream Kinase Analysis
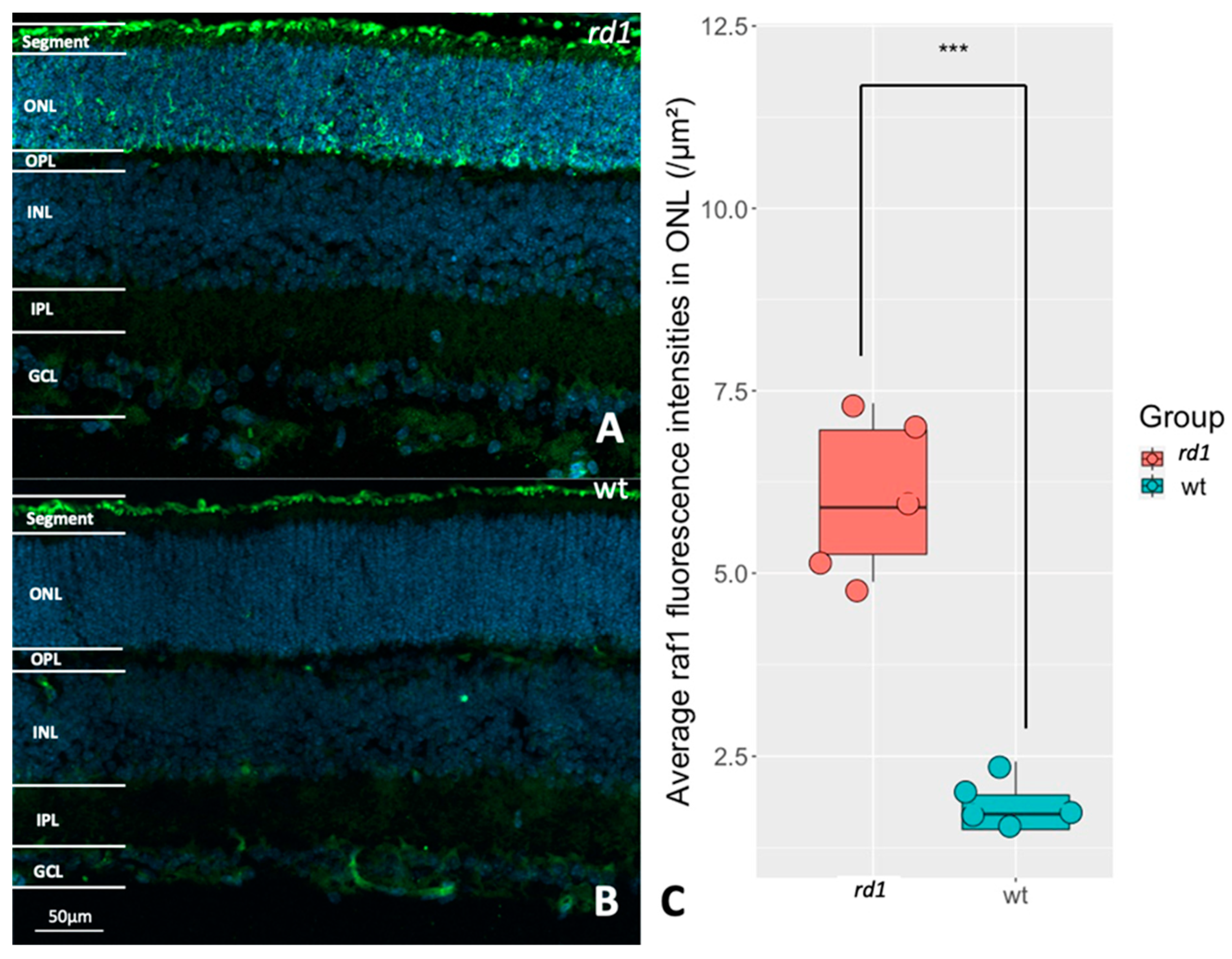
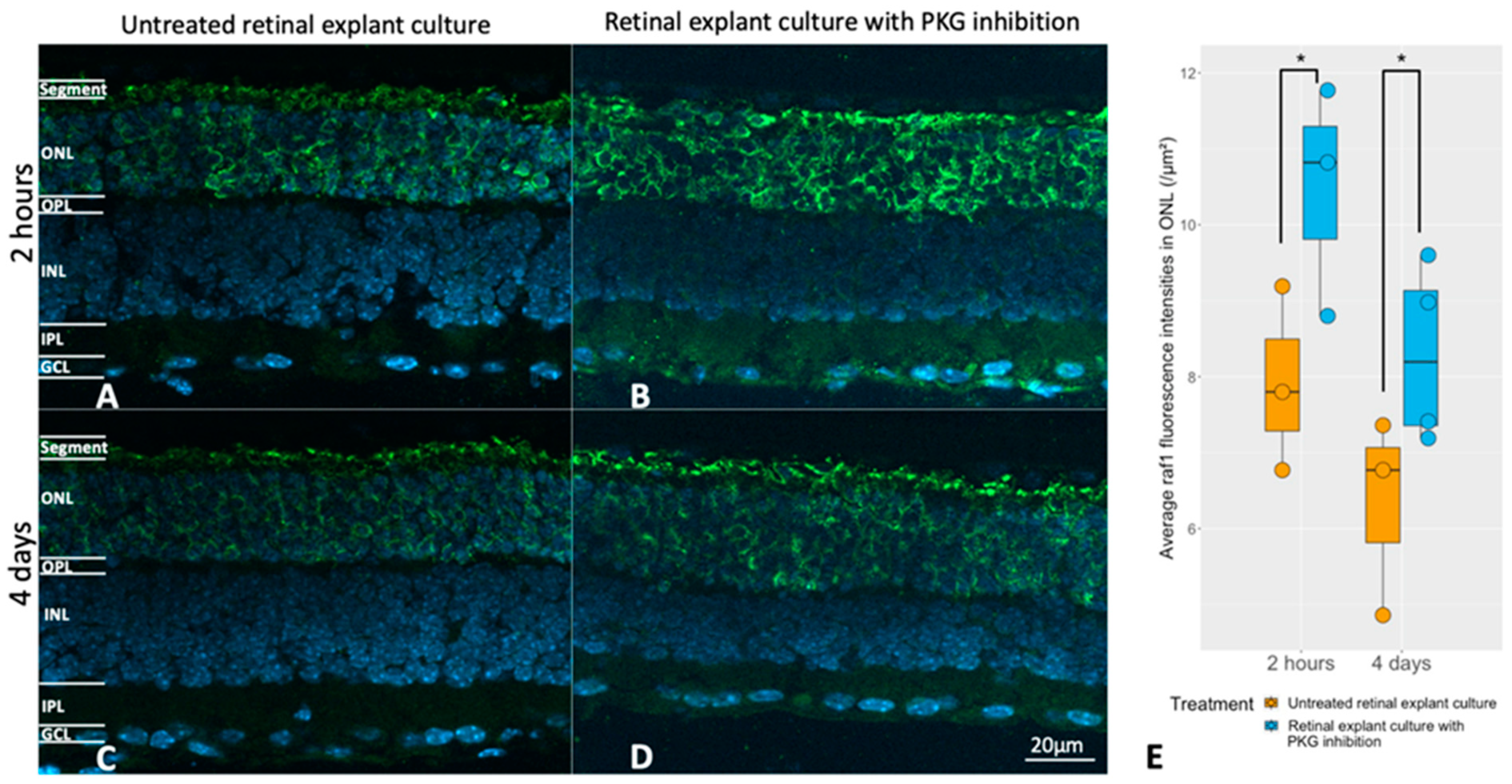
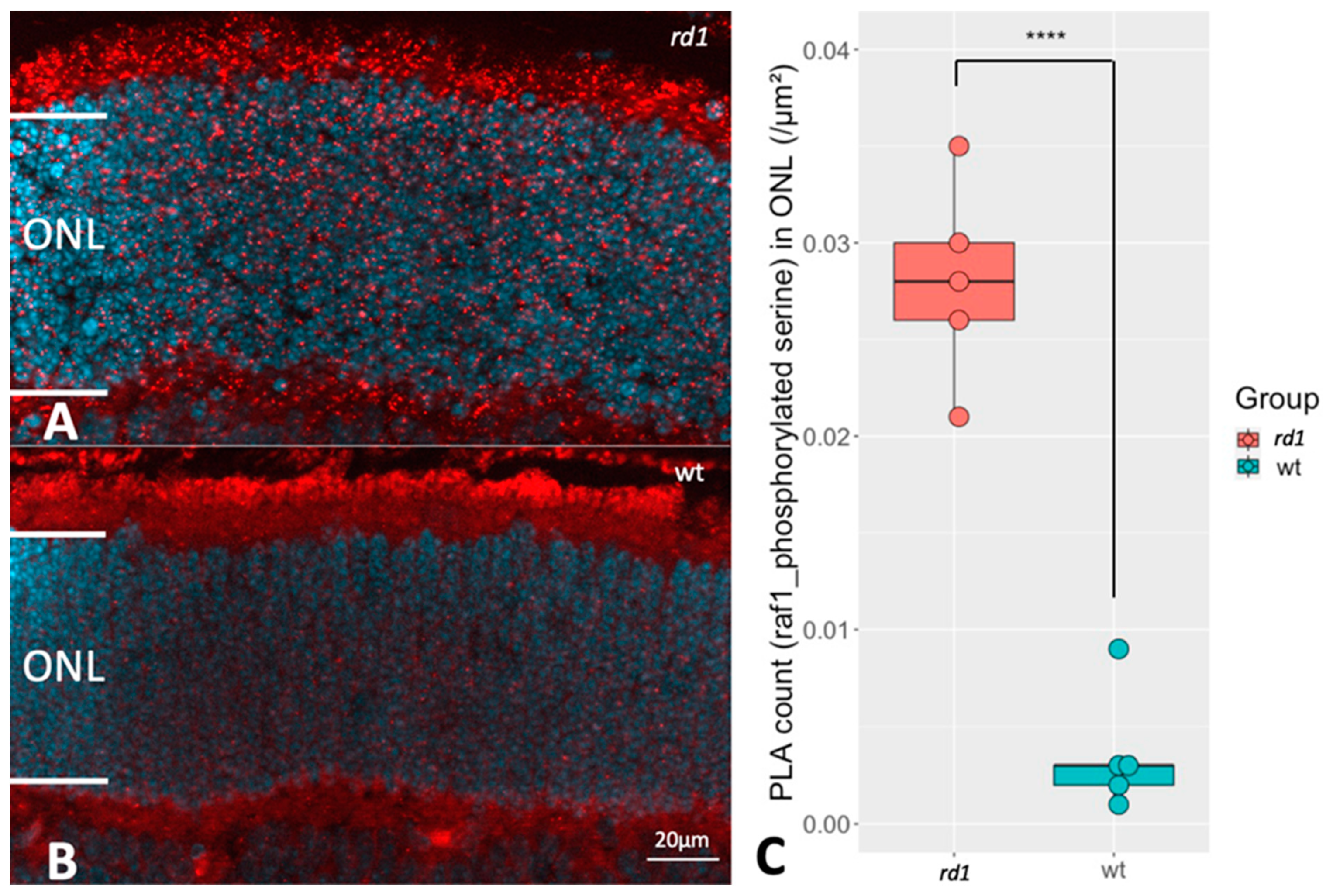
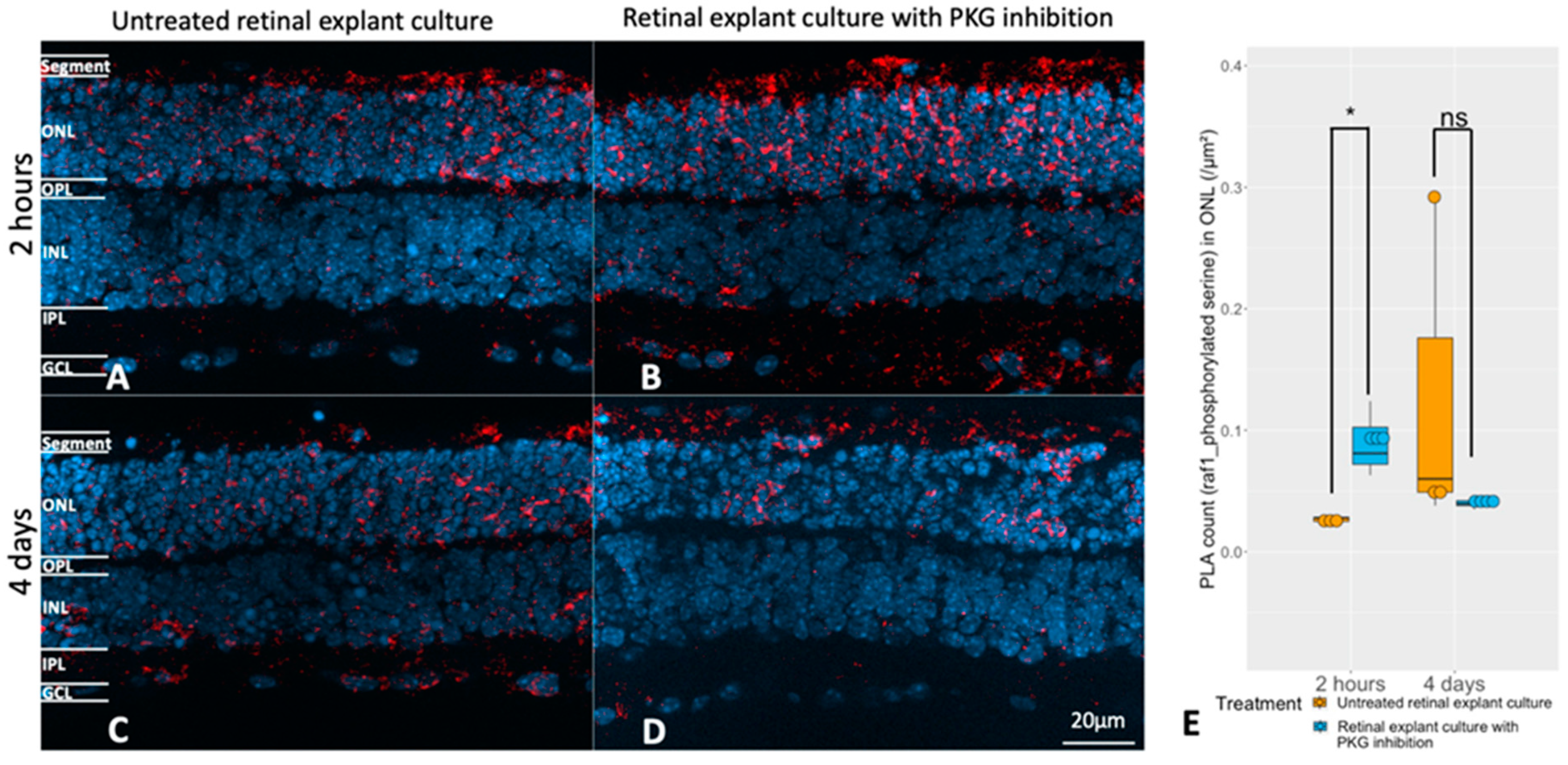
2.4. Selection of RAF-1 for Further Validation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Organotypic Retinal Explant Culture
4.3. Sample Preparation for MS
4.4. MS Acquisition and Analysis
4.5. Cryosection, Immunohistochemistry, and Proximity Ligation Assay (PLA)
4.6. Microscopy and Image Processing
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Silva, S.R.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Mahroo, O.A. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 2021, 82, 100898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, G. Recent Advances in Treatment of Retinitis Pigmentosa. Curr. Stem Cell Res. Ther. 2015, 10, 258–265. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Witters, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Mir, T.A. The mechanism of cone cell death in retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 62, 24–37. [Google Scholar] [CrossRef]
- Farber, D.B.; Lolley, R.N. Cyclic Guanosine Monophosphate: Elevation in Degenerating Photoreceptor Cells of the C3H Mouse Retina. Science 1974, 186, 449–451. [Google Scholar] [CrossRef]
- Pennesi, M.E.; Michaels, K.V.; Magee, S.S.; Maricle, A.; Davin, S.P.; Garg, A.K.; Gale, M.J.; Tu, D.C.; Wen, Y.; Erker, L.R.; et al. Long-Term Characterization of Retinal Degeneration in rd1andrd10Mice Using Spectral Domain Optical Coherence Tomography. Investig. Opthalmol. Vis. Sci. 2012, 53, 4644–4656. [Google Scholar] [CrossRef]
- Strettoi, E.; Pignatelli, V.; Rossi, C.; Porciatti, V.; Falsini, B. Remodeling of second-order neurons in the retina of rd/rd mutant mice. Vis. Res. 2003, 43, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Durand, F.; Hauck, S.; Van Veen, T.; Ueffing, M.; Ekström, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef]
