
Bromocriptine-QR Therapy Reduces Sympathetic Tone and Ameliorates a Pro-Oxidative/Pro-Inflammatory Phenotype in Peripheral Blood Mononuclear Cells and Plasma of Type 2 Diabetes Subjects

 ,
,
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics of Study Subjects
2.2. Bromocriptine-QR Effect on Plasma Levels of SNS Tone Markers, Pro-Oxidative Stress Markers, and Pro-Inflammatory Factors
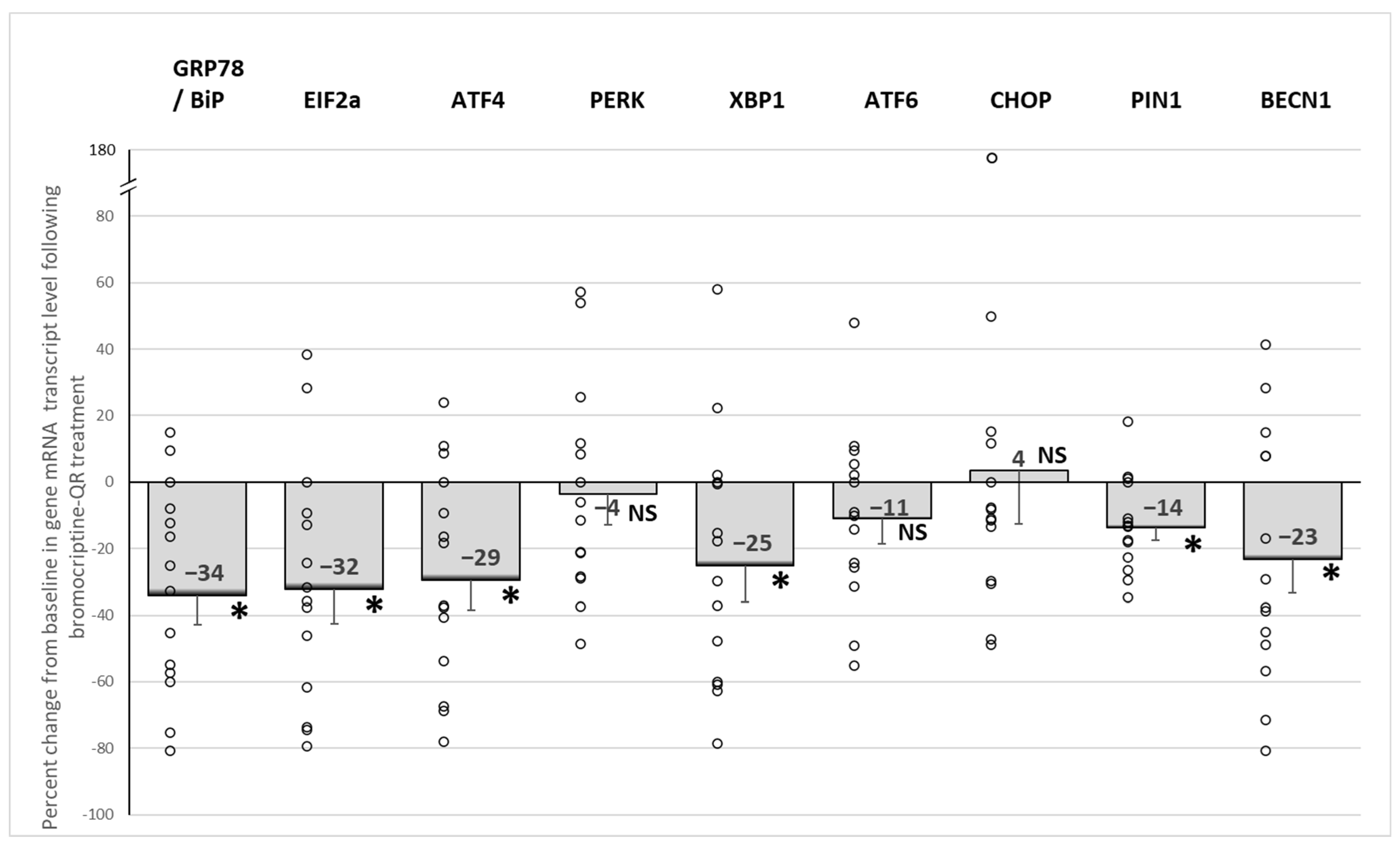
2.3. Effects of Bromocriptine-QR Therapy on mRNA Expression of PBMC ER Stress-Associated Unfolded Protein Response Genes
2.4. Effect of Bromocriptine-QR Therapy on mRNA Expression of PBMC Oxidative Stress Response (Anti-Oxidant) Genes
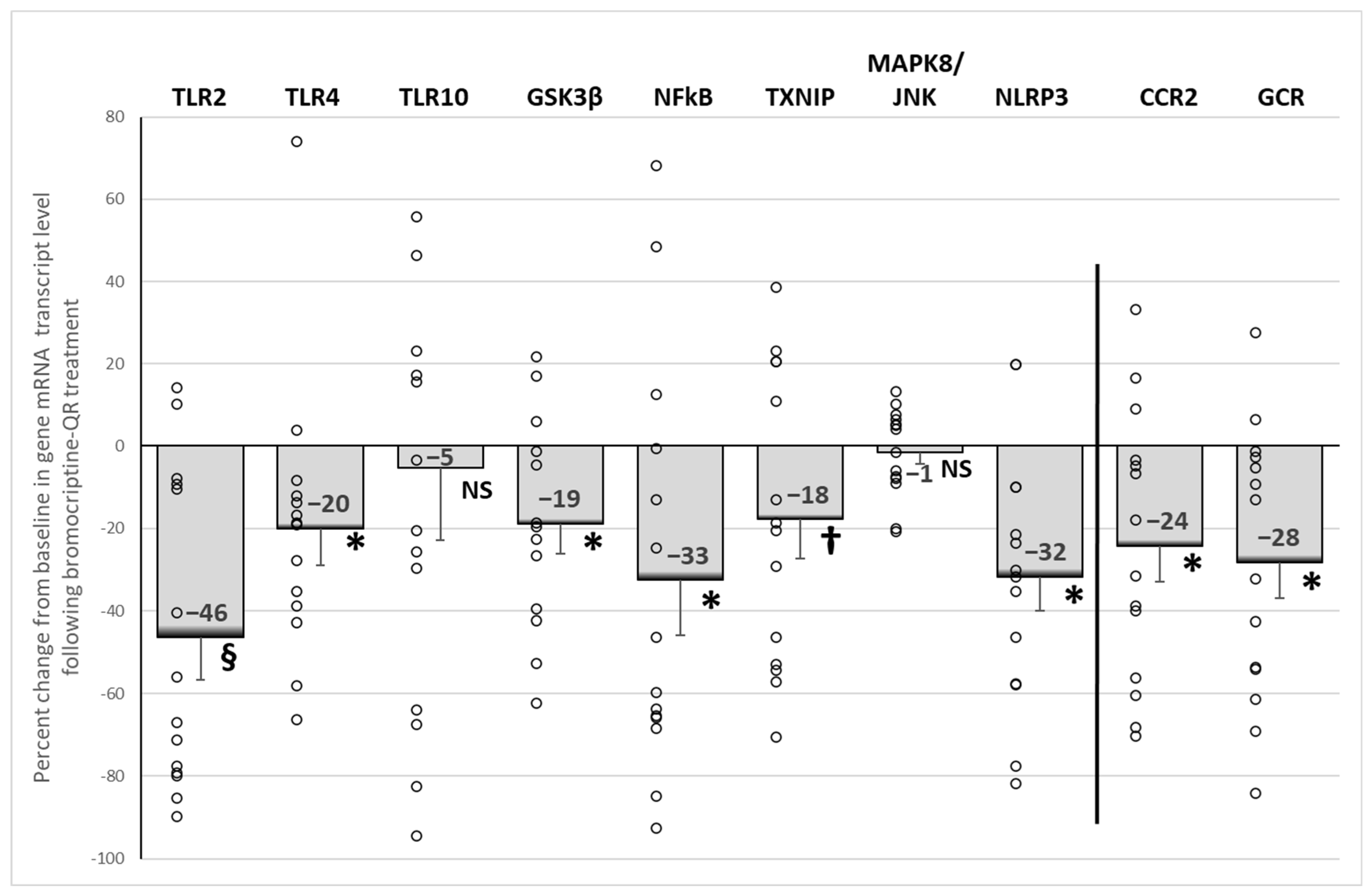
2.5. Effect of Bromocriptine-QR Therapy on mRNA Expression of PBMC Pro-Inflammatory Genes
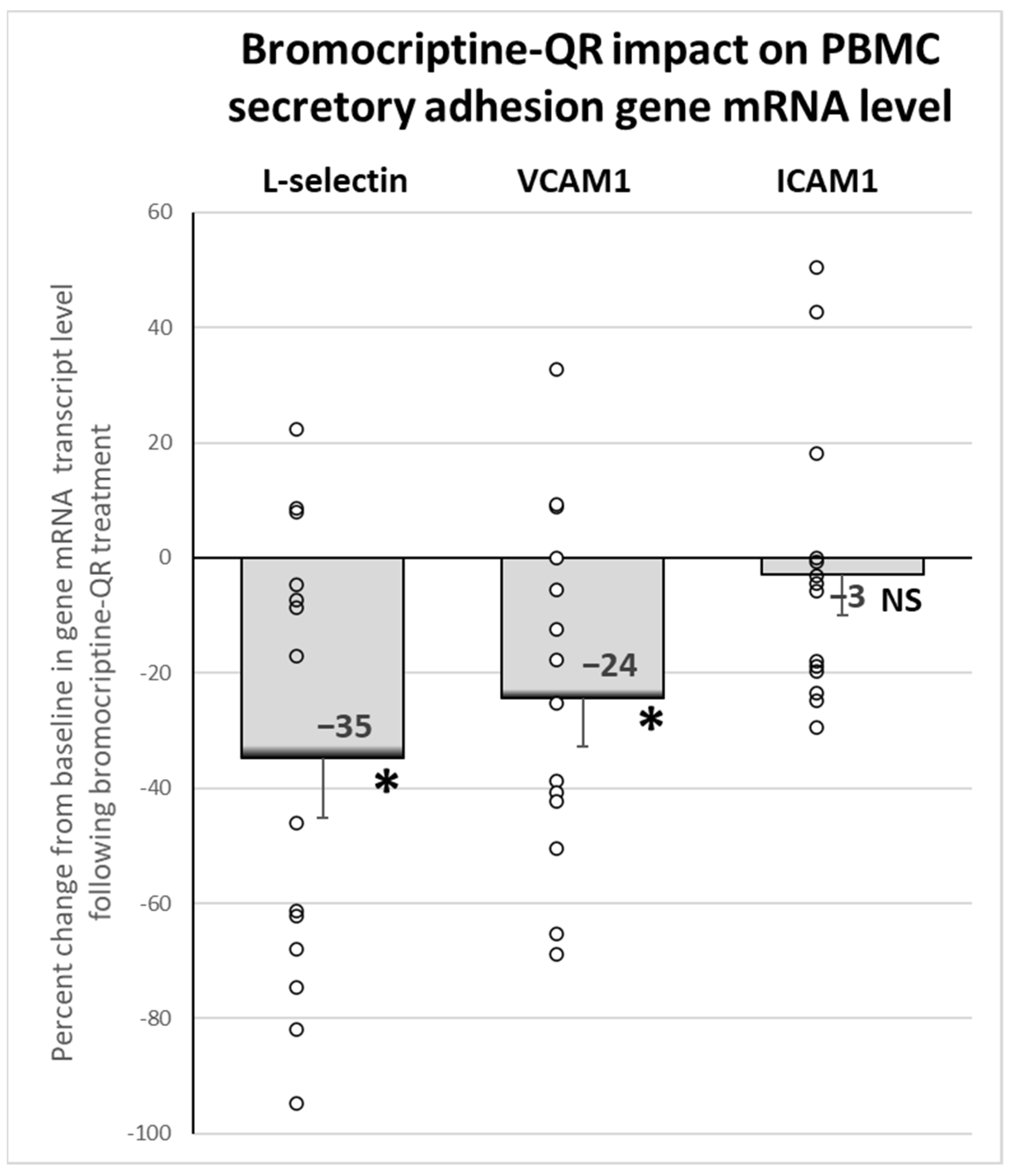
2.6. Effect of Bromocriptine-QR Therapy on mRNA Expression of PBMC Secretory Adhesion Molecule Genes
2.7. Adverse Effects
3. Discussion
4. Materials and Methods
4.1. Study Design and Therapeutic Intervention Regimen
4.2. Rationale for PBMC Analyses of Specific Gene mRNA Expression Levels as Assessment of the PO/PI State in T2D
4.3. Biochemical Assays of Blood Samples
4.3.1. Assessment of SNS Tone Markers (Norepinephrine, Normetanephrine)
4.3.2. Assessment of Plasma Oxidative Stress and Pro-Inflammatory Markers
4.4. Assessment of PBMC Pro-Oxidative/Pro-Inflammatory Status
4.4.1. PBMC Isolation
4.4.2. PBMC Gene Expression Analysis by Real-Time qPCR—mRNA Extraction and PCR Methods including Primers
ER Stress-Associated Unfolded Protein Response Genes
Oxidative Stress Response (Antioxidant) Genes
Pro-Inflammatory Genes
Secretory Adhesion Genes
Housekeeping Gene
4.5. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviations used in Figure 7 | |
| CNS | Central nervous system |
| DA | Dopaminergic |
| DAMP | Damage-associated molecular pattern |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| PBMC | Peripheral blood mononuclear cells |
| PI | Pro-inflammatory |
| ROS | Reactive oxygen species |
| SNS | Sympathetic nervous system |
| UPR | Unfolded protein response |
| Genes mentioned in Figure 7 | |
| ATF4 | Activating Transcription Factor 4 |
| ATF6 | Activating Transcription Factor 6 |
| BiP/GRP78 | Heat-Shock Protein Family A (Hsp70) Member 5 |
| CCR2 | C-C Motif Chemokine Receptor 2 |
| CHOP | DNA Damage Inducible Transcript 3 |
| EIF2α | Eukaryotic Translation Initiation Factor 2, Subunit 1 Alpha |
| GCR | Glucocorticoid receptor |
| GSK3β | Glycogen Synthase Kinase 3 Beta |
| ICAM1 | Intercellular Adhesion Molecule 1 |
| IRE1 | Endoplasmic Reticulum to Nucleus Signaling 1 |
| MAPK8/JNK | Mitogen-Activated Protein Kinase 8 |
| MCP1/CCL2 | Monocyte Chemoattractant Protein 1 |
| NFkB | NF-kappa-B p65 subunit |
| NLRP3 | NLR Family Pyrin Domain-Containing Protein 3 |
| PERK | Eukaryotic Translation Initiation Factor 2 Alpha Kinase 3 |
| TLR2 | Toll-like receptor 2 |
| TLR4 | Toll-like receptor 4 |
| TLR10 | Toll-like Receptor 10 |
| TXNIP | Thioredoxin Interacting Protein |
| VCAM1 | Vascular Cell Adhesion Molecule 1 |
| XBP1 | X-Box Binding Protein 1 functional variant |
References
- Raskin, A.; Cincotta, A.H. Bromocriptine-QR therapy for the management of type 2 diabetes mellitus: Developmental basis and therapeutic profile summary. Expert Rev. Endocrinol. Metab. 2016, 11, 113–148. [Google Scholar] [CrossRef] [PubMed]
- Cycloset [Package Insert]; VeroScience: Tiverton, RI, USA, 2020.
- Gaziano, J.M.; Cincotta, A.H.; O’Connor, C.M.; Ezrokhi, M.; Rutty, D.; Ma, Z.J.; Scranton, R.E. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 2010, 33, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Cincotta, A.H.; Vinik, A.; Blonde, L.; Bohannon, N.; Scranton, R. Effect of bromocriptine-QR (a quick-release formulation of bromocriptine mesylate) on major adverse cardiovascular events in type 2 diabetes subjects. J. Am. Heart Assoc. 2012, 1, e002279. [Google Scholar] [CrossRef] [PubMed]
- Chamarthi, B.; Gaziano, J.M.; Blonde, L.; Vinik, A.; Scranton, R.E.; Ezrokhi, M.; Rutty, D.; Cincotta, A.H. Timed Bromocriptine-QR Therapy Reduces Progression of Cardiovascular Disease and Dysglycemia in Subjects with Well-Controlled Type 2 Diabetes Mellitus. J. Diabetes Res. 2015, 2015, 157698. [Google Scholar] [CrossRef]
- Chamarthi, B.; Ezrokhi, M.; Rutty, D.; Cincotta, A.H. Impact of bromocriptine-QR therapy on cardiovascular outcomes in type 2 diabetes mellitus subjects on metformin. Postgrad. Med. 2016, 128, 761–769. [Google Scholar] [CrossRef]
- Franchi, F.; Lazzeri, C.; Barletta, G.; Ianni, L.; Mannelli, M. Centrally mediated effects of bromocriptine on cardiac sympathovagal balance. Hypertension 2001, 38, 123–129. [Google Scholar] [CrossRef]
- Mannelli, M.; Delitala, G.; De Feo, M.L.; Maggi, M.; Cuomo, S.; Piazzini, M.; Guazzelli, R.; Serio, M. Effects of different dopaminergic antagonists on bromocriptine-induced inhibition of norepinephrine release. J. Clin. Endocrinol. Metab. 1984, 59, 74–78. [Google Scholar] [CrossRef]
- Sowers, J.R. Dopaminergic control of circadian norepinephrine levels in patients with essential hypertension. J. Clin. Endocrinol. Metab. 1981, 53, 1133–1137. [Google Scholar] [CrossRef]
- Sowers, J.R.; Stern, N.; Nyby, M.D.; Jasberg, K.A. Dopaminergic regulation of circadian rhythms of blood pressure, renin and aldosterone in essential hypertension. Cardiovasc. Res. 1982, 16, 317–323. [Google Scholar] [CrossRef]
- Ezrokhi, M.; Zhang, Y.; Luo, S.; Cincotta, A.H. Time-of-Day-Dependent Effects of Bromocriptine to Ameliorate Vascular Pathology and Metabolic Syndrome in SHR Rats Held on High Fat Diet. Int. J. Mol. Sci. 2021, 22, 6142. [Google Scholar] [CrossRef]
- Huggett, R.J.; Burns, J.; Mackintosh, A.F.; Mary, D.A. Sympathetic neural activation in nondiabetic metabolic syndrome and its further augmentation by hypertension. Hypertension 2004, 44, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Huggett, R.J.; Hogarth, A.J.; Mackintosh, A.F.; Mary, D.A. Sympathetic nerve hyperactivity in non-diabetic offspring of patients with type 2 diabetes mellitus. Diabetologia 2006, 49, 2741–2744. [Google Scholar] [CrossRef] [PubMed]
- Pal, G.K.; Adithan, C.; Ananthanarayanan, P.H.; Pal, P.; Nanda, N.; Durgadevi, T.; Lalitha, V.; Syamsunder, A.N.; Dutta, T.K. Sympathovagal imbalance contributes to prehypertension status and cardiovascular risks attributed by insulin resistance, inflammation, dyslipidemia and oxidative stress in first degree relatives of type 2 diabetics. PLoS ONE 2013, 8, e78072. [Google Scholar] [CrossRef]
- Wulsin, L.R.; Horn, P.S.; Perry, J.L.; Massaro, J.M.; D’Agostino, R.B. Autonomic Imbalance as a Predictor of Metabolic Risks, Cardiovascular Disease, Diabetes, and Mortality. J. Clin. Endocrinol. Metab. 2015, 100, 2443–2448. [Google Scholar] [CrossRef] [PubMed]
- Seravalle, G.; Grassi, G. Sympathetic Nervous System, Hypertension, Obesity and Metabolic Syndrome. High Blood Press. Cardiovasc. Prev. 2016, 23, 175–179. [Google Scholar] [CrossRef]
- Thorp, A.A.; Schlaich, M.P. Relevance of Sympathetic Nervous System Activation in Obesity and Metabolic Syndrome. J. Diabetes Res. 2015, 2015, 341583. [Google Scholar] [CrossRef]
- Carnethon, M.R.; Golden, S.H.; Folsom, A.R.; Haskell, W.; Liao, D. Prospective investigation of autonomic nervous system function and the development of type 2 diabetes: The Atherosclerosis Risk in Communities study, 1987–1998. Circulation 2003, 107, 2190–2195. [Google Scholar] [CrossRef]
