
Rho/SRF Inhibitor Modulates Mitochondrial Functions
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
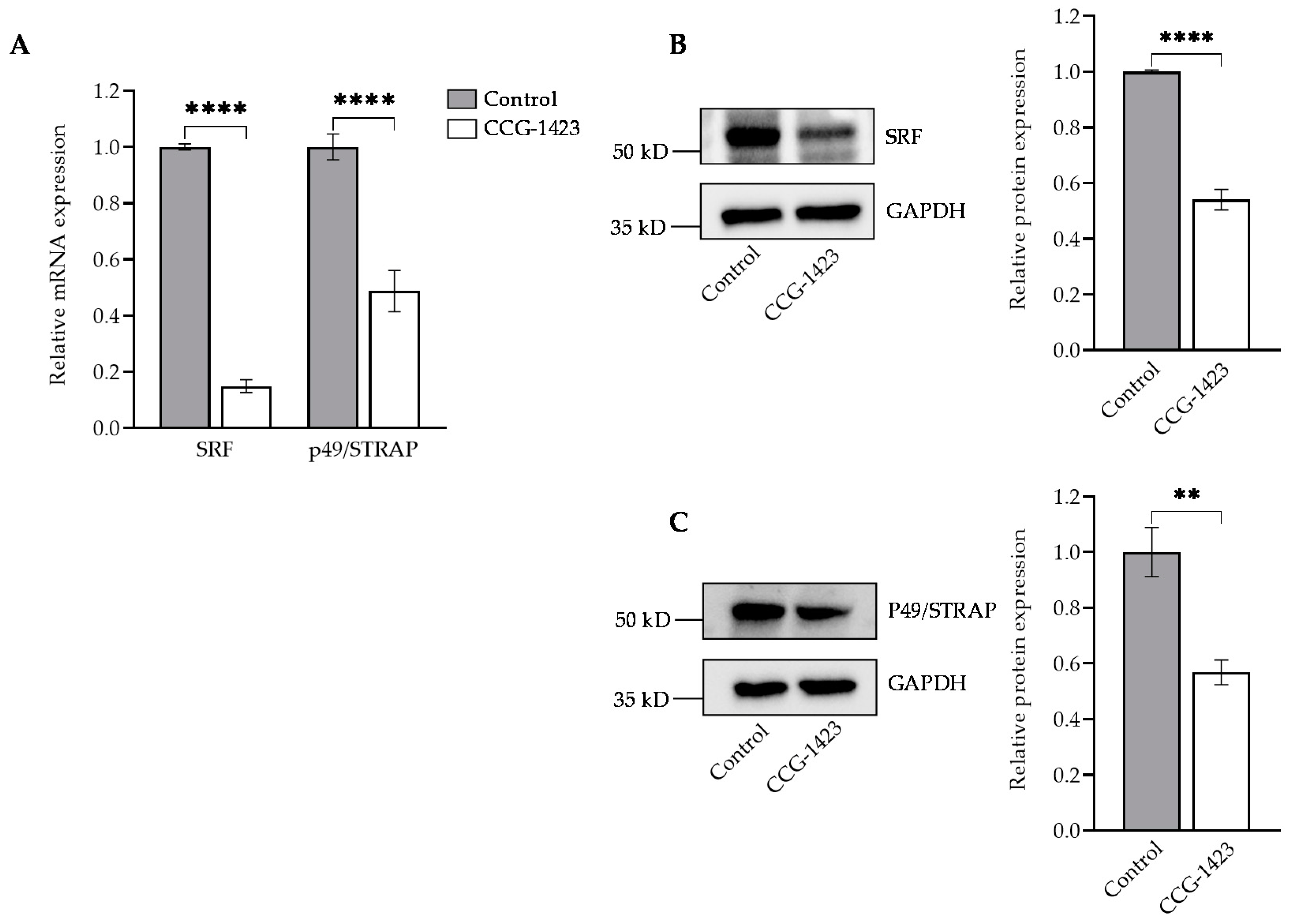
2.1. CCG-1423 Downregulates SRF, p49/STRAP Expression and Does Not Affect the Cell Viability
2.2. CCG-1423 Increases H4 Lysine-16 Acetylation but Decreases the Level of Acetylated-Lysine SRF
2.3. CCG-1423 Represses the Genes Involved in Mitochondrial Biogenesis, Fusion, and Fission
2.4. Actin Assembly and Mitochondrial Morphology
2.5. CCG-1423 Differentially Regulates Cellular Bioenergetics
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Proliferation Assay
4.2. Relative Quantification of Gene Expression
4.3. Immunoprecipitation and Western Blot Analysis
4.4. MitoTracker and Immunofluorescence Staining
4.5. Measurement of Mitochondrial Oxygen Consumption, Glycolytic and ATP Rate
4.6. High-Resolution Respirometry
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonald, O.G.; Wamhoff, B.R.; Hoofnagle, M.H.; Owens, G.K. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J. Clin. Investig. 2006, 116, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Miano, J.M. Serum response factor: Toggling between disparate programs of gene expression. J. Mol. Cell Cardiol. 2003, 35, 577–593. [Google Scholar] [CrossRef]
- Xie, L. MKL1/2 and ELK4 co-regulate distinct serum response factor (SRF) transcription programs in macrophages. BMC Genom. 2014, 15, 301. [Google Scholar] [CrossRef]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef]
- Miano, J.M.; Long, X.; Fujiwara, K. Serum response factor: Master regulator of the actin cytoskeleton and contractile apparatus. Am. J. Physiol. Cell Physiol. 2007, 292, C70–C81. [Google Scholar] [CrossRef]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Tsou, P.-S.; Haak, A.J.; Khanna, D.; Neubig, R.R. Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: Focus on Rho GTPase-regulated gene transcription. Am. J. Physiol. Cell Physiol. 2014, 307, C2–C13. [Google Scholar] [CrossRef]
- Sakai, N.; Chun, J.; Duffield, J.S.; Wada, T.; Luster, A.D.; Tager, A.M. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013, 27, 1830–1846. [Google Scholar] [CrossRef]
- Evelyn, C.R.; Wade, S.M.; Wang, Q.; Wu, M.; Iñiguez-Lluhí, J.A.; Merajver, S.D.; Neubig, R.R. CCG-1423: A small-molecule inhibitor of RhoA transcriptional signaling. Mol. Cancer Ther. 2007, 6, 2249–2260. [Google Scholar] [CrossRef]
- Hayashi, K.; Watanabe, B.; Nakagawa, Y.; Minami, S.; Morita, T. RPEL proteins are the molecular targets for CCG-1423, an inhibitor of Rho signaling. PLoS ONE 2014, 9, e89016. [Google Scholar] [CrossRef]