- Vighi, E.; Trifunovic, D.; Veiga-Crespo, P.; Rentsch, A.; Hoffmann, D.; Sahaboglu, A.; Strasser, T.; Kulkarni, M.; Bertolotti, E.; van den Heuvel, A.; et al. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2997–E3006. [Google Scholar] [CrossRef]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-Dependent Protein Kinases as Revealed by Gene Deletion. Physiol. Rev. 2006, 86, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Sellak, H.; Brown, F.; Lincoln, T. cGMP-dependent protein kinase and the regulation of vascular smooth muscle cell gene expression: Possible involvement of Elk-1 sumoylation. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1660–H1670. [Google Scholar] [CrossRef]
- Pilz, R.B.; Casteel, D.E. Regulation of Gene Expression by Cyclic GMP. Circ. Res. 2003, 93, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef]
- Butt, E.; Pöhler, D.; Genieser, H.-G.; Huggins, J.P.; Bucher, B. Inhibition of cyclic GMP-dependent protein kinase-mediated effects by (Rp)-8-bromo-PET-cyclic GMPS. Br. J. Pharmacol. 1995, 116, 3110–3116. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Draznin, M.B.; Murad, F. Sodium nitroprusside-induced protein phosphorylation in intact rat aorta is mimicked by 8-bromo cyclic GMP. Proc. Natl. Acad. Sci. USA 1982, 79, 6470–6474. [Google Scholar] [CrossRef] [PubMed]
- Schwede, F.; Maronde, E.; Genieser, H.-G.; Jastorff, B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther. 2000, 87, 199–226. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rasmussen, M.; Ekström, P. cGMP-PKG dependent transcriptome in normal and degenerating retinas: Novel insights into the retinitis pigmentosa pathology. Exp. Eye Res. 2021, 212, 108752. [Google Scholar] [CrossRef]