- Hu, A.; Jiao, X.; Gao, E.; Koch, W.J.; Sharifi-Azad, S.; Grunwald, Z.; Ma, X.L.; Sun, J.Z. Chronic beta-adrenergic receptor stimulation induces cardiac apoptosis and aggravates myocardial ischemia/reperfusion injury by provoking inducible nitric-oxide synthase-mediated nitrative stress. J. Pharmacol. Exp. Ther. 2006, 318, 469–475. [Google Scholar] [CrossRef]
- Bleeke, T.; Zhang, H.; Madamanchi, N.; Patterson, C.; Faber, J.E. Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ. Res. 2004, 94, 37–45. [Google Scholar] [CrossRef]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef]
- Bellinger, D.L.; Lorton, D. Sympathetic Nerve Hyperactivity in the Spleen: Causal for Nonpathogenic-Driven Chronic Immune-Mediated Inflammatory Diseases (IMIDs)? Int. J. Mol. Sci. 2018, 19, 1188. [Google Scholar] [CrossRef] [PubMed]
- Ahmari, N.; Santisteban, M.M.; Miller, D.R.; Geis, N.M.; Larkin, R.; Redler, T.; Denson, H.; Khoshbouei, H.; Baekey, D.M.; Raizada, M.K.; et al. Elevated bone marrow sympathetic drive precedes systemic inflammation in angiotensin II hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H279–H289. [Google Scholar] [CrossRef] [PubMed]
- Mravec, B. Chemical sympathectomy attenuates lipopolysaccharide-induced increase of plasma cytokine levels in rats pretreated by ACTH. J. Neuroimmunol. 2019, 337, 577086. [Google Scholar] [CrossRef] [PubMed]
- Fonkoue, I.T.; Marvar, P.J.; Norrholm, S.; Li, Y.; Kankam, M.L.; Jones, T.N.; Vemulapalli, M.; Rothbaum, B.; Bremner, J.D.; Le, N.A.; et al. Symptom severity impacts sympathetic dysregulation and inflammation in post-traumatic stress disorder (PTSD). Brain Behav. Immun. 2020, 83, 260–269. [Google Scholar] [CrossRef]
- Mishra, I.; Pullum, K.B.; Thayer, D.C.; Plummer, E.R.; Conkright, B.W.; Morris, A.J.; O’Hara, B.F.; Demas, G.E.; Ashley, N.T. Chemical sympathectomy reduces peripheral inflammatory responses to acute and chronic sleep fragmentation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R781–R789. [Google Scholar] [CrossRef]
- Kavelaars, A. Regulated expression of alpha-1 adrenergic receptors in the immune system. Brain Behav. Immun. 2002, 16, 799–807. [Google Scholar] [CrossRef]
- Nance, D.M.; Sanders, V.M. Autonomic innervation and regulation of the immune system (1987-2007). Brain Behav. Immun. 2007, 21, 736–745. [Google Scholar] [CrossRef]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve—An integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar]
- Abboud, F.M.; Singh, M.V. Autonomic regulation of the immune system in cardiovascular diseases. Adv. Physiol. Educ. 2017, 41, 578–593. [Google Scholar] [CrossRef]
- Moreira, H.G.; Lage, R.L.; Martinez, D.G.; Ferreira-Santos, L.; Rondon, M.; Negrao, C.E.; Nicolau, J.C. Sympathetic nervous activity in patients with acute coronary syndrome: A comparative study of inflammatory biomarkers. Clin. Sci. 2017, 131, 883–895. [Google Scholar] [CrossRef]
- Tian, X.; Guo, R.; Zhang, Y.; Xu, L.; Liu, X.; Hou, Y. Effects of the Sympathetic Nervous System on Regulatory T Cell and T Helper 1 Chemokine Expression in Patients with Acute Coronary Syndrome. Neuroimmunomodulation 2016, 23, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Fonkoue, I.T.; Le, N.A.; Kankam, M.L.; DaCosta, D.; Jones, T.N.; Marvar, P.J.; Park, J. Sympathoexcitation and impaired arterial baroreflex sensitivity are linked to vascular inflammation in individuals with elevated resting blood pressure. Physiol. Rep. 2019, 7, e14057. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.Z.; Wu, J.M.; Hu, G.M.; Gu, H.J.; Feng, Y.N.; Wang, S.X.; Cong, W.W.; Li, M.Z.; Xu, W.L.; Song, Y.; et al. alpha1-AR overactivation induces cardiac inflammation through NLRP3 inflammasome activation. Acta Pharmacol. Sin. 2020, 41, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.H.; Jenkins, N.T.; Padilla, J.; Parrish, A.R.; Fadel, P.J. Norepinephrine increases NADPH oxidase-derived superoxide in human peripheral blood mononuclear cells via alpha-adrenergic receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R1124–R1132. [Google Scholar] [CrossRef]
- Bellinger, D.L.; Millar, B.A.; Perez, S.; Carter, J.; Wood, C.; ThyagaRajan, S.; Molinaro, C.; Lubahn, C.; Lorton, D. Sympathetic modulation of immunity: Relevance to disease. Cell Immunol. 2008, 252, 27–56. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Souza-Neto, F.V.; Jimenez-Gonzalez, S.; Cachofeiro, V. Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest. Antioxidants 2021, 10, 406. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Katsiki, N. Oxidative Stress and Inflammation: Their Role in the Pathogenesis of Peripheral Artery Disease with or Without Type 2 Diabetes Mellitus. Curr. Vasc. Pharmacol. 2018, 16, 547–554. [Google Scholar] [CrossRef]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 193–196. [Google Scholar] [CrossRef]
- Halim, M.; Halim, A. The effects of inflammation, aging and oxidative stress on the pathogenesis of diabetes mellitus (type 2 diabetes). Diabetes Metab. Syndr. 2019, 13, 1165–1172. [Google Scholar] [CrossRef]
- Lv, S.; Wang, H.; Li, X. The Role of the Interplay between Autophagy and NLRP3 Inflammasome in Metabolic Disorders. Front. Cell Dev. Biol. 2021, 9, 634118. [Google Scholar] [CrossRef]
- Colak, Y.; Hasan, B.; Erkalma, B.; Tandon, K.; Zervos, X.; Menzo, E.L.; Erim, T. Pathogenetic mechanisms of nonalcoholic fatty liver disease and inhibition of the inflammasome as a new therapeutic target. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101710. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Abbate, A.; Toldo, S. NLRP3 Inflammasome Inhibitors in Cardiovascular Diseases. Molecules 2021, 26, 976. [Google Scholar] [CrossRef] [PubMed]
- Tsiotra, P.C.; Tsigos, C. Stress, the endoplasmic reticulum, and insulin resistance. Ann. N. Y. Acad. Sci. 2006, 1083, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Panzhinskiy, E.; Ren, J.; Nair, S. Protein tyrosine phosphatase 1B and insulin resistance: Role of endoplasmic reticulum stress/reactive oxygen species/nuclear factor kappa B axis. PLoS ONE 2013, 8, e77228. [Google Scholar] [CrossRef]
- Arnold, N.; Gori, T.; Schnabel, R.B.; Schulz, A.; Prochaska, J.H.; Zeller, T.; Binder, H.; Pfeiffer, N.; Beutel, M.; Espinola-Klein, C.; et al. Relation between Arterial Stiffness and Markers of Inflammation and Hemostasis—Data from the Population-based Gutenberg Health Study. Sci. Rep. 2017, 7, 6346. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Fatehi-Hassanabad, Z.; Chan, C.B.; Furman, B.L. Reactive oxygen species and endothelial function in diabetes. Eur. J. Pharmacol. 2010, 636, 8–17. [Google Scholar] [CrossRef]
- Alicka, M.; Marycz, K. The Effect of Chronic Inflammation and Oxidative and Endoplasmic Reticulum Stress in the Course of Metabolic Syndrome and Its Therapy. Stem. Cells Int. 2018, 2018, 4274361. [Google Scholar] [CrossRef]
- Toth, B.E.; Vecsernyes, M.; Zelles, T.; Kardar, K.; Nagy, G. Role of Peripheral and Brain-Derived Dopamine (DA) in Immune Regulation. Adv. Neuroimmune Biol. 2012, 3, 111–155. [Google Scholar] [CrossRef]
- Levite, M. Dopamine and T cells: Dopamine receptors and potent effects on T cells, dopamine production in T cells, and abnormalities in the dopaminergic system in T cells in autoimmune, neurological and psychiatric diseases. Acta Physiol. 2016, 216, 42–89. [Google Scholar] [CrossRef]
