
Spiropyran/Merocyanine Amphiphile in Various Solvents: A Joint Experimental–Theoretical Approach to Photophysical Properties and Self-Assembly



Abstract
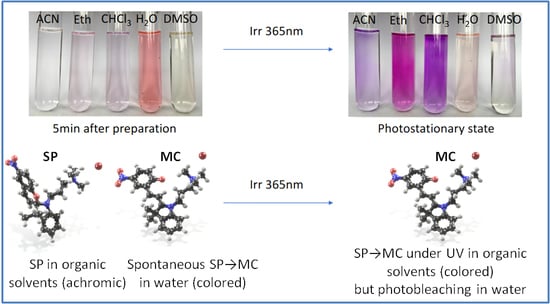
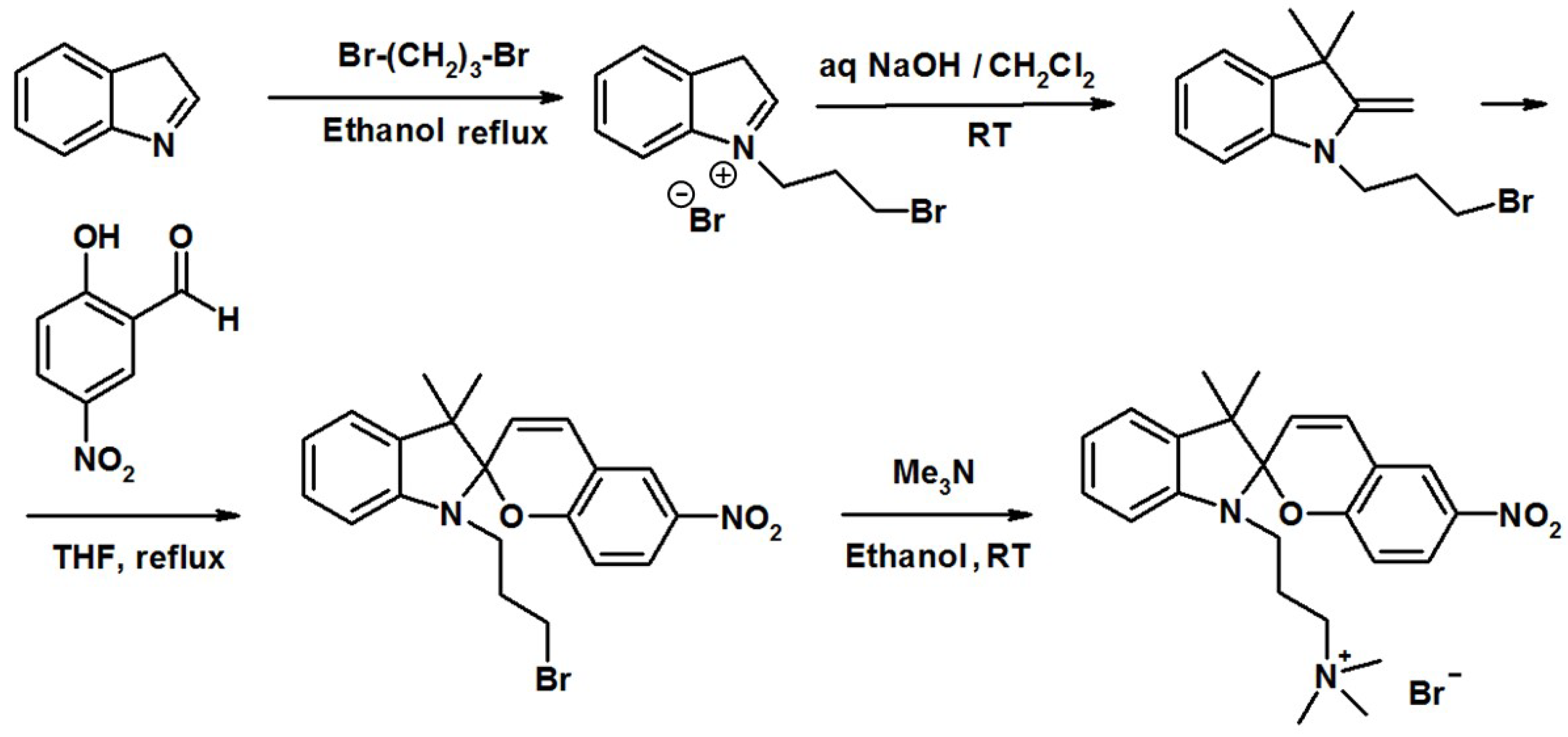
1. Introduction
2. Results
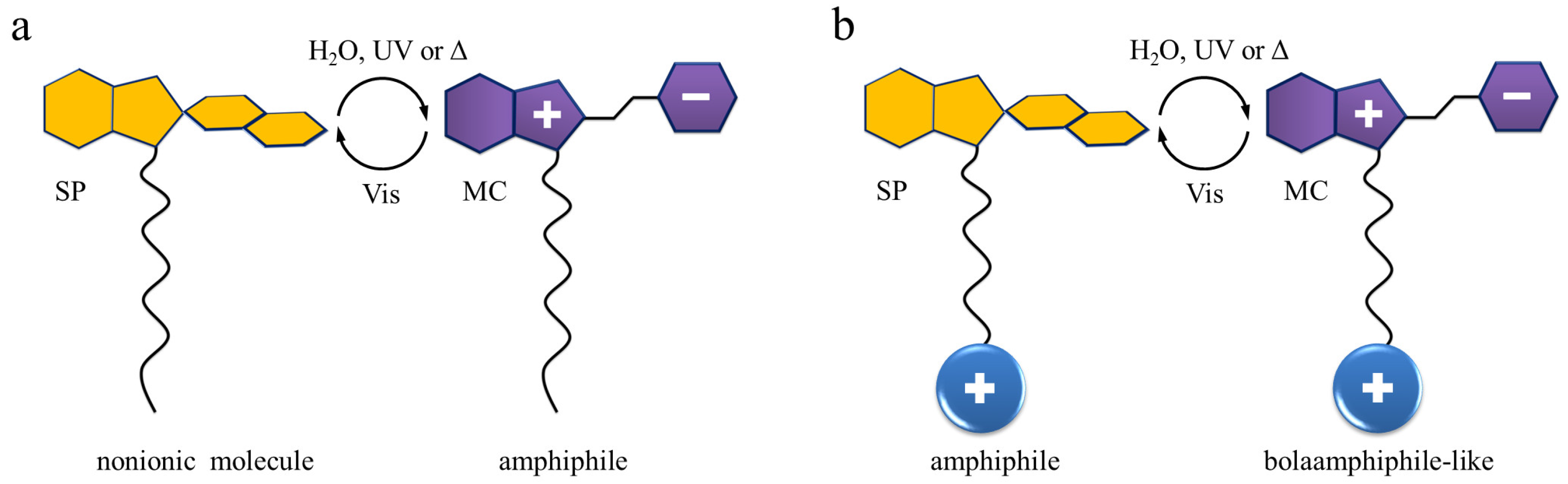
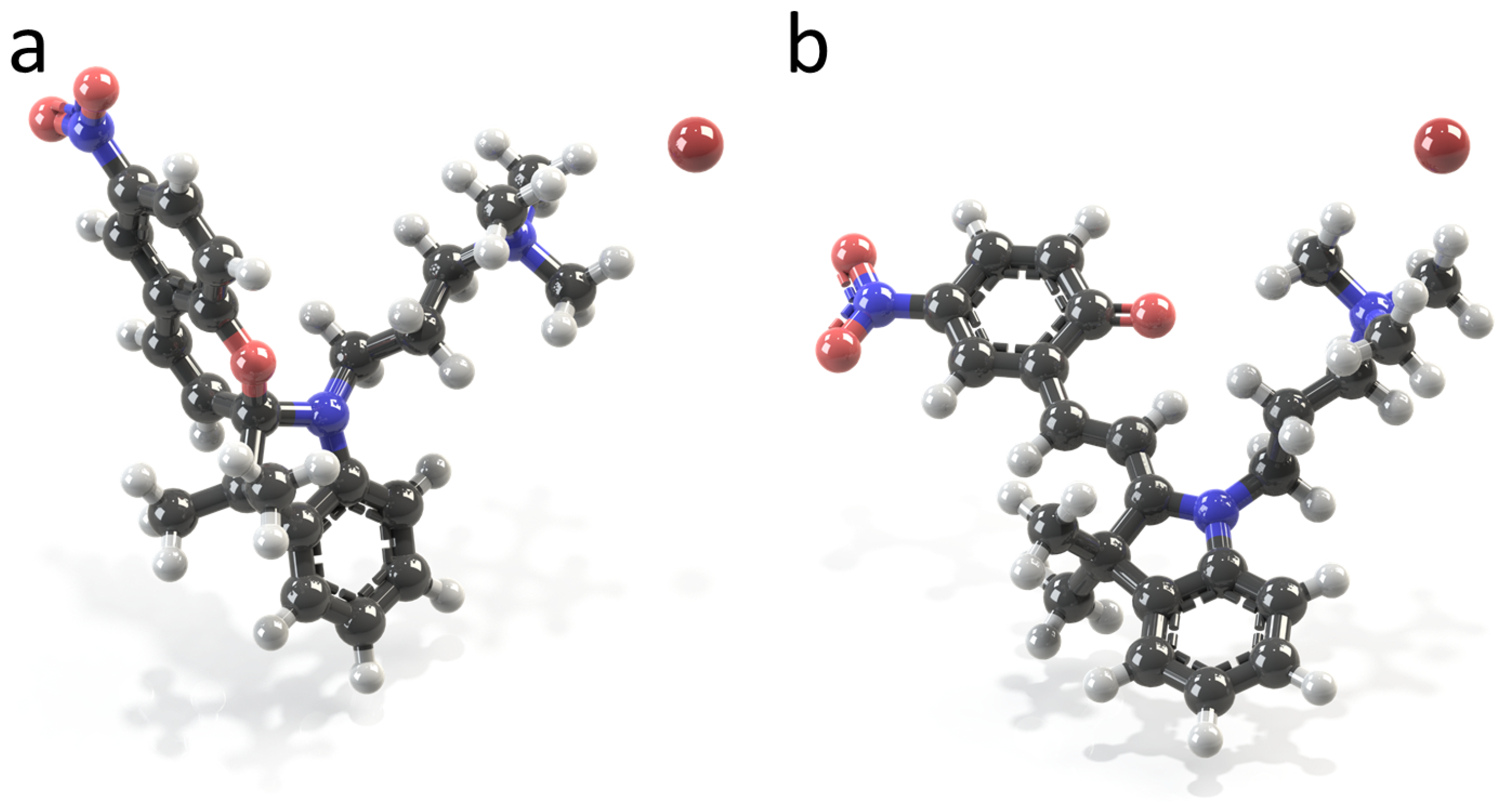
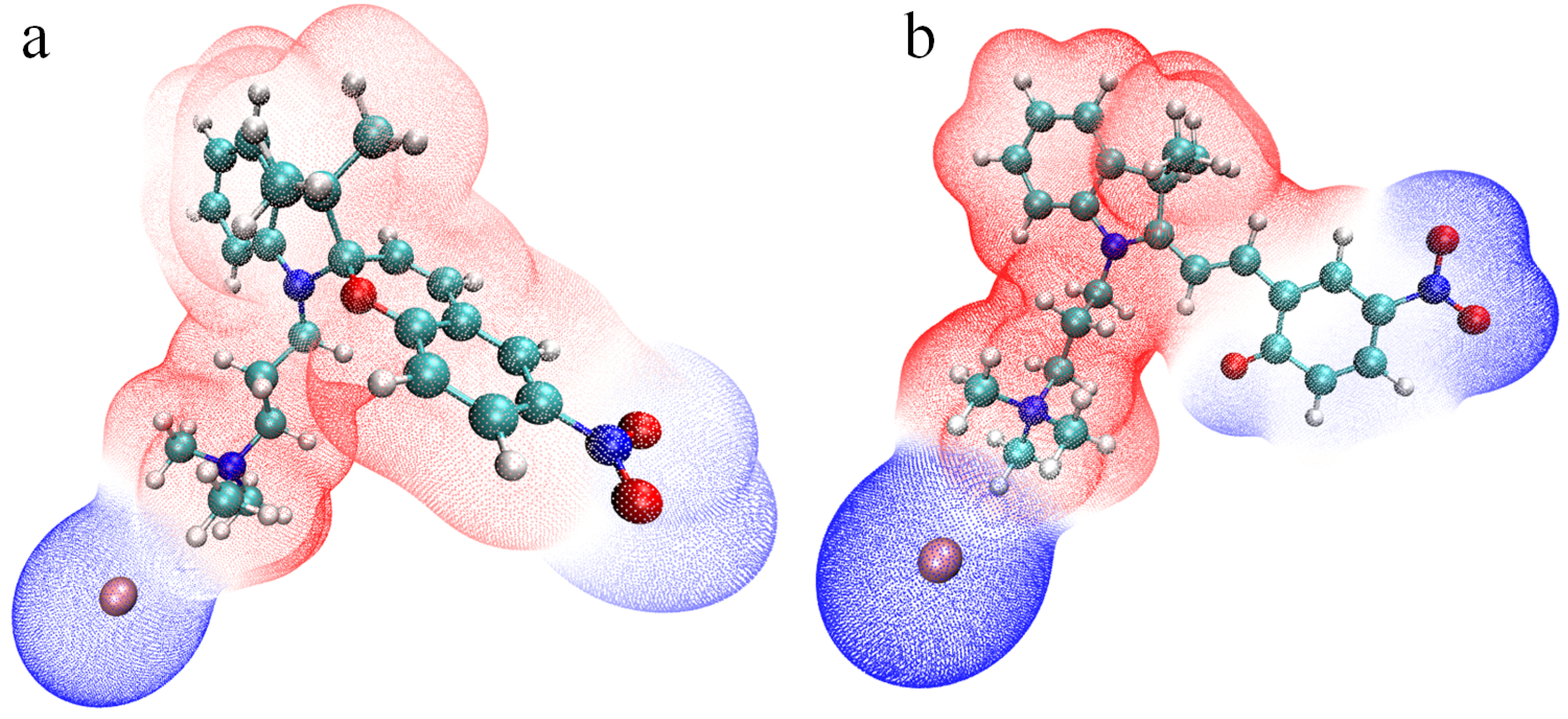
2.1. Molecular Properties
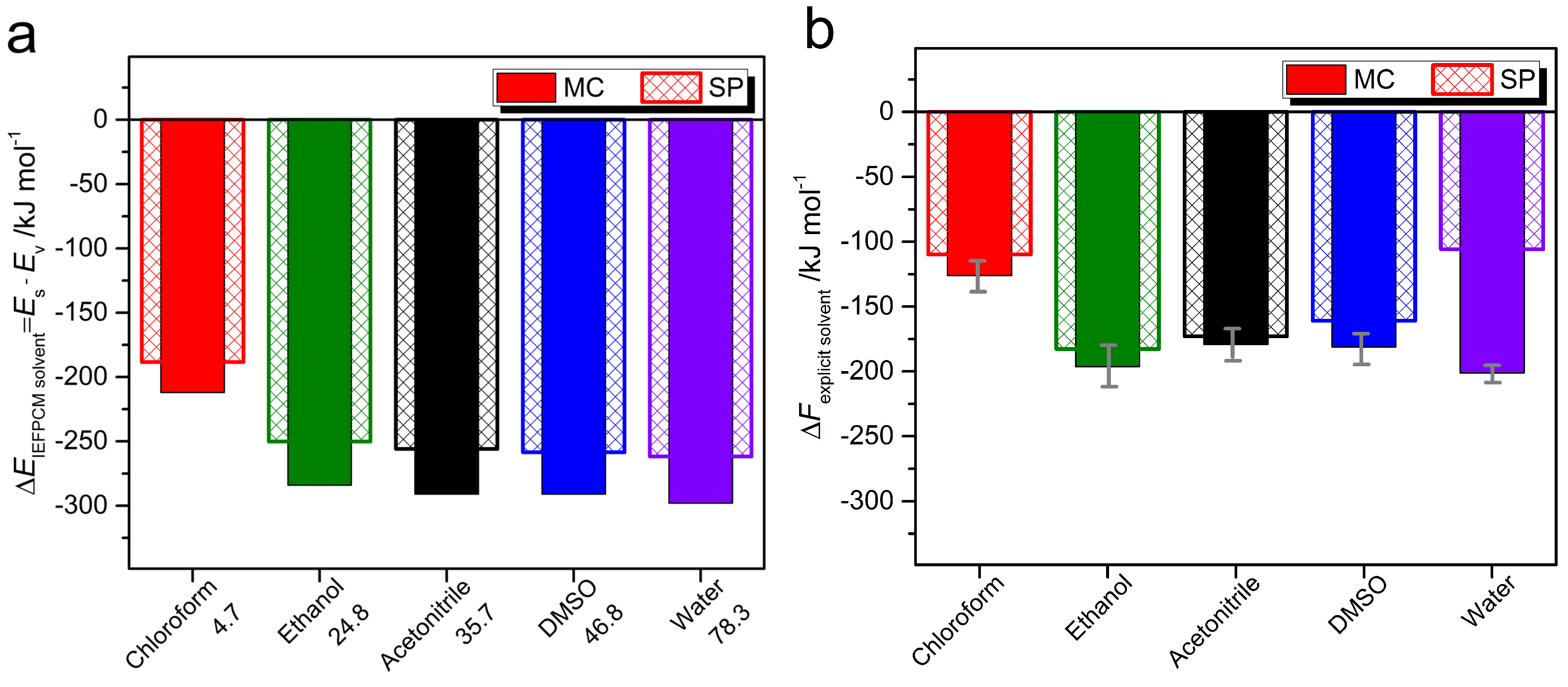
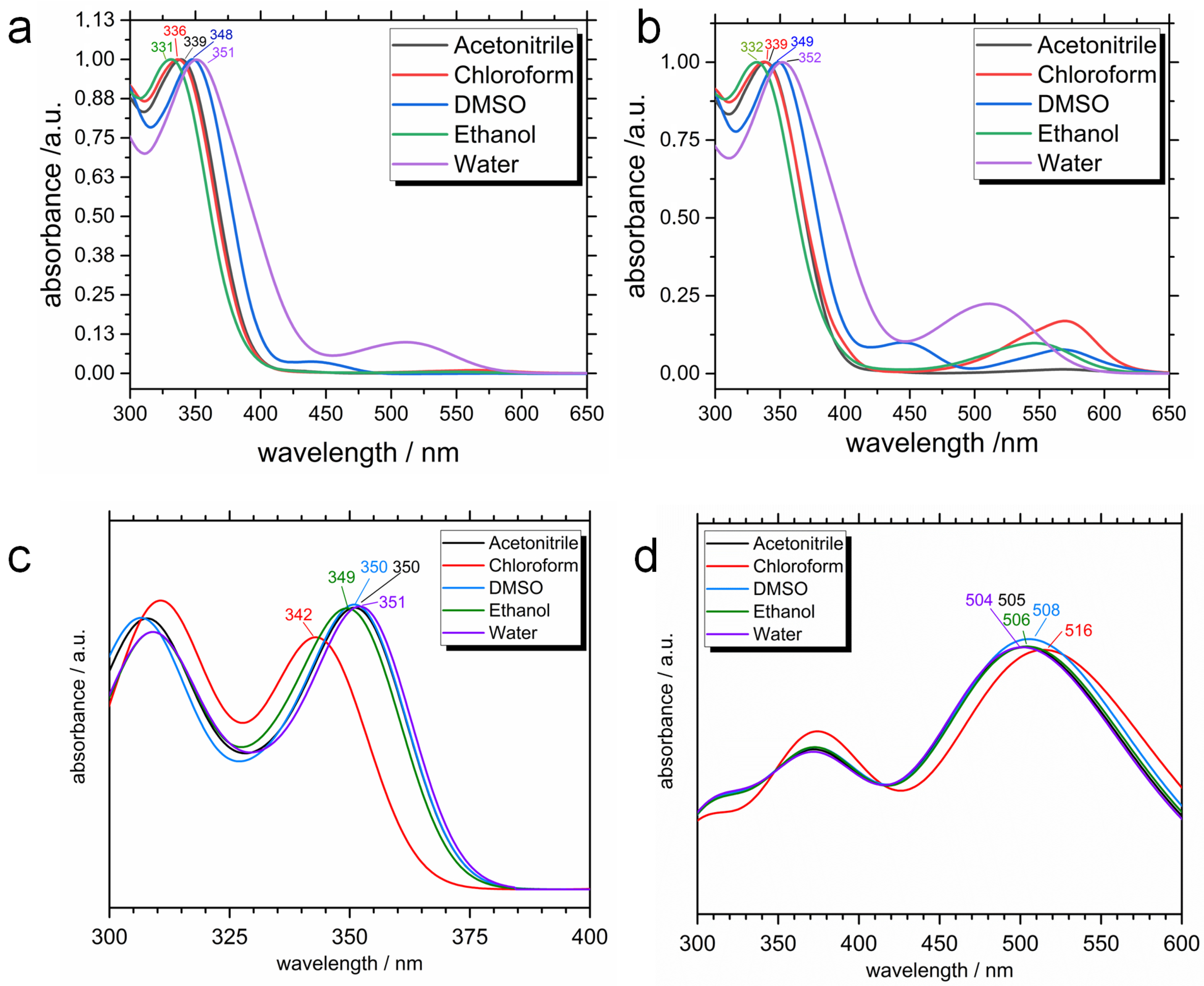
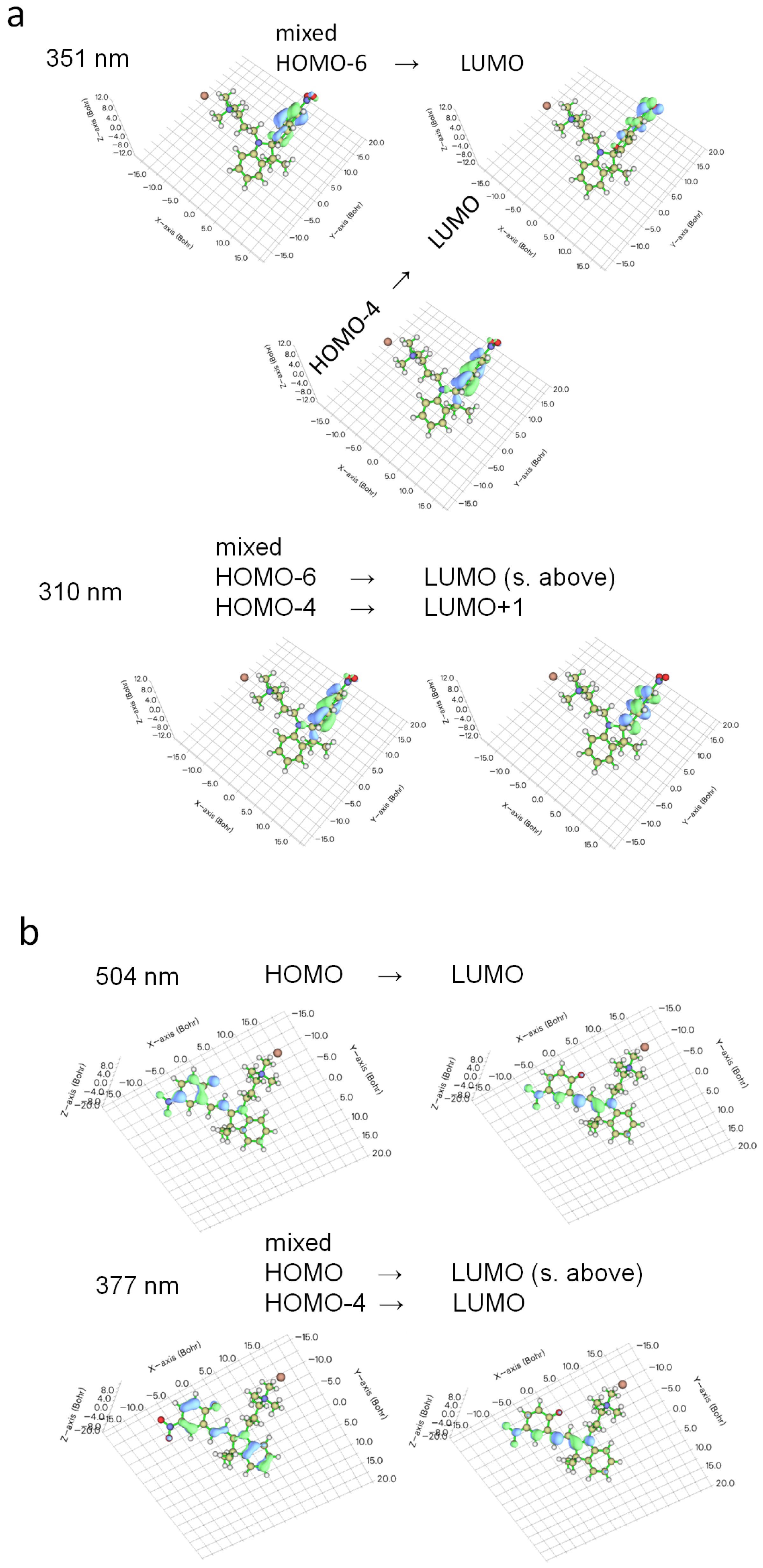
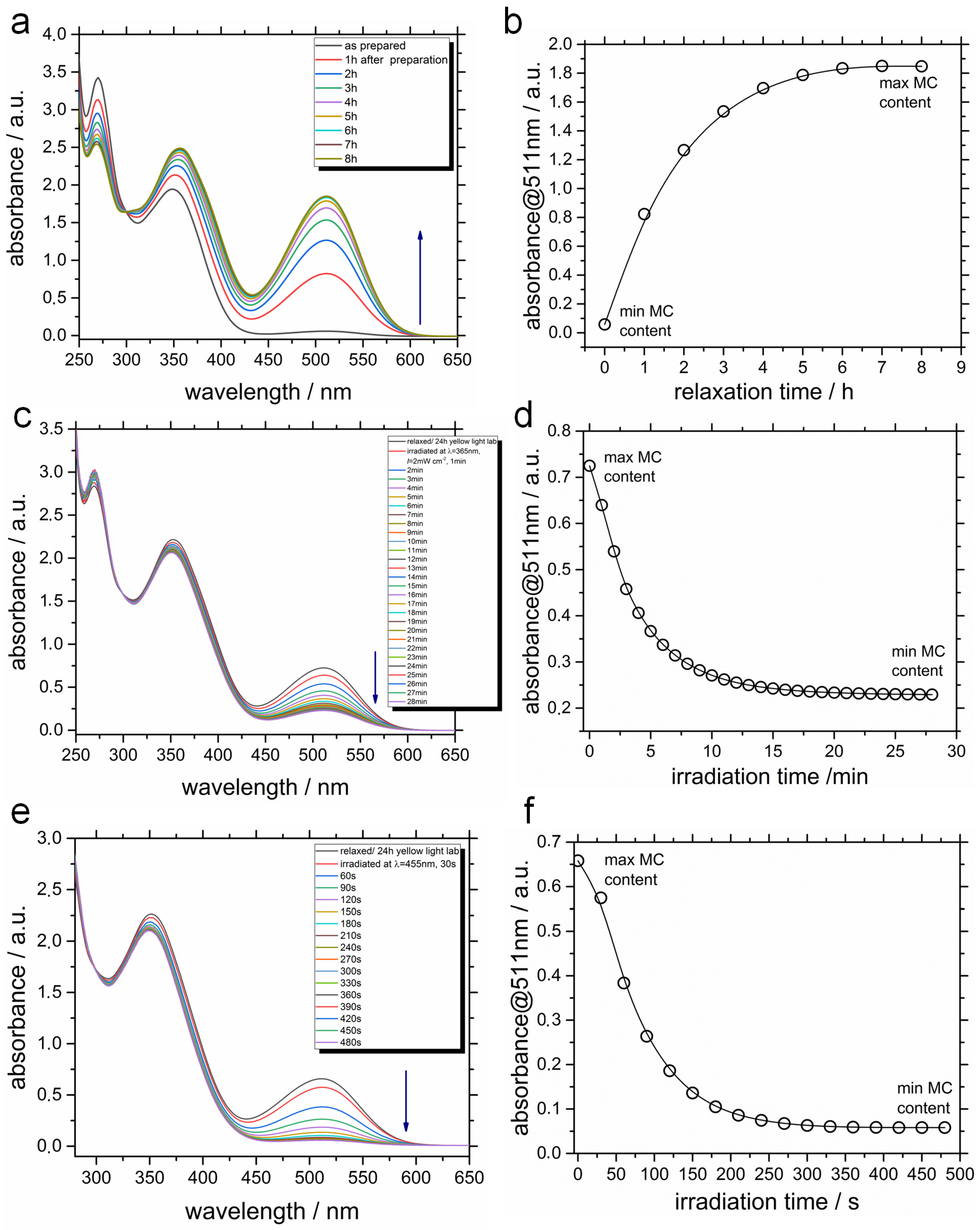
2.2. Photophysical Properties
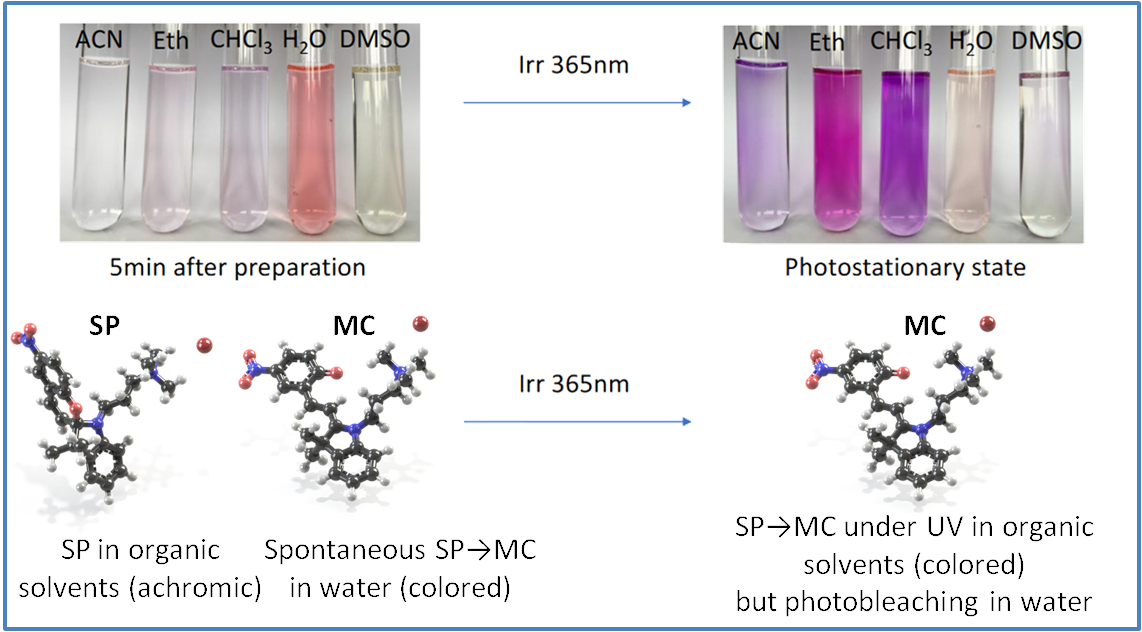
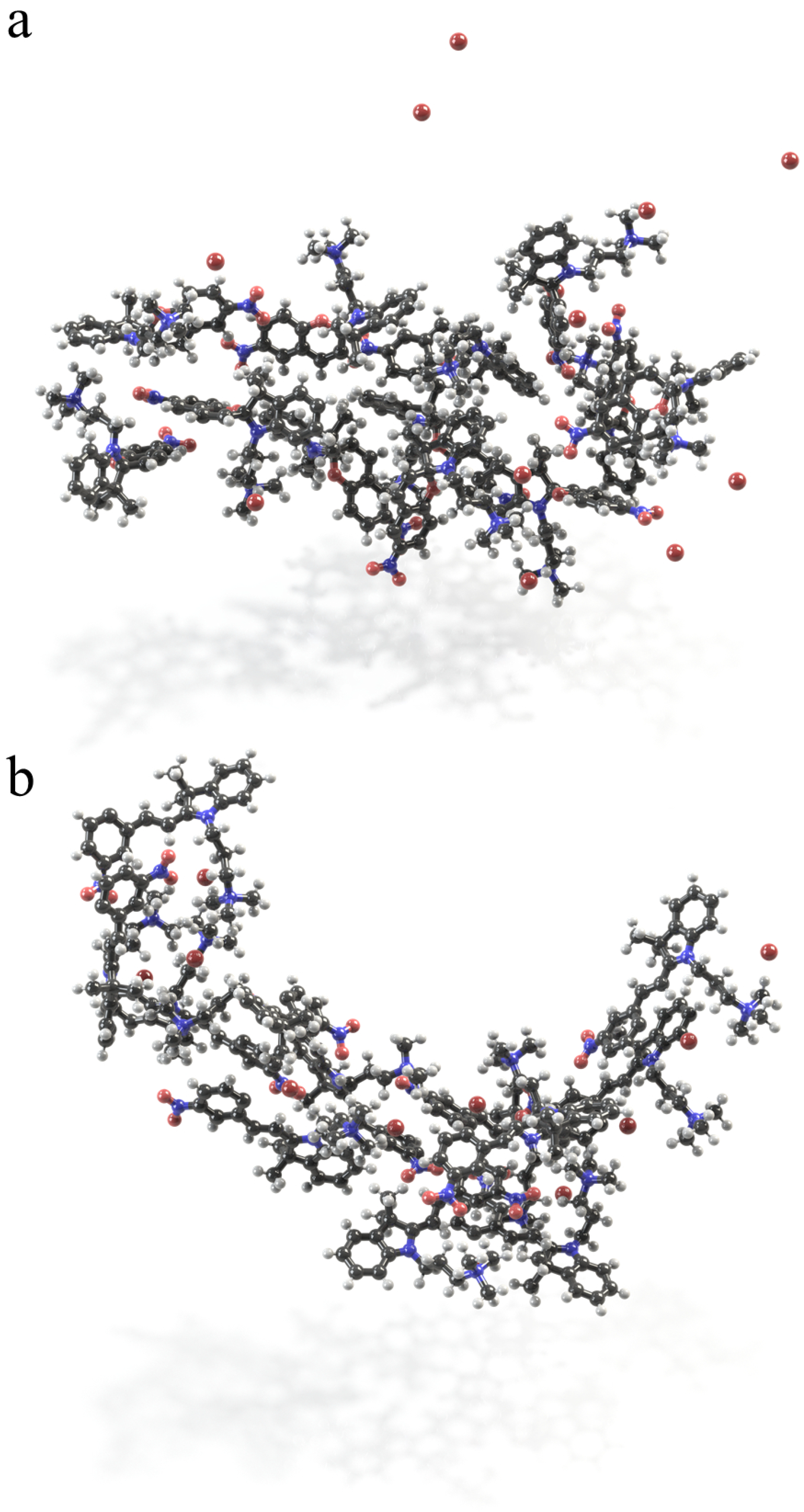
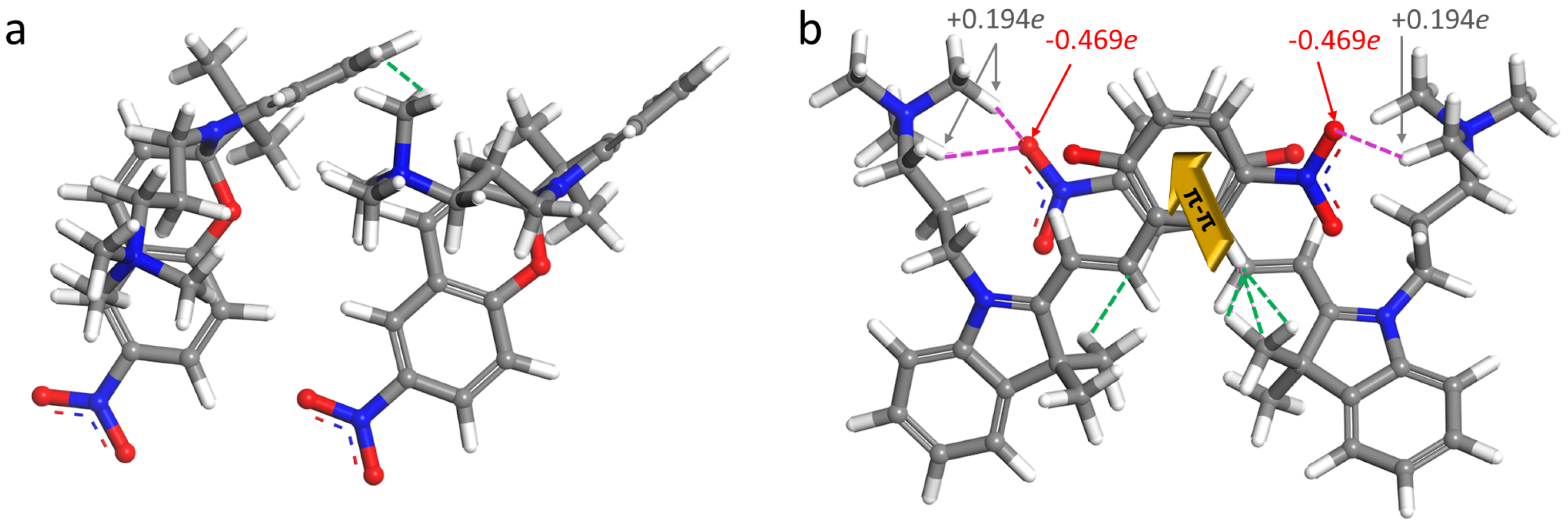
2.3. Self-Assembly in Water
3. Discussion
4. Methods and Materials
4.1. Materials
4.2. Methods
4.2.1. Time Resolved UV-Vis Measurements
4.2.2. DFT and Time-Dependent DFT Calculations
4.2.3. Thermodynamic Integration
4.2.4. Molecular Dynamics Simulation of Self-Assembly in Aqueous Phases
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SP | Spiropyran |
| MC | Merocyanine |
| DFT | Density functional theory |
| MD | Molecular dynamics |
| TI | Thermodynamic integration |
References
- Chen, S.; Costil, R.; Leung, F.K.-C.; Feringa, B.L. Self-Assembly of Photoresponsive Molecular Amphiphiles in Aqueous Media. Angew. Chem. 2021, 133, 11708–11731. [Google Scholar] [CrossRef]
- Volarić, J.; Szymanski, W.; Simeth, N.A.; Feringa, B.L. Molecular Photoswitches in Aqueous Environments. Chem. Soc. Rev. 2021, 50, 12377–12449. [Google Scholar] [CrossRef] [PubMed]
- Montagna, M.; Guskova, O. Photosensitive Cationic Azobenzene Surfactants: Thermodynamics of Hydration and the Complex Formation with Poly (methacrylic acid). Langmuir 2018, 34, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Hayashita, T.; Kurosawa, T.; Miyata, T.; Tanaka, K.; Igawa, M. Effect of Structural Variation within Cationic Azo-Surfactant upon Photoresponsive Function in Aqueous Solution. Colloid Polym. Sci. 1994, 272, 1611–1619. [Google Scholar] [CrossRef]
- Santer, S. Remote Control of Soft Nano-Objects by Light Using Azobenzene Containing Surfactants. J. Phys. D Appl. Phys. 2017, 51, 013002. [Google Scholar] [CrossRef]
- Sharma, A.; Jung, S.-H.; Lomadze, N.; Pich, A.; Santer, S.; Bekir, M. Adsorption Kinetics of a Photosensitive Surfactant Inside Microgels. Macromolecules 2021, 54, 10682–10690. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kishimoto, M.; Kondo, Y. Photoinduced Formation of Threadlike Micelles from Mixtures of a Cationic Surfactant and a Stilbene Amphiphile. J. Colloid Int. Sci. 2016, 470, 250–256. [Google Scholar] [CrossRef]
- Rossos, A.K.; Katsiaflaka, M.; Cai, J.; Myers, S.M.; Koenig, E.; Bucker, R.; Keskin, S.; Kassier, G.; Gengler, R.Y.; Miller, R.D.; et al. Photochromism of Amphiphilic Dithienylethenes as Langmuir–Schaefer Films. Langmuir 2018, 34, 10905–10912. [Google Scholar] [CrossRef]
- Zhao, L.; Seshadri, S.; Liang, X.; Bailey, S.J.; Haggmark, M.; Gordon, M.; Helgeson, M.E.; Read de Alaniz, J.; Luzzatto-Fegiz, P.; Zhu, Y. Depinning of Multiphase Fluid Using Light-and Photo-Responsive Surfactants. ACS Cent. Sci. 2022, 8, 235–245. [Google Scholar] [CrossRef]
