
The Role of CD28 and CD8+ T Cells in Keloid Development

Abstract
:1. Introduction
2. Result
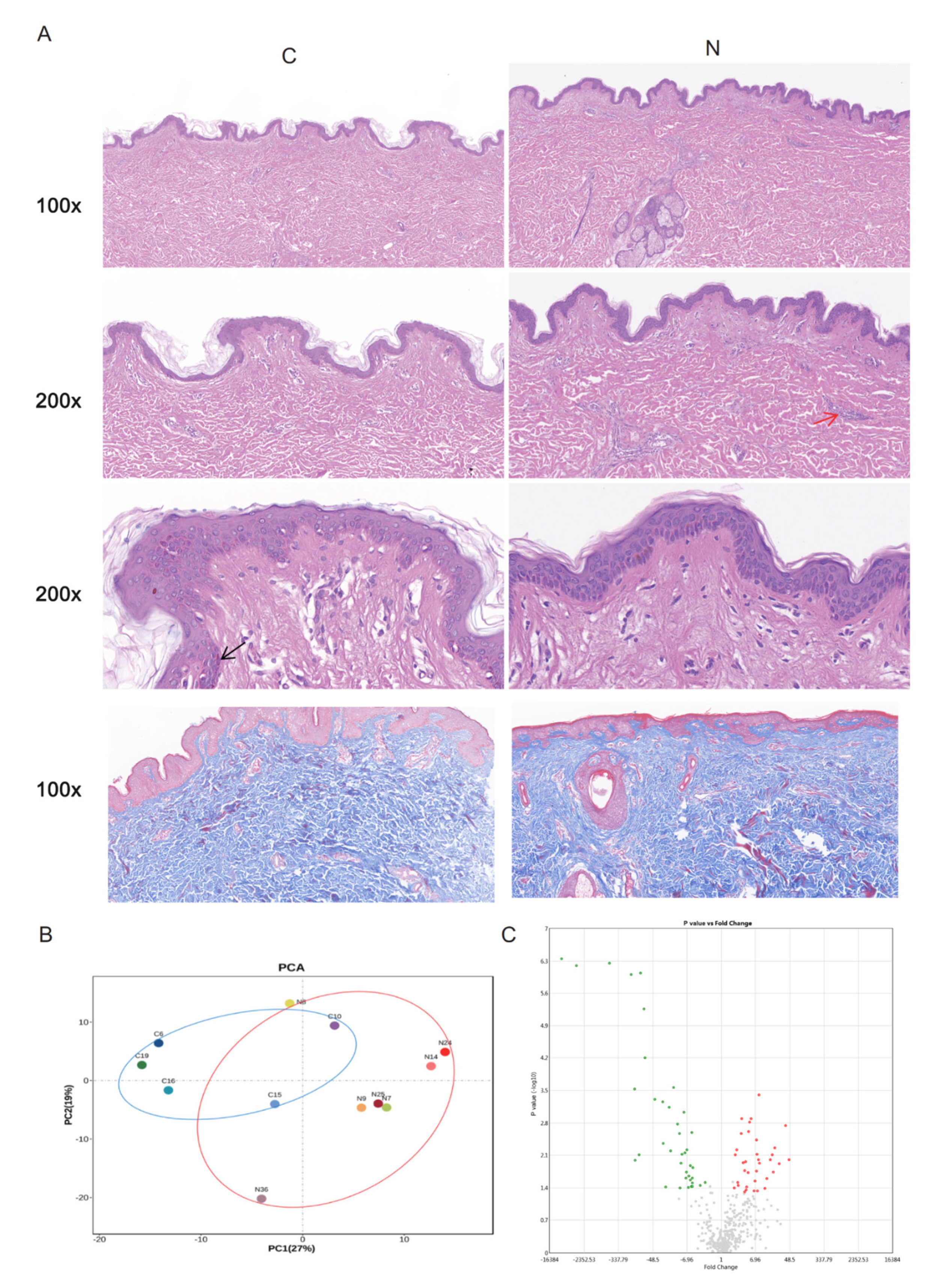
