1. Introduction
Essential cellular functions are executed and regulated by macromolecular complexes comprising many subunits, such as the ribosome, proteasome, replisome, and spliceosome. Their structures in functional cycles are often compositionally heterogeneous and conformationally dynamic, involving many reversibly associated components in cells. Visualizing the conformational continuum of highly dynamic megadalton complexes at high resolution is crucial for understanding their functional mechanisms and for guiding structure-based therapeutic development. The current approaches in cryogenic electron microscopy (cryo-EM) structure determination allow atomic-level visualization of major conformational states of these dynamic complexes [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12]. However, high-resolution structure determination of transient, nonequilibrium intermediates connecting their major states during their functional cycles has been prohibitory at large. To date, there has been a lack of appropriate approaches to visualizing the conformational space of large biomolecular complexes at high resolution.
The problem of structural heterogeneity in cryo-EM structure determination has been investigated with numerous computational approaches including maximum-likelihood-based classification and multivariate statistical analysis [
5,
6,
11,
13,
14,
15,
16,
17,
18]. As an extension of multivariate statistical analysis, several machine-learning approaches were recently proposed to estimate a continuous conformational distribution in the latent space [
11,
12,
19,
20,
21,
22]. The estimation of continuous conformational distributions does not guarantee an improvement of 3D classification accuracy nor warrant the identification of hidden conformational states of biological importance. These methods often trade off the reconstruction resolution for gaining an overall representation of the conformational landscape in the latent space. The insufficient resolution would preclude their potential applications in structure-based drug discovery.
The most widely used method so far to counteract structural heterogeneity in cryo-EM for resolution improvement is the hierarchical maximum-likelihood-based 3D classification due to its ease of application [
1,
23,
24]. To improve the resolution of major conformers, low-quality classes are often manually removed during iterations of curated hierarchical classification, inevitably causing an incomplete representation of the true conformational landscape [
25,
26]. The outcome of refined cryo-EM maps depends on user expertise in decision-making in class selection [
27], which can be biased by user experience and subjectivity. Importantly, a considerable portion of misclassified images can limit the achievable resolution of observed conformational states and result in missing states that could be biologically important.
In addition to the approaches characterizing global structural heterogeneity, other methods were proposed to analyze local structural dynamics or to improve the local resolution of flexible regions in cryo-EM reconstructions. These include the multi-body or focused refinement [
4,
28,
29], normal mode analysis [
30,
31], flexible refinement [
32,
33], non-uniform refinement [
34] and integrating traditional molecular dynamics (MD) simulations with or without use of deep learning [
35,
36]. Nonetheless, these methods were not designed to recover a complete picture of structural dynamics hidden in cryo-EM datasets.
The energy landscape is a statistical physical representation of the conformational space of a macromolecular complex and is the basis of the transition-state theory of chemical reaction dynamics [
37,
38]. The minimum-energy path (MEP) on the energy landscape theoretically represents the most probable trajectory of conformational transitions and can inform the activation energy for chemical reactions [
38,
39]. Previous studies have demonstrated the benefit of pseudo-energy landscape estimation in characterizing conformational variation of macromolecules from cryo-EM data [
40,
41,
42,
43,
44,
45,
46]. In these studies, single-particle images classified into the same conformation were assumed to have approximately similar free energy. Thus, the relative energy differences between distinct conformations could be computed by the ratio of particle numbers classified into different conformers using the Boltzmann relation, provided that the molecular system can be approximately described as a canonical ensemble. Because of inaccuracy in 3D classification, a potential violation of canonical ensemble approximation and possible compositional heterogeneity, such a procedure may be at best considered to yield a pseudo-energy landscape.
The combination of linear dimensionality reduction via 3D principal component analysis (PCA) and pseudo-energy landscape estimation has been demonstrated to be capable of capturing the overall conformational landscape [
41,
42,
43]. The benefit of using nonlinear dimensionality reduction and manifold embedding, such as diffusion map [
47], to replace PCA for estimating pseudo-energy landscapes has also been investigated at a limited resolution, with the assumption that the conformational changes can be discerned through a narrow angular aperture [
40,
44,
45]. This assumption restricts its potential applications to more complicated conformational dynamics.
Breaking the resolution barrier in visualizing the conformational space of highly dynamic complexes represents a formidable technical challenge. Achieving this goal may further leverage cryo-EM applications in both basic biomedical science and drug discovery, where high-resolution features of highly dynamic components may provide crucial mechanistic insights and guide more accurate structure-based ligand design. This would require a substantial improvement in 3D classification techniques that are optimized for achieving higher resolution. We hypothesize that a high-quality estimation of the pseudo-energy landscape could be potentially used to improve the 3D classification of highly heterogeneous cryo-EM datasets. If different conformers or continuous conformational changes can be sufficiently mapped and differentiated on a pseudo-energy landscape, the energetic visualization of the conformational continuum could then be used to discover previously ‘invisible’ conformers and achieve higher resolution for lowly populated, transient, or nonequilibrium states.
To explore these ideas in this study, we developed a novel machine learning framework named AlphaCryo4D that can break the existing limitations and enable 4D cryo-EM reconstruction of highly dynamic, lowly populated intermediates or transient states at the atomic level. We examined the general applicability of AlphaCryo4D in analyzing several large synthetic datasets over a wide range of signal-to-noise ratios (SNRs) and four experimental cryo-EM datasets of diverse sample behaviors in complex dynamics, including the human 26S proteasome [
25], malaria parasite 80S ribosome [
48], pre-catalytic spliceosome [
49] and bacterial ribosomal assembly intermediates [
50]. Our approach pushes the envelope of visualizing the conformational space of macromolecular complexes beyond the previously achieved scope toward the atomic level.
3. Discussion
One primary objective of the AlphaCryo4D development is to simultaneously improve reconstruction resolution and 3D classification accuracy without assumption on sample behaviors, such as conformational heterogeneity, continuity, or chemical composition. Importantly, AlphaCryo4D has recently been used to improve the state-resolving capacity of time-resolved cryo-EM analysis to yield more than a dozen intermediate conformers of USP14-regulated human proteasomes at atomic detail [
63]. Because time-resolved cryo-EM is often conducted on samples flash-frozen in the course of biochemical reactions, such samples often contain a much greater degree of compositional and conformational heterogeneity that may be intuitively pertinent to the functional, lowly populated intermediate states of the imaged systems. Thus, time-resolved cryo-EM in principle requires higher 3D classification accuracy to disentangle many coexisting, low-abundance conformers toward a high enough resolution [
63]. This is a major aspect limiting the general application of time-resolved cryo-EM, in which it is often a formidable challenge to make high-resolution sense of time-resolved datasets. Thus, combining a method of high accuracy in 3D classification with time-resolved cryo-EM may be practically desirable and present an emerging paradigm in visualizing functional complex dynamics [
63]. Several attempts have been made recently to characterize the so-called ‘continuous motion’ of protein complexes using cryo-EM data [
11,
12,
19,
20,
21,
22]. Nonetheless, it remains hypothetical that the conformational space of protein complex dynamics must be continuous. This may be approximately sensible in a classic picture of molecular mechanics but is not necessarily appropriate in the theoretical framework of quantum mechanics. Thus, a structural ensemble discovered by AlphaCryo4D is preferentially regarded as discrete approximations representing the conformational space of fundamentally unknown continuity.
This study further substantiates the notion that the improved accuracy of 3D classification is positively correlated with the resolution gain because misclassification reduces achievable resolution for reconstructing highly dynamic conformations. Because of extremely low SNR, most objective functions in image similarity measurement and machine learning algorithms tend to poorly perform or entirely fail on cryo-EM data. To date, no methods have included any implicit metrics or strategies for validating 3D classification accuracy in cryo-EM. We address this issue by introducing the M-fold particle shuffling method and the energy-based particle voting algorithm in AlphaCryo4D that automatically checks the reproducibility and robustness of the 3D classification. This allows users to objectively reject non-reproducible particles and permits a maximal number of particles to be assessed and classified in an integrative procedure based on uniform, objective criteria. Our extensive tests suggest that the particle-voting algorithm synergistically enhances the performance of deep manifold learning. Moreover, it can potentially rescue certain particles that are prone to be misclassified if processed only once by deep manifold learning, thus boosting the efficiency of particle usage without necessarily sacrificing the quality and homogeneity of selected particles. This mechanism simultaneously optimizes the usage of all available particles and their 3D classification accuracy for achieving higher resolution.
Our particle-shuffling and Bayesian resampling strategy are conceptually distinct from the stochastic bootstrap method previously proposed for estimating 3D variance maps [
65] or performing 3D PCA [
13] to detect conformational variability. One essential difference lies in that our resampling approach requires that only particles of similar or identical conformations are grouped together via likelihood-based similarity estimation instead of being combined or resampled in a stochastic manner [
1,
18]. Another difference is that the goal of
M-fold particle shuffling is to improve the quality of the reconstituted pseudo-energy landscape and to enable energy-based particle voting for the intrinsic cross-validation of 3D classification. Furthermore, AlphaCryo4D does not assume any prior knowledge regarding the conformational landscape, its continuity and topology, as well as chemical composition of the macromolecular system. Our approach allows multiple consensus models being used for optimizing the particle alignment during Bayesian resampling. This can theoretically deal with more complicated conformational dynamics when no stable core structure is available to guide the consensus alignment over a single model.
Using a pseudo-energy landscape to represent the conformational variation has theoretical roots in the physical chemistry of protein dynamics. Extensive research tools being built upon the energy landscape and transition-state theory allow comparison with other complementary approaches, such as single-molecule florescence microscopy, and nuclear magnetic resonance (NMR) and molecular dynamics (MD) simulation. Particularly, all-atom MD simulation with enhanced sampling techniques allows for the computation of the virtual energy landscape of kilodalton biomolecular systems over a limited timescale often up to a millisecond [
66,
67]. Due to the limited capacity of supercomputing resources, this approach is not yet commonly feasible for recapitulating large complex conformational changes of megadalton biomolecular systems in their functional actions. By contrast, AlphaCryo4D reconstitutes pseudo-energy landscape from experimental datasets of megadalton molecular weight, which may sample significantly larger conformational space over a functional course of much longer timescale up to minutes and even hours, when it is used in conjunction with time-resolved cryo-EM [
63]. With the continuous growth of supercomputing capacity, it might become possible in the future for the two approaches to be compared directly and cross-validated for the results from each other, given certain caveats being adequately dealt with, such as the appropriate calibration of energy scale and authentic estimation of energy resolution. Alternatively, part of the enhanced sampling techniques, such as the string method, has been integrated into the framework of AlphaCryo4D. Thus, it should be feasible to borrow other enhanced sampling techniques in the future development of AlphaCryo4D [
66,
67].
However, the primary goal of mapping pseudo-energy landscape in AlphaCryo4D is to improve the accuracy of 3D classification rather than the pursuit of the authenticity of characterizing free-energy landscape, owing to a limited energy resolution and non-intuitive dimensionality reduction used for the mapping procedure. In addition, the pseudo-energy landscape allows users to qualitatively examine kinetic relationships between the adjacent conformers and potentially discover new conformations. Quantitatively deriving biochemical kinetics from such a pseudo-energy landscape is not advised and may be error-prone. We stress that the reconstitution of an authentic free-energy landscape from cryo-EM data remains an open question rather than a solved one. While our method of mapping 3D volumes to manifolds increases the sampling grain in approximating the conformational space as compared to other methods directly mapping single particles to the latent space [
11,
12,
19], AlphaCryo4D trades off the fineness of sampling grain in visualizing conformational space for resolution gain. In nearly all cases we examined, AlphaCryo4D apparently still oversampled the conformational space at large in terms of finding those conformational states that can potentially achieve high resolution.
The key steps of AlphaCryo4D processing add about 20% of computational costs in addition to those commonly practiced steps, including initial consensus alignment and final high-resolution refinement (see
Section 4.15 below). We speculate that larger datasets are generally required to fully exploit the advantages and potentials of cryo-EM in solving heterogeneous structures, no matter which approach is used. The data size is expected to proportionally scale with and match the degree of conformational heterogeneity to visualize a full spectrum of conformational states at the atomic level. Thus, further optimization of the algorithmic design and code-executing efficiency via software engineering will help to reduce the computational costs and to make AlphaCryo4D more affordable for widespread applications.
Several limitations of AlphaCryo4D are in order. First, its outcomes depend on the success of the initial consensus alignment of all particles during particle shuffling and volume resampling. Certain conformational dynamics or heterogeneity can interfere with image alignment, in which case the performance of AlphaCryo4D will be restricted by alignment errors after consensus refinement. This is a common problem for all existing methods developed so far. Our tentative solution is to improve image alignment in a multi-reference refinement procedure. Failure in obtaining accurate alignment parameters can lead to futile, erroneous estimation of conformational heterogeneity in the subsequent steps no matter which methods are used.
Second, the use of t-SNE to visualize a pseudo-energy landscape poses both potential advantages and limitations. The t-SNE algorithm can well preserve the local topology of a pseudo-energy landscape but is not warranted to preserve global topology, which is nonetheless a common limitation for existing manifold learning techniques. The geometry of the estimated pseudo-energy landscape is inevitably influenced by the SNRs of cryo-EM data; lower SNRs cause dispersion of local energy wells and thus the overall geometrical distortion of the landscape (
Supplementary Figure S1). A higher level of noise introduces greater statistical bias in particle clustering into resampled 3D volumes and causes irreproducible particle ‘votes’. To alleviate this issue, the particle-voting algorithm is employed in the final 3D classification of the local areas of energy minima on a pseudo-energy landscape, which rejects particles with irreproducible votes.
Last, all existing approaches for visualizing a pseudo-energy landscape from cryo-EM data suffer from the ‘curse of dimensionality’, because the dimensions of reaction coordinates related to the conformational variation are expected to be very high in real space for a megadalton macromolecular complex that includes hundreds of thousands of atoms [
40,
41,
42,
43,
44,
45,
46]. Thus, the reduction of dimensions prior to the visualization of a pseudo-energy landscape is necessary. The reduced dimensions then lose intuitive physical meanings that differ from case to case. In all cases of experimental and simulated datasets in this study, the reduced dimensions by t-SNE appear to capture the most prominent conformational changes, analogous to the principal components computed by PCA. Thus, dimensionality reduction to 2D by t-SNE, as well as MEP searching on a 2D pseudo-energy landscape might limit its capability in disentangling complex dynamics governed by many reaction coordinates. Further investigations are required to understand how to map individual particles to pseudo-energy landscape without trading off reconstruction resolution, to estimate pseudo-energy landscape at higher dimensions, to preserve its global topology with high fidelity and to fully automatize MEP solution.