- Reifarth, M.; Bekir, M.; Bapolisi, A.M.; Titov, E.; Nußhardt, F.; Nowaczyk, J.; Grigoriev, D.; Sharma, A.; Saalfrank, P.; Santer, S.; et al. A Dual pH and Light-Responsive Spiropyrane-Based Surfactant: Investigations on its Switching Behavior and Remote Control over Emulsion Stability. Angew. Chem. 2022, 134, e202114687. [Google Scholar]
- Hammarson, M.; Nilsson, J.R.; Li, S.; Beke-Somfai, T.; Andreasson, J. Characterization of the Thermal and Photoinduced Reactions of Photochromic Spiropyrans in Aqueous Solution. J. Phys. Chem. B 2013, 117, 13561–13571. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Abbott, N.L. Using Light to Control Dynamic Surface Tensions of Aqueous Solutions of Water Soluble Surfactants. Langmuir 1999, 15, 4404–4410. [Google Scholar] [CrossRef]
- Feldmann, D.; Maduar, S.R.; Santer, M.; Lomadze, N.; Vinogradova, O.I.; Santer, S. Manipulation of Small Particles at Solid Liquid Interface: Light Driven Diffusioosmosis. Sci. Rep. 2016, 6, 36443. [Google Scholar] [CrossRef] [PubMed]
- Schnurbus, M.; Kabat, M.; Jarek, E.; Krzan, M.; Warszynski, P.; Braunschweig, B. Spiropyran Sulfonates for Photo- and pH-Responsive Air–Water Interfaces and Aqueous Foam. Langmuir 2020, 36, 6871–6879. [Google Scholar] [CrossRef] [PubMed]
- Baigl, D. Photo-Actuation of Liquids for Light-driven Microfluidics: State of the Art and Perspectives. Lab Chip 2012, 12, 3637–3653. [Google Scholar] [CrossRef] [PubMed]
- Majee, D.; Presolski, S. Dithienylethene-Based Photoswitchable Catalysts: State of the Art and Future Perspectives. ACS Catal. 2021, 11, 2244–2252. [Google Scholar] [CrossRef]
- Shinohara, M.; Ashikaga, Y.; Xu, W.; Kim, S.; Fukaminato, T.; Niidome, T.; Kurihara, S. Photochemical OFF/ON Cytotoxicity Switching by Using a Photochromic Surfactant with Visible Light Irradiation. ACS Omega 2022, 7, 6093–6098. [Google Scholar] [CrossRef]
- Sakai, H.; Ebana, H.; Sakai, K.; Tsuchiya, K.; Ohkubo, T.; Abe, M. Photo-Isomerization of Spiropyran-Modified Cationic Surfactants. J. Colloid Int. Sci. 2007, 316, 1027–1030. [Google Scholar] [CrossRef]
- Sakai, K.; Imaizumi, Y.; Oguchi, T.; Sakai, H.; Abe, M. Adsorption Characteristics of Spiropyran-Modified Cationic Surfactants at the Silica/Aqueous Solution Interface. Langmuir 2010, 26, 9283–9288. [Google Scholar] [CrossRef]
- Sakai, K.; Yamazaki, R.; Imaizumi, Y.; Endo, T.; Sakai, H.; Abe, M. Adsolubilization by a Photo-responsive Surfactant. Colloids Surf. A Physicochem. Eng. Asp. 2012, 410, 119–124. [Google Scholar] [CrossRef]
- Ohya, Y.; Okuyama, Y.; Fukunaga, A.; Ouchi, T. Photo-Sensitive Lipid Membrane Perturbation by a Single Chain Lipid Having Terminal Spiropyran Group. Supramol. Sci. 1998, 5, 21–29. [Google Scholar] [CrossRef]
- Kim, J.; Yun, H.; Lee, Y.J.; Lee, J.; Kim, S.-H.; Ku, K.H.; Kim, B.J. Photoswitchable Surfactant-Driven Reversible Shape- and Color-Changing Block Copolymer Particles. J. Am. Chem. Soc. 2021, 143, 13333–13341. [Google Scholar] [CrossRef]