- Mann, M.; Ong, S.-E.; Grønborg, M.; Steen, H.; Jensen, O.N.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002, 20, 261–268. [Google Scholar] [CrossRef]
- Power, M.; Das, S.; Schütze, K.; Marigo, V.; Ekström, P.; Paquet-Durand, F. Cellular mechanisms of hereditary photoreceptor degeneration—Focus on cGMP. Prog. Retin. Eye Res. 2020, 74, 100772. [Google Scholar] [CrossRef]
- Roignant, J.-Y.; Treisman, J.E. Exon Junction Complex Subunits Are Required to Splice Drosophila MAP Kinase, a Large Heterochromatic Gene. Cell 2010, 143, 238–250. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Meikle, S.; Peyssonnaux, C.; Grindlay, J.; Kaiser, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Eychène, A.; Kolch, W. A Raf-1 Mutant That Dissociates MEK/Extracellular Signal-Regulated Kinase Activation from Malignant Transformation and Differentiation but Not Proliferation. Mol. Cell. Biol. 2003, 23, 1983–1993. [Google Scholar] [CrossRef]
- Weibrecht, I.; Leuchowius, K.-J.; Clausson, C.-M.; Conze, T.; Jarvius, M.; Howell, W.M.; Kamali-Moghaddam, M.; Söderberg, O. Proximity ligation assays: A recent addition to the proteomics toolbox. Expert Rev. Proteom. 2010, 7, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, F.S.; Gallenga, C.; Bonifazzi, C.; Perri, P. A challenge to the striking genotypic heterogeneity of retinitis pigmentosa: A better understanding of the pathophysiology using the newest genetic strategies. Eye 2016, 30, 1542–1548. [Google Scholar] [CrossRef]
- Cehofski, L.J.; Mandal, N.; Honoré, B.; Vorum, H. Analytical platforms in vitreoretinal proteomics. Bioanalysis 2014, 6, 3051–3066. [Google Scholar] [CrossRef] [PubMed]
- Grønborg, M.; Kristiansen, T.Z.; Stensballe, A.; Andersen, J.S.; Ohara, O.; Mann, M.; Jensen, O.N.; Pandey, A. A Mass Spectrometry-based Proteomic Approach for Identification of Serine/Threonine-phosphorylated Proteins by Enrichment with Phospho-specific Antibodies. Mol. Cell. Proteom. 2002, 1, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Fíla, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2011, 43, 1025–1047. [Google Scholar] [CrossRef]
