
Placental Transcription Profiling in 6–23 Weeks’ Gestation Reveals Differential Transcript Usage in Early Development

 , , , , , , , ,
, , , , , , , ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Changes in the Placental Transcriptome from Early to Mid Gestation Are Highly Enriched for Genes Involved in Cell Migration and Transmembrane Signaling
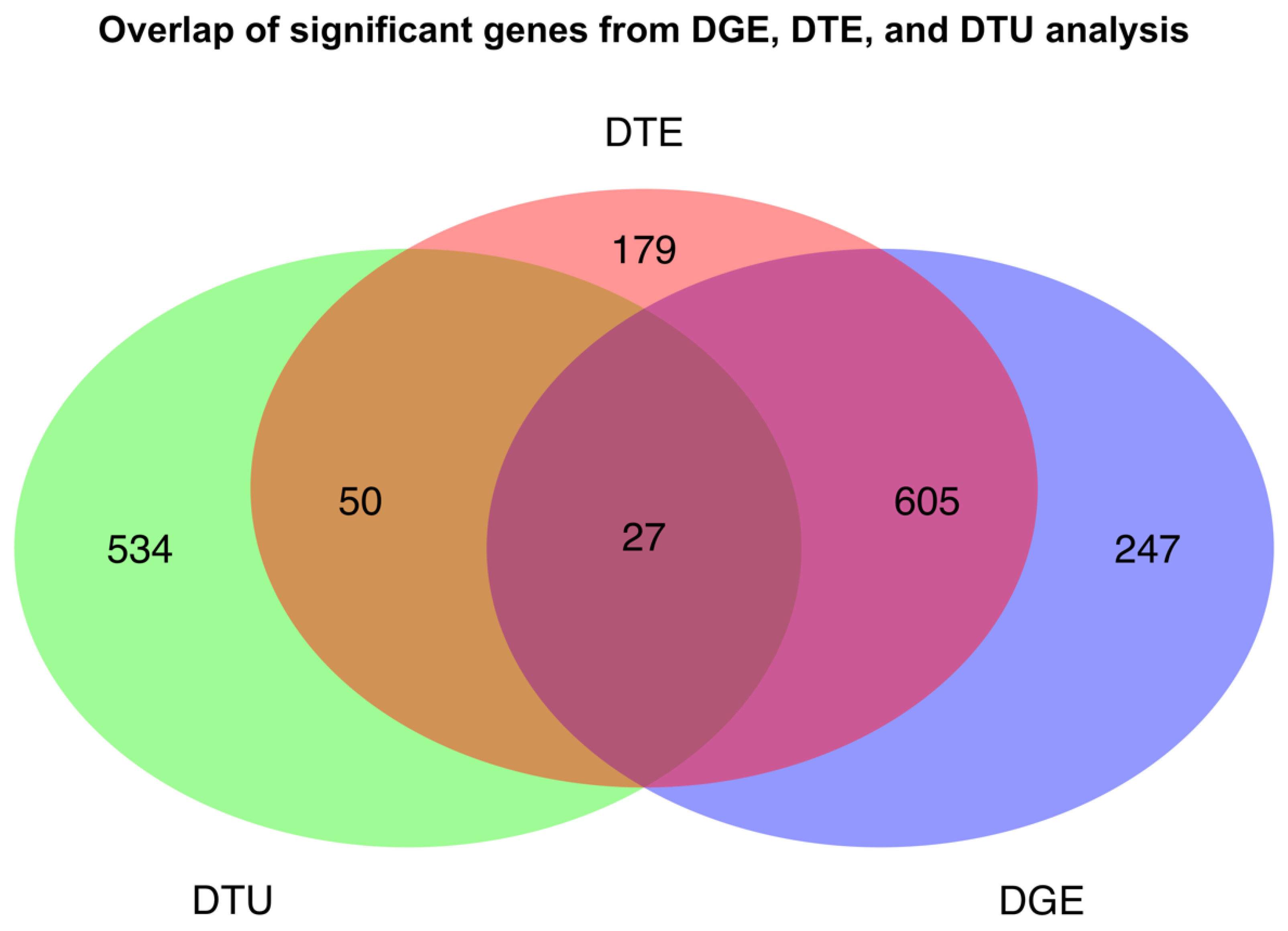
2.2. Variable Transcript Expression Masks Differential Gene Expression
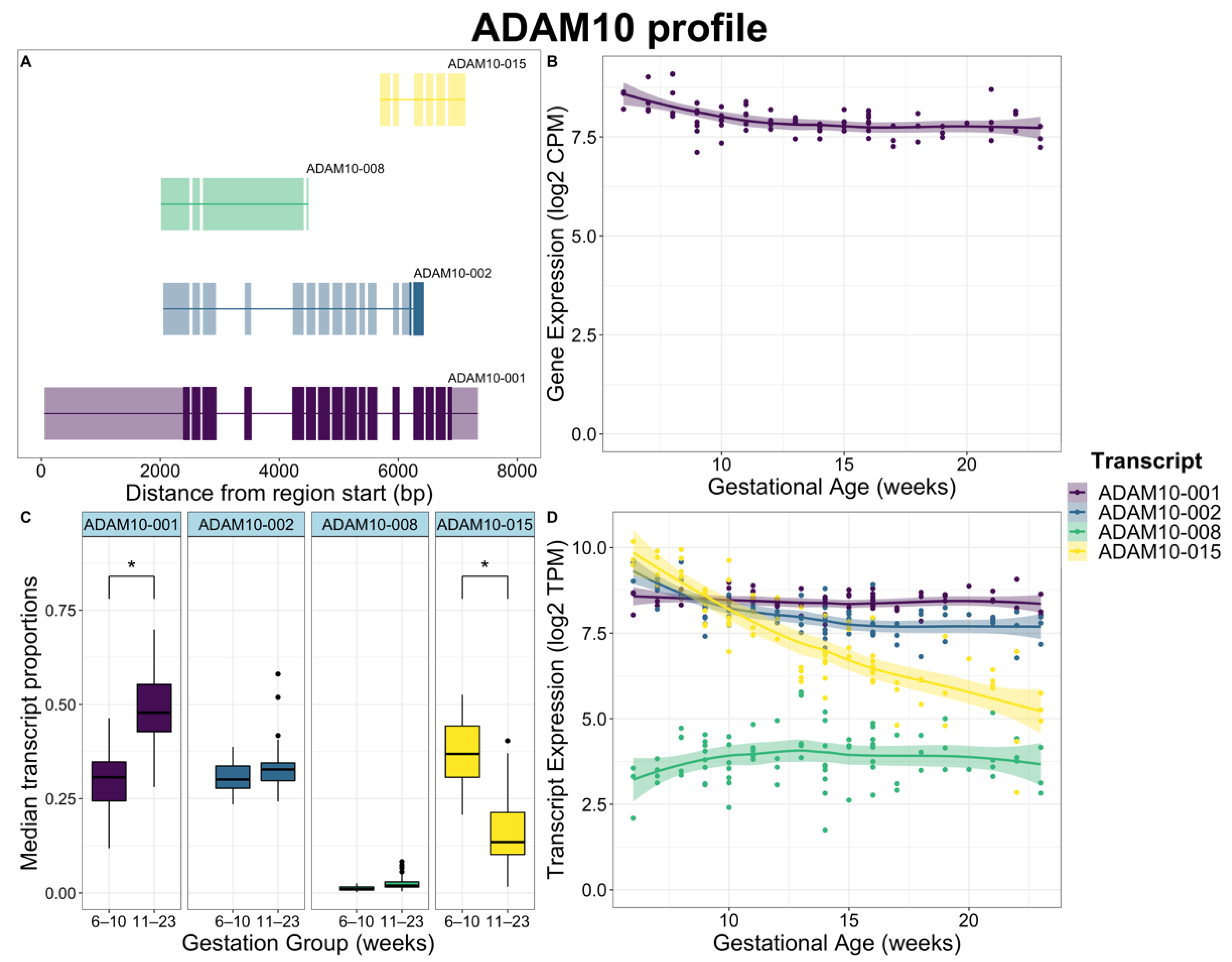
2.3. Global Differences in Transcript Usage Were Observed from Early to Mid Gestation
3. Discussion
4. Materials and Methods
4.1. Data Information and Ethics Statement
4.2. Data Processing
4.3. Differential Expression Analysis
4.4. Differential Transcript Usage
4.5. Gene Ontology Enrichment
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.T. IFPA Award in Placentology Lecture: Complicated interactions between genes and the environment in placentation, pregnancy outcome and long term health. Placenta 2010, 31, S47–S53. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, R.; Jauniaux, E.; Hustin, J. Maternal circulation in the first-trimester human placenta--myth or reality? Am. J. Obstet. Gynecol. 1997, 176, 695–705. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef]
- Burton, G.J.; Cindrova-Davies, T.; Yung, H.W.; Jauniaux, E. Oxygen and development of the human placenta. Reproduction 2020, 161, F53–F65. [Google Scholar] [CrossRef]
- Winn, V.D.; Haimov-Kochman, R.; Paquet, A.C.; Yang, Y.J.; Madhusudhan, M.S.; Gormley, M.; Feng, K.-T.V.; Bernlohr, D.A.; McDonagh, S.; Pereira, L.; et al. Gene expression profiling of the human maternal-fetal interface reveals dramatic changes between midgestation and term. Endocrinology 2007, 148, 1059–1079. [Google Scholar] [CrossRef]
- Sitras, V.; Fenton, C.; Paulssen, R.; Vårtun, Å.; Acharya, G. Differences in gene expression between first and third trimester human placenta: A microarray study. PLoS ONE 2012, 7, e33294. [Google Scholar] [CrossRef]
- Lim, Y.C.; Li, J.; Ni, Y.; Liang, Q.; Zhang, J.; Yeo, G.S.H.; Lyu, J.; Jin, S.; Ding, C. A complex association between DNA methylation and gene expression in human placenta at first and third trimesters. PLoS ONE 2017, 12, e0181155. [Google Scholar] [CrossRef]