In summary, the development of AlphaCryo4D and its applications in several important biomolecular systems have demonstrated the feasibility of atomic-level visualization of conformational space hidden in a non-equilibrium regime or corresponding to transient transitions between different metastable states, particularly for biomolecular systems that are too large or complex to be studied by all-atom MD simulations. Applications of this approach to important therapeutic targets in the future might unlock atomic views of cryptic or mobile sites for ligand discovery that otherwise remain invisible to alternative techniques. Our studies of AlphaCryo4D provide essential mechanistic insights into the computational problem of efficiently extracting atomic-level dynamics information from cryo-EM data. We expect that AlphaCryo4D will become an important steppingstone and inspire more efforts in leveraging machine learning algorithms to develop the next-generation cryo-EM pipelines that overcome the aforementioned limitations and push the envelope of visualizing functional dynamics of nonequilibrium biomolecular systems at the atomic level.
4. Materials and Methods
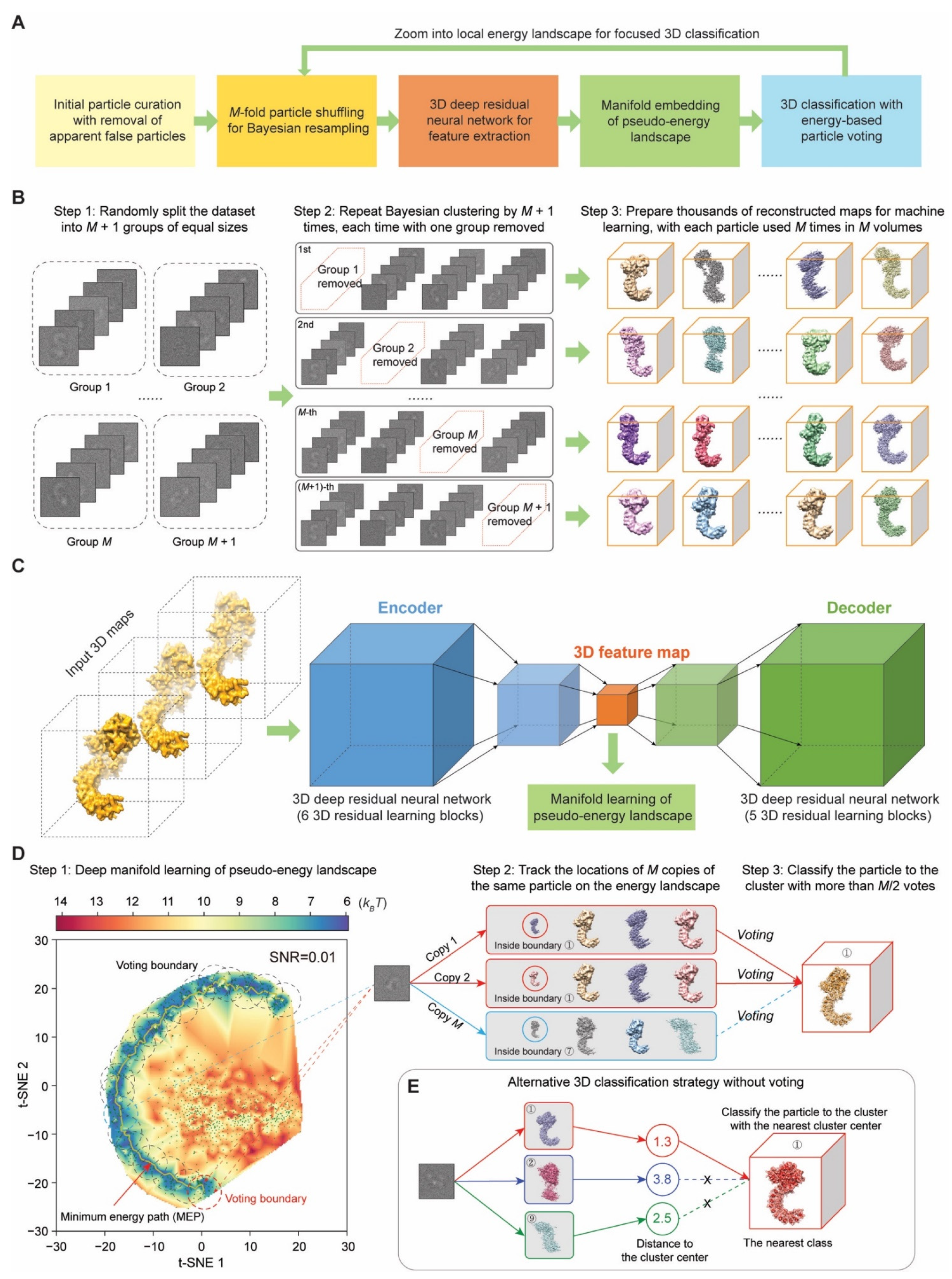
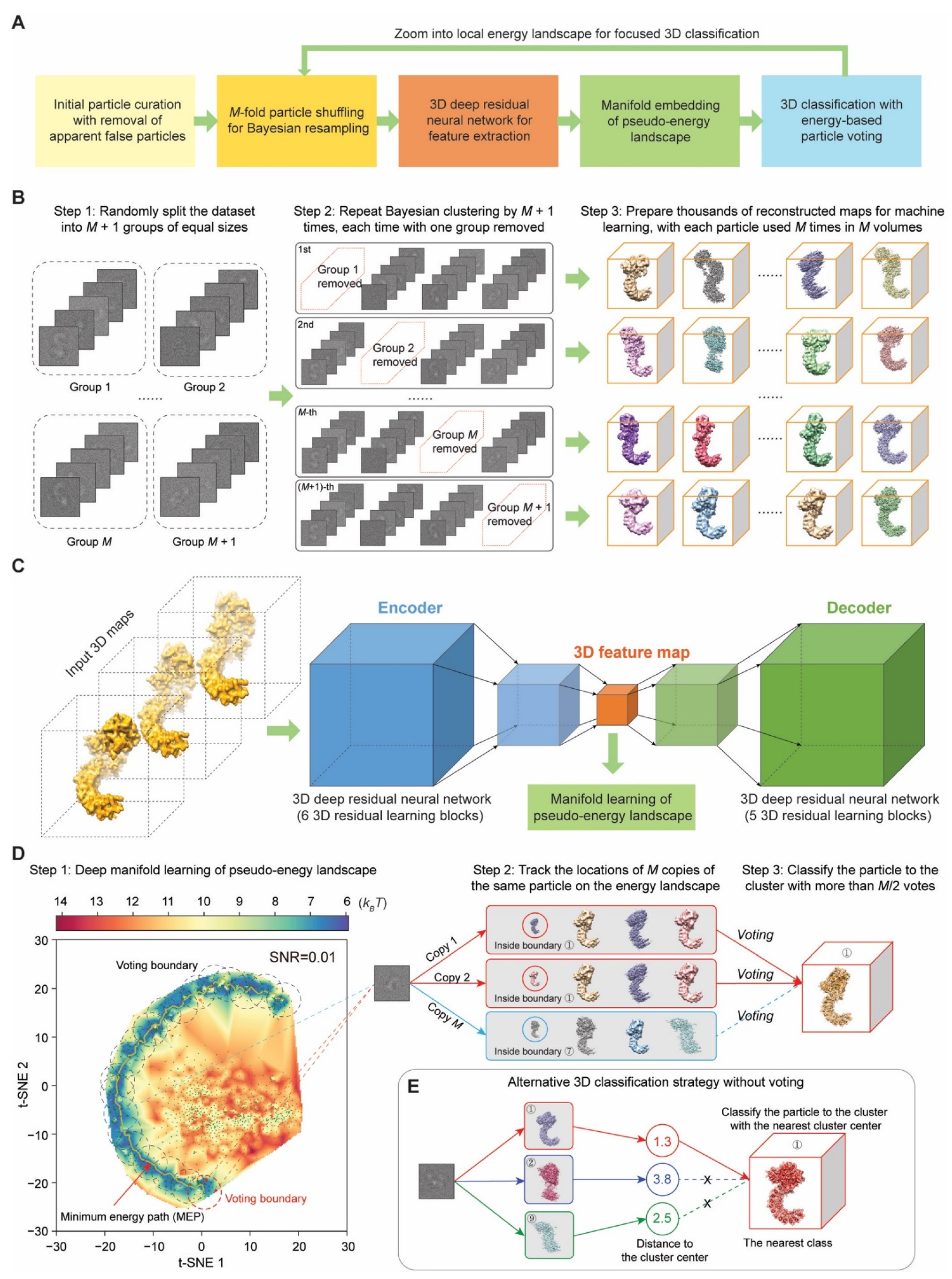
4.1. M-Fold Particle Shuffling and Bayesian Resampling
A key philosophy of the AlphaCryo4D design is to avoid subjective judgment on the particle quality and usability if it is not apparent false particles, such as ice contaminants, carbon edges and other obvious impurities. Deep-learning-based particle picking in DeepEM v1.0 (Peking University, Beijing, China) [
68] or other similarly performed software is favored for data preprocessing prior to AlphaCryo4D. To prepare particle datasets for AlphaCryo4D, an initial unsupervised 2D image classification and particle selection, preferentially conducted by the statistical manifold-learning-based algorithm in ROME v1.1.2 (Peking University, Beijing, China) [
5], is necessary to ensure that no apparent false particles are selected for further analysis and that the data have been collected under an optimal microscope alignment condition, such as optimized coma-free alignment. No particles should be discarded based on their structural appearance during this step if they are not apparent false particles. Any additional 3D classification should be avoided to pre-maturely reject particles prior to particle shuffling and volume resampling in the first step of AlphaCryo4D processing. Pre-maturely rejecting true particles via any 2D and 3D classification is expected to introduce subjective bias and impair the native conformational continuity and statistical integrity intrinsically existing in the dataset.
In raw cryo-EM data, 2D transmission images of biological macromolecules suffer from extremely heavy background noise, due to the use of low electron dose to avoid radiation damage. To tackle the conformational heterogeneity of the macromolecule sample of interest in the presence of heavy image noise, the particle shuffling and volume resampling procedure was devised to incorporate the Bayesian or maximum-likelihood-based 3D clustering in RELION v2.1 or v3.0 (MRC, Cambridge, UK) [
3]. To enable the particle-voting algorithm in the late stage of AlphaCryo4D, each particle is reused
M times during particle shuffling to resample a large number of 3D volumes (
Figure 1B). First, all particle images are aligned to the same frame of reference in a consensus 3D reconstruction and refinement in RELION [
3] or ROME [
5] to obtain the initial alignment parameters of three Euler angles and two translational shifts. Optimization for alignment accuracy should be pursued to avoid the error propagation to the subsequent steps in AlphaCryo4D. For a dataset with both compositional and conformational heterogeneity, coarsely classifying the dataset to a few 3D classes during initial image alignment, limiting the 3D alignment to a moderate resolution, such as 10 Å or 15 Å during a global orientational search, progressing to small enough angular steps in the final stage of consensus refinement, may be optionally exercised to optimize the initial 3D alignment. Failure of initial alignment of particles would lead to failure of all subsequent AlphaCryo4D analyses.
Next, based on the results of consensus alignment, in the particle-shuffling step, all particles were divided into M + 1 groups, and M was set to an odd number of at least 3. Then the whole dataset was shuffled M + 1 times by removing a different group from the dataset each time. Each shuffled dataset is classified into a large number (B) of 3D volumes, often tens to hundreds, by the maximum-likelihood 3D classification algorithm without further image alignment in RELION (with ‘skip-align’ option turned on). This is necessary because the alignment accuracy often degrades when the sizes of 3D classes decrease considerably. This step is repeated M + 1 times, each time on a shuffled dataset missing a different group among the M + 1 groups. Because of M-fold particle shuffling, the outcome of this entire process is expected to resample up to thousands of 3D volumes in total (i.e., B (M + 1) > 1000). Each particle is used and contributed to the 3D reconstructions of M volumes, which prepare it for the particle-voting algorithm to evaluate the robustness and reproducibility of each particle with respect to the eventual 3D classification.
For processing a large dataset including millions of single-particle images, it becomes infeasible even for a modern high-performance computing system to do the consensus alignment by including all particles once in a single run due to the limitations of supercomputer memory and the scalability of the alignment software. To tackle this issue, the whole dataset is randomly split into a number (D) of sub-datasets for batch processing, with each sub-dataset including about one to two hundred thousand particles, depending on the scale of the available supercomputing system. In this case, the initial reference should be used for the consensus alignment of different sub-datasets to minimize volume alignment errors in the later step. The total number of resulting resampled volumes becomes BD (M + 1) > 1000. This strategy can substantially reduce the supercomputer memory pressure and requirement. In each sub-dataset, all particles were divided into M + 1 groups and subject to the particle shuffling and volume resampling procedure as described above.
4.2. Deep Residual Autoencoder for 3D Feature Extraction
The resampled volumes may still suffer from reconstruction noises and errors due to variation of particle number, misclassification of conformers and limited alignment accuracy. Thus, we hypothesize that unsupervised deep learning could help capture the key features of structural variations in the resampled volumes and potentially enhance the quality of subsequently reconstituted pseudo-energy landscapes for improved 3D classification. To this end, a 3D autoencoder was constructed using a deep Fully Convolutional Network (FCN) composed of residual learning blocks [
51,
52]. The architecture of the 3D autoencoder consists of the encoder and the decoder, which are denoted as
and
, respectively (
Figure 1C). The relation between the output
and the input
of the network can be expressed as
, in which
is the input 3D density volume with the size of
N3, where
N is the box size of the density map in pixel units. For reconstruction of the 3D volumes and further optimization, the decoding output
should be in the same size and range with the input data
. In this way, the framework of FCN is established to restore the input volume, using the sigmoid function
as the activation function of the decoding layer to normalize the value of
into the range (0, 1). Meanwhile, all 3D density maps
are normalized with the following equation before input to the deep neural network:
where
and
are, respectively, the minimum and minimax value in all
.