- Jin, W.; Goldfine, A.B.; Boes, T.; Henry, R.R.; Ciaraldi, T.P.; Kim, E.-Y.; Emecan, M.; Fitzpatrick, C.; Sen, A.; Shah, A.; et al. Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J. Clin. Investig. 2011, 121, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Kuwahara, K.; Nakagawa, Y.; Takaoka, M.; Kinoshita, H.; Nakao, K.; Kuwabara, Y.; Yamada, Y.; Yamada, C.; Shibata, J.; et al. Reciprocal expression of MRTF-A and myocardin is crucial for pathological vascular remodelling in mice. EMBO J. 2012, 31, 4428–4440. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Rodansky, E.S.; Haak, A.J.; Larsen, S.D.; Neubig, R.R.; Higgins, P.D.R. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-β-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel Dis. 2014, 20, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Karnkowska, A.; Vacek, V.; Zubáčová, Z.; Treitli, S.C.; Petrželková, R.; Eme, L.; Novák, L.; Žárský, V.; Barlow, L.D.; Herman, E.K.; et al. A Eukaryote without a mitochondrial organelle. Curr. Biol. 2016, 26, 1274–1284. [Google Scholar] [CrossRef]
- Guo, Y.; Jardin, B.D.; Zhou, P.; Sethi, I.; Akerberg, B.N.; Toepfer, C.N.; Ai, Y.; Li, Y.; Ma, Q.; Guatimosim, S.; et al. Hierarchical and Stage-Specific Regulation of Murine Cardiomyocyte Maturation by Serum Response Factor. Nat. Commun. 2018, 9, 3837. [Google Scholar] [CrossRef]
- Rugowska, A.; Starosta, A.; Konieczny, P. Epigenetic modifications in muscle regeneration and progression of duchenne muscular dystrophy. Clin. Epigenet. 2021, 13, 13. [Google Scholar] [CrossRef]
- Mack, C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef]
- Arif, M.; Selvi, B.R.; Kundu, T.K. Lysine acetylation: The tale of a modification from transcription regulation to metabolism. Chembiochem 2010, 11, 1501–1504. [Google Scholar] [CrossRef]
- Millar, C.B.; Kurdistani, S.K.; Grunstein, M. Acetylation of yeast histone H4 lysine 16: A switch for protein interactions in heterochromatin and euchromatin. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 193–200. [Google Scholar] [CrossRef]
- Schratt, G.; Philippar, U.; Berger, J.; Schwarz, H.; Heidenreich, O.; Nordheim, A. Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J. Cell Biol. 2002, 156, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.D.; Martin, K.M.; Rickman, C.; Metcalfe, J.C.; Kemp, P.R. Increased actin polymerization reduces the inhibition of serum response factor activity by Yin Yang 1. Biochem. J. 2002, 364, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Azhar, G.; Zhong, Y.; Wei, J.Y. Identification of a novel serum response factor cofactor in cardiac gene regulation. J. Biol. Chem. 2004, 279, 55626–55632. [Google Scholar] [CrossRef]
- Zhang, X.; Azhar, G.; Helms, S.; Zhong, Y.; Wei, J.Y. Identification of a subunit of NADH-dehydrogenase as a P49/STRAP-binding protein. BMC Cell Biol. 2008, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Williams, E.D.; Azhar, G.; Rogers, S.C.; Wei, J.Y. Does P49/STRAP, a SRF-binding protein (SRFBP1), modulate cardiac mitochondrial function in aging? Exp. Gerontol. 2016, 82, 150–159. [Google Scholar] [CrossRef]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res. 2014, 114, 626–636. [Google Scholar] [CrossRef]