- Breen, J.; McAninch, D.; Jankovic-Karasoulos, T.; McCullough, D.; Smith, M.D.; Bogias, K.J.; Wan, Q.; Choudhry, A.; Hin, N.; Pederson, S.M.; et al. Temporal placental genome wide expression profiles reflect three phases of utero-placental blood flow during early to mid human gestation. Indigenous Genomics, Telethon Kids Institute (Adelaide Office): Adelaide, SA, Australia, 2020. manuscript in preparation.
- Gu, Y.; Sun, J.; Groome, L.J.; Wang, Y. Differential miRNA expression profiles between the first and third trimester human placentas. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E836–E843. [Google Scholar] [CrossRef]
- Smith, M.D.; Pillman, K.; Jankovic-Karasoulos, T.; McAninch, D.; Wan, Q.; Bogias, K.J.; McCullough, D.; Bianco-Miotto, T.; Breen, J.; Roberts, C.T. Large-scale transcriptome-wide profiling of microRNAs in human placenta and maternal plasma at early to mid gestation. RNA Biol. 2021, 18, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Buckberry, S.; Bianco-Miotto, T.; Roberts, C.T. Imprinted and X-linked non-coding RNAs as potential regulators of human placental function. Epigenetics 2014, 9, 81–89. [Google Scholar] [CrossRef] [PubMed]
- McAninch, D.; Roberts, C.T.; Bianco-Miotto, T. Mechanistic Insight into Long Noncoding RNAs and the Placenta. Int. J. Mol. Sci. 2017, 18, 1371. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Yi, L.; Pimentel, H.; Bray, N.L.; Pachter, L. Gene-level differential analysis at transcript-level resolution. Genome Biol. 2018, 19, 53. [Google Scholar] [CrossRef]
- Roberts, H.J.; Hu, S.; Qiu, Q.; Leung, P.C.K.; Caniggia, I.; Gruslin, A.; Tsang, B.; Peng, C. Identification of novel isoforms of activin receptor-like kinase 7 (ALK7) generated by alternative splicing and expression of ALK7 and its ligand, Nodal, in human placenta. Biol. Reprod. 2003, 68, 1719–1726. [Google Scholar] [CrossRef][Green Version]
- Yang, W.; Ahn, H.; Hinrichs, M.; Torry, R.J.; Torry, D.S. Evidence of a novel isoform of placenta growth factor (PlGF-4) expressed in human trophoblast and endothelial cells. J. Reprod. Immunol. 2003, 60, 53–60. [Google Scholar] [CrossRef]
- Monk, D.; Sanches, R.; Arnaud, P.; Apostolidou, S.; Hills, F.A.; Abu-Amero, S.; Murrell, A.; Friess, H.; Reik, W.; Stanier, P.; et al. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum. Mol. Genet. 2006, 15, 1259–1269. [Google Scholar] [CrossRef]