- Moo, J.G.S.; Presolski, S.; Pumera, M. Photochromic Spatiotemporal Control of Bubble-propelled Micromotors by a Spiropyran Molecular Switch. ACS Nano 2016, 10, 3543–3552. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, J.; Shen, K.; Wang, G.; Li, Y.; Zhao, D. Synthesis of Novel Photochromic Spiropyran Dyes Containing Quaternary Ammonium Salt or Cinnamoyl Moiety and Their Properties as Photoinitiators. J. Appl. Polym. Sci. 2012, 126, 30–37. [Google Scholar] [CrossRef]
- Holden, D.A.; Ringsdorf, H.; Deblauwe, V.; Smets, G. Photosensitive Monolayers. Studies of Surface-Active Spiropyrans at the Air-Water Interface. J. Phys. Chem. 1984, 88, 716–720. [Google Scholar]
- Tangso, K.J.; Fong, W.-K.; Darwish, T.; Kirby, N.; Boyd, B.J.; Hanley, T.L. Novel Spiropyran Amphiphiles and Their Application as Light-responsive Liquid Crystalline Components. J. Phys. Chem. B 2013, 117, 10203–10210. [Google Scholar] [PubMed]
- Liu, C.; Yang, D.; Jin, Q.; Zhang, L.; Liu, M. A Chiroptical Logic Circuit Based on Self-assembled Soft Materials Containing Amphiphilic Spiropyran. Adv. Mater. 2016, 28, 1644–1649. [Google Scholar]
- Kwangmettatam, S.; Kudernac, T. A Light-Fuelled Reversible Expansion of Spiropyran-Based Vesicles in Water. Chem. Comm. 2018, 54, 5311–5314. [Google Scholar] [CrossRef]
- Khairutdinov, R.F.; Hurst, J.K. A Photocontrol of Ion Permeation through Bilayer Membranes Using an Amphiphilic Spiropyran. Langmuir 2001, 17, 6881–6886. [Google Scholar]
- Tong, R.; Hemmati, H.D.; Langer, R.; Kohane, D.S. Photoswitchable Nanoparticles for Triggered Tissue Penetration and Drug Delivery. J. Am. Chem. Soc. 2012, 134, 8848–8855. [Google Scholar]
- Matsumoto, M.; Nakazawa, T.; Mallia, V.A.; Tamaoki, N.; Azumi, R.; Sakai, H.; Abe, M. Thermal Hysteresis in the Photoresponsivity of a Langmuir Film of Amphiphilic Spiropyran. J. Am. Chem. Soc. 2004, 126, 1006–1007. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, M.; Li, G.; Gu, Z.; Sato, O.; Einaga, Y. Magnetic Vesicles of Amphiphilic Spiropyran Containing Iron Oxide Particles on a Solid State Substrate. Chem. Mater. 2003, 15, 4756–4760. [Google Scholar] [CrossRef]
- Koryako, N.E.; Ivakhnenko, D.A.; Ivakhnenko, A.A.; Lyubimov, A.V.; Zaichenko, N.L.; Lyubimova, G.V.; Arslanov, V.V.; Shokurov, A.V.; Raitman, O.A. Negative Photochromism and Luminescent Properties of Amphiphilic Spiropyran in Solutions and at the Interface. Prot. Met. Phys. Chem. Surf. 2019, 55, 1118–1123. [Google Scholar] [CrossRef]
- Ivakhnenko, D.A.; Shokurov, A.V.; Lyubimova, G.V.; Zaichenko, N.L.; Arslanov, V.V.; Raitman, O.A. Photochromic Transformations of Amphiphilic Spiropyran in Acetonitrile Solutions and at the Air/Water Interface. Russ. Chem. Bull. 2018, 67, 2266–2270. [Google Scholar] [CrossRef]
- Zhang, Y.; Ng, M.; Hong, E.Y.-H.; Chan, A.K.-W.; Wu, N.M.-W.; Chan, M.H.-Y.; Wu, L.; Yam, V.W.-W. Synthesis and Photoswitchable Amphiphilicity and Self-Assembly Properties of Photochromic Spiropyran Derivatives. J. Mater. Chem. C 2020, 8, 13676–13685. [Google Scholar] [CrossRef]
- Tazuke, S.; Kurihara, S.; Yamaguchi, H.; Ikeda, T. Photochemically Triggered Physical Amplification of Photoresponsiveness. J. Phys. Chem. 1987, 91, 249–251. [Google Scholar] [CrossRef]