- Kizhatil, K.; Sandhu, N.K.; Peachey, N.S.; Bennett, V. Ankyrin-B is required for coordinated expression of beta-2-spectrin, the Na/K-ATPase and the Na/Ca exchanger in the inner segment of rod photoreceptors. Exp. Eye Res. 2009, 88, 57–64. [Google Scholar] [CrossRef]
- Nag, T.C.; Kathpalia, P.; Wadhwa, S. Microtubule alterations may destabilize photoreceptor integrity: Age-related microtubule changes and pattern of expression of MAP-2, Tau and hyperphosphorylated Tau in aging human photoreceptor cells. Exp. Eye Res. 2020, 198, 108153. [Google Scholar] [CrossRef]
- Grossman, G.H.; Beight, C.D.; Ebke, L.A.; Pauer, G.J.; Hagstrom, S.A. Interaction of Tubby-Like Protein-1 (Tulp1) and Microtubule-Associated Protein (MAP) 1A and MAP1B in the Mouse Retina. Adv Exp Med. Biol. 2014, 801, 511–518. [Google Scholar] [CrossRef]
- Zhou, J.; Ekström, P. A Potential Role of Cyclic Dependent Kinase 1 (CDK1) in Late Stage of Retinal Degeneration. Cells 2022, 11, 2143. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Eychene, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. The role of RAF-1 kinase in diabetic retinopathy. Expert Opin. Ther. Targets 2011, 15, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Meikle, S.; Yazici, Z.A.; Eulitz, M.; Kolch, W. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J. 2002, 21, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S.; Bal, A.K. Comparative light and electron microscopic study of retinal histogenesis in normal and rd mutant mice. Z. Anat. Entwickl. 1973, 142, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2014, 43, D512–D520. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top 20 after 2 h | Top 20 after 4 Days | Appearing in Both | |||||