- Felger, J.C.; Treadway, M.T. Inflammation Effects on Motivation and Motor Activity: Role of Dopamine. Neuropsychopharmacology 2017, 42, 216–241. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C. The Role of Dopamine in Inflammation-Associated Depression: Mechanisms and Therapeutic Implications. Curr. Top Behav. Neurosci. 2017, 31, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Li, Z.; Haroon, E.; Woolwine, B.J.; Jung, M.Y.; Hu, X.; Miller, A.H. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry 2016, 21, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.; Ribeiro, L. Dopaminergic Pathways in Obesity-Associated Inflammation. J. Neuroimmune. Pharmacol. 2020, 15, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.; Lima, M.; Marino, F.; Cosentino, M.; Ribeiro, L. Dopaminergic Receptors and Tyrosine Hydroxylase Expression in Peripheral Blood Mononuclear Cells: A Distinct Pattern in Central Obesity. PLoS ONE 2016, 11, e0147483. [Google Scholar] [CrossRef]
- Tolstanova, G.; Deng, X.; Ahluwalia, A.; Paunovic, B.; Prysiazhniuk, A.; Ostapchenko, L.; Tarnawski, A.; Sandor, Z.; Szabo, S. Role of Dopamine and D2 Dopamine Receptor in the Pathogenesis of Inflammatory Bowel Disease. Dig. Dis. Sci. 2015, 60, 2963–2975. [Google Scholar] [CrossRef]
- Han, X.; Li, B.; Ye, X.; Mulatibieke, T.; Wu, J.; Dai, J.; Wu, D.; Ni, J.; Zhang, R.; Xue, J.; et al. Dopamine D2 receptor signalling controls inflammation in acute pancreatitis via a PP2A-dependent Akt/NF-kappaB signalling pathway. Br. J. Pharmacol. 2017, 174, 4751–4770. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Wu, J.; Manaenko, A.; Yang, P.; Tang, J.; Fu, W.; Zhang, J.H. Activation of Dopamine D2 Receptor Suppresses Neuroinflammation through alphaB-Crystalline by Inhibition of NF-kappaB Nuclear Translocation in Experimental ICH Mice Model. Stroke 2015, 46, 2637–2646. [Google Scholar] [CrossRef]
- Balkowiec-Iskra, E.; Kurkowska-Jastrzebska, I.; Joniec, I.; Ciesielska, A.; Muszynska, A.; Przybylkowski, A.; Czlonkowska, A.; Czlonkowski, A. MPTP-induced central dopamine depletion exacerbates experimental autoimmune encephalomyelitis (EAE) in C57BL mice. Inflamm. Res. 2007, 56, 311–317. [Google Scholar] [CrossRef]
- Kawano, M.; Takagi, R.; Saika, K.; Matsui, M.; Matsushita, S. Dopamine regulates cytokine secretion during innate and adaptive immune responses. Int. Immunol. 2018, 30, 591–606. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Sugino, Y.; Shibagaki, F.; Yamamuro, A.; Ishimaru, Y.; Maeda, S. Dopamine attenuates lipopolysaccharide-induced expression of proinflammatory cytokines by inhibiting the nuclear translocation of NF-kappaB p65 through the formation of dopamine quinone in microglia. Eur. J. Pharmacol. 2020, 866, 172826. [Google Scholar] [CrossRef]
- Liu, A.; Ding, S. Anti-inflammatory Effects of Dopamine in Lipopolysaccharide (LPS)-stimulated RAW264.7 Cells via Inhibiting NLRP3 Inflammasome Activation. Ann. Clin. Lab. Sci. 2019, 49, 353–360. [Google Scholar] [PubMed]
- Shimojo, G.; Joseph, B.; Shah, R.; Consolim-Colombo, F.M.; De Angelis, K.; Ulloa, L. Exercise activates vagal induction of dopamine and attenuates systemic inflammation. Brain Behav. Immun. 2019, 75, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Romero, M.C.; Delgado-Cortes, M.J.; Sarmiento, M.; de Pablos, R.M.; Espinosa-Oliva, A.M.; Arguelles, S.; Bandez, M.J.; Villaran, R.F.; Maurino, R.; Santiago, M.; et al. Peripheral inflammation increases the deleterious effect of CNS inflammation on the nigrostriatal dopaminergic system. Neurotoxicology 2012, 33, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhao, H.; Zhang, W.; Liu, B.; Liu, Y.; Guo, Y.; Nie, L. Overexpression of conserved dopamine neurotrophic factor (CDNF) in astrocytes alleviates endoplasmic reticulum stress-induced cell damage and inflammatory cytokine secretion. Biochem. Biophys. Res. Commun. 2013, 435, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.E. Bromocriptine treatment of systemic lupus erythematosus. Lupus 2001, 10, 762–768. [Google Scholar] [CrossRef]
- Alvarez-Nemegyei, J.; Cobarrubias-Cobos, A.; Escalante-Triay, F.; Sosa-Munoz, J.; Miranda, J.M.; Jara, L.J. Bromocriptine in systemic lupus erythematosus: A double-blind, randomized, placebo-controlled study. Lupus 1998, 7, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; Liuqin, L.; Hao, L.; Shiwen, Y.; Zhongping, Z.; Dongying, C.; Fan, L.; Hanshi, X.; Xiuyan, Y.; Yujin, Y. The effects of bromocriptine on preventing postpartum flare in systemic lupus erythematosus patients from South China. J. Immunol. Res. 2015, 2015, 316965. [Google Scholar] [CrossRef]
- Jara, L.J.; Cruz-Cruz, P.; Saavedra, M.A.; Medina, G.; Garcia-Flores, A.; Angeles, U.; Miranda-Limon, J.M. Bromocriptine during pregnancy in systemic lupus erythematosus: A pilot clinical trial. Ann. N. Y. Acad. Sci. 2007, 1110, 297–304. [Google Scholar] [CrossRef]
- Palestine, A.G.; Muellenberg-Coulombre, C.G.; Kim, M.K.; Gelato, M.C.; Nussenblatt, R.B. Bromocriptine and low dose cyclosporine in the treatment of experimental autoimmune uveitis in the rat. J. Clin. Investig. 1987, 79, 1078–1081. [Google Scholar] [CrossRef]
- Figueroa, F.; Carrion, F.; Martinez, M.E.; Rivero, S.; Mamani, I.; Gonzalez, G. Effects of bromocriptine in patients with active rheumatoid arthritis. Rev. Med. Chil. 1998, 126, 33–41. [Google Scholar] [PubMed]
- Hilfiker-Kleiner, D.; Struman, I.; Hoch, M.; Podewski, E.; Sliwa, K. 16-kDa prolactin and bromocriptine in postpartum cardiomyopathy. Curr. Heart Fail Rep. 2012, 9, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Haghikia, A.; Berliner, D.; Vogel-Claussen, J.; Schwab, J.; Franke, A.; Schwarzkopf, M.; Ehlermann, P.; Pfister, R.; Michels, G.; et al. Bromocriptine for the treatment of peripartum cardiomyopathy: A multicentre randomized study. Eur. Heart J. 2017, 38, 2671–2679. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Schwab, J.; Vogel-Claussen, J.; Berliner, D.; Pfeffer, T.; Konig, T.; Zwadlo, C.; Moulig, V.A.; Franke, A.; Schwarzkopf, M.; et al. Bromocriptine treatment in patients with peripartum cardiomyopathy and right ventricular dysfunction. Clin. Res. Cardiol. 2019, 108, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Moron, E.; Abad-Jimenez, Z.; Maranon, A.M.; Iannantuoni, F.; Escribano-Lopez, I.; Lopez-Domenech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Szpigel, A.; Hainault, I.; Carlier, A.; Venteclef, N.; Batto, A.F.; Hajduch, E.; Bernard, C.; Ktorza, A.; Gautier, J.F.; Ferre, P.; et al. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia 2018, 61, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Xia, C.Q.; Butfiloski, E.; Clare-Salzler, M. Effect of high glucose on cytokine production by human peripheral blood immune cells and type I interferon signaling in monocytes: Implications for the role of hyperglycemia in the diabetes inflammatory process and host defense against infection. Clin. Immunol. 2018, 195, 139–148. [Google Scholar] [CrossRef]
- Li, H.; Peng, W.; Jian, W.; Li, Y.; Li, Q.; Li, W.; Xu, Y. ROCK inhibitor fasudil attenuated high glucose-induced MCP-1 and VCAM-1 expression and monocyte-endothelial cell adhesion. Cardiovasc. Diabetol. 2012, 11, 65. [Google Scholar] [CrossRef]
- Akhter, N.; Madhoun, A.; Arefanian, H.; Wilson, A.; Kochumon, S.; Thomas, R.; Shenouda, S.; Al-Mulla, F.; Ahmad, R.; Sindhu, S. Oxidative Stress Induces Expression of the Toll-Like Receptors (TLRs) 2 and 4 in the Human Peripheral Blood Mononuclear Cells: Implications for Metabolic Inflammation. Cell Physiol. Biochem. 2019, 53, 1–18. [Google Scholar] [CrossRef]