- Kaiser, C.; Halbritter, T.; Heckel, A.; Wachtveitl, J. Proton-Transfer Dynamics of Photoacidic Merocyanines in Aqueous Solution. Chem. Eur. J. 2021, 27, 9160–9173. [Google Scholar] [CrossRef]
- Gan, M.; Xiao, T.; Liu, Z.; Wang, Y. Layered Photochromic Films Stacked from Spiropyran-Modified Montmorillonite Nanosheets. RSC Adv. 2019, 9, 12325–12330. [Google Scholar] [CrossRef]
- Jonsson, F.; Beke-Somfai, T.; Andreasson, J.; Norden, B. Interactions of a Photochromic Spiropyran with Liposome Model Membranes. Langmuir 2013, 29, 2099–2103. [Google Scholar] [CrossRef]
- Hammarson, M.; Andersson, J.; Li, S.; Lincoln, P.; Andréasson, J. Molecular AND-logic for Dually Controlled Activation of a DNA-binding Spiropyran. Chem. Commun. 2010, 46, 7130–7132. [Google Scholar] [CrossRef]
- Nilsson, J.R.; Li, S.; Önfelt, B.; Andréasson, J. Light-Induced Cytotoxicity of a Photochromic Spiropyran. Chem. Commun. 2011, 47, 11020–11022. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Li, S.; Lincoln, P.; Andréasson, J. Photoswitched DNA-Binding of a Photochromic Spiropyran. J. Am. Chem. Soc. 2008, 130, 11836–11837. [Google Scholar] [CrossRef] [PubMed]
- Wimberger, L.; Prasad, S.K.K.; Peeks, M.D.; Andréasson, J.; Schmidt, T.W.; Beves, J.E. Large, Tunable, and Reversible pH Changes by Merocyanine Photoacids. J. Am. Chem. Soc. 2021, 143, 20758–20768. [Google Scholar] [CrossRef] [PubMed]
- Wimberger, L.; Andréasson, J.; Beves, J.E. Basic-to-Acidic Reversible pH Switching with a Merocyanine Photoacid. Chem. Commun. 2022, 58, 5610–5613. [Google Scholar] [CrossRef]
- Halbritter, T.; Kaiser, C.; Wachtveitl, J.; Heckel, A. Pyridine–Spiropyran Derivative as a Persistent, Reversible Photoacid in Water. J. Org. Chem. 2017, 82, 8040–8047. [Google Scholar] [CrossRef]
- Périllat, V.J.; Berton, C.; Pezzato, C. The Effect of Temperature on the Photoacidity of Merocyanine Photoacids in Water. Mater. Today Chem. 2022, 25, 100918. [Google Scholar] [CrossRef]
- Hughes, J.R.; Miller, A.S.; Wallace, C.E.; Vemuri, G.N.; Iovine, P.M. Biomedically Relevant Applications of Bolaamphiphiles and Bolaamphiphile-Containing Materials. Front. Chem. 2021, 8, 604151. [Google Scholar] [CrossRef]
- Aldaz, C.R.; Wiley, T.E.; Miller, N.A.; Abeyrathna, N.; Liao, Y.; Zimmerman, P.M.; Sension, R.J. Experimental and Theoretical Characterization of Ultrafast Water-Soluble Photochromic Photoacids. J. Phys. Chem. B 2021, 125, 4120–4131. [Google Scholar] [CrossRef]
- Sheng, Y.; Leszczynski, J.; Garcia, A.A.; Rosario, R.; Gust, D.; Springer, J. Comprehensive Theoretical Study of the Conversion Reactions of Spiropyrans: Substituent and Solvent Effects. J. Phys. Chem. B. 2004, 108, 16233–16243. [Google Scholar] [CrossRef]
- Zhai, G.; Shao, S.; Wu, S.; Lei, Y.; Dou, Y. Detailed Molecular Dynamics of the Photochromic Reaction of Spiropyran: A Semiclassical Dynamics Study. Int. J. Photoenergy 2014, 2014, 541791. [Google Scholar] [CrossRef]
- Zhai, G.-H.; Yang, P.; Wu, S.-M.; Lei, Y.-B.; Dou, Y.-S. A Semiclassical Molecular Dynamics of the Photochromic Ring-opening Reaction of Spiropyran. Chin. Chem. Lett. 2014, 25, 727–731. [Google Scholar] [CrossRef]