The distance between the decoded maps and the input volumes can be used for constructing the loss function to train the 3D kernels and bias of the networks. The value distribution of the encoded 3D feature maps
is expected to be an abstract, numerical representation of the underlying structures in the volume data, which may not necessarily have any intuitive real-space physical meanings. The neural network can extract such abstract information in the prediction step, with no restriction on the feature maps
in the expression of training loss. The loss function is then formulated as:
where
denotes the weights and bias of the network, and
is the L2 norm regularization coefficient. As the feature of the complex structure is difficult to be learned from the 3D volume data, the value of
is set to 0.001 in default to prevent overfitting in the applications to experimental datasets. However, it is set to 0 in the tests on the simulated datasets (see below).
To improve the learning capacity of the 3D autoencoder, residual learning blocks containing 3D convolutional and transposed convolutional layers are employed in the encoder and decoder, respectively. In each residual learning block, a convolutional layer followed by a Batch Normalization (BN) [
69] layer and an activation layer appears twice as a basic mapping, which is added with the input to generate the output. The mathematical expression of the
th block can be shown as:
where
and
represent the input and the output of the block, respectively.
denotes the basic mapping of the
th block parameterized by
, and
is the sequential operation of convolution or transposed convolution
, BN
and activation
. The rectified linear unit (ReLU) function is used in all the activation layers but the last one. In addition, the output of mapping function
must have the same dimension as the input
. If this is not the case, the input
must be rescaled along with
using a convolutional or transposed convolutional transformation
with an appropriate stride value, the parameters of which can be updated in the training step.
4.3. Autoencoder Training
To analyze a large number of volumes, we carefully trained the 3D deep residual autoencoder to obtain suitable kernels and biases. First, parallel computation with multiple GPUs has been implemented to reduce the training time. Then the parameters of the network are optimized by the stochastic gradient descent Adam (Adaptive moment estimation) algorithm, in which the gradients of the objective function with respect to the parameters can be calculated by the chain rule. Moreover, the learning rate is reduced to one-tenth when the loss function does not decrease in three epochs based on the initial value of 0.01. After training about 50 epochs, the best model is picked to execute the task of structural feature extraction. Using the unsupervised 3D autoencoder, the feature maps encoding the structural discrepancy among the 3D volume data can be extracted automatically without any human intervention.
4.4. Autoencoder Hyperparameters
The recommended hyperparameters for the deep residual autoencoder architecture are provided in
Table 1. Although we expand the residual network (ResNet) into 3D, we keep the original design rules of ResNet for constructing the 3D autoencoder [
51,
52]. The first and last convolutional layers have 5 × 5 × 5 filters (kernels). The remaining convolutional layers have 3 × 3 × 3 filters. Because the cubic filter in the convolutional layer is computationally expensive, only one 3D filter is used in each of the last three convolutional layers in the encoder and of the first two and last transposed convolutional layers in the decoder. To accommodate a large volume input, two filters are used in the first three convolutional layers and in the third and fourth transposed convolutional layers. Downsampling is directly performed by convolutional layers with a stride of 2. The output dimension of encoding layers is set to be a quarter of the input dimension to avoid over-compressing the feature maps. We employed six 3D convolutional layers in the encoder and five transposed convolutional layers in the decoder based on the expected tradeoff between the learning accuracy and training cost, which is roughly comparable to a 2D ResNet with more than 200 layers with respect to the training cost. Lastly, one should randomly inspect some feature maps to ensure that all hyperparameter setting works as expected. If a feature map clearly shows no structural correspondence to its input volume and exhibits strong noise or abnormal features, it might signal the existence of overfitting. In this case, a non-zero value of
in the L2 norm regularization should be empirically examined to avoid overfitting.
4.5. Manifold Embedding of Pseudo-Energy Landscape
To prepare for the pseudo-energy landscape reconstitution, each resampled 3D volume was concatenated with its 3D feature map learned by the 3D autoencoder to form an expanded higher-dimensional data point. Each expanded data point is either low-pass filtered at a given resolution (5 Å in default setting) or standardized by the z-score method prior to t-SNE processing:
in which
and
denote the mean and standard deviation of the 3D volume voxels
with the box size of
, respectively. Similarly,
and
are the mean and standard deviation of the 3D feature map voxels
with the box size of
, respectively. All the input data points were then embedded onto a low-dimensional manifold via the t-SNE algorithm by preserving the geodesic relationships among all high-dimensional data [
53]. During manifold embedding, it is assumed that the pairwise similarities in the high dimensional data space and low dimensional latent space follow a Gaussian distribution and a student’s t-distribution, respectively. To find the low-dimensional latent variable of each data point, the relative entropy, also called the Kullback-Leibler (KL) divergence, is used as an objective function measuring the distance between the similarity distribution
in the data space and
in the latent space:
which is minimized by the gradient descent algorithm with the momentum method [
70]. The hyperparameter of t-SNE may be tuned to better preserve both local and global geodesic distances (
https://distill.pub/2016/misread-tsne/, accessed on 2 August 2022).
The idea of using concatenated data composed of both volumes and their feature maps for manifold embedding is similar to the design philosophy of deep residual learning, in which the input and feature output of a residual learning block are added together to be used as input of the next residual learning block. Such a design has been demonstrated to improve learning accuracy and reduce the performance degradation issues when the neural network goes much deeper [
51,
52]. Although the 3D feature maps or the resampled volumes alone can be used for manifold embedding, both appear to be inferior in 3D classification accuracy (
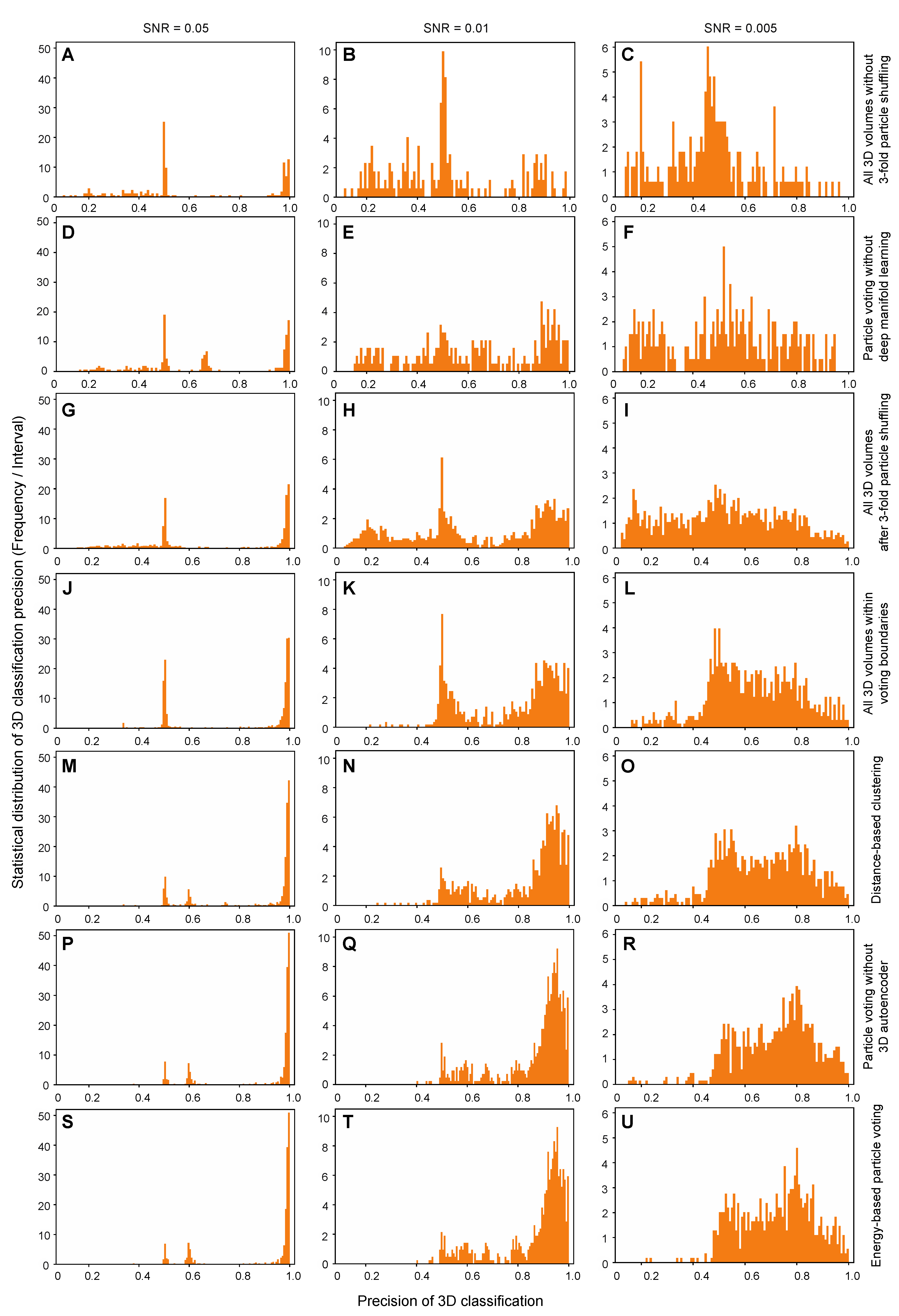
Figure 5). The concatenated format of input data for manifold learning is potentially beneficial to the applications in those challenging scenarios, such as visualizing a reversibly associated small ubiquitin protein (~8.6 kDa) of low occupancy by the focused AlphaCryo4D classification [
26].
After the manifold embedding by t-SNR, each 3D volume is mapped to a low-dimensional data point in the learned manifold. The coordinate system, in which the low-dimensional representation of the manifold is embedded, is used for reconstructing a pseudo-energy landscape. The difference in the Gibbs free energy Δ
G between two states with classified particle numbers of
Ni and
Nj is defined by the Boltzmann relation
Ni/
Nj = exp(−Δ
G/
kBT). Thus, the free energy of each volume can be estimated using its corresponding particle number:
where
denotes the free energy difference of the data point with the classified particle number of
against a common reference energy level,
is the Boltzmann constant and
is the temperature in Kelvin. The pseudo-energy landscape was plotted by interpolation of the free energy difference in areas with sparse data. We suggest that linear interpolation is used for the pseudo-energy landscape with loosely sampled areas to avoid overfitting by polynomial or quadratic interpolation. For densely sampled pseudo-energy landscapes, polynomial interpolation could give rise to a smoother pseudo-energy landscape that is easier to be tackled by the string method for MEP solution (see below).
The coordinate system of the embedded manifold output by t-SNE is inherited by the reconstitution of pseudo-energy landscape as reaction coordinates. They do not have an intuitive physical meaning of length scale in real space. However, they can be viewed as transformed, rescaled, reprojected coordinates from the real-space reaction coordinates along which the most prominent structural changes can be observed. Alternatively, they can be intuitively understood as being similar to transformed, reprojected principal components (PCs) in principle component analysis (PCA). In the case of the 26S proteasome, the two most prominent motions are the rotation of the lid relative to the base and the intrinsic motion of the AAA-ATPase motor [
25], which can be approximately projected to two reaction coordinates. In real space, both motions are notably complex and in fact are characterized by a considerable number of degrees of freedom, which are partially defined by the solved conformers.
4.6. String Method for Finding Minimum Energy Path (MEP)
The string method is an effective algorithm to find the MEP on the potential energy surface [
54]. To extract the dynamic information implicated in the experimental pseudo-energy landscape, an improved and simplified version of the string method has been previously developed [
55]. Along the MEP on the pseudo-energy landscape, the local minima of interest could be defined as 3D clustering centers to guide the particle-voting algorithm for 3D classification to generate high-resolution cryo-EM density maps (
Figure 1D and
Figure 2C). The objective of the MEP identification in energy barrier-crossing events lies in finding a curve
having the same tangent direction as the gradient of energy surface
. It can be expressed as:
where
denotes the component of
perpendicular to the path
. To optimize this objective function, two computational steps referred to as the ‘evolution of the transition path’ and ‘reparameterization of the string’, are iterated until convergence within a given precision threshold.
After initialization with the starting and ending points, in the step of evolution of the transition path, the positions of interval points along the transition path were updated according to the gradient of the free energy at the
th iteration:
with
being the
th intermediate point along the transition path at the
th iteration (
denoting the temporary values), and
the learning rate.
In the step of reparameterization of the string, the values of positions
were interpolated onto a uniform mesh with a constant number of points. Prior to interpolation, the normalized length
along the path was calculated as:
Given a set of data points , the linear interpolation function was next used to generate the new values of positions at the uniform grid points . The iteration was terminated when the relative difference became small enough.
4.7. String Method Parameters
The string method of searching for a rational MEP on the pseudo-energy landscape can only guarantee the solution of local optimum and is not capable of ensuring a solution of global optimum [
55]. The outcome of the string method depends on the initialization of the starting and ending points of the MEP on the pseudo-energy landscape. If there are too many local minima to sample along a long MEP, the string method could retrieve an MEP solution that partly misses some local energy minima by going off the pathway. In this case, the search for an expected MEP solution can be divided into several segments of shorter MEPs connecting one another, with each MEP being defined by a closer pair of starting and ending points, which travels through a smaller number of energy minima. Another parameter affecting the MEP solution is the step size being set to explore the MEP on pseudo-energy landscapes. Although a default value of 0.1 empirically recommended may work in many cases, it may be helpful to tune the step size according to the quality of the pseudo-energy landscape. The value of step size can be decreased if the computed path runs out of the pseudo-energy landscape and can be increased if the path updates too slowly during iterative searching by the string method.
4.8. Energy-Based Particle Voting Algorithm
The particle-voting algorithm was designed to conduct 3D classification, particle quality control, reproducibility test and particle selection in an integrative manner. The particle-voting algorithm mainly involves two steps (
Figure 1D). First, we count the number of votes for each particle mapped within each voting boundary. The particles voted for a given voting boundary define a new 3D cluster approximately centered on a local energy minimum of the pseudo-energy landscape. One vote is rigorously mapped to one copy of the particle used in reconstructing a 3D volume and to no more than one 3D cluster on the pseudo-energy landscape where the corresponding volume is located. Thus, each particle can have
M votes cast for no more than
M 3D clusters. If the vote is mapped outside of any 3D cluster boundary, it becomes an ‘empty vote’ with no cluster label. Each non-empty vote is thus labeled for both its particle identify and corresponding cluster identity. For each pair of particles and cluster, we compute the total number (
V) of votes that the cluster receives from the same particle. Each particle is then assigned and classified to the 3D cluster that receives
V >
M/2 votes from this particle (
Figure 1D). Note that after particle voting, each particle is assigned no more than once to a 3D class, with its redundant particle copies removed from this class. This strategy only retains the particles that can reproducibly vote for a 3D cluster corresponding to a homogeneous conformation, while abandoning those non-reproducible particles with divergent, inconsistent votes.