|---|---|---|---|---|---|---|---|
| Gene Symbol | Protein Name | Fold Change | Gene Symbol | Protein Name | Fold Change | Gene Symbol | Protein Name |
| CDC27 | cell division cycle 27 | −2.894 | EPN1 | epsin 1 | −3.188 | PKN1 | protein kinase N1 |
| AUTS2 | activator of transcription and developmental regulator | −2.638 | PKN1 | protein kinase N1 | −2.668 | CIC | capicua transcriptional repressor |
| DMXL2 | Dmx like 2 | −2.150 | PITPNM1 | phosphatidylinositol transfer protein membrane-associated 1 | −2.640 | TOP2B | DNA topoisomerase II beta |
| PICALM | phosphatidylinositol binding clathrin assembly protein | −2.100 | MAP7D1 | MAP7 domain containing 1 | −2.530 | CCDC88A | coiled-coil domain containing 88A |
| TRAF7 | TNF receptor-associated factor 7 | −2.100 | MTMR2 | myotubularin related protein 2 | −2.225 | PBRM1 | polybromo 1 |
| PARD3B | par-3 family cell polarity regulator beta | −2.069 | GRK1 | G protein-coupled receptor kinase 1 | −2.180 | ||
| AMPH | amphiphysin | −2.051 | PCBP1 | poly(rC) binding protein 1 | −2.144 | ||
| ATG16L1 | autophagy related 16 like 1 | −2.006 | JPT1 | Jupiter microtubule associated homolog 1 | −2.115 | ||
| RNF34 | ring finger protein 34 | −1.980 | EPB41 | Protein 4.1 isoform 2 | −2.023 | ||
| ACIN1 | apoptotic chromatin condensation inducer 1 | −1.934 | GPHN | Gephyrin | −1.836 | ||
| SYN3 | synapsin III | −1.759 | PPP6R2 | protein phosphatase 6 regulatory subunit 2 | −1.826 | ||
| DPY30 | dpy-30 histone methyltransferase complex regulatory subunit | −1.759 | FLNB | filamin B | −1.762 | ||
| ZNF687 | zinc finger protein 687 | −1.747 | RNF20 | ring finger protein 20 | −1.747 | ||
| CTBP2 | C-terminal binding protein 2 | −1.736 | TSC22D1 | TSC22 domain family member 1 | −1.602 | ||
| CCDC88A | coiled-coil domain containing 88A | −1.696 | LIN37 | lin-37 DREAM MuvB core complex component | −1.595 | ||