- Wang, P.-X.; Bai, F.-Q.; Zhang, Z.-X.; Wang, Y.-P.; Wang, J.; Zhang, H.-X. The Theoretical Study of Substituent and Charge Effects in the Conformational Transformation Process of Molecular Machine Unit Spiropyran. Org. Electron. 2017, 45, 33–41. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, G.; Zheng, H.; Zhan, K.; Liu, B.; Zhao, L. Tracking of the Molecular Geometrical Changes in the Primary Event of Photoinduced Ring-opening Reactions of a Spiropyran Model in Gas Phase. Mol. Phys. 2021, 119, e1814971. [Google Scholar] [CrossRef]
- Granucci, G.; Padula, G. Photoisomerization Dynamics of Spiropyran: A Surface-Hopping Investigation. J. Chem. Phys. 2021, 154, 124312. [Google Scholar] [CrossRef]
- Liu, F.; Morokuma, K. Multiple Pathways for the Primary Step of the Spiropyran Photochromic Reaction: A CASPT2/CASSCF Study. J. Am. Chem. Soc. 2013, 135, 10693–10702. [Google Scholar] [CrossRef]
- Murugan, N.A.; Chakrabarti, S.; Ågren, H. Solvent Dependence of Structure, Charge Distribution, and Absorption Spectrum in the Photochromic Merocyanine–Spiropyran Pair. J. Phys. Chem. B 2011, 115, 4025–4032. [Google Scholar] [CrossRef]
- Balasubramanian, G.; Schulte, J.; Müller-Plathe, F.; Böhm, M.C. Structural and Thermochemical Properties of a Photoresponsive Spiropyran and Merocyanine Pair: Basis Set and Solvent Dependence in Density Functional Predictions. Chem. Phys. Lett. 2012, 554, 60–66. [Google Scholar] [CrossRef]
- Eilmes, A. Spiropyran to Merocyanine Conversion: Explicit versus Implicit Solvent Modeling. J. Chem. Phys. A 2013, 117, 2629–2635. [Google Scholar] [CrossRef]
- Singh, R.; Böhm, M.C.; Balasubramanian, G. Energetic and Structural Properties of Different Conformations of Merocyanine and Its Protonated Forms. Chem. Phys. Lett. 2015, 633, 287–291. [Google Scholar] [CrossRef]
- Matczyszyn, K.; Olesiak-Banska, J.; Nakatani, K.; Yu, P.; Murugan, N.A.; Zalesny, R.; Roztoczynska, A.; Bednarska, J.; Bartkowiak, W.; Kongsted, J.; et al. One-and Two-Photon Absorption of a Spiropyran–Merocyanine System: Experimental and Theoretical Studies. J. Phys. Chem. B 2015, 119, 1515–1522. [Google Scholar] [CrossRef]
- Bae, S.Y.; Arnold, B.R. Characterization of Solvatochromic Probes: Simulation of Merocyanine 540 Absorption Spectra in Binary Solvent Mixtures and Pure Solvent Systems. J. Phys. Org. Chem. 2004, 17, 187–193. [Google Scholar] [CrossRef]
- Kortekaas, L.; Browne, W.R. The Evolution of Spiropyran: Fundamentals and Progress of an Extraordinarily Versatile Photochrome. Chem. Soc. Rev. 2019, 48, 3406–3424. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.A.; Rodier, A.R.; Stanley, J.A.; Douglas, N.A.; Li, X.; Brittain, W.J. A Study of the Spiropyran–Merocyanine System Using Ion Mobility-Mass Spectrometry: Experimental Support for the Cisoid Conformation. Chem. Commun. 2014, 50, 3424–3426. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, L.; Chen, J.; Jacquemin, D.; Browne, W.R. Proton-Stabilized Photochemically Reversible E/Z Isomerization of Spiropyrans. J. Phys. Chem. B 2018, 122, 6423–6430. [Google Scholar] [CrossRef]