- Ikeda, T.; Sun, L.; Tsuruoka, N.; Ishigaki, Y.; Yoshitomi, Y.; Yoshitake, Y.; Yonekura, H. Hypoxia down-regulates sFlt-1 (sVEGFR-1) expression in human microvascular endothelial cells by a mechanism involving mRNA alternative processing. Biochem. J. 2011, 436, 399–407. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. [version 2; peer review: 2 approved]. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Matthes, K.L.; Nowicka, M.; Law, C.W.; Robinson, M.D. Isoform prefiltering improves performance of count-based methods for analysis of differential transcript usage. Genome Biol. 2016, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Soneson, C.; Patro, R. Swimming downstream: Statistical analysis of differential transcript usage following Salmon quantification. F1000Research 2018, 7, 952. [Google Scholar] [CrossRef]
- Majewska, M.; Lipka, A.; Paukszto, L.; Jastrzebski, J.P.; Szeszko, K.; Gowkielewicz, M.; Lepiarczyk, E.; Jozwik, M.; Majewski, M.K. Placenta transcriptome profiling in intrauterine growth restriction (IUGR). Int. J. Mol. Sci. 2019, 20, 1510. [Google Scholar] [CrossRef] [PubMed]
- Ruano, C.S.M.; Apicella, C.; Jacques, S.; Gascoin, G.; Gaspar, C.; Miralles, F.; Méhats, C.; Vaiman, D. Alternative splicing in normal and pathological human placentas is correlated to genetic variants. Hum. Genet. 2021, 140, 827–848. [Google Scholar] [CrossRef] [PubMed]
- Georgiadou, D.; Boussata, S.; Keijser, R.; Janssen, D.A.M.; Afink, G.B.; van Dijk, M. Knockdown of splicing complex protein PCBP2 reduces extravillous trophoblast differentiation through transcript switching. Front. Cell Dev. Biol. 2021, 9, 671806. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gonzàlez-Porta, M.; Santos, S.; Brazma, A.; Marioni, J.C.; Aebersold, R.; Venkitaraman, A.R.; Wickramasinghe, V.O. Impact of alternative splicing on the human proteome. Cell Rep. 2017, 20, 1229–1241. [Google Scholar] [CrossRef]
- Nucci, M.; Poon, L.C.; Demirdjian, G.; Darbouret, B.; Nicolaides, K.H. Maternal serum placental growth factor isoforms 1 and 2 at 11–13, 20–24 and 30–34 weeks’ gestation in late-onset pre-eclampsia and small for gestational age neonates. Fetal Diagn. Ther. 2014, 35, 249–257. [Google Scholar] [CrossRef]
- Regnault, T.R.H.; Orbus, R.J.; de Vrijer, B.; Davidsen, M.L.; Galan, H.L.; Wilkening, R.B.; Anthony, R.V. Placental expression of VEGF, PlGF and their receptors in a model of placental insufficiency-intrauterine growth restriction (PI-IUGR). Placenta 2002, 23, 132–144. [Google Scholar] [CrossRef]
- Thomas, C.P.; Andrews, J.I.; Liu, K.Z. Intronic polyadenylation signal sequences and alternate splicing generate human soluble Flt1 variants and regulate the abundance of soluble Flt1 in the placenta. FASEB J. 2007, 21, 3885–3895. [Google Scholar] [CrossRef]