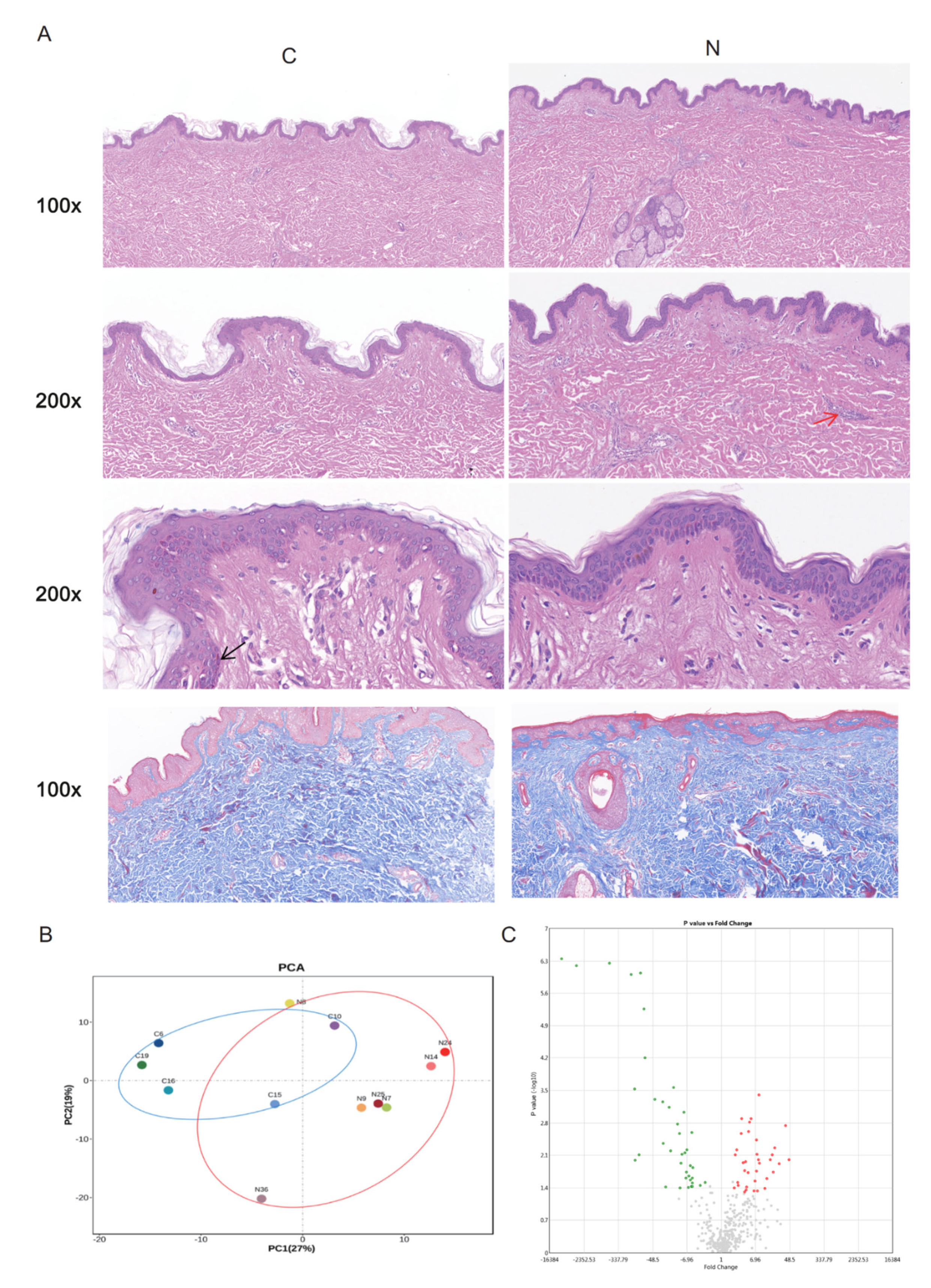
2.1. DEGs between Groups N and C
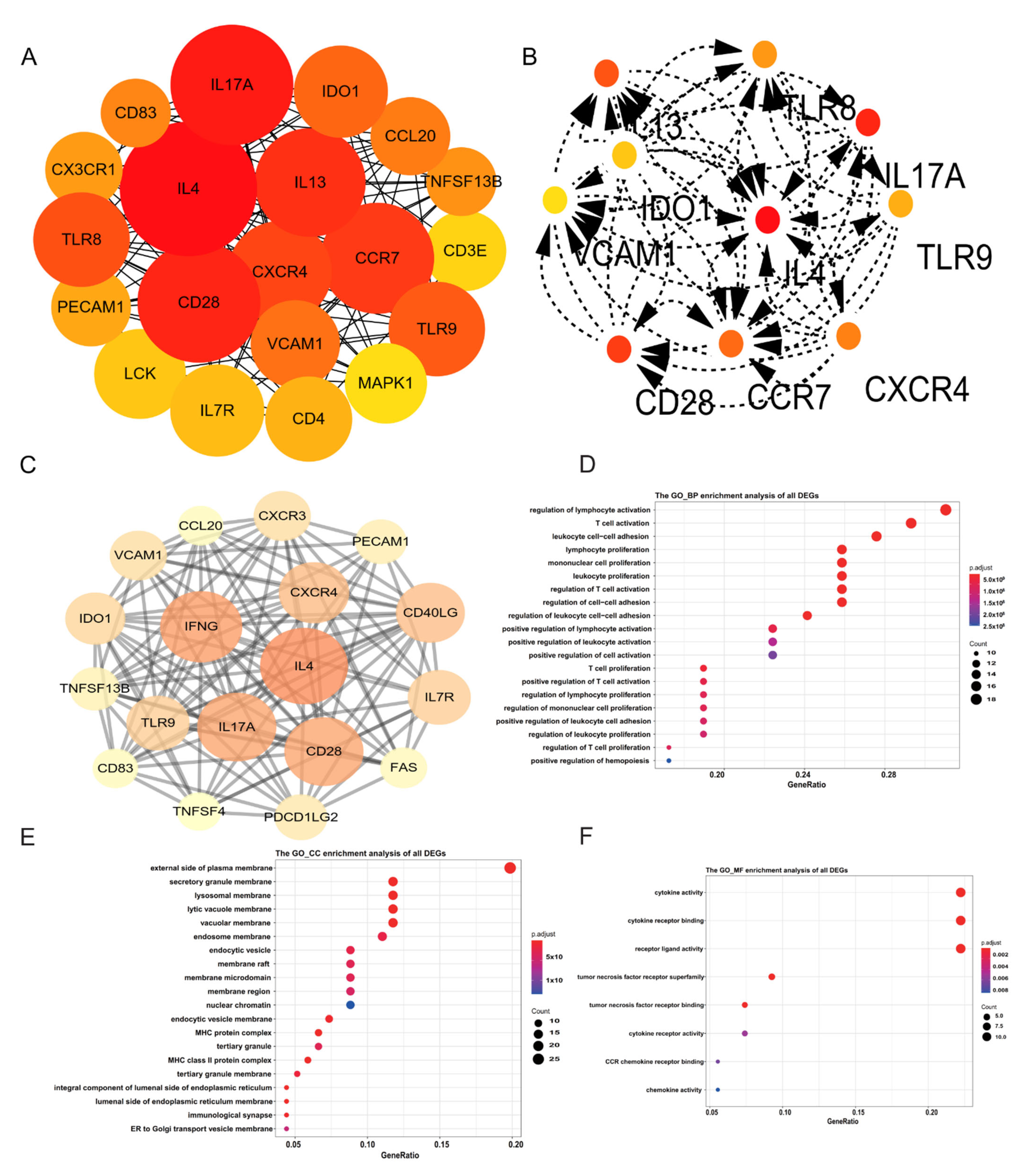