- Viana, R.B.; da Silva, A.B.F.; Pimentel, A.S. Infrared Spectroscopy of Anionic, Cationic, and Zwitterionic Surfactants. Adv. Phys. Chem. 2012, 2012, 903272. [Google Scholar] [CrossRef]
- Seiler, V.S.; Tumanov, N.; Robeyns, K.; Wouters, J.; Champagne, B.; Leyssens, T.A. Structural Analysis of Spiropyran and Spirooxazine Compounds and Their Polymorphs. Crystals 2017, 7, 84. [Google Scholar] [CrossRef]
- Pugachev, A.D.; Tkachev, V.V.; Ozhogin, I.V.; Lukyanova, M.B.; Aldoshin, S.M.; Minkin, V.I.; Mukhanov, E.L.; Metelitsa, A.V.; Stankevich, N.V.; Lukyanov, B.S. Structures of Spiropyrans Exhibiting Photochromic Properties in the Solid State. Russ. Chem. Bull. Int. Ed. 2021, 70, 2090–2099. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.; Tang, Y.; Zhao, F.; Song, X.; Li, E. Detailed Investigation on a Negative Photochromism Spiropyran. J. Photochem. Photobiol. A Chem. 1995, 90, 117–123. [Google Scholar] [CrossRef]
- Barachevsky, V.A. Negative Photochromism in Organic Systems. Rev. J. Chem. 2017, 7, 334–371. [Google Scholar] [CrossRef]
- Kinashi, K.; Nakamura, S.; Imamura, M.; Ishida, K.; Ueda, Y. The Mechanism for Negative Photochromism of Spiropyran in Silica. J. Phys. Org. Chem. 2012, 25, 462–466. [Google Scholar] [CrossRef]
- Funasako, Y.; Miyazaki, H.; Sasaki, T.; Goshima, K.; Inokichu, M. Synthesis, Photochromic Properties, and Crystal Structures of Salts Containing a Pyridinium-Fused Spiropyran: Positive and Negative Photochromism in the Solution and Solid State. J. Phys. Chem. B 2020, 124, 7251–7257. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2014. [Google Scholar]
- Abdel-Mottaleb, M.S.A.; Ale, S.N. A New Approach for Studying Bund Rupture/Closure of a Spiro Benzopyran Photochromic Materials: Reactivty Descriptors Derived from Frontier Orbitals and DFT Computed Electrostatic Potential Energy Surface Maps. Int. J. Photoenergy 2016, 2016, 6765805. [Google Scholar] [CrossRef]
- Kovalenko, O.; Reguero, M. Why Thermal Isomerization of the Chromic Switch Spiropyran-Merocyanine is Enhance in Polar Protic Solvents. A Computational Study of the Reaction Mechanism. Phys. Scr. 2020, 95, 055402. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Koch, M.; Saphiannikova, M.; Santer, S.; Guskova, O. Photoisomers of Azobenzene Star with a Flat Core: Theoretical Insights into Multiple States from DFT and MD Perspective. J. Phys. Chem. B 2017, 121, 8854–8867. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA; Oxford, UK, 1997. [Google Scholar]
- Piard, J. Influence of the Solvent on the Thermal Back Reaction of One Spiropyran. J. Chem. Educ. 2014, 91, 2105–2111. [Google Scholar] [CrossRef]
- He, J.; Yang, Y.; Li, Y.; He, Z.; Chen, Y.; Wang, Z.; Zhao, H.; Jiang, G. Multiple Anti-Counterfeiting Guarantees from Simple Spiropyran Derivatives with Solid Photochromism and Mechanochromism. Cell Rep. Phys. Sci. 2021, 2, 100643. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Sabey, C.J.; Fernando, G.F. Photoresponsive Polymers: An Investigation of their Photoinduced Temperature Changes During Photoviscosity Measurements. Polymer 2007, 48, 255–263. [Google Scholar] [CrossRef]