- Shia, W.-J.; Pattenden, S.G.; Workman, J.L. Histone H4 Lysine 16 Acetylation Breaks the Genome’s Silence. Genome Biol. 2006, 7, 217. [Google Scholar] [CrossRef]
- Esterbauer, H.; Oberkofler, H.; Krempler, F.; Patsch, W. Human peroxisome proliferator activated receptor gamma coactivator 1 (PPARGC1) gene: CDNA sequence, genomic organization, chromosomal localization, and tissue expression. Genomics 1999, 62, 98–102. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 Coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef]
- Pipes, G.C.T.; Creemers, E.E.; Olson, E.N. The myocardin family of transcriptional coactivators: Versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006, 20, 1545–1556. [Google Scholar] [CrossRef]
- Gau, D.; Veon, W.; Capasso, T.L.; Bottcher, R.; Shroff, S.; Roman, B.L.; Roy, P. Pharmacological intervention of MKL/SRF signaling by CCG-1423 impedes endothelial cell migration and angiogenesis. Angiogenesis 2017, 20, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Lisabeth, E.M.; Kahl, D.; Gopallawa, I.; Haynes, S.E.; Misek, S.A.; Campbell, P.L.; Dexheimer, T.S.; Khanna, D.; Fox, D.A.; Jin, X.; et al. identification of pirin as a molecular target of the CCG-1423/CCG-203971 series of antifibrotic and antimetastatic compounds. ACS Pharmacol. Transl. Sci. 2019, 2, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yu, H. Global burden of cancer. Yale J. Biol. Med. 2006, 79, 85–94. [Google Scholar] [PubMed]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef]
- Nakajima, M.; Hayashi, K.; Egi, Y.; Katayama, K.; Amano, Y.; Uehata, M.; Ohtsuki, M.; Fujii, A.; Oshita, K.; Kataoka, H.; et al. Effect of Wf-536, a NOVEL ROCK inhibitor, against metastasis of B16 melanoma. Cancer Chemother. Pharmacol. 2003, 52, 319–324. [Google Scholar] [CrossRef]
- Wei, L.; Surma, M.; Shi, S.; Lambert-Cheatham, N.; Shi, J. Novel insights into the roles of Rho kinase in cancer. Arch. Immunol. Ther. Exp. 2016, 64, 259–278. [Google Scholar] [CrossRef]
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Dalton, K.M.; Lochmann, T.L.; Floros, K.V.; Calbert, M.L.; Kurupi, R.; Stein, G.T.; McClanaghan, J.; Murchie, E.; Egan, R.K.; Greninger, P.; et al. Catastrophic ATP loss underlies a metabolic combination therapy tailored for MYCN-amplified neuroblastoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2009620118. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Rogers, S.C.; Zhang, X.; Azhar, G.; Wei, J.Y. Elevated oxygen consumption rate in response to acute low-glucose stress: Metformin restores rate to normal level. Exp. Gerontol. 