| SMPD3 | sphingomyelin phosphodiesterase 3 | −1.666 | CIC | capicua transcriptional repressor | −1.480 | ||
| NKTR | natural killer cell triggering receptor | −1.583 | SLC20A2 | solute carrier family 20 member 2 | −1.416 | ||
| NONO | non-POU domain containing octamer binding | −1.554 | PRKG1 | protein kinase cGMP-dependent 1 | −1.387 | ||
| RBM8A | RNA binding motif protein 8A | −1.509 | ARHGAP21 | Rho GTPase activating protein 21 | −1.385 | ||
| SNX2 | sorting nexin 2 | −1.500 | PABIR1 | PP2A Aalpha (PPP2R1A) and B55A (PPP2R2A) interacting phosphatase regulator 1 | −1.356 | ||
| Top 20 after 2 h | Top 20 after 4 Days | Appearing in Both | |||||
|---|---|---|---|---|---|---|---|
| Gene Symbol | Protein Name | Fold Change | Gene Symbol | Protein Name | Fold Change | Gene Symbol | Protein Name |
| HNRNPUL1 | heterogeneous nuclear ribonucleoprotein U like 1 | 3.343 | MAP1B | microtubule-associated protein 1B | 3.674 | HNRNPUL1 | heterogeneous nuclear ribonucleoprotein U like 1 |
| EEF1B2 | eukaryotic translation elongation factor 1 beta 2 | 2.593 | LGALS12 | galectin 12 | 2.279 | PALM2AKAP2 | PALM2 and AKAP2 fusion |
| HNRNPU | heterogeneous nuclear ribonucleoprotein U | 2.242 | HNRNPUL1 | heterogeneous nuclear ribonucleoprotein U like 1 | 2.138 | DLG1 | discs large MAGUK scaffold protein 1 |
| GTPBP1 | GTP binding protein 1 | 2.165 | NR5A1 | nuclear receptor subfamily 5 group A member 1 | 2.128 | SEPTIN4 | septin 4 |
| MAP4 | microtubule associated protein 4 | 2.161 | GTF2F1 | general transcription factor IIF subunit 1 | 2.115 | MAP1B | microtubule-associated protein 1B |
| VPS53 | VPS53 subunit of GARP complex | 2.083 | SRRM2 | serine/arginine repetitive matrix 2 | 2.041 | HNRNPU | heterogeneous nuclear ribonucleoprotein U |
| NES | nestin | 1.725 | RBSN | rabenosyn, RAB effector | 2.006 | MCM7 | minichromosome maintenance complex component 7 |