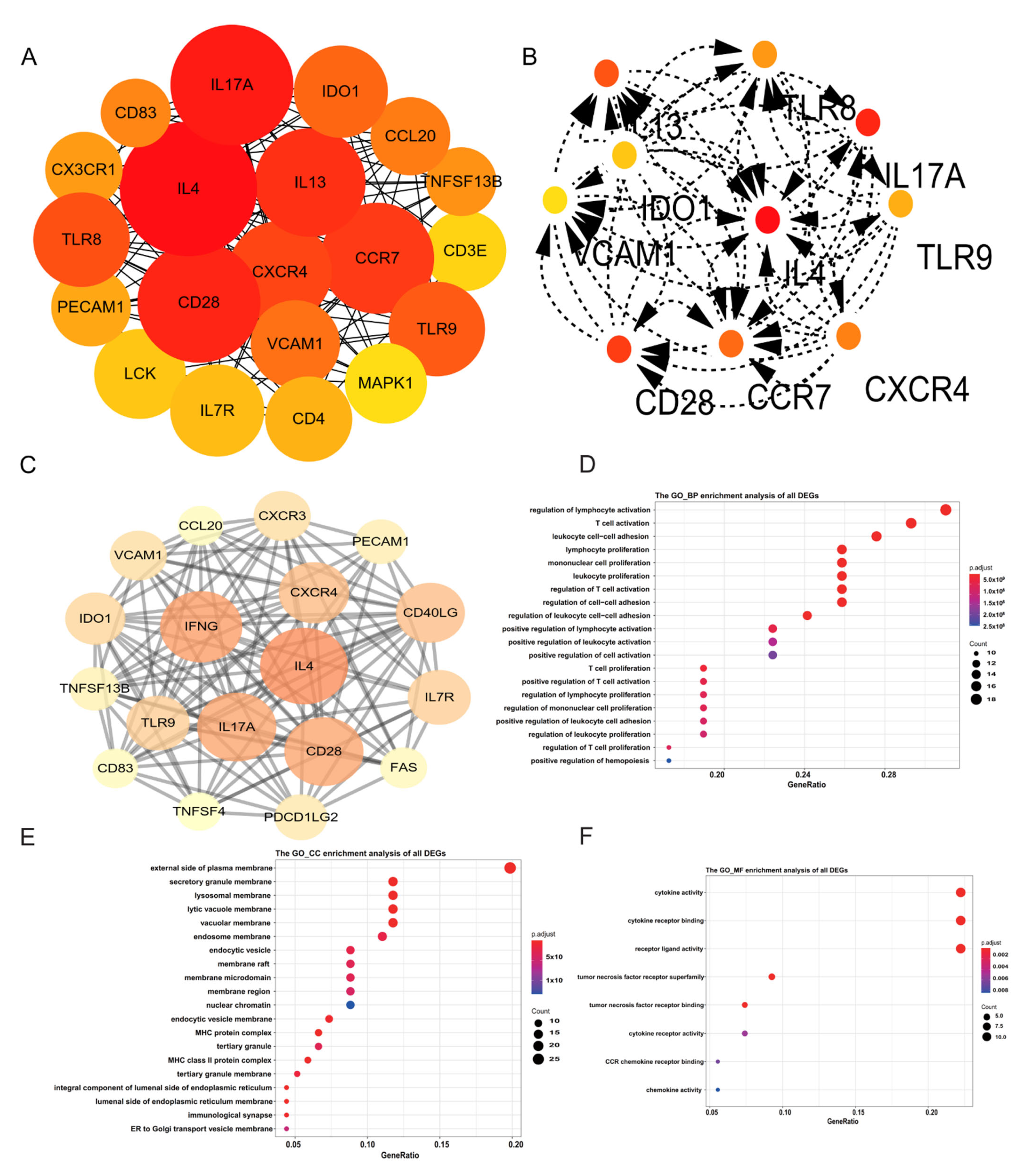
2.2. Identification of Hub Genes
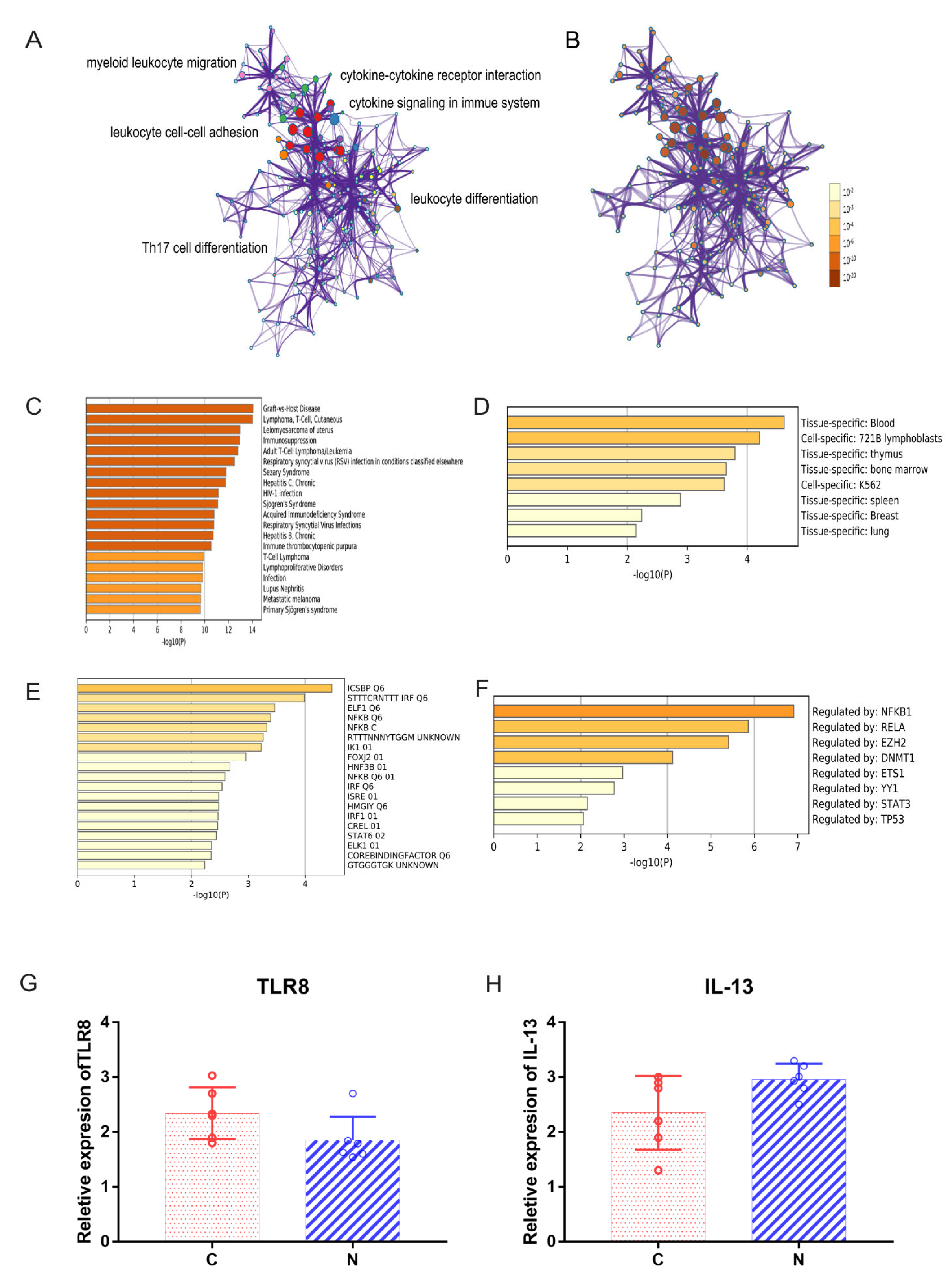