- Schumann, S.; Buck, V.U.; Classen-Linke, I.; Wennemuth, G.; Grümmer, R. Claudin-3, claudin-7, and claudin-10 show different distribution patterns during decidualization and trophoblast invasion in mouse and human. Histochem. Cell Biol. 2015, 144, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Mwaikambo, B.R.; Yang, C.; Chemtob, S.; Hardy, P. Hypoxia up-regulates CD36 expression and function via hypoxia-inducible factor-1- and phosphatidylinositol 3-kinase-dependent mechanisms. J. Biol. Chem. 2009, 284, 26695–26707. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Coma, S.; Shimizu, A.; Klagsbrun, M. Hypoxia induces tumor and endothelial cell migration in a semaphorin 3F- and VEGF-dependent manner via transcriptional repression of their common receptor neuropilin 2. Cell Adh. Migr. 2011, 5, 266–275. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- Heydarian, M.; McCaffrey, T.; Florea, L.; Yang, Z.; Ross, M.M.; Zhou, W.; Maynard, S.E. Novel splice variants of sFlt1 are upregulated in preeclampsia. Placenta 2009, 30, 250–255. [Google Scholar] [CrossRef]
- Palmer, K.R.; Kaitu’u-Lino, T.J.; Hastie, R.; Hannan, N.J.; Ye, L.; Binder, N.; Cannon, P.; Tuohey, L.; Johns, T.G.; Shub, A.; et al. Placental-Specific sFLT-1 e15a Protein Is Increased in Preeclampsia, Antagonizes Vascular Endothelial Growth Factor Signaling, and Has Antiangiogenic Activity. Hypertension 2015, 66, 1251–1259. [Google Scholar] [CrossRef]
- Jin, X.; Qiao, L.; Fan, H.; Liao, C.; Zheng, J.; Wang, W.; Ma, X.; Yang, M.; Sun, X.; Zhao, W. Long non-coding RNA MSC-AS1 facilitates the proliferation and glycolysis of gastric cancer cells by regulating PFKFB3 expression. Int. J. Med. Sci. 2021, 18, 546–554. [Google Scholar] [CrossRef]
- Han, W.; Yu, G.; Meng, X.; Hong, H.; Zheng, L.; Wu, X.; Zhang, D.; Yan, B.; Ma, Y.; Li, X.; et al. Potential of C1QTNF1-AS1 regulation in human hepatocellular carcinoma. Mol. Cell. Biochem. 2019, 460, 37–51. [Google Scholar] [CrossRef]
- Gonzàlez-Porta, M.; Frankish, A.; Rung, J.; Harrow, J.; Brazma, A. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol. 2013, 14, R70. [Google Scholar] [CrossRef]
- Vitting-Seerup, K.; Sandelin, A. The landscape of isoform switches in human cancers. Mol. Cancer Res. 2017, 15, 1206–1220. [Google Scholar] [CrossRef] [PubMed]