2015, 70, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Shiota, J.; Yamada, T.; Ishikawa, M.; Ihara, D.; Fukuchi, M.; Tsuda, M.; Tabuchi, A. Rho signaling inhibitor, CCG-1423, inhibits axonal elongation and dendritic complexity of rat cortical neurons. Biochem. Biophys. Res. Commun. 2017, 492, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, B.; Minami, S.; Ishida, H.; Yoshioka, R.; Nakagawa, Y.; Morita, T.; Hayashi, K. Stereospecific inhibitory effects of CCG-1423 on the cellular events mediated by myocardin-related transcription factor A. PLoS ONE 2015, 10, e0136242. [Google Scholar] [CrossRef]
- Gineitis, D.; Treisman, R. Differential usage of signal transduction pathways defines two types of serum response factor target gene. J. Biol. Chem. 2001, 276, 24531–24539. [Google Scholar] [CrossRef]
- Holst, B.; Holliday, N.D.; Bach, A.; Elling, C.E.; Cox, H.M.; Schwartz, T.W. Common structural basis for constitutive activity of the ghrelin receptor family. J. Biol. Chem. 2004, 279, 53806–53817. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Shore, P.; Sharrocks, A.D.; Davis, R.J. Integration of MAP kinase signal transduction pathways at the serum response element. Science 1995, 269, 403–407. [Google Scholar] [CrossRef]
- Connelly, J.T.; Mishra, A.; Gautrot, J.E.; Watt, F.M. Shape-induced terminal differentiation of human epidermal stem cells requires P38 and is regulated by histone acetylation. PLoS ONE 2011, 6, e27259. [Google Scholar] [CrossRef]
- Gräff, J.; Tsai, L.-H. Histone acetylation: Molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 2013, 14, 97–111. [Google Scholar] [CrossRef]
- Alberts, A.S.; Geneste, O.; Treisman, R. Activation of SRF-regulated chromosomal templates by Rho-family GTPases requires a signal that also induces H4 hyperacetylation. Cell 1998, 92, 475–487. [Google Scholar] [CrossRef]
- Huang, M.; Lou, D.; Charli, A.; Kong, D.; Jin, H.; Zenitsky, G.; Anantharam, V.; Kanthasamy, A.; Wang, Z.; Kanthasamy, A.G. Mitochondrial dysfunction–induced H3K27 hyperacetylation perturbs enhancers in Parkinson’s disease. JCI Insight 2021, 6, e138088. [Google Scholar] [CrossRef] [PubMed]
- Kopinski, P.K.; Janssen, K.A.; Schaefer, P.M.; Trefely, S.; Perry, C.E.; Potluri, P.; Tintos-Hernandez, J.A.; Singh, L.N.; Karch, K.R.; Campbell, S.L.; et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc. Natl. Acad. Sci. USA 2019, 116, 16028–16035. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.-G. Mitochondrial transcription factor A regulates MtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism—Emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Chen, L.; Qin, Y.; Liu, B.; Gao, M.; Li, A.; Li, X.; Gong, G. PGC-1α-Mediated Mitochondrial Quality Control: Molecular Mechanisms and Implications for Heart Failure. Front. Cell Dev. Biol. 2022, 10, 871357. [Google Scholar] [CrossRef]