4.9. Alternative Strategy of 3D Clustering without Particle Voting
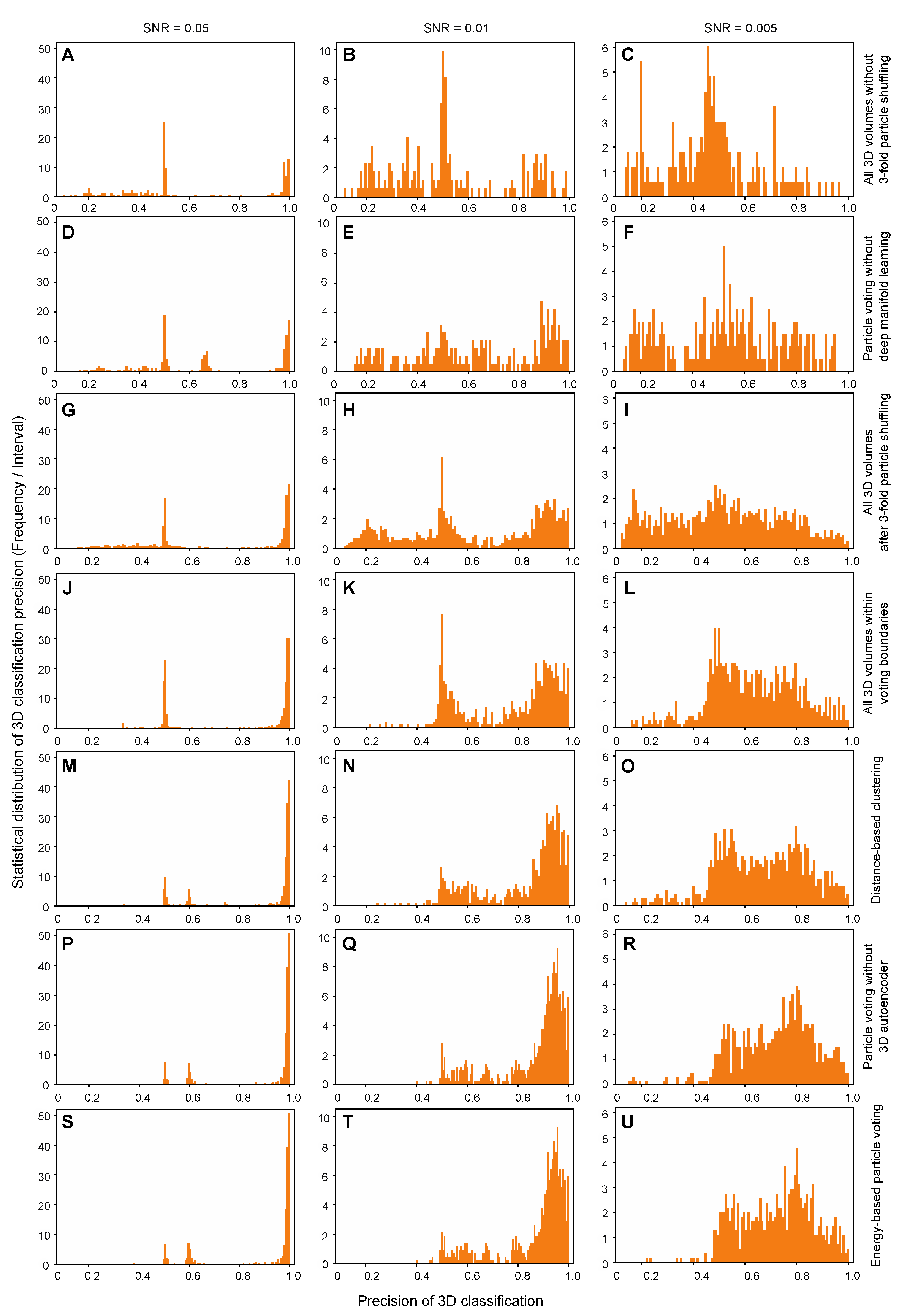
Because the particle-voting algorithm imposes strong constraints on the reproducibility of particle classification by deep manifold learning, some 3D classes might be assigned with a small number of particles that are insufficient to support high-resolution reconstruction. To remedy this limitation, an alternative, distance-based classification algorithm was devised to replace the particle-voting algorithm when there are not enough particles to gain the advantage of particle voting (
Figure 1E). In this method, the distances of all
M copies of each particle to all 3D cluster centers on the pseudo-energy landscape are measured and ranked. Then, the particle is classified into the 3D cluster of the shortest distance. A threshold could also be manually preset to remove particles that are too far away from any of the cluster centers. The distance-based classification method can keep more particles, but it ignores the potential issue of irreproducibility of low-quality particles. Thus, it is proven to be less accurate in 3D classification (
Figure 5M–O). In other words, it trades off the classification accuracy and class homogeneity to gain more particles, which is expected to be potentially useful for small datasets or small classes. By contrast, the energy-based particle-voting algorithm imposes a more stringent constraint to select particles of high reproducibility during classification, resulting in higher quality and homogeneity in the classified particles, which is superior to the distance-based classification method (
Figure 5S–U).
4.10. Practical Consideration of Particle Shuffling and Voting Parameters
The parameter M determines the degree of implicit cross-validation of classification reproducibility, as well as the sampling densities of the pseudo-energy landscape. To establish reproducibility for 3D classification, M should be no less than 3. The variation of M is not supposed to change the pseudo-energy landscape because it multiplies on both denominator and enumerator in the Boltzmann relation and is canceled in the ratio of particle densities in computing the free-energy differences. Increasing the M value will allow more volumes to be resampled, which then leads to a higher sampling density in computing the manifold of the pseudo-energy landscape and potentially enhances its reconstitution quality. The default classification threshold M/2 is the minimal number of votes for verifying the reproducibility of 3D classification. However, a higher threshold, such as 2M/3, will give rise to more stringent criteria in cross-validation, with a tradeoff of voting out more particles. The particle voting algorithm does not entirely eradicate misclassified particles. However, increasing either M or the classification threshold could theoretically have a similar impact and improve the conformational homogeneity, because it gets less probable for a particle to be misclassified to the same cluster more times.
Several considerations may be applied to the choice of M and to set up particle shuffling and voting. First, to obtain a high-quality pseudo-energy landscape, a thousand or more resampled volumes are expected for datasets of more than 1–2 million particles. Second, the average particle number per volume is expected to be no less than 5000 or more to ensure that majority of volumes include sufficient particles for quality reconstructions. The expected particle number per volume must increase if the average SNR per particle is decreased. It may need to reach 10,000–20,000 or more for small proteins or lower SNR datasets. Third, for a dataset of moderate size, M can be adjusted to a higher value to mitigate the lack of image data for resampling.
4.11. Data-Processing Workflow of AlphaCryo4D
The following summarizes the complete procedure of AlphaCryo4D, with the detailed Algorithm 1 rationale explained in the previous subsections.
| Algorithm 1 AlphaCryo4D |
Input: Single-particle cryo-EM dataset after initial particle rejection of apparent false particles.
Output: Pseudo-energy landscape, MEP, 3D class assignment of each particle.
Step 1. Resample many 3D volumes through particle shuffling, consensus alignment and Bayesian clustering.Split the particle dataset randomly to many sub-datasets, if necessary, in case of a large dataset (e.g., >150,000 particle images), for batch processing of particle shuffling and volume resampling. Otherwise, skip this step if the dataset is small enough (e.g., <150,000 particle images). For each sub-dataset, conduct a consensus alignment to generate initial parameters of Euler angles and translations in RELION v2.1/v3.0 (MRC, Cambridge, UK) or ROME v1.1.2 (Peking University, Beijing, China) with one or more consensus models. Divide each sub-dataset into M + 1 groups, shuffle the sub-dataset M + 1 times and each time take a different group out of the shuffled sub-dataset, giving rise to M + 1 shuffled sub-datasets all with different collections of particles. Conduct 3D Bayesian classification on all the M + 1 shuffled sub-datasets to generate hundreds of 3D volumes, making each particle contribute to M different volumes. Execute steps (2) to (4) using the same initial model (low-pass filtered at 60-Å) for all sub-datasets.
Step 2. Extract 3D feature maps of all volume data with the 3D deep residual autoencoder.Align all 3D volumes and adjust them to share a common frame of reference. Initialize the hyper-parameters of the 3D autoencoder ( Table 1). Train the neural network with the 3D volume data to minimize the mean square error between the decoding layer and the input by the Adam algorithm of an initial learning rate of 0.01. Extract the 3D feature maps of all volumes from the encoding layer.
Step 3. Embed the volume data to two-dimensional manifolds through the t-SNE algorithm, compute the pseudo-energy landscape and find the MEP.Calculate the pairwise similarities between volumes using their feature-map-expanded volume vectors, and randomly initialize the low-dimensional points. Minimize the Kullback–Leibler divergence by the Momentum algorithm to generate 2D manifold embeddings with t-SNE. Compute the pseudo-energy landscape from the manifold using the Boltzmann relation. Initialize searching of the MEP with a straight line between given starting and ending points. Find the optimal MEP solution using the string method.
Step 4. Classify all particles through the energy-based particle-voting algorithm.Sample the clustering centers along the MEP and calculate the recommended clustering radius. Define the local energy minima as the 3D clustering centers and their corresponding cluster boundary for particle voting. For each particle, cast a labeled vote for a 3D class when a volume containing one of the M particle copies is located within the voting boundary. Count the number of votes of each particle with respect to each 3D class and assign the particle to the 3D class that receives more than M/2 votes from this particle. Refine each 3D density map separately to high resolution in RELION v2.1/v3.0 (MRC, Cambridge, UK) or cryoSPARC v2.9+ (Structura Biotechnology Inc., Toronto, ON, Canada) using particles classified into the same 3D classes.
|
4.12. Software Implementation
This section provides a brief account of the implementation of the AlphaCryo4D prototype system. The source code is freely available at
http://github.com/alphacryo4d/alphacryo4d/ (accessed on 2 August 2022). The main programs were implemented in the Python language. Some auxiliary scripts were written in the Shell language due to its convenience. The implementation of AlphaCryo4D includes four modules, named
Resample,
DeepFeature,
ManifoldLandscape and
ParticleVoting.
Table 2 summarizes all major scripts and key arguments in each module.
The programs of Bayesian resampling are provided in the folder of
Resample. The kernel programs in Bayesian resampling include
randsf.py,
resample.py and
bigdata.py (
Table 2). In algorithm testing, RELION v2.1 or v3.0 (MRC, Cambridge, UK) is used in the consensus refinement and initial class assignment [
3]. To speed up numerical operations, the package NumPy is called to perform mathematical calculations in the manner of vectorization. In addition, files in the
mrc format are processed by the library
mrcfile. If necessary, memory-mapped files are written into hard disks to break the memory limit at the cost of more running time when processing big data.
The programs of feature extraction by 3D deep residual autoencoder are provided in the folder of
DeepFeature. The core programs to construct the 3D deep residual autoencoder and extract features of dynamics are
run_resnet.py and
run_predict.py (
Table 2). The 3D deep residual neural network is built, trained and predicted by
Keras with the backend of TensorFlow v1.15.4 (Alphabet Inc., Mountain View, CA, USA) [
71]. The package TensorFlow can support the parallel computing of GPU to accelerate the computation, where the running environment with Compute Unified Device Architecture (CUDA) is required. Once the training of the 3D residual autoencoder is completed, the best and final network model can be saved to files of the
hdf5 format for further analysis.
The folder of
ManifoldLandscape includes the programs
tsne_rd.py and
string_method.py to map the manifold embedding of 3D volumes for energy landscape estimation (
Table 2). For t-SNE running in the step of manifold mapping, the package
scikit-learn is imported to call the API of manifold learning. Besides, both the low-dimensional embedding and the energy landscape are plotted by the package
matplotlib, which can export figures of
png or
pdf format. The optional calculation of the string method can generate a
npy file of clustering centers along the MEP with the user-defined parameters.
The code in the folder of
ParticleVoting is utilized for particle assignment in the final 3D classification using either energy-based particle voting (
Figure 1D) or distance-based classification (
Figure 1E). The core programs include
clustering.py and
post_and_f.sh for energy-based particle voting,
post_or_parallel.sh and
dedup.sh for distance-based classification (
Table 2). Different from particle voting, the strategy of distance-based classification can retain more particle images in final 3D classes by assigning each particle to its nearest energy well without any particles being voted out. Most operations of particle assignment to 3D classes are performed by reading and writing the
star files of particle images in parallel.
In addition, the computation of AlphaCryo4D generates multiple intermediate files and resulting output files in each module.
Table 3 summarizes the Input/Output (I/O) convention of each module. The inputs and outputs of four modules are introduced with their data file formats to meet the demand for specific analysis. For compatibility with other operations in Python, most intermediate files are saved in the format of
npy. AlphaCryo4D also exports the
star files of particle images of each conformational state to be readily processed by RELION [
3].
To configure the runtime environment for AlphaCryo4D, Python 3.7 (Python Software Foundation, Wilmington, DE, USA) is needed with the available computing environment and dependencies. The necessary dependent packages installed by conda and pip can be configured by the files
EnvConda.txt and
EnvPip.txt provided in the source code, respectively. Installation of RELION v2.1 or v3.0 and EMAN2 v2.91 (Baylor College of Medicine, Houston, TX, USA) is required to run through certain steps before and after AlphaCryo4D classification (
Table 3) [
3,
72].