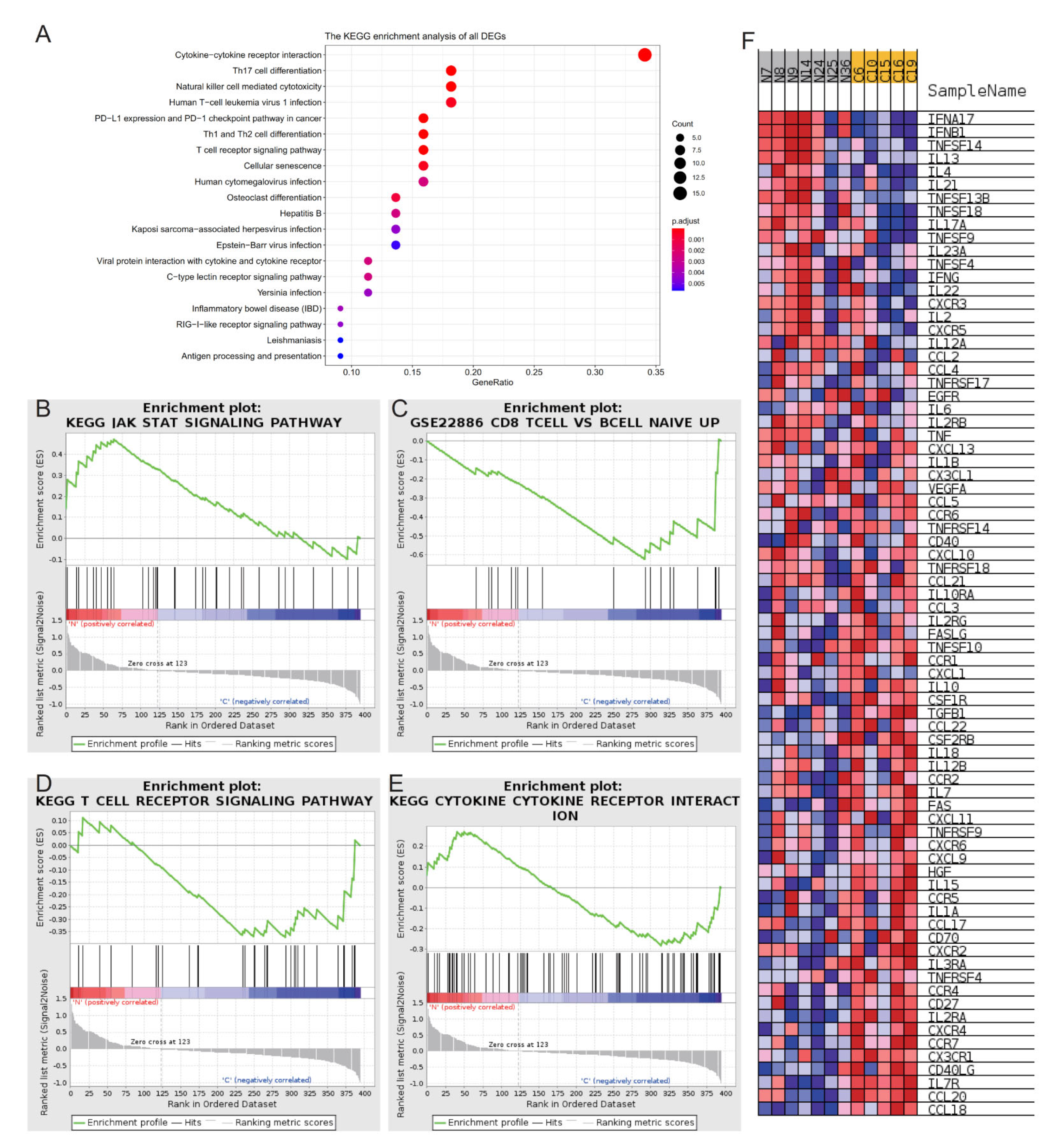
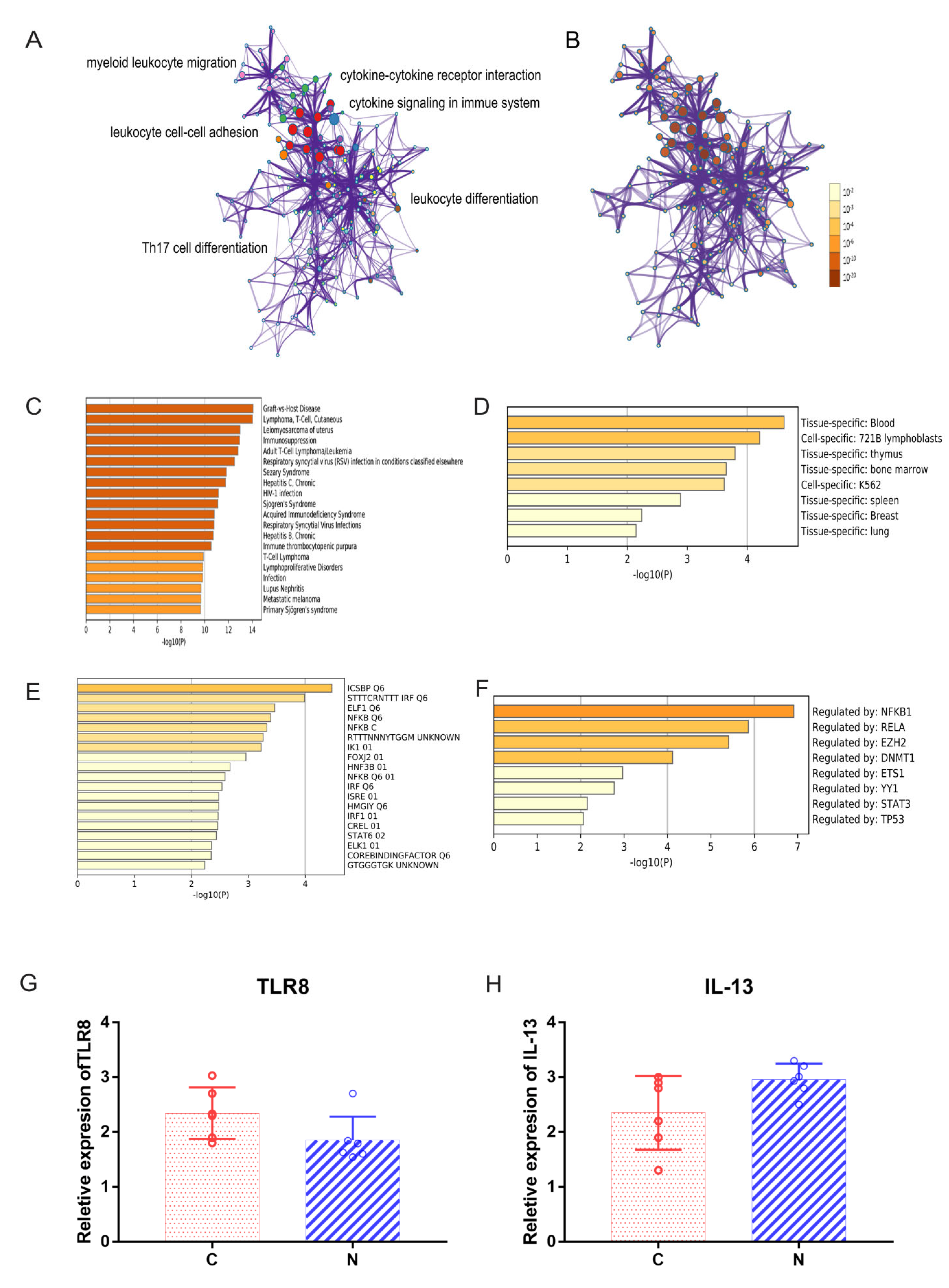
2.3. Functional Annotation of DEGs