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Investing. 2013, 123, 1068–1081. [Google Scholar] [CrossRef]
- Connelly, J.T.; Gautrot, J.E.; Trappmann, B.; Tan, D.W.-M.; Donati, G.; Huck, W.T.S.; Watt, F.M. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat. Cell Biol. 2010, 12, 711–718. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.C.; Zhang, X.; Azhar, G.; Luo, S.; Wei, J.Y. Exposure to high or low glucose levels accelerates the appearance of markers of endothelial cell senescence and induces dysregulation of nitric oxide synthase. J. Gerontol. Ser. A 2013, 68, 1469–1481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ameer, F.S.; Azhar, G.; Wei, J.Y. Alternative splicing increases sirtuin gene family diversity and modulates their subcellular localization and function. IJMS 2021, 22, 473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Azhar, G.; Wei, J.Y. The expression of MicroRNA and MicroRNA clusters in the aging heart. PLoS ONE 2012, 7, e34688. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial pathways and respiratory control. In An Introduction to OXPHOS Analysis, 5th ed.; Bioenergetics Communications: Axams, Austria, 2020. [Google Scholar] [CrossRef]
- Tobacyk, J.; Kc, G.; MacMillan-Crow, L.A. Overexpression of MnSOD protects against cold storage-induced mitochondrial injury but not against OMA1-Dependent OPA1 proteolytic processing in rat renal proximal tubular cells. Antioxidants 2021, 10, 1272. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. In Mitochondrial Bioenergetics; Palmeira, C.M., Moreno, A.J., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 810, pp. 25–58. ISBN 978-1-61779-381-3. [Google Scholar]






 = inhibition.
= inhibition.
= inhibition.
= inhibition.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patyal, P.; Nguyen, B.; Zhang, X.; Azhar, G.; Ameer, F.S.; Verma, A.; Crane, J.; KC, G.; Che, Y.; Wei, J.Y. Rho/SRF Inhibitor Modulates Mitochondrial Functions. Int. J. Mol. Sci. 2022, 23, 11536. https://doi.org/10.3390/ijms231911536
Patyal P, Nguyen B, Zhang X, Azhar G, Ameer FS, Verma A, Crane J, KC G, Che Y, Wei JY. Rho/SRF Inhibitor Modulates Mitochondrial Functions. International Journal of Molecular Sciences. 2022; 23(19):11536. https://doi.org/10.3390/ijms231911536
Chicago/Turabian StylePatyal, Pankaj, Bachkhoa Nguyen, Xiaomin Zhang, Gohar Azhar, Fathima S. Ameer, Ambika Verma, Jasmine Crane, Grishma KC, Yingni Che, and Jeanne Y. Wei. 2022. "Rho/SRF Inhibitor Modulates Mitochondrial Functions" International Journal of Molecular Sciences 23, no. 19: 11536. https://doi.org/10.3390/ijms231911536
APA StylePatyal, P., Nguyen, B., Zhang, X., Azhar, G., Ameer, F. S., Verma, A., Crane, J., KC, G., Che, Y., & Wei, J. Y. (2022). Rho/SRF Inhibitor Modulates Mitochondrial Functions. International Journal of Molecular Sciences, 23(19), 11536. https://doi.org/10.3390/ijms231911536