- Yasunari, K.; Watanabe, T.; Nakamura, M. Reactive oxygen species formation by polymorphonuclear cells and mononuclear cells as a risk factor of cardiovascular diseases. Curr. Pharm. Biotechnol. 2006, 7, 73–80. [Google Scholar] [CrossRef]
- Takahashi, M. Cell-Specific Roles of NLRP3 Inflammasome in Myocardial Infarction. J. Cardiovasc. Pharmacol. 2019, 74, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, S.; Chang, S.; Ren, D.; Shali, S.; Li, C.; Yang, H.; Huang, Z.; Ge, J. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-kappaB/NLRP3 inflammasome signaling pathway. J. Mol. Cell. Cardiol. 2020, 142, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Komura, T.; Sakai, Y.; Honda, M.; Takamura, T.; Matsushima, K.; Kaneko, S. CD14+ monocytes are vulnerable and functionally impaired under endoplasmic reticulum stress in patients with type 2 diabetes. Diabetes 2010, 59, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Lopez, I.; de Maranon, A.M.; Iannantuoni, F.; Lopez-Domenech, S.; Abad-Jimenez, Z.; Diaz, P.; Sola, E.; Apostolova, N.; Rocha, M.; Victor, V.M. The Mitochondrial Antioxidant SS-31 Modulates Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy in Type 2 Diabetes. J. Clin. Med. 2019, 8, 1322. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Forte, M.; Raffa, S. Circulating Leukocytes and Oxidative Stress in Cardiovascular Diseases: A State of the Art. Oxid. Med. Cell Longev. 2019, 2019, 2650429. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 2010, 16, 396–399. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Hummasti, S.; Hotamisligil, G.S. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ. Res. 2010, 107, 579–591. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Flamment, M.; Hajduch, E.; Ferre, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Targeting endoplasmic reticulum stress in metabolic disease. Expert. Opin. Ther. Targets 2013, 17, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kouri, G.; Wollheim, C.B. ER stress and SREBP-1 activation are implicated in beta-cell glucolipotoxicity. J. Cell Sci. 2005, 118, 3905–3915. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, K.D.; Alejandro, E.U.; Luciani, D.S.; Kalynyak, T.B.; Hu, X.; Li, H.; Lin, Y.; Townsend, R.R.; Polonsky, K.S.; Johnson, J.D. Carboxypeptidase E mediates palmitate-induced beta-cell ER stress and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 8452–8457. [Google Scholar] [CrossRef]
- McAlpine, C.S.; Bowes, A.J.; Werstuck, G.H. Diabetes, hyperglycemia and accelerated atherosclerosis: Evidence supporting a role for endoplasmic reticulum (ER) stress signaling. Cardiovasc. Hematol. Disord. Drug Targets 2010, 10, 151–157. [Google Scholar] [CrossRef]
- Mozzini, C.; Garbin, U.; Stranieri, C.; Pasini, A.; Solani, E.; Tinelli, I.A.; Cominacini, L.; Fratta Pasini, A.M. Endoplasmic reticulum stress and Nrf2 repression in circulating cells of type 2 diabetic patients without the recommended glycemic goals. Free Radic. Res. 2015, 49, 244–252. [Google Scholar] [CrossRef]
- Rajan, S.; Shankar, K.; Beg, M.; Varshney, S.; Gupta, A.; Srivastava, A.; Kumar, D.; Mishra, R.K.; Hussain, Z.; Gayen, J.R.; et al. Chronic hyperinsulinemia reduces insulin sensitivity and metabolic functions of brown adipocyte. J. Endocrinol. 2016, 230, 275–290. [Google Scholar] [CrossRef]
- Tampakakis, E.; Tabit, C.E.; Holbrook, M.; Linder, E.A.; Berk, B.D.; Frame, A.A.; Breton-Romero, R.; Fetterman, J.L.; Gokce, N.; Vita, J.A.; et al. Intravenous Lipid Infusion Induces Endoplasmic Reticulum Stress in Endothelial Cells and Blood Mononuclear Cells of Healthy Adults. J. Am. Heart Assoc. 2016, 5, e002574. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox. Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Lenin, R.; Sankaramoorthy, A.; Mohan, V.; Balasubramanyam, M. Altered immunometabolism at the interface of increased endoplasmic reticulum (ER) stress in patients with type 2 diabetes. J. Leukoc. Biol. 2015, 98, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.T.; Holtby-Ottenhof, S.; Shi, Y.; Damjanovic, S.; Sharma, A.M.; Werstuck, G.H. Metabolic syndrome and acute hyperglycemia are associated with endoplasmic reticulum stress in human mononuclear cells. Obes. (Silver Spring) 2012, 20, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Alatrach, M.; Agyin, C.; Adams, J.; Chilton, R.; Triplitt, C.; DeFronzo, R.A.; Cersosimo, E. Glucose lowering and vascular protective effects of cycloset added to GLP-1 receptor agonists in patients with type 2 diabetes. Endocrinol. Diabetes Metab. 2018, 1, e00034. [Google Scholar] [CrossRef] [PubMed]
- Victor, P.; Sarada, D.; Ramkumar, K.M. Crosstalk between endoplasmic reticulum stress and oxidative stress: Focus on protein disulfide isomerase and endoplasmic reticulum oxidase 1. Eur. J. Pharmacol. 2021, 892, 173749. [Google Scholar] [CrossRef]
- Delaunay-Moisan, A.; Appenzeller-Herzog, C. The antioxidant machinery of the endoplasmic reticulum: Protection and signaling. Free Radic. Biol. Med. 2015, 83, 341–351. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Miani, M.; Colli, M.L.; Ladriere, L.; Cnop, M.; Eizirik, D.L. Mild endoplasmic reticulum stress augments the proinflammatory effect of IL-1beta in pancreatic rat beta-cells via the IRE1alpha/XBP1s pathway. Endocrinology 2012, 153, 3017–3028. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef]
- Boyce, M.; Py, B.F.; Ryazanov, A.G.; Minden, J.S.; Long, K.; Ma, D.; Yuan, J. A pharmacoproteomic approach implicates eukaryotic elongation factor 2 kinase in ER stress-induced cell death. Cell Death Differ. 2008, 15, 589–599. [Google Scholar] [CrossRef]
- Kong, F.J.; Ma, L.L.; Guo, J.J.; Xu, L.H.; Li, Y.; Qu, S. Endoplasmic reticulum stress/autophagy pathway is involved in diabetes-induced neuronal apoptosis and cognitive decline in mice. Clin. Sci. 2018, 132, 111–125. [Google Scholar] [CrossRef]
- Ning, B.; Zhang, Q.; Wang, N.; Deng, M.; Fang, Y. beta-Asarone Regulates ER Stress and Autophagy via Inhibition of the PERK/CHOP/Bcl-2/Beclin-1 Pathway in 6-OHDA-Induced Parkinsonian Rats. Neurochem. Res. 2019, 44, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Esnault, S.; Shen, Z.J.; Malter, J.S. Pinning down signaling in the immune system: The role of the peptidyl-prolyl isomerase Pin1 in immune cell function. Crit. Rev. Immunol. 2008, 28, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B. Role of the Nrf2 signaling system in health and disease. Clin. Genet. 2014, 86, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Fratta Pasini, A.M.; Ferrari, M.; Stranieri, C.; Vallerio, P.; Mozzini, C.; Garbin, U.; Zambon, G.; Cominacini, L. Nrf2 expression is increased in peripheral blood mononuclear cells derived from mild-moderate ex-smoker COPD patients with persistent oxidative stress. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 1733–1743. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Hennig, P.; Garstkiewicz, M.; Grossi, S.; Di Filippo, M.; French, L.E.; Beer, H.D. The Crosstalk between Nrf2 and Inflammasomes. Int. J. Mol. Sci. 2018, 19, 562. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Volkert, M.R.; Crowley, D.J. Preventing Neurodegeneration by Controlling Oxidative Stress: The Role of OXR1. Front. Neurosci. 2020, 14, 611904. [Google Scholar] [CrossRef]
- Rebrin, I.; Sohal, R.S. Pro-oxidant shift in glutathione redox state during aging. Adv. Drug Deliv. Rev. 2008, 60, 1545–1552. [Google Scholar] [CrossRef]