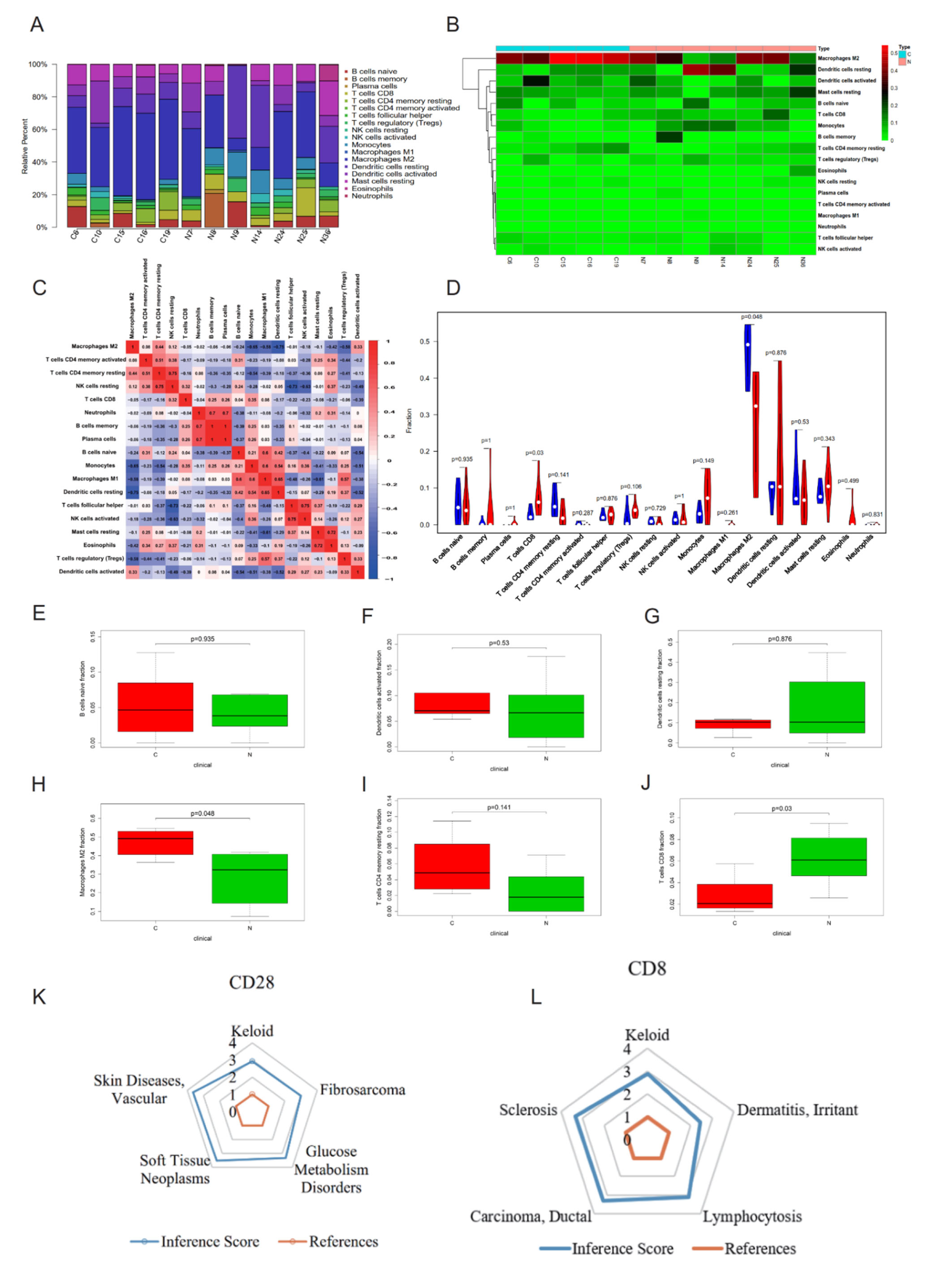
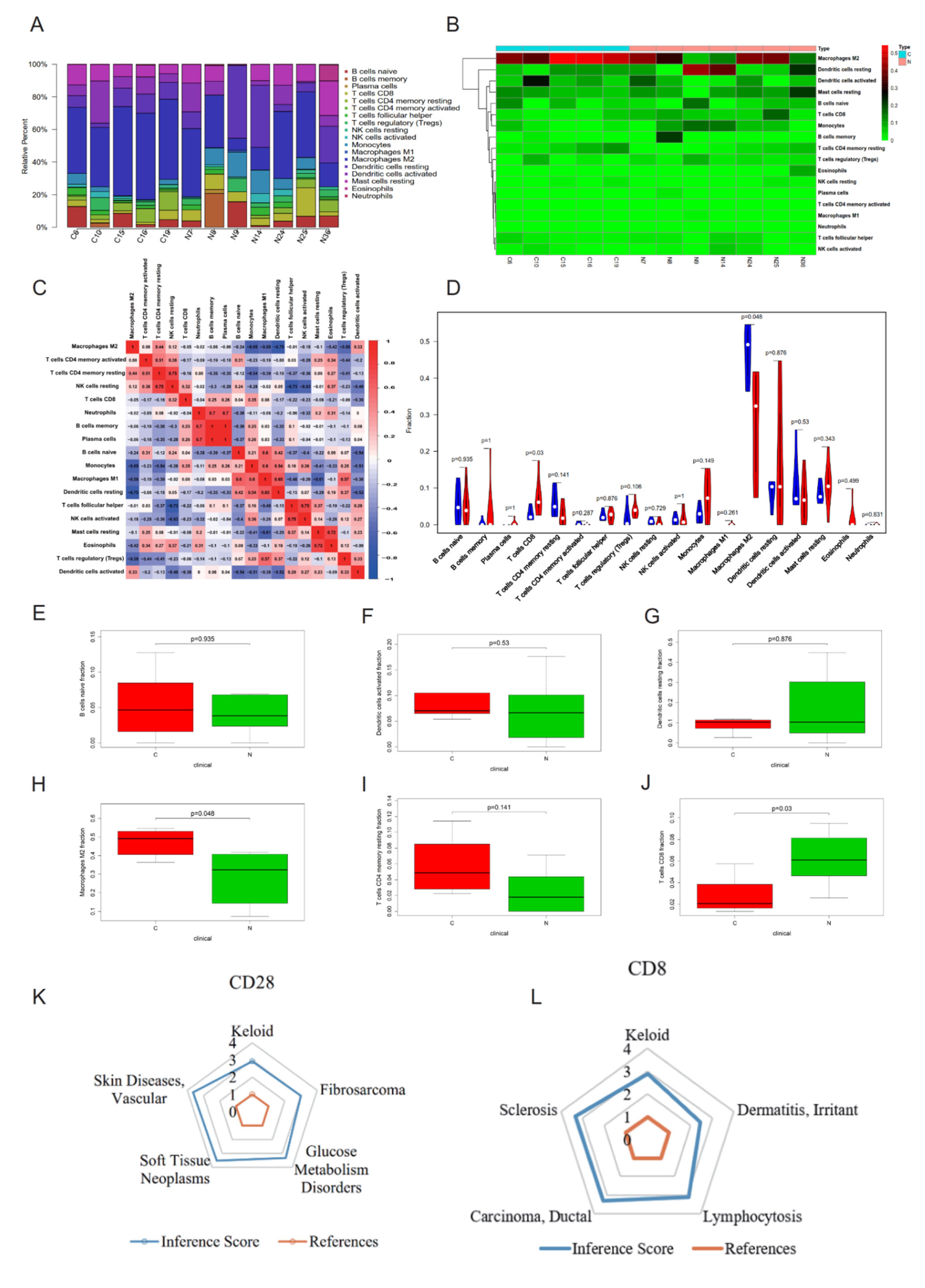
2.4. CIBERSORT Immune Cell Analysis and CTD Analysis of Hub Genes CD28 and CD8