| LARP1 | La ribonucleoprotein 1, translational regulator | 1.650 | TPD52L2 | TPD52 like 2 | 1.946 | ATF7IP | activating transcription factor 7 interacting protein |
| CDK13 | cyclin-dependent kinase 13 | 1.621 | VSX2 | visual system homeobox 2 | 1.945 | MAP4 | microtubule-associated protein 4 |
| AFDN | afadin, adherens junction formation factor | 1.323 | RBM12 | RNA binding motif protein 12 | 1.915 | ANK2 | ankyrin 2 |
| PXN | paxillin | 1.284 | DZIP3 | DAZ interacting zinc finger protein 3 | 1.897 | SRRM1 | serine and arginine repetitive matrix 1 |
| RCOR1 | REST corepressor 1 | 1.275 | ABLIM1 | actin binding LIM protein 1 | 1.842 | KIAA0930 | KIAA0930 |
| RUNX1T1 | RUNX1 partner transcriptional co-repressor 1 | 1.230 | PITPNM2 | phosphatidylinositol transfer protein membrane-associated 2 | 1.827 | CCDC88A | coiled-coil domain containing 88A |
| HTATSF1 | HIV-1 Tat specific factor 1 | 1.199 | ELF2 | E74 like ETS transcription factor 2 | 1.778 | CTBP2 | C-terminal binding protein 2 |
| ZC3H6 | zinc finger CCCH-type containing 6 | 1.143 | BICRAL | BRD4 interacting chromatin remodeling complex associated protein like | 1.771 | MAP2 | microtubule-associated protein 2 |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase | 1.127 | PALM2AKAP2 | PALM2 and AKAP2 fusion | 1.761 | RERE | arginine-glutamic acid dipeptide repeats |
| BASP1 | brain abundant membrane attached signal protein 1 | 1.122 | DNAJC5 | DnaJ heat shock protein family (Hsp40) member C5 | 1.707 | ||
| PLCL2 | phospholipase C like 2 | 1.086 | PACS2 | phosphofurin acidic cluster sorting protein 2 | 1.705 | ||
| SPHK2 | sphingosine kinase 2 | 1.012 | CCNL1 | cyclin L1 | 1.650 | ||
| SEPTIN4 | septin 4 | 0.987 | EIF4EBP1 | eukaryotic translation initiation factor 4E binding protein 1 | 1.647 | ||