- Rahman, I.; Biswas, S.K.; Jimenez, L.A.; Torres, M.; Forman, H.J. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid. Redox Signal 2005, 7, 42–59. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Gall, T.; Balla, G.; Balla, J. Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress—A New Insight into the Pathophysiology of Vascular Diseases. Int. J. Mol. Sci. 2019, 20, 3675. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef]
- Upmacis, R.K.; Crabtree, M.J.; Deeb, R.S.; Shen, H.; Lane, P.B.; Benguigui, L.E.; Maeda, N.; Hajjar, D.P.; Gross, S.S. Profound biopterin oxidation and protein tyrosine nitration in tissues of ApoE-null mice on an atherogenic diet: Contribution of inducible nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2878–H2887. [Google Scholar] [CrossRef]
- Ishii, M.; Shimizu, S.; Wajima, T.; Hagiwara, T.; Negoro, T.; Miyazaki, A.; Tobe, T.; Kiuchi, Y. Reduction of GTP cyclohydrolase I feedback regulating protein expression by hydrogen peroxide in vascular endothelial cells. J. Pharmacol. Sci. 2005, 97, 299–302. [Google Scholar] [CrossRef]
- Rocha, M.; Diaz-Morales, N.; Rovira-Llopis, S.; Escribano-Lopez, I.; Banuls, C.; Hernandez-Mijares, A.; Diamanti-Kandarakis, E.; Victor, V.M. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Diabetes. Curr. Pharm. Des. 2016, 22, 2640–2649. [Google Scholar] [CrossRef]
- Liu, H.; Cao, M.M.; Wang, Y.; Li, L.C.; Zhu, L.B.; Xie, G.Y.; Li, Y.B. Endoplasmic reticulum stress is involved in the connection between inflammation and autophagy in type 2 diabetes. Gen. Comp. Endocrinol. 2015, 210, 124–129. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, Y.; Yang, T.; Song, M.; Wang, C.; Yao, Y.; Fan, H. Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors. Front. Mol. Neurosci. 2019, 12, 210. [Google Scholar] [CrossRef]
- Busillo, J.M.; Azzam, K.M.; Cidlowski, J.A. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J. Biol. Chem. 2011, 286, 38703–38713. [Google Scholar] [CrossRef]
- Yeager, M.P.; Pioli, P.A.; Collins, J.; Barr, F.; Metzler, S.; Sites, B.D.; Guyre, P.M. Glucocorticoids enhance the in vivo migratory response of human monocytes. Brain Behav. Immun. 2016, 54, 86–94. [Google Scholar] [CrossRef]
- Penton-Rol, G.; Cota, M.; Polentarutti, N.; Luini, W.; Bernasconi, S.; Borsatti, A.; Sica, A.; LaRosa, G.J.; Sozzani, S.; Poli, G.; et al. Up-regulation of CCR2 chemokine receptor expression and increased susceptibility to the multitropic HIV strain 89.6 in monocytes exposed to glucocorticoid hormones. J. Immunol. 1999, 163, 3524–3529. [Google Scholar]
- Lv, S.; Li, X.; Wang, H. The Role of the Effects of Endoplasmic Reticulum Stress on NLRP3 Inflammasome in Diabetes. Front. Cell Dev. Biol. 2021, 9, 663528. [Google Scholar] [CrossRef]
- Ruan, Y.; Zeng, J.; Jin, Q.; Chu, M.; Ji, K.; Wang, Z.; Li, L. Endoplasmic reticulum stress serves an important role in cardiac ischemia/reperfusion injury (Review). Exp. Ther. Med. 2020, 20, 268. [Google Scholar] [CrossRef]
- Kim, S.; Joe, Y.; Surh, Y.J.; Chung, H.T. Differential Regulation of Toll-Like Receptor-Mediated Cytokine Production by Unfolded Protein Response. Oxid. Med. Cell Longev. 2018, 2018, 9827312. [Google Scholar] [CrossRef]
- Kim, S.; Joe, Y.; Kim, H.J.; Kim, Y.S.; Jeong, S.O.; Pae, H.O.; Ryter, S.W.; Surh, Y.J.; Chung, H.T. Endoplasmic reticulum stress-induced IRE1alpha activation mediates cross-talk of GSK-3beta and XBP-1 to regulate inflammatory cytokine production. J. Immunol. 2015, 194, 4498–4506. [Google Scholar] [CrossRef]
- Dragomir, E.; Simionescu, M. Monocyte chemoattractant protein-1—A major contributor to the inflammatory process associated with diabetes. Arch Physiol. Biochem. 2006, 112, 239–244. [Google Scholar] [CrossRef]
- Troseid, M.; Seljeflot, I.; Arnesen, H. The role of interleukin-18 in the metabolic syndrome. Cardiovasc. Diabetol. 2010, 9, 11. [Google Scholar] [CrossRef]
- Hung, J.; McQuillan, B.M.; Chapman, C.M.; Thompson, P.L.; Beilby, J.P. Elevated interleukin-18 levels are associated with the metabolic syndrome independent of obesity and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1268–1273. [Google Scholar] [CrossRef]
- Santalahti, K.; Maksimow, M.; Airola, A.; Pahikkala, T.; Hutri-Kahonen, N.; Jalkanen, S.; Raitakari, O.T.; Salmi, M. Circulating Cytokines Predict the Development of Insulin Resistance in a Prospective Finnish Population Cohort. J. Clin. Endocrinol. Metab. 2016, 101, 3361–3369. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-18 and the pathogenesis of inflammatory diseases. Semin. Nephrol. 2007, 27, 98–114. [Google Scholar] [CrossRef]
- Niu, J.; Kolattukudy, P.E. Role of MCP-1 in cardiovascular disease: Molecular mechanisms and clinical implications. Clin. Sci. 2009, 117, 95–109. [Google Scholar] [CrossRef]
- Franca, C.N.; Izar, M.C.O.; Hortencio, M.N.S.; do Amaral, J.B.; Ferreira, C.E.S.; Tuleta, I.D.; Fonseca, F.A.H. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin. Sci. 2017, 131, 1215–1224. [Google Scholar] [CrossRef]
- Panee, J. Monocyte Chemoattractant Protein 1 (MCP-1) in obesity and diabetes. Cytokine 2012, 60, 1–12. [Google Scholar] [CrossRef]
- Xia, Y.; Frangogiannis, N.G. MCP-1/CCL2 as a therapeutic target in myocardial infarction and ischemic cardiomyopathy. Inflamm. Allergy Drug Targets 2007, 6, 101–107. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Peiro, C.; Lorenzo, O.; Carraro, R.; Sanchez-Ferrer, C.F. IL-1beta Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus. Front. Pharmacol. 2017, 8, 363. [Google Scholar] [CrossRef]
- Bujak, M.; Frangogiannis, N.G. The role of IL-1 in the pathogenesis of heart disease. Arch Immunol. Ther. Exp. (Warsz.) 2009, 57, 165–176. [Google Scholar] [CrossRef]
- Dourado, M.; Cavalcanti, F.; Vilar, L.; Cantilino, A. Relationship between Prolactin, Chronic Kidney Disease, and Cardiovascular Risk. Int. J. Endocrinol. 2020, 2020, 9524839. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, Y.; Liu, E. C-reactive protein and cardiovascular disease: From animal studies to the clinic (Review). Exp. Ther. Med. 2020, 20, 1211–1219. [Google Scholar] [CrossRef]
- Xu, Y.; Whitmer, K. C-reactive protein and cardiovascular disease in people with diabetes: High-sensitivity CRP testing can help assess risk for future cardiovascular disease events in this population. Am. J. Nurs. 2006, 106, 66–72. [Google Scholar] [CrossRef]
- Parrinello, C.M.; Lutsey, P.L.; Ballantyne, C.M.; Folsom, A.R.; Pankow, J.S.; Selvin, E. Six-year change in high-sensitivity C-reactive protein and risk of diabetes, cardiovascular disease, and mortality. Am. Heart J. 2015, 170, 380–389. [Google Scholar] [CrossRef]
- Mallard, A.R.; Hollekim-Strand, S.M.; Ingul, C.B.; Coombes, J.S. High day-to-day and diurnal variability of oxidative stress and inflammation biomarkers in people with type 2 diabetes mellitus and healthy individuals. Redox. Rep. 2020, 25, 64–69. [Google Scholar] [CrossRef]
- Kanabrocki, E.L.; Murray, D.; Hermida, R.C.; Scott, G.S.; Bremner, W.F.; Ryan, M.D.; Ayala, D.E.; Third, J.L.; Shirazi, P.; Nemchausky, B.A.; et al. Circadian variation in oxidative stress markers in healthy and type II diabetic men. Chronobiol. Int. 2002, 19, 423–439. [Google Scholar] [CrossRef]
- Maio, R.; Perticone, M.; Sciacqua, A.; Tassone, E.J.; Naccarato, P.; Bagnato, C.; Iannopollo, G.; Sesti, G.; Perticone, F. Oxidative stress impairs endothelial function in nondipper hypertensive patients. Cardiovasc. Ther. 2012, 30, 85–92. [Google Scholar] [CrossRef]