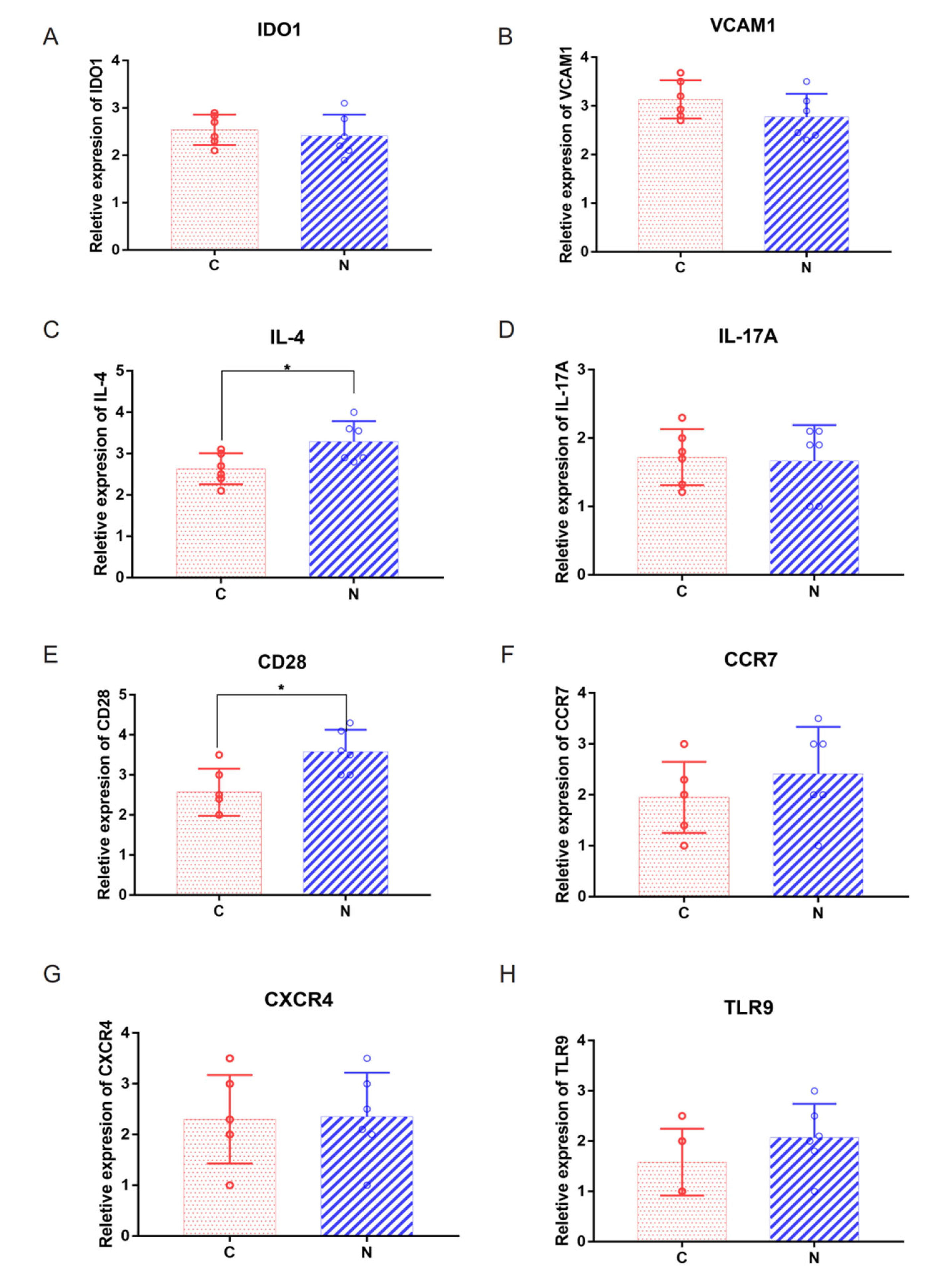
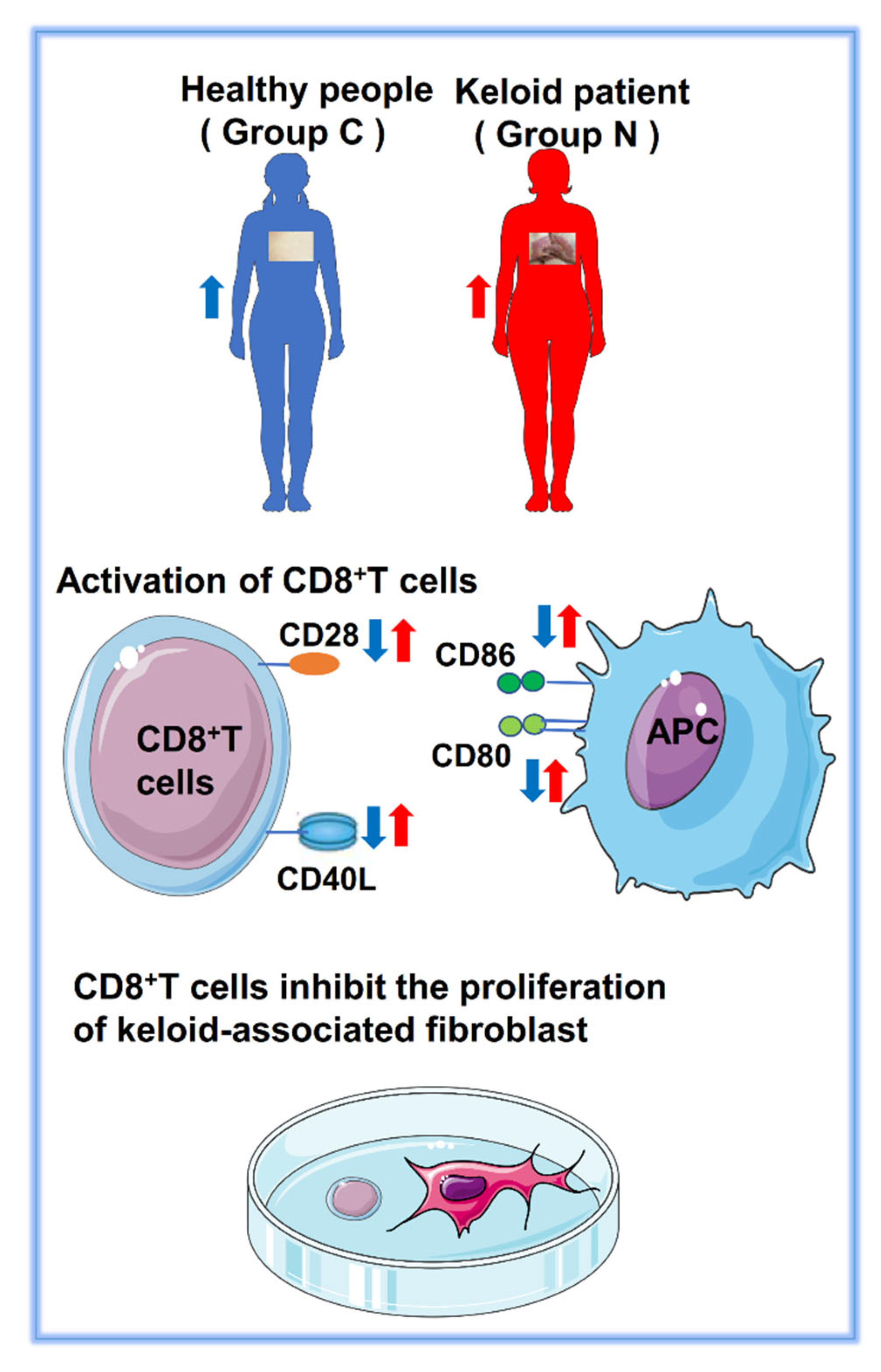
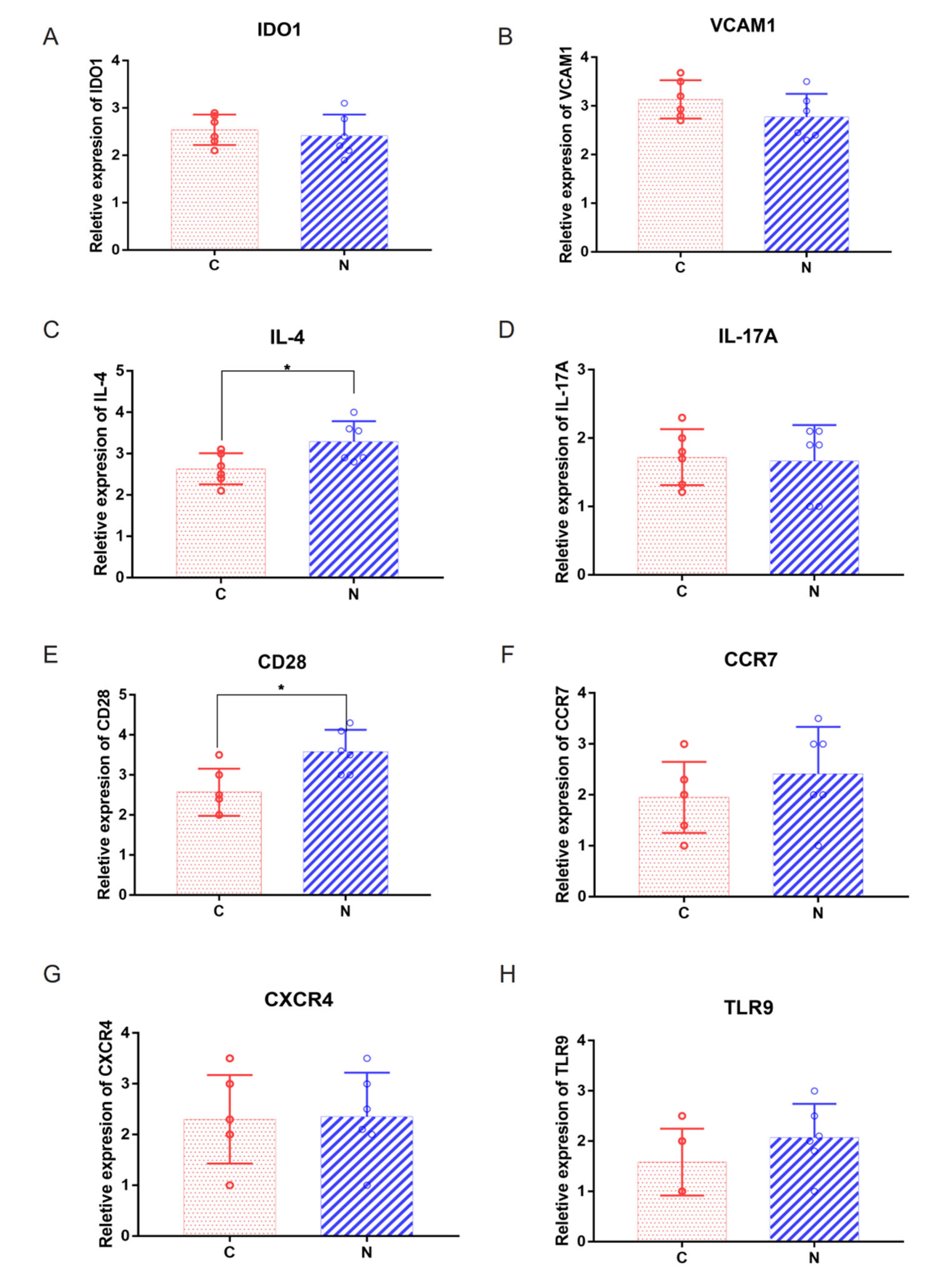