- Tyer, N.W., Jr.; Becker, R.S. Photochromic Spiropyrans. I. Absorption Spectra and Evaluation of the π-Electron Orthogonality of the Constituent Halves. J. Am. Chem. Soc. 1970, 92, 1289–1294. [Google Scholar] [CrossRef]
- Favaro, G.; Masetti, F.; Mazzucato, U.; Ottavi, G.; Allegrini, P.; Malatesta, V. Photochromism, Thermochromism and Solvatochromism of Some Spiro [indolinoxazine]-Photomerocyanine Systems: Effects of Structure and Solvent. J. Chem. Soc. Faraday Trans. 1994, 90, 333–338. [Google Scholar] [CrossRef]
- da Costa Duarte, R.; da Silveira Santos, F.; Bercini de Araújo, B.; Cercena, R.; Brondani, D.; Zapp, E.; Bruno Gonçalves, P.F.; Severo Rodembush, F.; Gonxcxalves Dal-Bó, A. Synthesis of a 5-Carboxy Indole-Based Spiropyran Fluorophore: Thermal, Electrochemical, Photophysical and Bovine Serum Albumin Interaction Investigations. Chemosensors 2020, 8, 31. [Google Scholar] [CrossRef]
- Tirri, B.; Mazzone, G.; Ottochian, A.; Gomar, J.; Raucci, U.; Adamo, C.; Ciofini, I. A Combined Monte Carlo/DFT approach to simulate UV-vis Spectra of Molecules and Aggregates: Merocyanine Dyes as a Case Study. J. Comp. Chem. 2021, 42, 1054–1063. [Google Scholar] [CrossRef]
- Kim, H.-J.; Jung, I.-S.; Jung, S.; Kim, D.; Minami, D.; Byun, S.; Choi, T.; Shin, J.; Yun, S.; Heo, C.-J.; et al. Harnessing Intramolecular Chalcogen–Chalcogen Bonding in Merocyanines for Utilization in High-Efficiency Photon-to-Current Conversion Optoelectronics. ACS Appl. Mater. Interfaces 2021, 14, 4360–4370. [Google Scholar] [CrossRef]
- Klajn, R. Spiropyran-Based Dynamic Materials. Chem. Soc. Rev. 2014, 43, 148–184. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Itoh, M.; Hirai, T. Thermal Isomerization of Spiropyran to Merocyanine in Aqueous Media and Its Application to Colorimetric Temperature Indication. Phys. Chem. Chem. Phys. 2010, 12, 13737–13745. [Google Scholar] [CrossRef]
- Filipová, L.; Kohagen, M.; Štacko, P.; Muchova, E.; Slavicek, P.; Klán, P. Photoswitching of Azobenzene-Based Reverse Micelles above and at Subzero Temperatures As Studied by NMR and Molecular Dynamics Simulations. Langmuir 2017, 33, 2306–2317. [Google Scholar] [CrossRef]
- Kasyanenko, N.; Lysyakova, L.; Ramazanov, R.; Nesterenko, A.; Yaroshevich, I.; Titov, E.; Alexeev, G.; Lezov, A.; Unksov, I. Conformational and Phase Transitions in DNA—Photosensitive Surfactant Solutions: Experiment and Modeling. Biopolymers 2015, 103, 109–122. [Google Scholar] [CrossRef]
- Arya, P.; Jelken, J.; Lomadze, N.; Santer, S.; Bekir, M. Kinetics of Photo-Isomerization of Azobenzene Containing Surfactants. J. Chem. Phys. 2020, 152, 024904. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N.; Mitchell, D.; Ninham, B.W. Theory of Self-assembly of Hydrocarbon Amphiphiles into Micelles and Bilayers. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1976, 72, 1525–1568. [Google Scholar] [CrossRef]