- Avogaro, A.; Pagnin, E.; Calo, L. Monocyte NADPH oxidase subunit p22(phox) and inducible hemeoxygenase-1 gene expressions are increased in type II diabetic patients: Relationship with oxidative stress. J. Clin. Endocrinol. Metab. 2003, 88, 1753–1759. [Google Scholar] [CrossRef][Green Version]
- Farah, R.; Shurtz-Swirski, R.; Lapin, O. Intensification of oxidative stress and inflammation in type 2 diabetes despite antihyperglycemic treatment. Cardiovasc. Diabetol. 2008, 7, 20. [Google Scholar] [CrossRef]
- Huang, X.; Sun, M.; Li, D.; Liu, J.; Guo, H.; Dong, Y.; Jiang, L.; Pan, Q.; Man, Y.; Wang, S.; et al. Augmented NADPH oxidase activity and p22phox expression in monocytes underlie oxidative stress of patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2011, 91, 371–380. [Google Scholar] [CrossRef]
- Cortez-Espinosa, N.; Garcia-Hernandez, M.H.; Reynaga-Hernandez, E.; Cortes-Garcia, J.D.; Corral-Fernandez, N.E.; Rodriguez-Rivera, J.G.; Bravo-Ramirez, A.; Gonzalez-Amaro, R.; Portales-Perez, D.P. Abnormal expression and function of Dectin-1 receptor in type 2 diabetes mellitus patients with poor glycemic control (HbA1c > 8%). Metabolism 2012, 61, 1538–1546. [Google Scholar] [CrossRef]
- He, L.; Wong, C.K.; Cheung, K.K.; Yau, H.C.; Fu, A.; Zhao, H.L.; Leung, K.M.; Kong, A.P.; Wong, G.W.; Chan, P.K.; et al. Anti-inflammatory effects of exendin-4, a glucagon-like peptide-1 analog, on human peripheral lymphocytes in patients with type 2 diabetes. J. Diabetes Investig. 2013, 4, 382–392. [Google Scholar] [CrossRef]
- Mandal, L.K.; Choudhuri, S.; Dutta, D.; Mitra, B.; Kundu, S.; Chowdhury, I.H.; Sen, A.; Chatterjee, M.; Bhattacharya, B. Oxidative stress-associated neuroretinal dysfunction and nitrosative stress in diabetic retinopathy. Can. J. Diabetes 2013, 37, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Lenin, R.; Maria, M.S.; Agrawal, M.; Balasubramanyam, J.; Mohan, V.; Balasubramanyam, M. Amelioration of glucolipotoxicity-induced endoplasmic reticulum stress by a “chemical chaperone” in human THP-1 monocytes. Exp. Diabetes Res. 2012, 2012, 356487. [Google Scholar] [CrossRef] [PubMed]
- Chamarthi, B.; Vinik, A.; Ezrokhi, M.; Cincotta, A.H. Circadian-timed quick-release bromocriptine lowers elevated resting heart rate in patients with type 2 diabetes mellitus. Endocrinol. Diabetes Metab. 2020, 3, e00101. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.N.; Kvakan, H.; Luft, F.C. Immune-related effects in hypertension and target-organ damage. Curr. Opin. Nephrol. Hypertens. 2011, 20, 113–117. [Google Scholar] [CrossRef]
- Wrigley, B.J.; Lip, G.Y.; Shantsila, E. The role of monocytes and inflammation in the pathophysiology of heart failure. Eur. J. Heart Fail. 2011, 13, 1161–1171. [Google Scholar] [CrossRef]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid. Med. Cell Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef]
- Guo, Z.; Yu, S.; Chen, X.; Ye, R.; Zhu, W.; Liu, X. NLRP3 Is Involved in Ischemia/Reperfusion Injury. CNS Neurol. Disord. Drug Targets 2016, 15, 699–712. [Google Scholar] [CrossRef]
- Gao, J.; Guo, J.; Li, H.; Bai, S.; Li, H.; Wu, B.; Wang, L.; Xi, Y.; Tian, Y.; Yang, G.; et al. Involvement of dopamine D2 receptors activation in ischemic post-conditioning-induced cardioprotection through promoting PKC-epsilon particulate translocation in isolated rat hearts. Mol. Cell Biochem. 2013, 379, 267–276. [Google Scholar] [CrossRef]
- Li, H.; Wei, C.; Gao, J.; Bai, S.; Li, H.; Zhao, Y.; Li, H.; Han, L.; Tian, Y.; Yang, G.; et al. Mediation of dopamine D2 receptors activation in post-conditioning-attenuated cardiomyocyte apoptosis. Exp. Cell Res. 2014, 323, 118–130. [Google Scholar] [CrossRef]
- Li, H.Z.; Guo, J.; Gao, J.; Han, L.P.; Jiang, C.M.; Li, H.X.; Bai, S.Z.; Zhang, W.H.; Li, G.W.; Wang, L.N.; et al. Role of dopamine D2 receptors in ischemia/reperfusion induced apoptosis of cultured neonatal rat cardiomyocytes. J. Biomed. Sci. 2011, 18, 18. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Hicks, C.A.; Ward, M.A.; Cardwell, G.P.; Reymann, J.M.; Allain, H.; Bentue-Ferrer, D. Dopamine D2 receptor agonists protect against ischaemia-induced hippocampal neurodegeneration in global cerebral ischaemia. Eur. J. Pharmacol. 1998, 352, 37–46. [Google Scholar] [CrossRef]
- Narkar, V.; Kunduzova, O.; Hussain, T.; Cambon, C.; Parini, A.; Lokhandwala, M. Dopamine D2-like receptor agonist bromocriptine protects against ischemia/reperfusion injury in rat kidney. Kidney Int. 2004, 66, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhang, Y.; Ezrokhi, M.; Li, Y.; Tsai, T.H.; Cincotta, A.H. Circadian peak dopaminergic activity response at the biological clock pacemaker (suprachiasmatic nucleus) area mediates the metabolic responsiveness to a high-fat diet. J. Neuroendocrinol. 2018, 30, e12563. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Luo, J.; Cincotta, A.H. Suprachiasmatic nuclei monoamine metabolism of glucose tolerant versus intolerant hamsters. Neuroreport 1999, 10, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Luo, J.; Meier, A.H.; Cincotta, A.H. Dopaminergic neurotoxin administration to the area of the suprachiasmatic nuclei induces insulin resistance. Neuroreport 1997, 8, 3495–3499. [Google Scholar] [CrossRef]
- Luo, S.; Ezrokhi, M.; Cominos, N.; Tsai, T.H.; Stoelzel, C.R.; Trubitsyna, Y.; Cincotta, A.H. Experimental dopaminergic neuron lesion at the area of the biological clock pacemaker, suprachiasmatic nuclei (SCN) induces metabolic syndrome in rats. Diabetol. Metab. Syndr. 2021, 13, 11. [Google Scholar] [CrossRef]
- Luo, S.; Liang, Y.; Cincotta, A.H. Intracerebroventricular administration of bromocriptine ameliorates the insulin-resistant/glucose-intolerant state in hamsters. Neuroendocrinology 1999, 69, 160–166. [Google Scholar] [CrossRef]
- Stoelzel, C.R.; Zhang, Y.; Cincotta, A.H. Circadian-timed dopamine agonist treatment reverses high-fat diet-induced diabetogenic shift in ventromedial hypothalamic glucose sensing. Endocrinol. Diabetes Metab. 2020, 3, e00139. [Google Scholar] [CrossRef]
- Ezrokhi, M.; Luo, S.; Trubitsyna, Y.; Cincotta, A.H. Neuroendocrine and metabolic components of dopamine agonist amelioration of metabolic syndrome in SHR rats. Diabetol. Metab. Syndr. 2014, 6, 104. [Google Scholar] [CrossRef]
- Bahler, L.; Verberne, H.J.; Brakema, E.; Tepaske, R.; Booij, J.; Hoekstra, J.B.; Holleman, F. Bromocriptine and insulin sensitivity in lean and obese subjects. Endocr. Connect. 2016, 5, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Aziz, I.S.; McMahon, A.M.; Friedman, D.; Rabinovich-Nikitin, I.; Kirshenbaum, L.A.; Martino, T.A. Circadian influence on inflammatory response during cardiovascular disease. Curr. Opin. Pharmacol. 2020, 57, 60–70. [Google Scholar] [CrossRef]
- Denis, R.G.; Joly-Amado, A.; Cansell, C.; Castel, J.; Martinez, S.; Delbes, A.S.; Luquet, S. Central orchestration of peripheral nutrient partitioning and substrate utilization: Implications for the metabolic syndrome. Diabetes Metab. 2014, 40, 191–197. [Google Scholar] [CrossRef]
- Gupta, A.K.; Ravussin, E.; Johannsen, D.L.; Stull, A.J.; Cefalu, W.T.; Johnson, W.D. Endothelial Dysfunction: An Early Cardiovascular Risk Marker in Asymptomatic Obese Individuals with Prediabetes. Br. J. Med. Med. Res. 2012, 2, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; Palm, I.F.; La Fleur, S.E.; Scheer, F.A.; Perreau-Lenz, S.; Ruiter, M.; Kreier, F.; Cailotto, C.; Buijs, R.M. SCN outputs and the hypothalamic balance of life. J. Biol. Rhythms. 2006, 21, 458–469. [Google Scholar] [CrossRef]