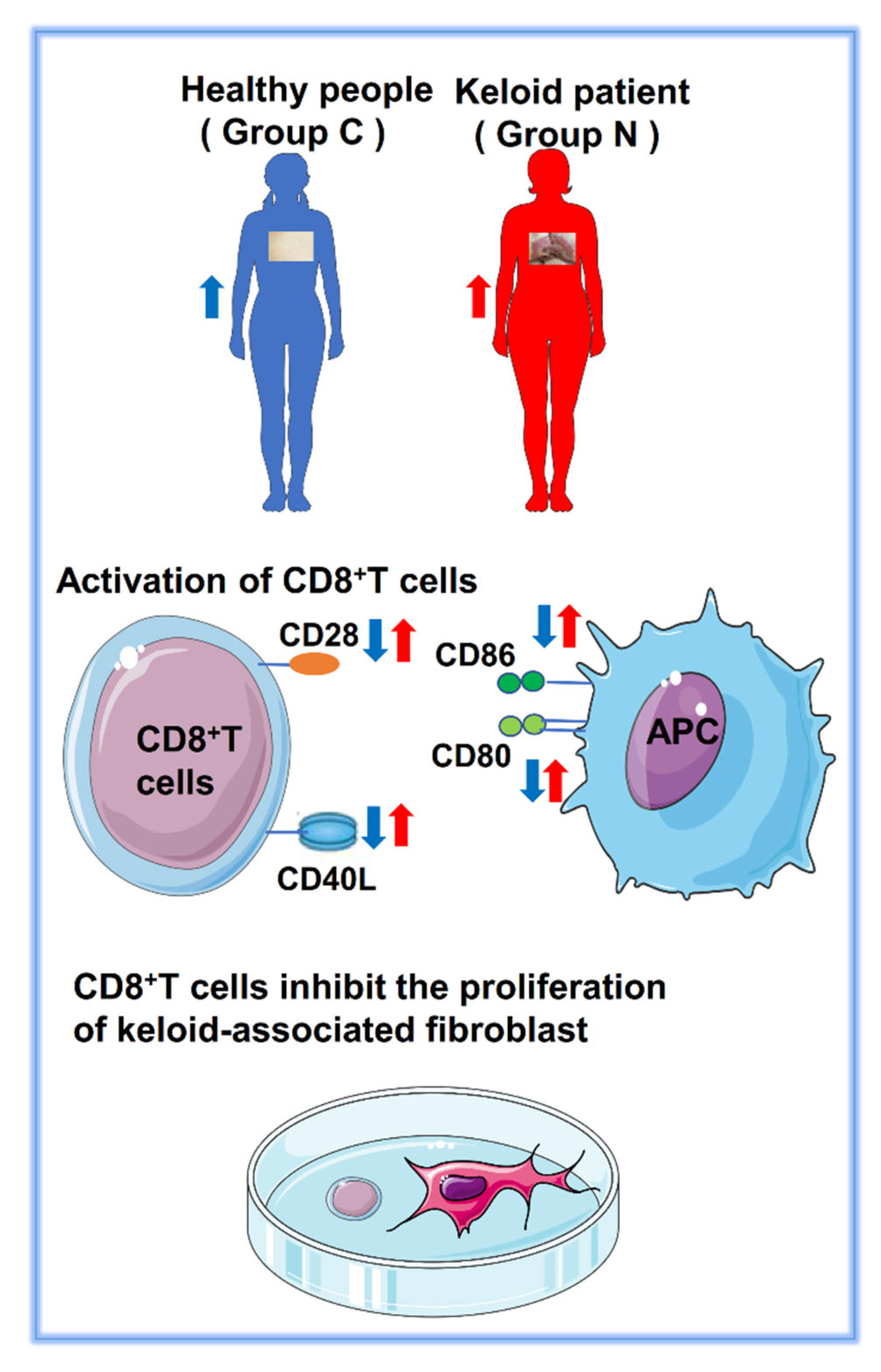
2.5. Abnormal Expression of Costimulatory Signaling Molecules