- Modrek, B.; Lee, C. A genomic view of alternative splicing. Nat. Genet. 2002, 30, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Caniggia, I.; Mostachfi, H.; Winter, J.; Gassmann, M.; Lye, S.J.; Kuliszewski, M.; Post, M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J. Clin. Investig. 2000, 105, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Caolo, V.; Swennen, G.; Chalaris, A.; Wagenaar, A.; Verbruggen, S.; Rose-John, S.; Molin, D.G.M.; Vooijs, M.; Post, M.J. ADAM10 and ADAM17 have opposite roles during sprouting angiogenesis. Angiogenesis 2015, 18, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Raikwar, N.S.; Liu, K.Z.; Thomas, C.P. N-terminal cleavage and release of the ectodomain of Flt1 is mediated via ADAM10 and ADAM 17 and regulated by VEGFR2 and the Flt1 intracellular domain. PLoS ONE 2014, 9, e112794. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Wang, G.; Zhu, Z.; Huang, Y.; Gu, H.; Ni, X. Increased ADAM10 expression in preeclamptic placentas is associated with decreased expression of hydrogen sulfide production enzymes. Placenta 2015, 36, 947–950. [Google Scholar] [CrossRef]
- Palmer, K.R.; Tong, S.; Kaitu’u-Lino, T.J. Placental-specific sFLT-1: Role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol. Hum. Reprod. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Gutwein, P.; Mechtersheimer, S.; Riedle, S.; Stoeck, A.; Gast, D.; Joumaa, S.; Zentgraf, H.; Fogel, M.; Altevogt, D.P. ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J. 2003, 17, 292–294. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Hamilton, T.K.; Li, X.; Cotechini, T.; Miles, E.A.; Siemens, D.R.; Graham, C.H. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: Role of nitric oxide. Cancer Res. 2011, 71, 7433–7441. [Google Scholar] [CrossRef]
- Lewis, B.P.; Green, R.E.; Brenner, S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. USA 2003, 100, 189–192. [Google Scholar] [CrossRef]
- de Lima Morais, D.A.; Harrison, P.M. Large-scale evidence for conservation of NMD candidature across mammals. PLoS ONE 2010, 5, e11695. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Wang, Y.; Huang, H.; Yu, W.; Bai, M.; Zhang, L.; Bryan, B.A.; Wang, Y.; Luo, J.; Li, D.; et al. GPR126 protein regulates developmental and pathological angiogenesis through modulation of VEGFR2 receptor signaling. J. Biol. Chem. 2014, 289, 34871–34885. [Google Scholar] [CrossRef] [PubMed]