- Kudo, T.; Horikawa, K.; Shibata, S. Circadian rhythms in the CNS and peripheral clock disorders: The circadian clock and hyperlipidemia. J. Pharmacol. Sci. 2007, 103, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Leproult, R.; Holmback, U.; Van Cauter, E. Circadian misalignment augments markers of insulin resistance and inflammation, independently of sleep loss. Diabetes 2014, 63, 1860–1869. [Google Scholar] [CrossRef]
- Maemura, K.; Takeda, N.; Nagai, R. Circadian rhythms in the CNS and peripheral clock disorders: Role of the biological clock in cardiovascular diseases. J. Pharmacol. Sci. 2007, 103, 134–138. [Google Scholar] [CrossRef]
- Man, A.W.C.; Li, H.; Xia, N. Circadian Rhythm: Potential Therapeutic Target for Atherosclerosis and Thrombosis. Int. J. Mol. Sci. 2021, 22, 676. [Google Scholar] [CrossRef]
- Manfredini, R.; Fabbian, F.; Manfredini, F.; Salmi, R.; Gallerani, M.; Bossone, E. Chronobiology in aortic diseases—“Is this really a random phenomenon?”. Prog. Cardiovasc. Dis. 2013, 56, 116–124. [Google Scholar] [CrossRef]
- Maywood, E.S.; O’Neill, J.; Wong, G.K.; Reddy, A.B.; Hastings, M.H. Circadian timing in health and disease. Prog. Brain Res. 2006, 153, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Perez-Torres, I.; Manzano-Pech, L.; Rubio-Ruiz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef] [PubMed]
- Prasai, M.J.; George, J.T.; Scott, E.M. Molecular clocks, type 2 diabetes and cardiovascular disease. Diab. Vasc. Dis. Res. 2008, 5, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Reilly, D.F.; Westgate, E.J.; FitzGerald, G.A. Peripheral circadian clocks in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Scheer, F.A.; Hilton, M.F.; Mantzoros, C.S.; Shea, S.A. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc. Natl. Acad. Sci. USA 2009, 106, 4453–4458. [Google Scholar] [CrossRef]
- Shimokawa, H. Reactive oxygen species in cardiovascular health and disease: Special references to nitric oxide, hydrogen peroxide, and Rho-kinase. J. Clin. Biochem. Nutr. 2020, 66, 83–91. [Google Scholar] [CrossRef]
- Shinozaki, K.; Ayajiki, K.; Kashiwagi, A.; Masada, M.; Okamura, T. Malfunction of vascular control in lifestyle-related diseases: Mechanisms underlying endothelial dysfunction in the insulin-resistant state. J. Pharmacol. Sci. 2004, 96, 401–405. [Google Scholar] [CrossRef]
- Young, M.E.; Bray, M.S. Potential role for peripheral circadian clock dyssynchrony in the pathogenesis of cardiovascular dysfunction. Sleep Med. 2007, 8, 656–667. [Google Scholar] [CrossRef]
- Cincotta, A. Hypothalamic role in the insulin resistance syndrome. In Insulin Resistance and Insulin Resistance Syndrome; Hansen, B., Shafrir, E., Eds.; Taylor and Francis: London, UK, 2002; pp. 271–312. [Google Scholar]
- Liang, Y.; Lubkin, M.; Sheng, H.; Scislowski, P.W.; Cincotta, A.H. Dopamine agonist treatment ameliorates hyperglycemia, hyperlipidemia, and the elevated basal insulin release from islets of ob/ob mice. Biochim. Biophys. Acta 1998, 1405, 1–13. [Google Scholar] [CrossRef]
- Cincotta, A.H.; MacEachern, T.A.; Meier, A.H. Bromocriptine redirects metabolism and prevents seasonal onset of obese hyperinsulinemic state in Syrian hamsters. Am. J. Physiol. 1993, 264, E285–E293. [Google Scholar] [CrossRef]
- Lech, K.; Ackermann, K.; Revell, V.L.; Lao, O.; Skene, D.J.; Kayser, M. Dissecting Daily and Circadian Expression Rhythms of Clock-Controlled Genes in Human Blood. J. Biol. Rhythms. 2016, 31, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Crnko, S.; Schutte, H.; Doevendans, P.A.; Sluijter, J.P.G.; van Laake, L.W. Minimally Invasive Ways of Determining Circadian Rhythms in Humans. Physiology (Bethesda) 2021, 36, 7–20. [Google Scholar] [CrossRef] [PubMed]
- James, F.O.; Cermakian, N.; Boivin, D.B. Circadian rhythms of melatonin, cortisol, and clock gene expression during simulated night shift work. Sleep 2007, 30, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Boivin, D.B.; James, F.O.; Wu, A.; Cho-Park, P.F.; Xiong, H.; Sun, Z.S. Circadian clock genes oscillate in human peripheral blood mononuclear cells. Blood 2003, 102, 4143–4145. [Google Scholar] [CrossRef]
- Umpierrez, G.E.; Gonzalez, A.; Umpierrez, D.; Pimentel, D. Diabetes mellitus in the Hispanic/Latino population: An increasing health care challenge in the United States. Am. J. Med. Sci. 2007, 334, 274–282. [Google Scholar] [CrossRef]
- Wenzel, P. Monocytes as immune targets in arterial hypertension. Br. J. Pharmacol. 2019, 176, 1966–1977. [Google Scholar] [CrossRef]
- Frodermann, V.; Nahrendorf, M. Macrophages and Cardiovascular Health. Physiol. Rev. 2018, 98, 2523–2569. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Swirski, F.K.; Robbins, C.S. Monocyte fate in atherosclerosis. Arter. Thromb. Vasc. Biol. 2015, 35, 272–279. [Google Scholar] [CrossRef]
- Luft, F.C.; Dechend, R.; Muller, D.N. Immune mechanisms in angiotensin II-induced target-organ damage. Ann. Med. 2012, 44 (Suppl. 1), S49–S54. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bezsonov, E.E.; Popkova, T.V.; Starodubova, A.V.; Orekhov, A.N. Immunity in Atherosclerosis: Focusing on T and B Cells. Int. J. Mol. Sci. 2021, 22, 8379. [Google Scholar] [CrossRef]
- Flores-Gomez, D.; Bekkering, S.; Netea, M.G.; Riksen, N.P. Trained Immunity in Atherosclerotic Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2021, 41, 62–69. [Google Scholar] [CrossRef]
- Su, J.; Zhou, L.; Kong, X.; Yang, X.; Xiang, X.; Zhang, Y.; Li, X.; Sun, L. Endoplasmic reticulum is at the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in the pathogenesis of diabetes mellitus. J. Diabetes Res. 2013, 2013, 193461. [Google Scholar] [CrossRef]
- Salvado, L.; Palomer, X.; Barroso, E.; Vazquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 2015, 26, 438–448. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int. J. Obes. 2008, 32 (Suppl. 7), S52–S54. [Google Scholar] [CrossRef]
- Sabroe, I.; Parker, L.C.; Dower, S.K.; Whyte, M.K. The role of TLR activation in inflammation. J. Pathol. 2008, 214, 126–135. [Google Scholar] [CrossRef]
- Ospelt, C.; Gay, S. TLRs and chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 495–505. [Google Scholar] [CrossRef]
- Said, E.A.; Tremblay, N.; Al-Balushi, M.S.; Al-Jabri, A.A.; Lamarre, D. Viruses Seen by Our Cells: The Role of Viral RNA Sensors. J. Immunol. Res. 2018, 2018, 9480497. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef]
- Wong, S.K.; Chin, K.Y.; Ima-Nirwana, S. Toll-like Receptor as a Molecular Link between Metabolic Syndrome and Inflammation: A Review. Curr. Drug Targets 2019, 20, 1264–1280. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, Y.; Liu, C.; Youn, J.Y.; Cai, H. Toll-Like Receptor 2 (TLR2) Knockout Abrogates Diabetic and Obese Phenotypes While Restoring Endothelial Function via Inhibition of NOX1. Diabetes 2021, 70, 2107–2119. [Google Scholar] [CrossRef]
- Sharma, A.; Choi, J.S.Y.; Stefanovic, N.; Al-Sharea, A.; Simpson, D.S.; Mukhamedova, N.; Jandeleit-Dahm, K.; Murphy, A.J.; Sviridov, D.; Vince, J.E.; et al. Specific NLRP3 Inhibition Protects Against Diabetes-Associated Atherosclerosis. Diabetes 2021, 70, 772–787. [Google Scholar] [CrossRef]
- Holley, A.E.; Cheeseman, K.H. Measuring free radical reactions in vivo. Br. Med. Bull. 1993, 49, 494–505. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef]