As an example, given the properly configured environment, the Python program, such as run_resnet.py in the folder DeepFeature, can be applied using the sample command like:
python DeepFeature/run_resnet.py --batchsize 8 --validationsize 200 --regularization 0.001 --data data_dl.npy --gpu 0,1,2,3
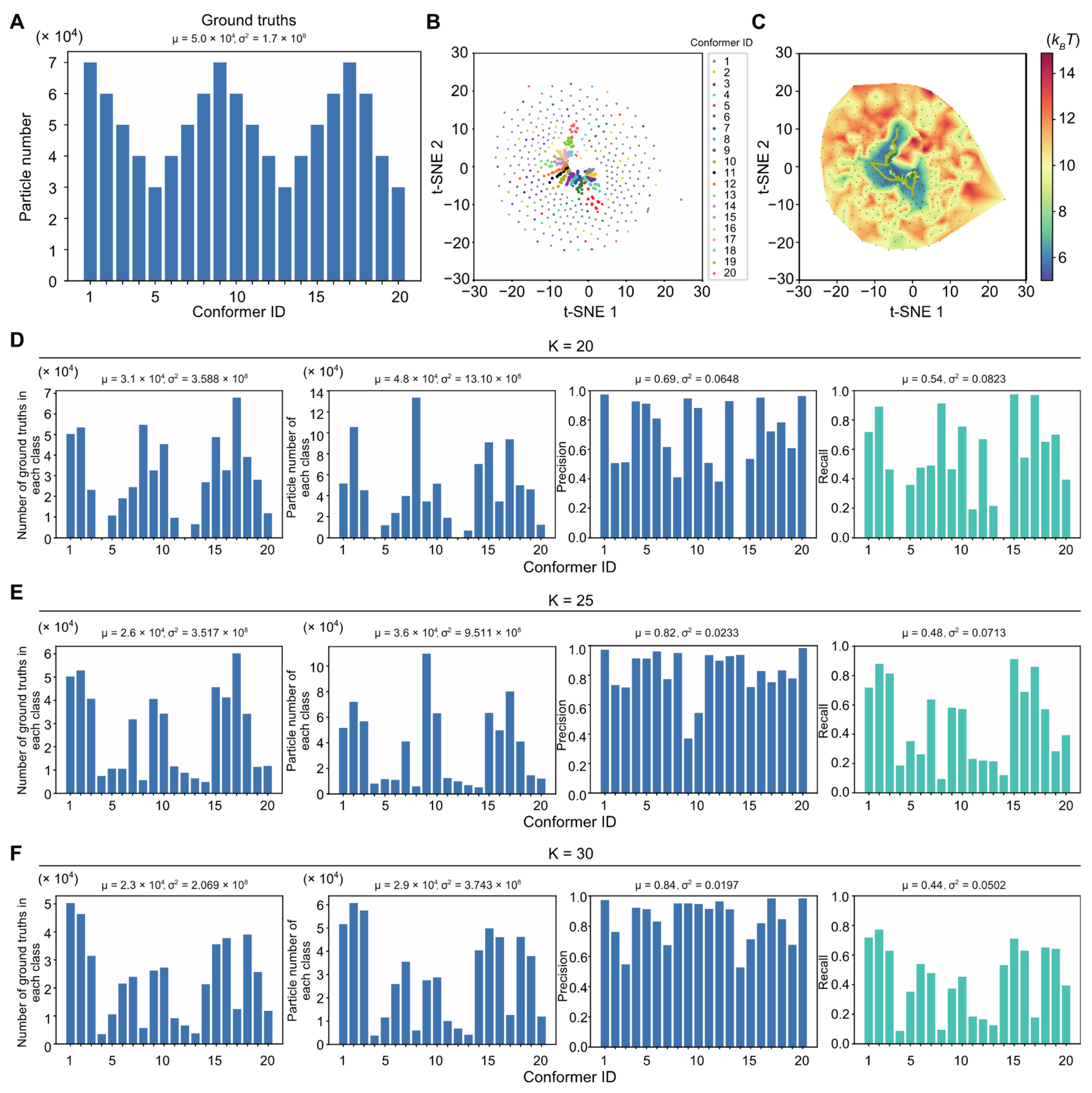
4.13. Blind Assessments with Simulated Datasets
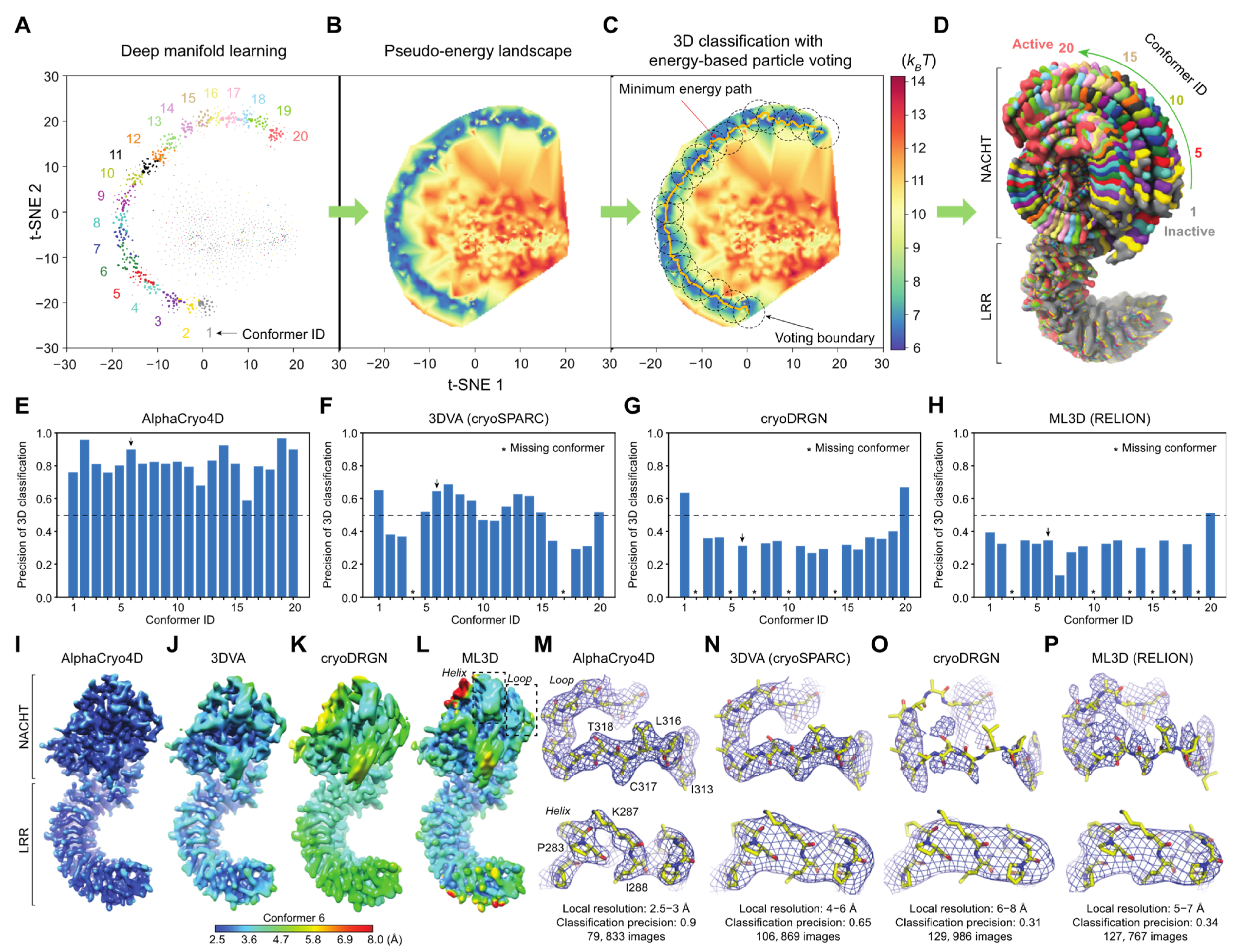
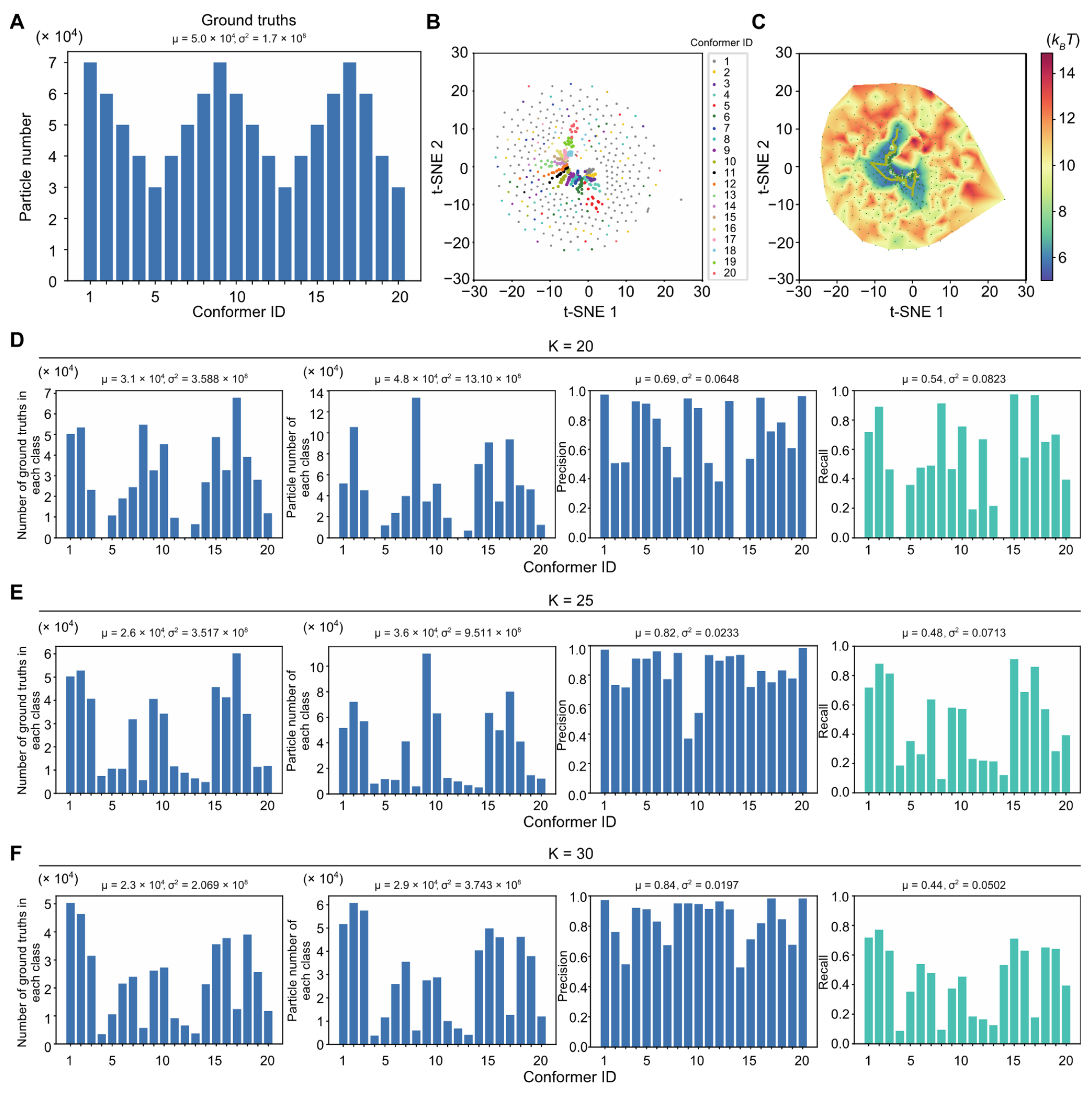
Three simulated large datasets with the SNRs of 0.05, 0.01 and 0.005, each including 2 million particle images, were employed to benchmark AlphaCryo4D and to compare its performance with alternative methods. For each synthetic dataset, the particles were computationally simulated by projecting the 20 3D density maps calculated from 20 hypothetical atomic models emulating continuous inter-domain rotation of the NLRP3 inflammasome protein. The 20 atomic models were interpolated between the inactive NLRP3 structure and its hypothetical active state, which was generated through homology modeling using the activated NLRC4 structure (
Figure 2D) [
56]. The 20 atomic models represent sequential intermediate conformations during a continuous rotation in its NATCH domain against its LRR domain over an angular range of 90°. The NATCH domain in each conformation is thus rotated 4.5° over its immediate predecessor in the conformational continuum sequence; 100,000 simulated single-particle images per conformational state were generated with random defocus values in the range of −0.5 to −3.0 μm, resulting in 2 million single-particles for each dataset of a given SNR. The defocus values were not blind to all comparative tests, due to difficulty in determining accurate defocus values from single particle images of very low SNRs. The pixel size of the simulated image was set to the same as the pixel size (0.84 Å) of the real experimental dataset of the NLRP3-NEK7 complex [
56]. To emulate realistic circumstances in cryo-EM imaging, Gaussian noises, random Euler angles covering half a sphere and random in-plane translational shifts from −5.0 to 5.0 pixels were then applied to every particle image.
Each simulated heterogeneous NLRP3 dataset was analyzed separately by AlphaCryo4D and used to characterize the performance and robustness of AlphaCryo4D against the variation of SNRs. In the step of particle shuffling and volume resampling, 2,000,000 particles in the dataset of any given SNR were divided randomly into 10 sub-datasets for batch processing. The orientation of each particle was determined in the initial 3D consensus alignment in RELION, which did not change in the subsequent 3D classification. In this step, the maximum number of iterations of the 3D alignment was set up as 30, with the initial reference low-pass filtered to 60 Å. Three-fold particle shuffling (indicated as × 3 below) was conducted on each sub-dataset for volume resampling. The first round of maximum-likelihood 3D classification divided the input shuffled particle sub-dataset into five classes, each of these classes was then further classified into eight classes. This procedure was separately executed on all shuffled particle sub-datasets. The particle shuffling and volume resampling generated 1372, 1489, and 1587 volumes by the datasets with SNRs of 0.05, 0.01 and 0.005, respectively. These volume data were used as inputs for deep residual autoencoder to compute low-dimensional manifolds and pseudo-energy landscapes (
Figure 1D and
Figure 2A–D and
Supplementary Figures S1A,B). After searching the MEP on the pseudo-energy landscapes by the string method, 20 cluster centers along the MEP were defined by the local energy minima along the MEP by the approximately equal geodesic distance between adjacent minima, which represent potentially different conformations of the macromolecule (
Supplementary Figure S1A–C). The particle-voting algorithm was applied in every cluster to determine the final particle sets for all 3D classes. For validation of the methodology and investigation of 3D classification improvement, we labeled each resampled 3D volume with the conformational state that held the maximum proportion of particles in the class and computed its 3D classification precision as the ratio of the particle number belonging to the labeled class versus the total particle number in the volume (
Figure 2E and
Figure 3).