- Monteuuis, G.; Wong, J.J.L.; Bailey, C.G.; Schmitz, U.; Rasko, J.E.J. The changing paradigm of intron retention: Regulation, ramifications and recipes. Nucleic Acids Res. 2019, 47, 11497–11513. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.E.; Catrinacio, C.; Ropolo, A.; Rivarola, V.A.; Vaccaro, M.I. A novel HIF-1α/VMP1-autophagic pathway induces resistance to photodynamic therapy in colon cancer cells. Photochem. Photobiol. Sci. 2017, 16, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.-H.; Hsieh, T.-T.; Chen, S.-F.; Li, M.-J.; Yeh, Y.-L. Autophagy in the human placenta throughout gestation. PLoS ONE 2013, 8, e83475. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [PubMed]
- Gebai, A.; Gorelik, A.; Li, Z.; Illes, K.; Nagar, B. Structural basis for the activation of acid ceramidase. Nat. Commun. 2018, 9, 1621. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.; Garandeau, D.; Pandiani, C.; Gaudel, C.; Bille, K.; Nottet, N.; Garcia, V.; Colosetti, P.; Pagnotta, S.; Bahadoran, P.; et al. Lysosomal acid ceramidase ASAH1 controls the transition between invasive and proliferative phenotype in melanoma cells. Oncogene 2019, 38, 1282–1295. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics–FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 September 2020).
- Ward, C.M.; To, T.-H.; Pederson, S.M. ngsReports: A Bioconductor package for managing FastQC reports and other NGS related log files. Bioinformatics 2020, 36, 2587–2588. [Google Scholar] [CrossRef]
- Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Malik, L.; Sarkar, H.; Zakeri, M.; Almodaresi, F.; Soneson, C.; Love, M.I.; Kingsford, C.; Patro, R. Alignment and mapping methodology influence transcript abundance estimation. Genome Biol. 2020, 21, 239. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Hicks, S.C.; Irizarry, R.A. Quantro: A data-driven approach to guide the choice of an appropriate normalization method. Genome Biol. 2015, 16, 117. [Google Scholar] [CrossRef]
- Hicks, S.C.; Okrah, K.; Paulson, J.N.; Quackenbush, J.; Irizarry, R.A.; Bravo, H.C. Smooth quantile normalization. Biostatistics 2018, 19, 185–198. [Google Scholar] [CrossRef]
- Lun, A.T.L.; Chen, Y.; Smyth, G.K. It’s DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR. Methods Mol. Biol. 2016, 1418, 391–416. [Google Scholar] [PubMed]
- Ward, J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 1.0.12. 2019. Available online: https://cran.r-project.org/package=pheatmap (accessed on 31 March 2022).