- Boss, A.; Kao, C.H.; Murray, P.M.; Marlow, G.; Barnett, M.P.; Ferguson, L.R. Human Intervention Study to Assess the Effects of Supplementation with Olive Leaf Extract on Peripheral Blood Mononuclear Cell Gene Expression. Int. J. Mol. Sci. 2016, 17, 2019. [Google Scholar] [CrossRef]
- Liew, C.C.; Ma, J.; Tang, H.C.; Zheng, R.; Dempsey, A.A. The peripheral blood transcriptome dynamically reflects system wide biology: A potential diagnostic tool. J. Lab. Clin. Med. 2006, 147, 126–132. [Google Scholar] [CrossRef]
- Olsen, K.S.; Skeie, G.; Lund, E. Whole-Blood Gene Expression Profiles in Large-Scale Epidemiological Studies: What Do They Tell? Curr. Nutr. Rep. 2015, 4, 377–386. [Google Scholar] [CrossRef]
- Restaino, R.M.; Deo, S.H.; Parrish, A.R.; Fadel, P.J.; Padilla, J. Increased monocyte-derived reactive oxygen species in type 2 diabetes: Role of endoplasmic reticulum stress. Exp. Physiol. 2017, 102, 139–153. [Google Scholar] [CrossRef]
- Degasperi, G.R.; Denis, R.G.; Morari, J.; Solon, C.; Geloneze, B.; Stabe, C.; Pareja, J.C.; Vercesi, A.E.; Velloso, L.A. Reactive oxygen species production is increased in the peripheral blood monocytes of obese patients. Metabolism 2009, 58, 1087–1095. [Google Scholar] [CrossRef]
- Jialal, I.; Devaraj, S.; Adams-Huet, B.; Chen, X.; Kaur, H. Increased cellular and circulating biomarkers of oxidative stress in nascent metabolic syndrome. J. Clin. Endocrinol. Metab. 2012, 97, E1844–E1850. [Google Scholar] [CrossRef]
- Navarro-Gonzalez, J.F.; Mora-Fernandez, C.; Muros de Fuentes, M.; Garcia-Perez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Melo, E.P.; Chambers, J.E.; Avezov, E. Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum. Cells 2021, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef] [PubMed]
- Ko, R.; Lee, S.Y. Glycogen synthase kinase 3beta in Toll-like receptor signaling. BMB Rep. 2016, 49, 305–310. [Google Scholar] [CrossRef]
- Wang, H.; Brown, J.; Martin, M. Glycogen synthase kinase 3: A point of convergence for the host inflammatory response. Cytokine 2011, 53, 130–140. [Google Scholar] [CrossRef]
- Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Therapeutic regulation of the NLRP3 inflammasome in chronic inflammatory diseases. Arch. Pharm. Res. 2021, 44, 16–35. [Google Scholar] [CrossRef]
- Fusco, R.; Siracusa, R.; Genovese, T.; Cuzzocrea, S.; Di Paola, R. Focus on the Role of NLRP3 Inflammasome in Diseases. Int. J. Mol. Sci. 2020, 21, 4223. [Google Scholar] [CrossRef] [PubMed]
- Padron, J.G.; Saito Reis, C.A.; Kendal-Wright, C.E. The Role of Danger Associated Molecular Patterns in Human Fetal Membrane Weakening. Front. Physiol. 2020, 11, 602. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Ji, N.; Qi, Z.; Wang, Y.; Yang, X.; Yan, Z.; Li, M.; Ge, Q.; Zhang, J. Pyroptosis: A New Regulating Mechanism in Cardiovascular Disease. J. Inflamm. Res. 2021, 14, 2647–2666. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 2006, 83, 447S–455S. [Google Scholar] [CrossRef] [PubMed]
- Gracie, J.A.; Robertson, S.E.; McInnes, I.B. Interleukin-18. J. Leukoc. Biol. 2003, 73, 213–224. [Google Scholar] [CrossRef]
- Dhawan, V.; Mahajan, N.; Jain, S. Role of C-C chemokines in Takayasu’s arteritis disease. Int. J. Cardiol. 2006, 112, 105–111. [Google Scholar] [CrossRef]
- Morel, J.C.; Park, C.C.; Woods, J.M.; Koch, A.E. A novel role for interleukin-18 in adhesion molecule induction through NF kappa B and phosphatidylinositol (PI) 3-kinase-dependent signal transduction pathways. J. Biol. Chem. 2001, 276, 37069–37075. [Google Scholar] [CrossRef]
- Khan, A.I.; Landis, R.C.; Malhotra, R. L-Selectin ligands in lymphoid tissues and models of inflammation. Inflammation 2003, 27, 265–280. [Google Scholar] [CrossRef]
- Gautier, E.L.; Jakubzick, C.; Randolph, G.J. Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1412–1418. [Google Scholar] [CrossRef]
- Barlic, J.; Murphy, P.M. Chemokine regulation of atherosclerosis. J. Leukoc. Biol. 2007, 82, 226–236. [Google Scholar] [CrossRef]
- O’Connor, T.; Borsig, L.; Heikenwalder, M. CCL2-CCR2 Signaling in Disease Pathogenesis. Endocr. Metab. Immune. Disord. Drug Targets 2015, 15, 105–118. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Grechko, A.V.; Myasoedova, V.A.; Orekhov, A.N. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp. Mol. Pathol. 2018, 104, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Schober, A. Chemokines in vascular dysfunction and remodeling. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1950–1959. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Usarek, E.; Baranczyk-Kuzma, A.; Kazmierczak, B.; Gajewska, B.; Kuzma-Kozakiewicz, M. Validation of qPCR reference genes in lymphocytes from patients with amyotrophic lateral sclerosis. PLoS ONE 2017, 12, e0174317. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Mean ± SEM |
|---|---|
| Age | 57 ± 9 years |
| Baseline HbA1c | 8.3 ± 0.3%, all subjects had baseline HbA1c >7.5% |
| Duration of diabetes | 10.2 ± 5.6 years |
| Body weight | 88 ± 13 kg |
| BMI | 33.4 ± 4.4 kg/m2, |
| Fasting plasma glucose | 145 ± 3 mg/dL |
| Fasting plasma insulin | 20.0 ± 2.1 μU/mL |
| Fasting plasma C-peptide | 4.8 ± 0.3 ng/mL |
| Fasting FFA | 530 ± 12 μmol/L |
| Heart rate | 74 ± 8 bpm |
| Systolic blood pressure | 134 ± 4 mm Hg |
| Diastolic blood pressure | 78 ± 3 mm Hg |
| Mean arterial blood pressure | 97 ± 5 mm Hg |
| Sex | 11 females (8 post-menopausal), 4 males |
| Concomitant diabetes medications | |
| Subjects on liraglutide | 1.2–1.8 mg/day; n = 15 |
| Subjects on metformin | n = 12 |
| Subjects on insulin glargine, low dose | n = 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cincotta, A.H.; Cersosimo, E.; Alatrach, M.; Ezrokhi, M.; Agyin, C.; Adams, J.; Chilton, R.; Triplitt, C.; Chamarthi, B.; Cominos, N.; et al. Bromocriptine-QR Therapy Reduces Sympathetic Tone and Ameliorates a Pro-Oxidative/Pro-Inflammatory Phenotype in Peripheral Blood Mononuclear Cells and Plasma of Type 2 Diabetes Subjects. Int. J. Mol. Sci. 2022, 23, 8851. https://doi.org/10.3390/ijms23168851
Cincotta AH, Cersosimo E, Alatrach M, Ezrokhi M, Agyin C, Adams J, Chilton R, Triplitt C, Chamarthi B, Cominos N, et al. Bromocriptine-QR Therapy Reduces Sympathetic Tone and Ameliorates a Pro-Oxidative/Pro-Inflammatory Phenotype in Peripheral Blood Mononuclear Cells and Plasma of Type 2 Diabetes Subjects. International Journal of Molecular Sciences. 2022; 23(16):8851. https://doi.org/10.3390/ijms23168851
Chicago/Turabian StyleCincotta, Anthony H., Eugenio Cersosimo, Mariam Alatrach, Michael Ezrokhi, Christina Agyin, John Adams, Robert Chilton, Curtis Triplitt, Bindu Chamarthi, Nicholas Cominos, and et al. 2022. "Bromocriptine-QR Therapy Reduces Sympathetic Tone and Ameliorates a Pro-Oxidative/Pro-Inflammatory Phenotype in Peripheral Blood Mononuclear Cells and Plasma of Type 2 Diabetes Subjects" International Journal of Molecular Sciences 23, no. 16: 8851. https://doi.org/10.3390/ijms23168851
APA StyleCincotta, A. H., Cersosimo, E., Alatrach, M., Ezrokhi, M., Agyin, C., Adams, J., Chilton, R., Triplitt, C., Chamarthi, B., Cominos, N., & DeFronzo, R. A. (2022). Bromocriptine-QR Therapy Reduces Sympathetic Tone and Ameliorates a Pro-Oxidative/Pro-Inflammatory Phenotype in Peripheral Blood Mononuclear Cells and Plasma of Type 2 Diabetes Subjects. International Journal of Molecular Sciences, 23(16), 8851. https://doi.org/10.3390/ijms23168851