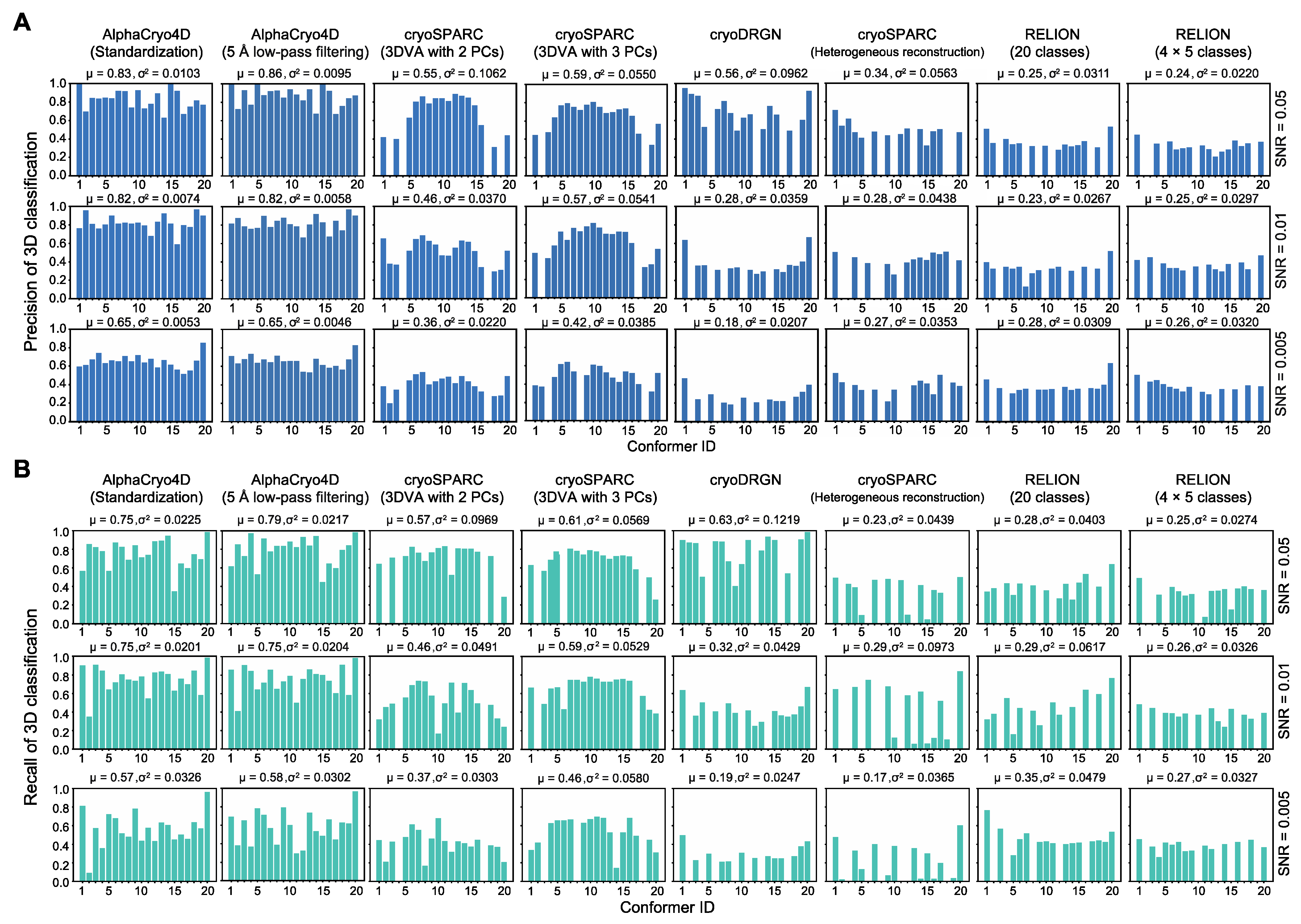
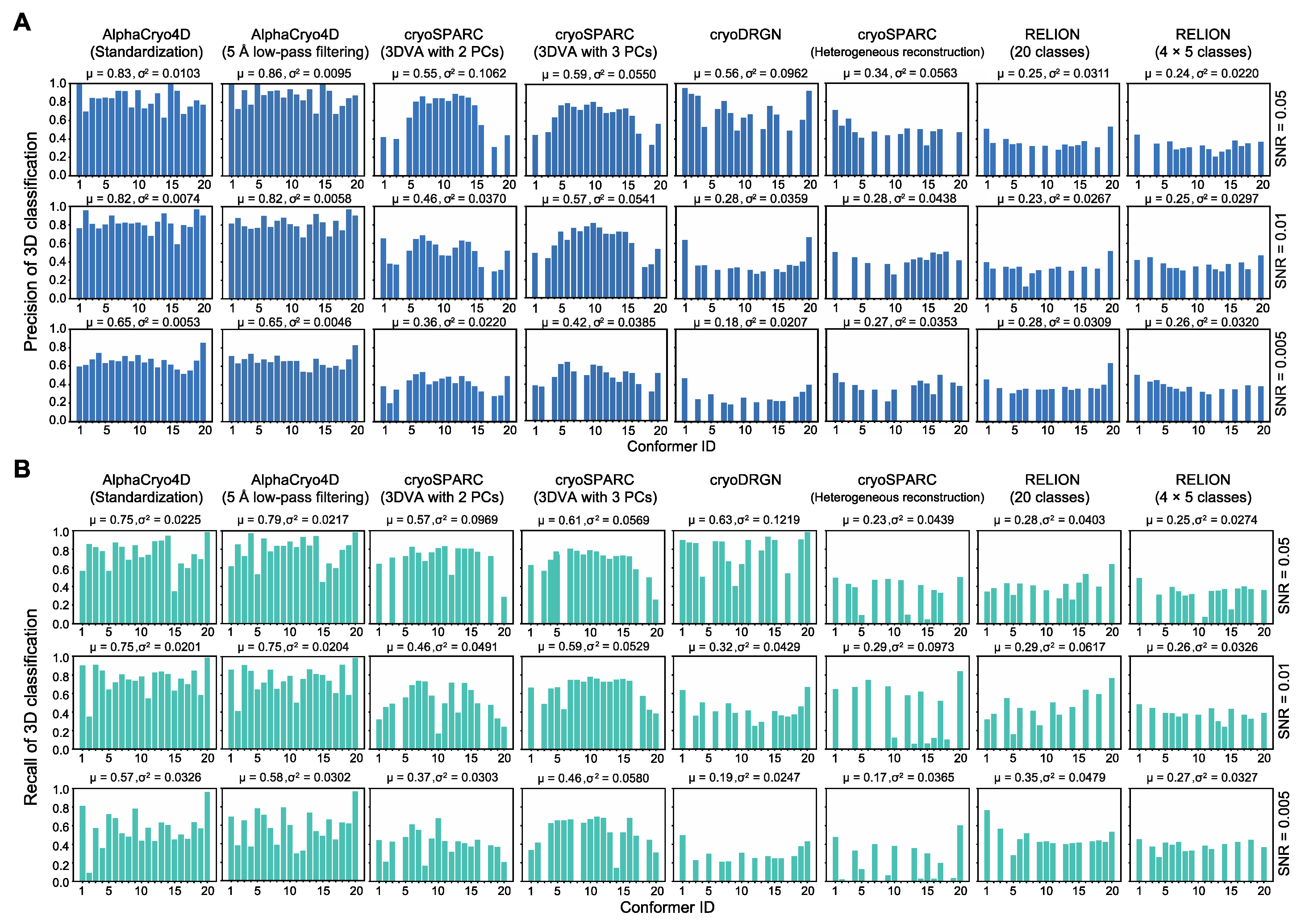
4.14. Comparisons with Alternative Methods
Using 3D classification precision as a benchmark indicator, the performance of AlphaCryo4D preprocessed by standardization or 5 Å low-pass filtering was compared with several other methods: (1) the conventional maximum-likelihood-based 3D (ML3D) classification in RELION v3.0 (MRC, Cambridge, UK) [
3,
4,
6] with and without a hierarchical strategy, (2) the conventional heterogeneous reconstruction in cryoSPARC v2.9+ (Structura Biotechnology Inc., Toronto, ON, Canada) [
2], (3) the 3DVA algorithm with two and three principal components (PCs) in cryoSPARC v2.9+ [
11] and (4) the deep generative model-based cryoDRGN v0.3.1 (Massachusetts Institute of Technology, Cambridge, MA, USA) [
12]. A total of 24 comparative tests by these alternative methods have been conducted blindly using the three simulated datasets, which include 6 million images in total. In all our comparative tests on AlphaCryo4D and the alternative methods, the ground-truth information of particle orientations, in-plane translations and conformational identities were completely removed from the methods being tested and were not used for any steps of data processing or training. The ground-truth conformational identities of particles were only used as validation references to compute the 3D classification precisions of the blind testing results.
For the tests using ML3D in RELION, we classified all particles directly into 20 classes and hierarchically into 4 × 5 classes, which first divided the dataset into four classes, with each class further classified into five sub-classes (
Figure 3). To generate the initial model for RELION, 20 ground truth density maps were averaged and low-pass filtered to 60 Å prior to ML3D; 30 iterations of ML3D classifications were then performed. For testing the conventional discrete ab initio heterogeneous reconstructions in cryoSPARC, each synthetic dataset was directly classified into 20 classes without providing any low-resolution reference model. For comparison with the 3DVA algorithm in cryoSPARC, we first conducted the blind consensus alignment of the entire dataset to find the orientation of each particle. Then the alignment and the mask generated from the consensus reconstruction were used as inputs into the 3DVA calculation, with the number of orthogonal principal modes being set to 2 or 3 in 3D classification. The 3D variability display module in the cluster mode was used to analyze the results of 3D classification. For blind tests with cryoDRGN, a default 8D latent variable model was trained for 25 epochs. The encoder and decoder architectures were 256 × 3, as recommended by the cryoDRGN developers [
12]. The particle alignment parameters prior to cryoDRGN training were obtained by the same blind consensus refinement in RELION used for other parallel tests. The metadata of 3D classification precisions as well as the 3D density maps from all the algorithms applied to the three simulated datasets were collected to conduct the statistical analysis (
Figure 2,
Figure 3,
Figure 4 and
Figure 5).
4.15. Computational Costs
Although the computational cost of AlphaCryo4D is potentially higher than the conventional approach, it does not appear to increase drastically and likely falls in an affordable range, while reducing the average cost of computation per conformational state (
Table 4). In a nutshell, we can have a brief comparison of the computational efficiency of the simulated 2 million image dataset with an SNR of 0.01. In the step of 3D data resampling, we split the dataset into 20 subsets, which contained 100,000 particles each. The 3D consensus alignment of all 2,000,000 particles costs about 75 h using eight Tesla V100 GPUs interconnected with the high-speed NVLink data bridge in an NVIDIA DGX-1 supercomputing system. Within each subset, the 3D Bayesian classification for one leave-one-group-out dataset cost about 2.5 h using 320 CPU cores (Intel Xeon Gold 6142, 2.6 GHz, 16-core chip), so the total time spent in one subset was about 10 h using 320 CPU cores in an Intel processor-based HPC cluster. In addition, it spent about 3 h extracting features via a deep neural network using eight Tesla V100 GPUs of the NVIDIA DGX-1 system. In comparison to about 160 h cost in traditional classification methods, this approach costs about 213 h using eight Tesla V100 GPUs and 320 CPU cores. As shown in
Table 4, the majority of the computational cost was spent on the steps of consensus alignment and high-resolution refinement, but not on the steps of machine learning which adds overall only ~20% of the complete data processing time. We have recently optimized the ML3D code-executing efficiency for CPU-based clusters in a new version of ROME v1.1.2 [
5] to speed up the consensus alignment by three- to six-fold, which will be described in detail elsewhere. Taken together, the computational cost of AlphaCryo4D is well justified, considering the output of more high-resolution conformers yielded by the procedures. In fact, in a recent study combining AlphaCryo4D with time-resolved cryo-EM [
63], the actual data processing time was comparable to a similarly large dataset [
25], while obtaining twice more high-resolution conformers.