- Nowicka, M.; Robinson, M.D. DRIMSeq: A Dirichlet-multinomial framework for multivariate count outcomes in genomics. F1000Research 2016, 5, 1356. [Google Scholar] [CrossRef]
- Van den Berge, K.; Soneson, C.; Robinson, M.D.; Clement, L. stageR: A general stage-wise method for controlling the gene-level false discovery rate in differential expression and differential transcript usage. Genome Biol. 2017, 18, 151. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Rainer, J.; Gatto, L.; Weichenberger, C.X. ensembldb: An R package to create and use Ensembl-based annotation resources. Bioinformatics 2019, 35, 3151–3153. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | Ontology | * DE in Term | FDR |
|---|---|---|---|---|
| GO:0005886 | plasma MEMBRANE | CC | 637 (21.0%) | |
| GO:0031226 | intrinsic component of plasma MEMBRANE | CC | 237 (28.4%) | |
| GO:0005887 | integral component of plasma MEMBRANE | CC | 228 (28.9%) | 1.34 × 10−28 |
| GO:0016021 | integral component of MEMBRANE | CC | 559 (19.9%) | |
| GO:0004888 | transmembrane signaling receptor ACTIVITY | MF | 137 (31.6%) | |
| GO:0005102 | signaling receptor BINDING | MF | 220 (24.7%) | |
| GO:0016477 | cell MIGRATION | BP | 238 (23.8%) | 6.55 × 10−18 |
| GO:0070887 | CELLULAR RESPONSE to chemical stimulus | BP | 414 (20.0%) | |
| GO:0006952 | defense response | BP | 232 (23.7%) | 3.52 × 10−17 |
| GO:0071310 | CELLULAR RESPONSE to organic substance | BP | 342 (20.7%) | 3.29 × 10−16 |
| GO:0005539 | glycosaminoglycan BINDING | MF | 59 (44.7%) | |
| GO:0030334 | REGULATION of cell MIGRATION | BP | 168 (25.8%) | |
| GO:0048018 | receptor ligand ACTIVITY | MF | 68 (39.8%) | |
| GO:0007166 | cell surface receptor signaling pathway | BP | 355 (20.1%) | |
| GO:0051270 | REGULATION of cellular component movement | BP | 180 (24.8%) | |
| GO:2000145 | REGULATION of cell motility | BP | 171 (25.1%) | |
| GO:0030546 | signaling receptor activator ACTIVITY | MF | 69 (38.5%) | |
| GO:0008201 | heparin BINDING | MF | 48 (49.0%) | |
| GO:0006935 | chemotaxis | BP | 109 (29.9%) | |
| GO:0031982 | vesicle | CC | 509 (18.1%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogias, K.J.; Pederson, S.M.; Leemaqz, S.; Smith, M.D.; McAninch, D.; Jankovic-Karasoulos, T.; McCullough, D.; Wan, Q.; Bianco-Miotto, T.; Breen, J.; et al. Placental Transcription Profiling in 6–23 Weeks’ Gestation Reveals Differential Transcript Usage in Early Development. Int. J. Mol. Sci. 2022, 23, 4506. https://doi.org/10.3390/ijms23094506
Bogias KJ, Pederson SM, Leemaqz S, Smith MD, McAninch D, Jankovic-Karasoulos T, McCullough D, Wan Q, Bianco-Miotto T, Breen J, et al. Placental Transcription Profiling in 6–23 Weeks’ Gestation Reveals Differential Transcript Usage in Early Development. International Journal of Molecular Sciences. 2022; 23(9):4506. https://doi.org/10.3390/ijms23094506
Chicago/Turabian StyleBogias, Konstantinos J., Stephen M. Pederson, Shalem Leemaqz, Melanie D. Smith, Dale McAninch, Tanja Jankovic-Karasoulos, Dylan McCullough, Qianhui Wan, Tina Bianco-Miotto, James Breen, and et al. 2022. "Placental Transcription Profiling in 6–23 Weeks’ Gestation Reveals Differential Transcript Usage in Early Development" International Journal of Molecular Sciences 23, no. 9: 4506. https://doi.org/10.3390/ijms23094506
APA StyleBogias, K. J., Pederson, S. M., Leemaqz, S., Smith, M. D., McAninch, D., Jankovic-Karasoulos, T., McCullough, D., Wan, Q., Bianco-Miotto, T., Breen, J., & Roberts, C. T. (2022). Placental Transcription Profiling in 6–23 Weeks’ Gestation Reveals Differential Transcript Usage in Early Development. International Journal of Molecular Sciences, 23(9), 4506. https://doi.org/10.3390/ijms23094506