| Proteins with Decreased Phosphorylation after PKG Inhibition for 2 h/Proteins with Decreased Phosphorylation after PKG Inhibition for 4 Days | Proteins with Increased Phosphorylation after PKG Inhibition for 2 h/Proteins with Increased Phosphorylation after PKG Inhibition for 4 Days | Proteins with Decreased Phosphorylation after PKG Inhibition for 2 h/Proteins with Increased Phosphorylation after PKG Inhibition for 4 Days | Proteins with Increased Phosphorylation after PKG Inhibition for 2 h/Proteins with Decreased Phosphorylation after PKG Inhibition for 4 Days |
|---|---|---|---|
| CDK1 | CSNK1E | CDK2 | MAPK3 |
| PRKACA | MAP2K7 | PRKAA2 | MAPK1 |
| MAP2K4 | MAP3K7 | PRKCE | |
| MAP3K5 | CDK7 | ||
| CSNK2A1 | |||
| CSNK2A2 | |||
| CSNK1E | |||
| BCR | |||
| MAPK8 | |||
| MTOR | |||
| MAPK14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Welinder, C.; Ekström, P. The Phosphoproteome of the Rd1 Mouse Retina, a Model of Inherited Photoreceptor Degeneration, Changes after Protein Kinase G Inhibition. Int. J. Mol. Sci. 2023, 24, 9836. https://doi.org/10.3390/ijms24129836
Zhou J, Welinder C, Ekström P. The Phosphoproteome of the Rd1 Mouse Retina, a Model of Inherited Photoreceptor Degeneration, Changes after Protein Kinase G Inhibition. International Journal of Molecular Sciences. 2023; 24(12):9836. https://doi.org/10.3390/ijms24129836
Chicago/Turabian StyleZhou, Jiaming, Charlotte Welinder, and Per Ekström. 2023. "The Phosphoproteome of the Rd1 Mouse Retina, a Model of Inherited Photoreceptor Degeneration, Changes after Protein Kinase G Inhibition" International Journal of Molecular Sciences 24, no. 12: 9836. https://doi.org/10.3390/ijms24129836
APA StyleZhou, J., Welinder, C., & Ekström, P. (2023). The Phosphoproteome of the Rd1 Mouse Retina, a Model of Inherited Photoreceptor Degeneration, Changes after Protein Kinase G Inhibition. International Journal of Molecular Sciences, 24(12), 9836. https://doi.org/10.3390/ijms24129836