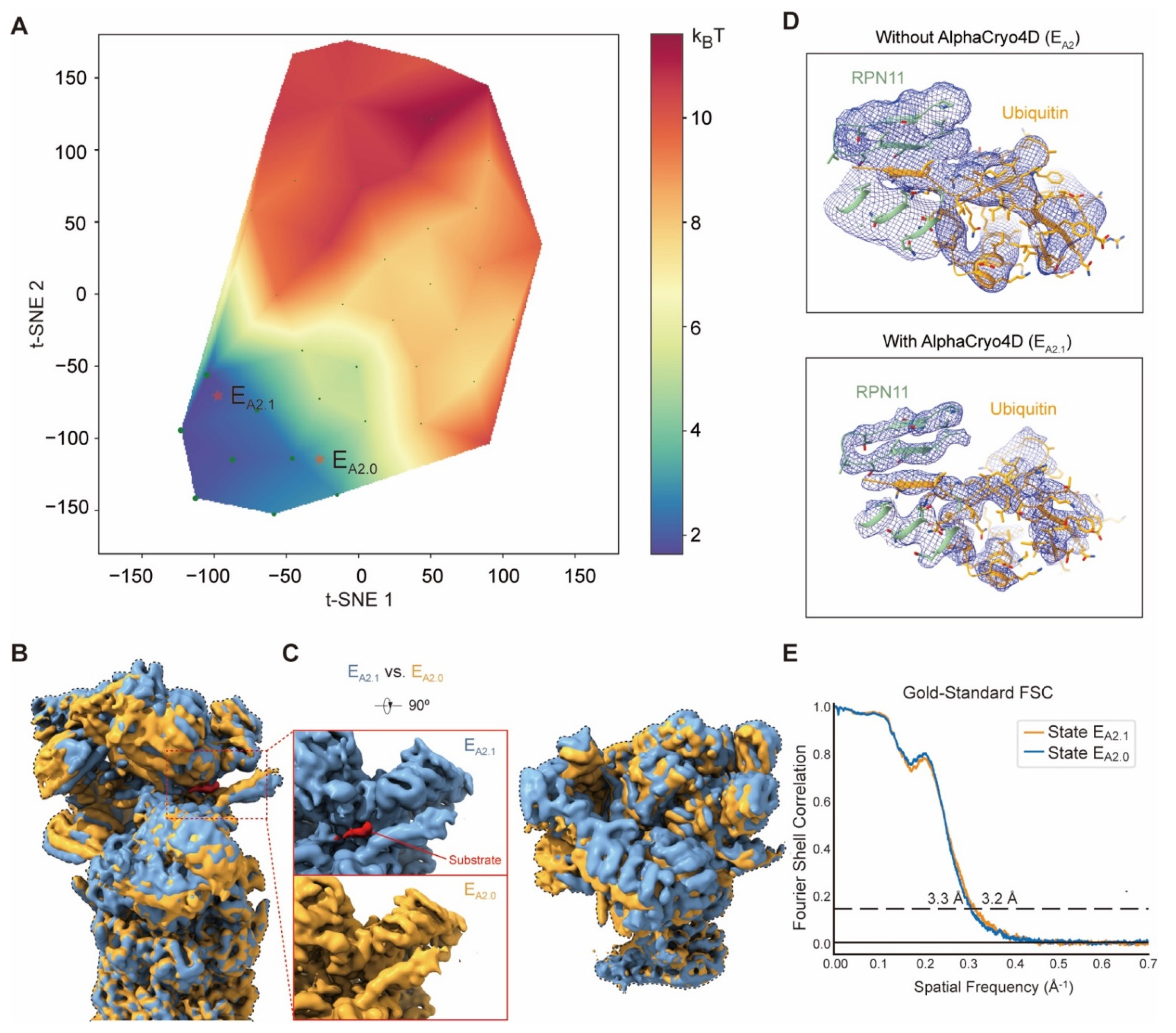
4.16. Applications to the Experimental Cryo-EM Dataset of the Human 26S Proteasome
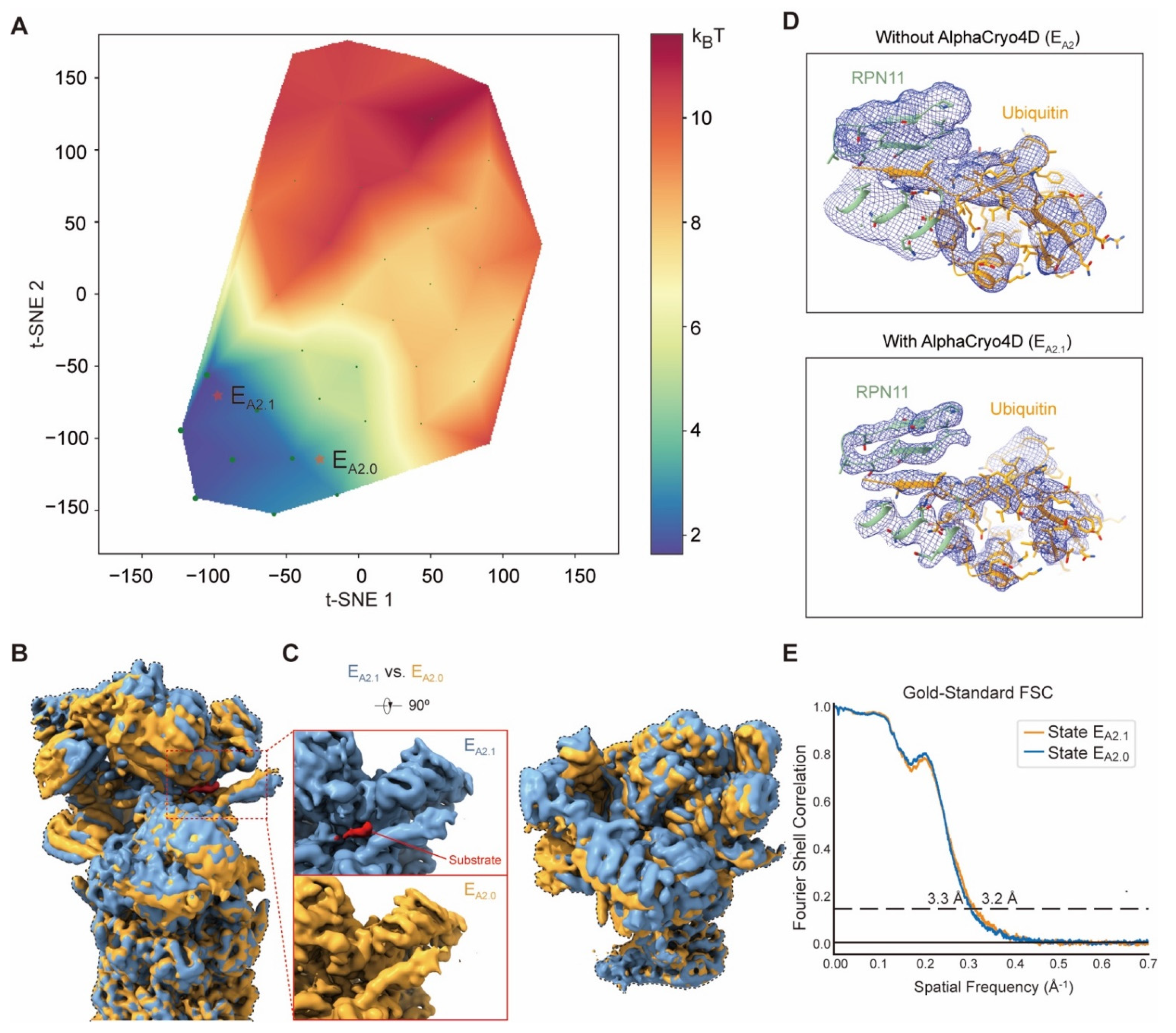
The substrate-engaged human 26S proteasome dataset (EMPIAR-10669) [
25] includes 3,254,352 RP-CP particles (combined with particle images from both the doubly capped and singly capped proteasomes) in total, with the super-resolution counting mode pixel size of 0.685 Å and the undecimated box size of 600 × 600 pixels. The substrates of the 26S proteasome first appear in the state E
A2. In this focused study, 147,108 particles of E
A2 were utilized for computing the specific pseudo-energy landscape with 40 volumes. The alignment of these particles was firstly refined with a 19S mask.
M = 3 particle shuffling was then conducted to resample the 3D volumes with a soft mask of 19S. By clustering on the pseudo-energy landscape and particle voting, two classes containing 99,043 and 47,389 particles were generated for high-resolution refinement using RELION v3.0, which were designated state E
A2.1 and E
A2.0, respectively. The density map of state E
A2.0 exhibits a conformation identical to state E
A2 in the previous report [
25].
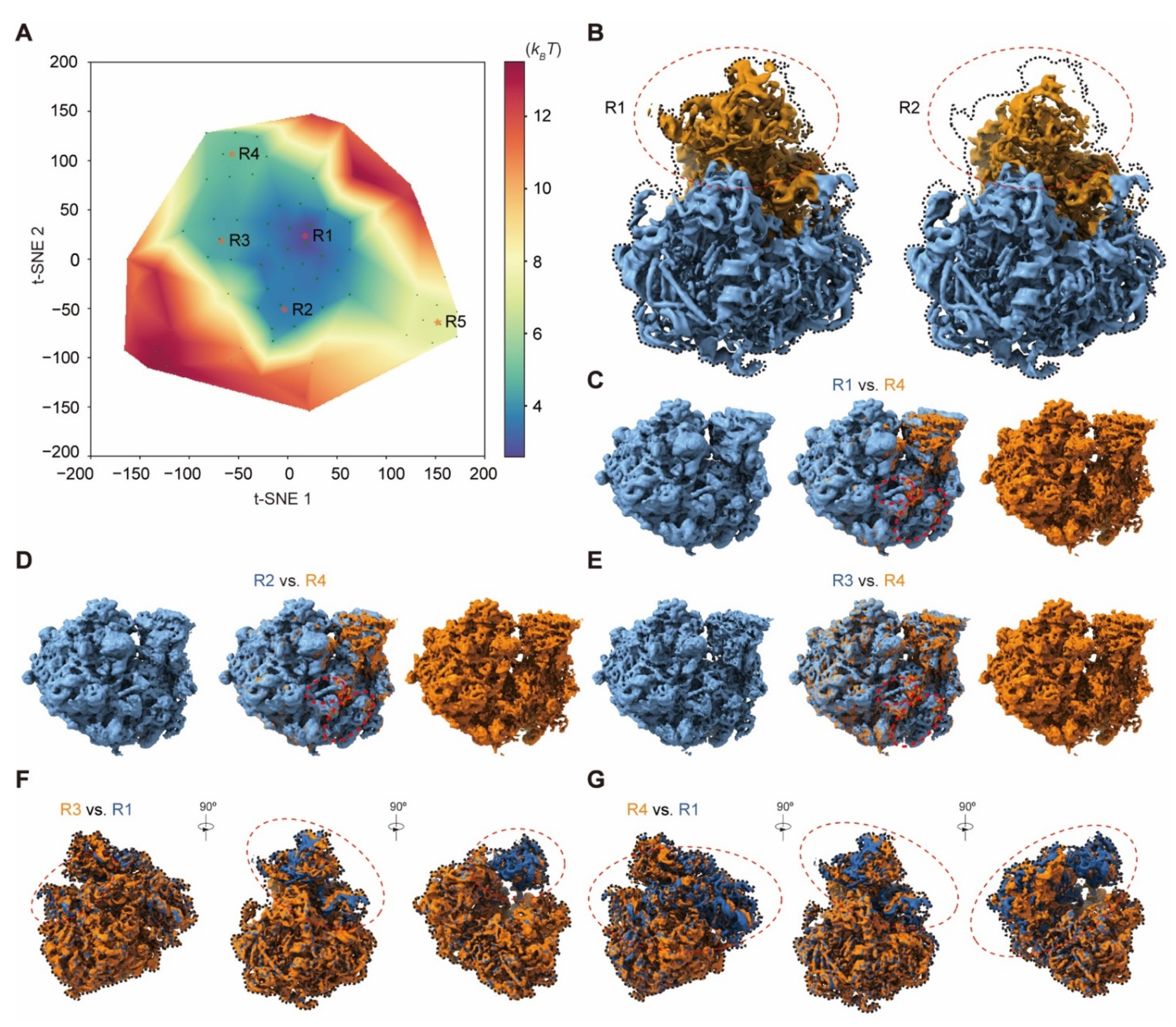
4.17. Applications to the Experimental Cryo-EM Dataset of the Pf80S Ribosome
The Pf80S ribosome dataset (EMPIAR-10028) contains 105,247 particle images. First, the alignment of all particles was refined with a global mask in RELION. To resample enough 3D volumes for the pseudo-energy landscape, we set M = 7 in the particle shuffling step, resulting in 79 volumes in total with a global mask and 15 Å resolution limit in the expectation step. Moreover, the regularization coefficient was set to 0.001 when training the 3D residual network with this small dataset. All volumes were then low-pass filtered at 5 Å prior to manifold embedding. Five clusters were obtained from the pseudo-energy landscape, which had 66,035, 21,482, 9922, 6424, and 657 particle images, respectively, after voting. All these clusters were refined independently using RELION v3.0.
4.18. Applications to the Experimental Cryo-EM Dataset of the Yeast Pre-Catalytic Spliceosome
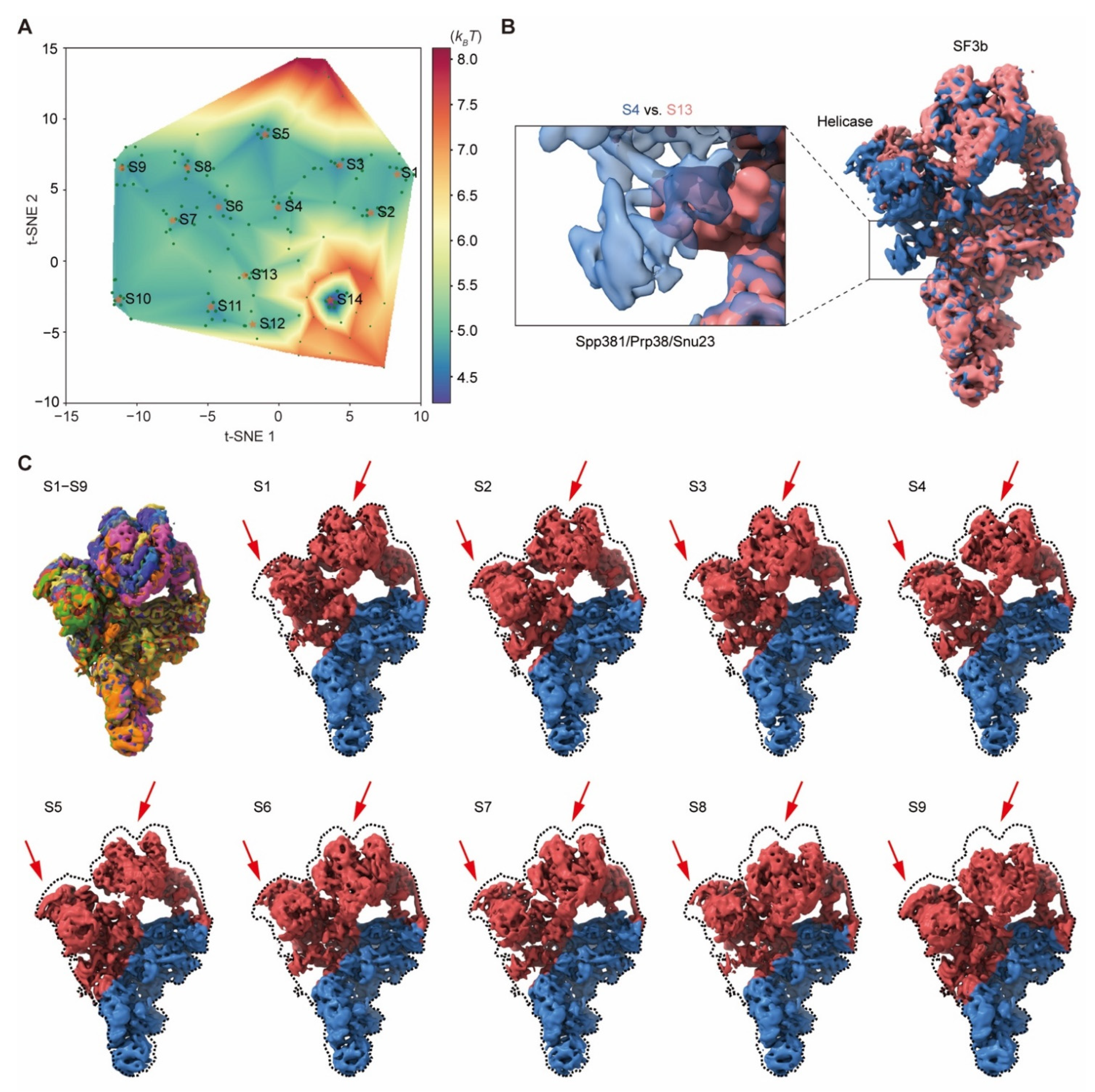
The pre-catalytic spliceosome dataset (EMPIAR-10180) with the particle number of 327,490 shows high conformational dynamics. The consensus alignment of these particles in the original dataset was used in the first step. M = 7 particle shuffling was utilized to resample 160 volumes with a soft global mask and 15 Å resolution limit in the expectation step. The regularization coefficient of 0.001 was set to train the 3D Autoencoder with this dataset. All volumes were then low-pass filtered at 5 Å prior to manifold embedding. Based on the pseudo-energy landscape, we obtained 14 classes with the particle number of 15,749, 17,703, 27,522, 13,820, 20,841, 14,989, 16,377, 22,047, 25,049, 21,327, 18,949, 6157, 9790 and 36,878 after voting. Each class was refined independently using RELION v3.0.