- Zhang, Y.; Ng, M.; Chan, M.H.-Y.; Wu, N.M.-W.; Wu, L.; Yam, V.W.-W. Synthesis and Characterization of Photochromic Triethylene Glycol-Containing Spiropyrans and Their Assembly in Solution. Org. Chem. Front. 2021, 8, 3047–3058. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systèmes, Materials Studio 9.0; Dassault Systèmes: San Diego, CA, USA, 2014.
- Fagan, A.; Bartkowski, M.; Giordani, S. Spiropyran-Based Drug Delivery Systems. Front. Chem. 2021, 9, 720087. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Wodak, S.J.; Janin, J. Analytical Approximation to the Accessible Surface Area of Proteins. Proc. Natl. Acad. Sci. USA 1980, 77, 1736–1740. [Google Scholar] [CrossRef]
- Guskova, O.A.; Varanasi, S.R.; Sommer, J.-U. C60-Dyad Aggregates: Self-organized Structures in Aqueous Solutions. J. Chem. Phys. 2014, 141, 144303. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An Approach to Computing Electrostatic Charges for Molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Marcus, Y. The Properties of Solvents; Wiley Series in Solution Chemistry; John Wiley & Sons Ltd.: Chichester, UK, 1998; Volume 4. [Google Scholar]
- Prager, S.; Burghardt, I.; Dreuw, A. Ultrafast CSpiro–O Dissociation via a Conical Intersection Drives Spiropyran to Merocyanine Photoswitching. J. Phys. Chem. A 2014, 118, 1339–1349. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Dielectric Permittivity | MC Form, E | SP Form, E = E – E |
|---|---|---|---|
| chloroform | 4.7113 | 0 | +58.02 |
| ethanol | 24.852 | 0 | +67.52 |
| acetonitrile | 35.688 | 0 | +68.77 |
| dimethyl sulfoxide | 46.826 | 0 | +68.76 |
| water | 78.3553 | 0 | +70.21 |
| Solvent | (g cm) at 25 C [102] | (g cm) | (g cm) |
|---|---|---|---|
| chloroform | 1.4830 | 1.200 | 1.321(MC) 1.340(SP) |
| ethanol | 0.7848 | 0.500 | 0.743(MC) 0.742(SP) |
| acetonitrile | 0.7860 | 0.500 | 0.741(MC) 0.743(SP) |
| dimethyl sulfoxide | 1.0958 | 0.800 | 1.032(MC) 1.042(SP) |
| water | 0.9974 | 0.800 | 0.980(MC) 0.982(SP) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savchenko, V.; Lomadze, N.; Santer, S.; Guskova, O. Spiropyran/Merocyanine Amphiphile in Various Solvents: A Joint Experimental–Theoretical Approach to Photophysical Properties and Self-Assembly. Int. J. Mol. Sci. 2022, 23, 11535. https://doi.org/10.3390/ijms231911535
Savchenko V, Lomadze N, Santer S, Guskova O. Spiropyran/Merocyanine Amphiphile in Various Solvents: A Joint Experimental–Theoretical Approach to Photophysical Properties and Self-Assembly. International Journal of Molecular Sciences. 2022; 23(19):11535. https://doi.org/10.3390/ijms231911535
Chicago/Turabian StyleSavchenko, Vladyslav, Nino Lomadze, Svetlana Santer, and Olga Guskova. 2022. "Spiropyran/Merocyanine Amphiphile in Various Solvents: A Joint Experimental–Theoretical Approach to Photophysical Properties and Self-Assembly" International Journal of Molecular Sciences 23, no. 19: 11535. https://doi.org/10.3390/ijms231911535
APA StyleSavchenko, V., Lomadze, N., Santer, S., & Guskova, O. (2022). Spiropyran/Merocyanine Amphiphile in Various Solvents: A Joint Experimental–Theoretical Approach to Photophysical Properties and Self-Assembly. International Journal of Molecular Sciences, 23(19), 11535. https://doi.org/10.3390/ijms231911535