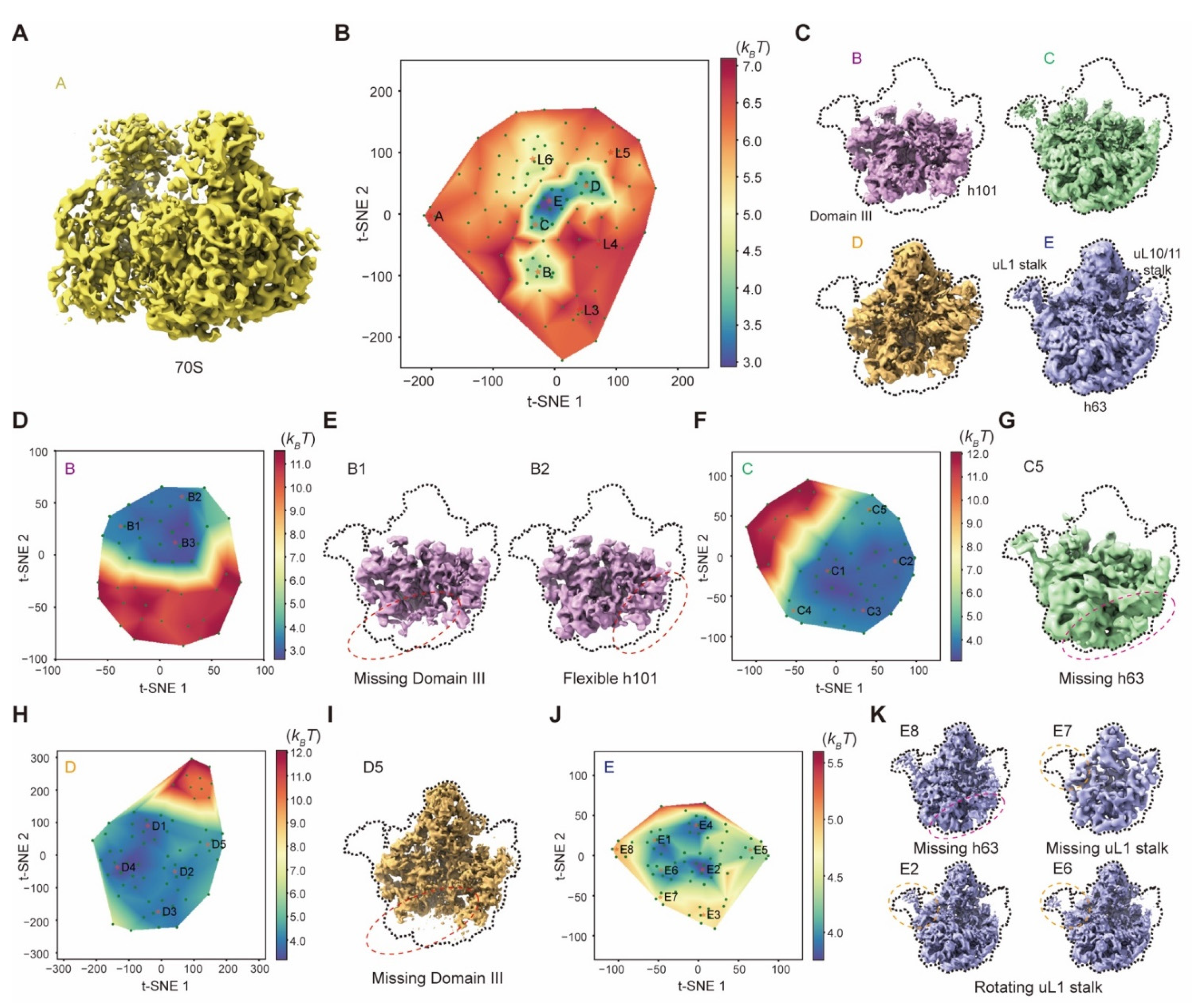
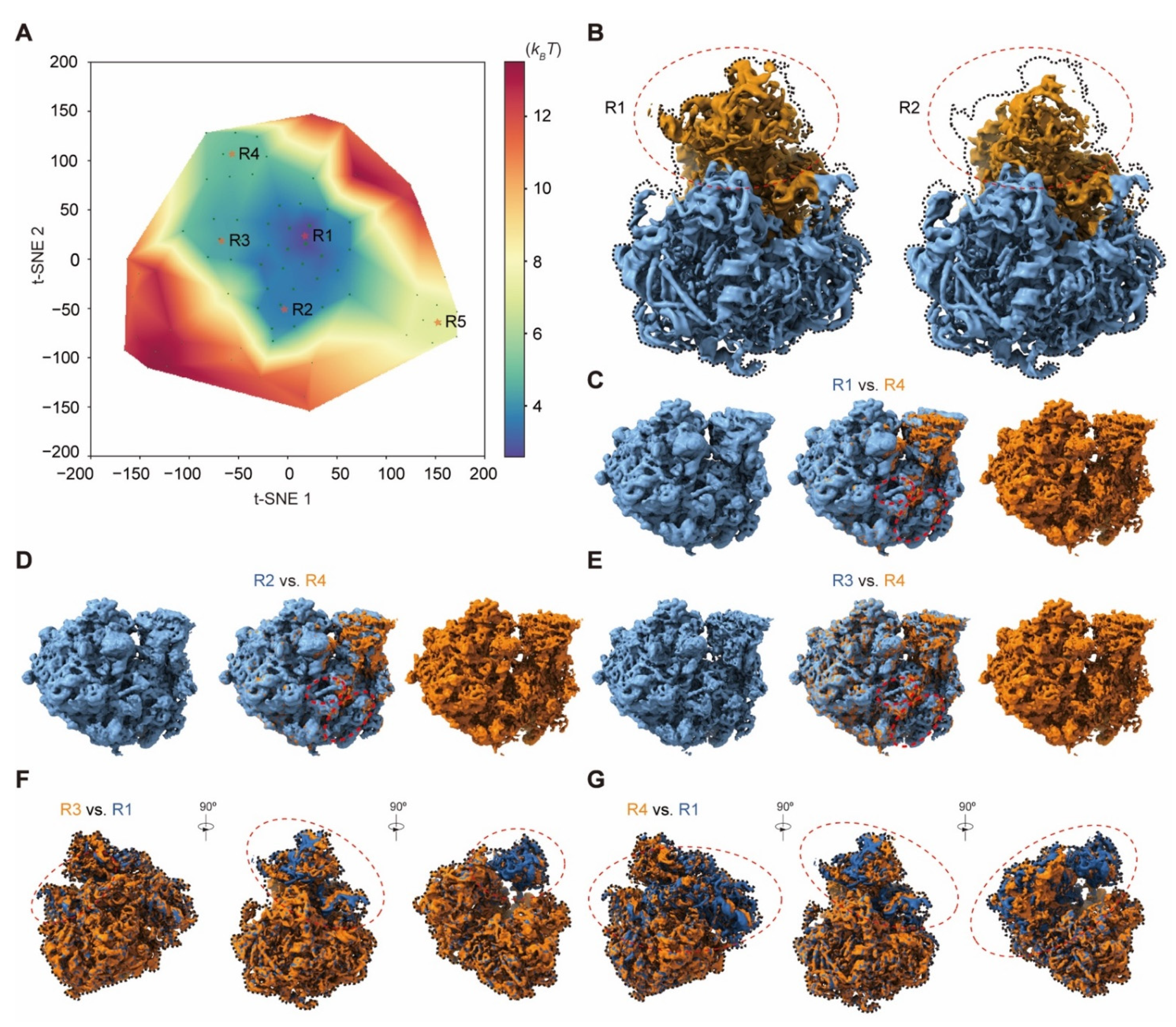
4.19. Applications to the Experimental Cryo-EM Dataset of the Bacterial 50S Ribosomal Intermediates
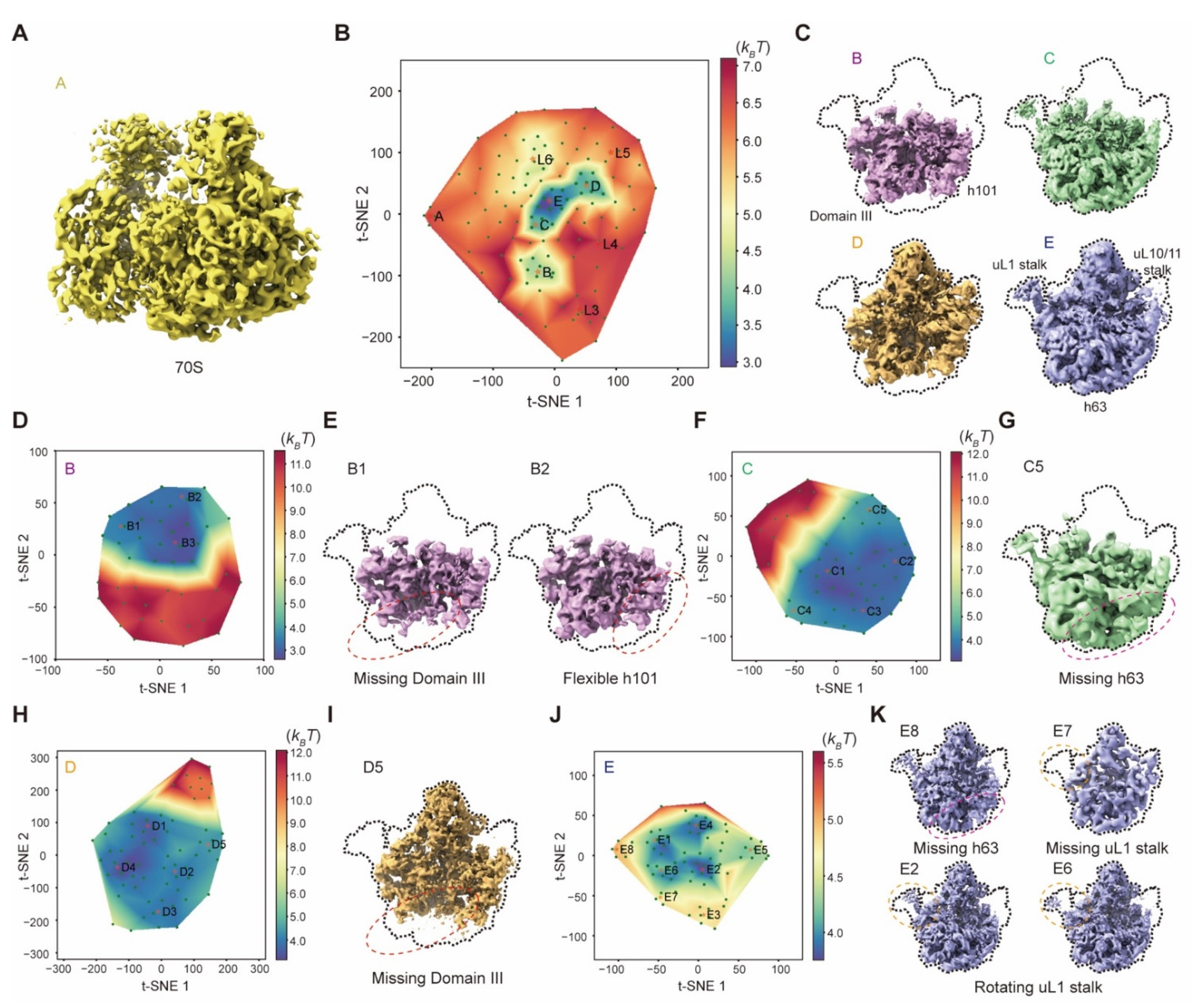
The 131,899 particle images of the 50S ribosomal large subunit (EMPIAR-10076) are highly heterogeneous. We refined the alignment of all particles with a global mask of the 50S using RELION v3.0. In the first step, 119 resampled volumes were utilized to calculate the pseudo-energy landscape. M = 7 particle shuffling was conducted to generate these volumes without any mask. The regularization coefficient of 0.001 was set when training the deep neural network. All volumes were then low-pass filtered at 5 Å prior to manifold embedding. Then the first pseudo-energy landscape resulted in nine clusters with the particle number of 766 (A), 15,129 (B), 1236, 322, 670, 22,037, 25,445 (D), 33,976 (E) and 25,115 (C), respectively. For the clusters of states B–E, four zoom-in pseudo-energy landscapes were then computed to discover more sub-states with 46, 56, 66 and 72 resampled volumes, respectively. In the zoom-in pseudo-energy landscape calculation, M = 7 was set in particle shuffling for resampling 3D volumes with a global mask and 20 Å resolution limit in the expectation step. Then the regularization coefficient of 0.001 was used to train the deep residual network. All volumes were then low-pass filtered at 5 Å prior to manifold embedding. After particle voting, the pseudo-energy landscape of state B resulted in three sub-clusters with particle numbers of 3494, 3600 and 7803. The state C pseudo-energy landscape resulted in five sub-clusters with the particle numbers of 2444, 7442, 711, 7633 and 5557. The state D pseudo-energy landscape resulted in five sub-clusters with the particle numbers of 6231, 8785, 2123, 3718 and 4092. Moreover, the pseudo-energy landscape of state E resulted in eight sub-clusters with the particle numbers of 4783, 1588, 5891, 3620, 7352, 5265, 357 and 2241. All these clusters were refined using RELION v3.0.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}