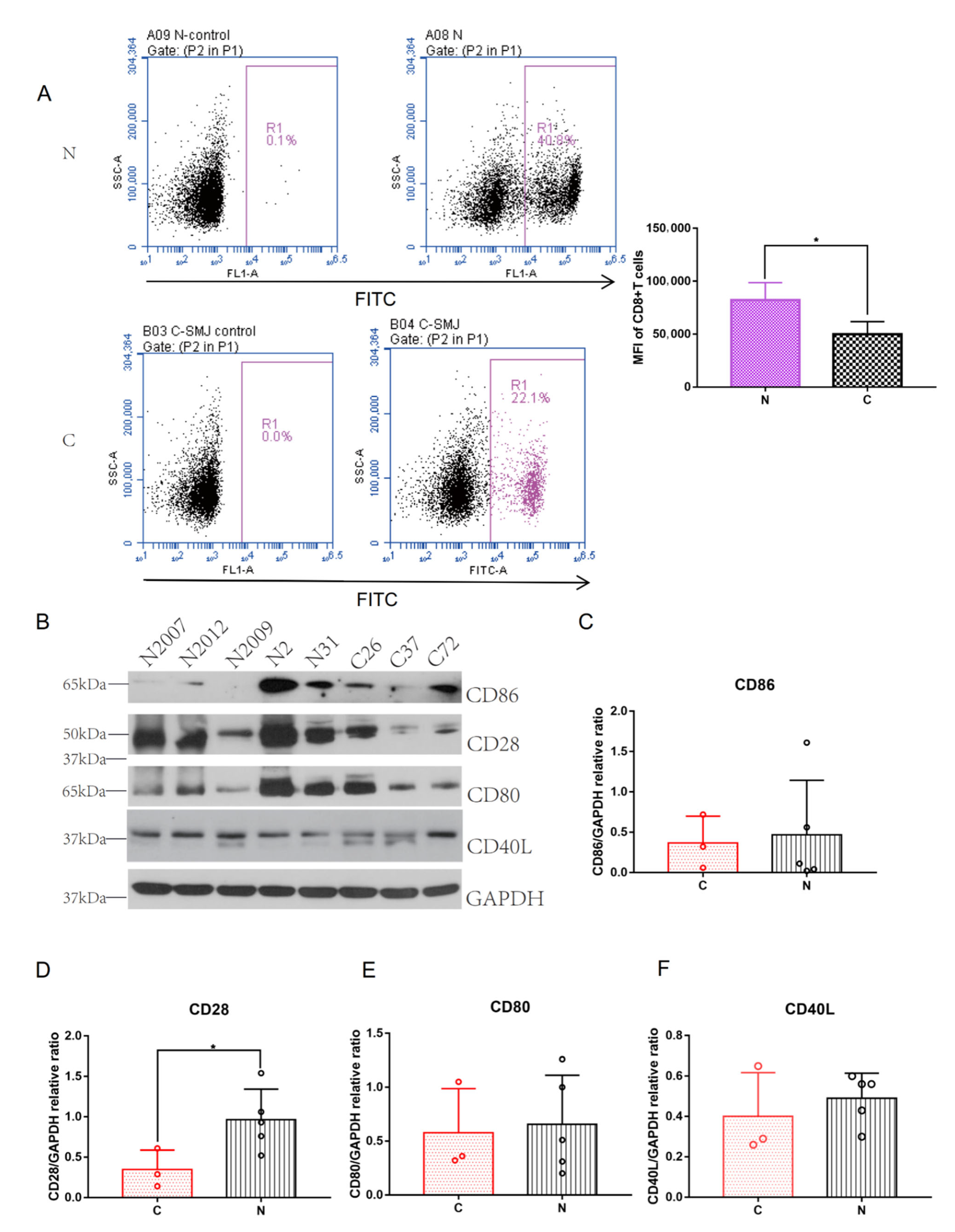
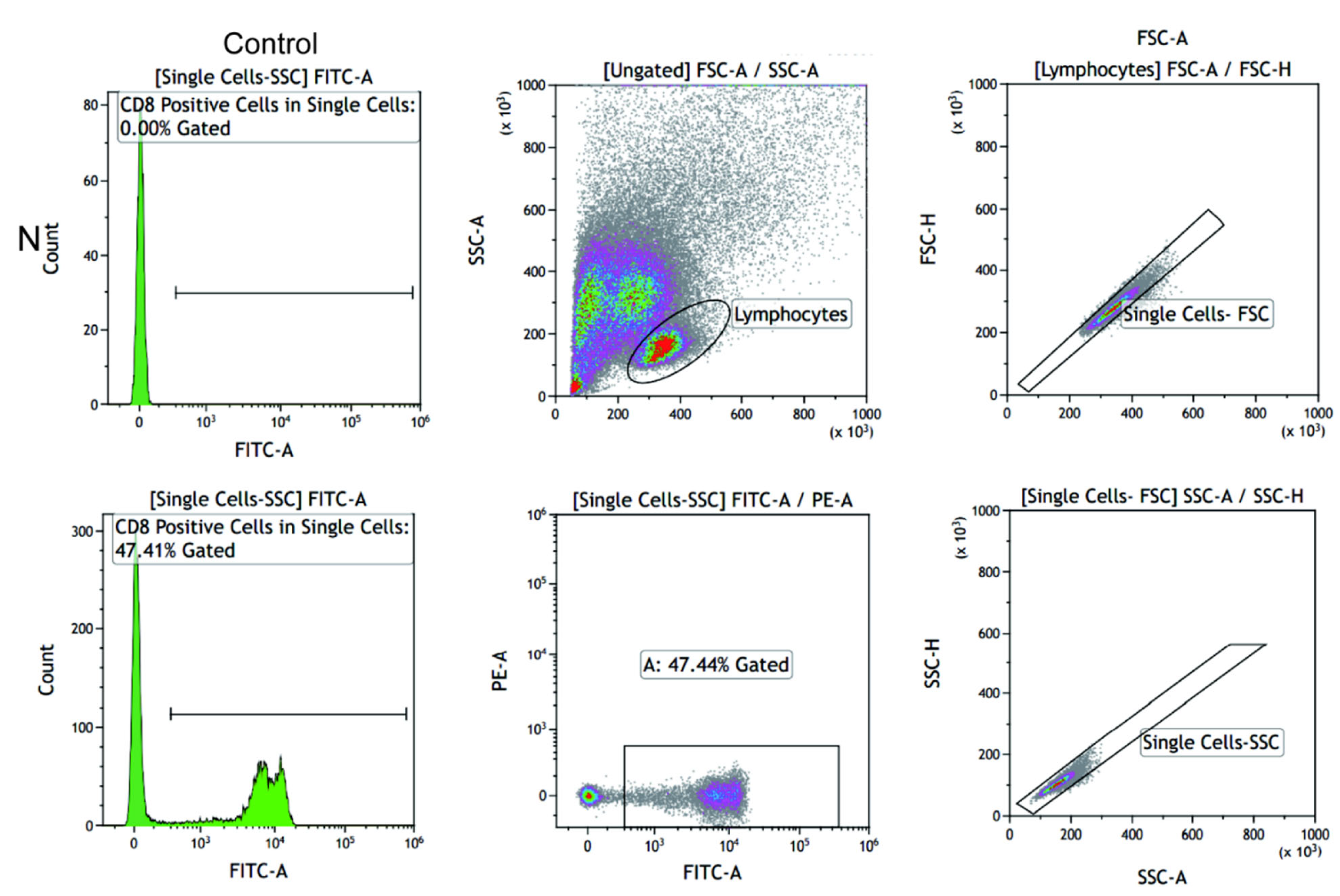
2.6. CD28 Expressed on the Surface of CD8+ T Cells
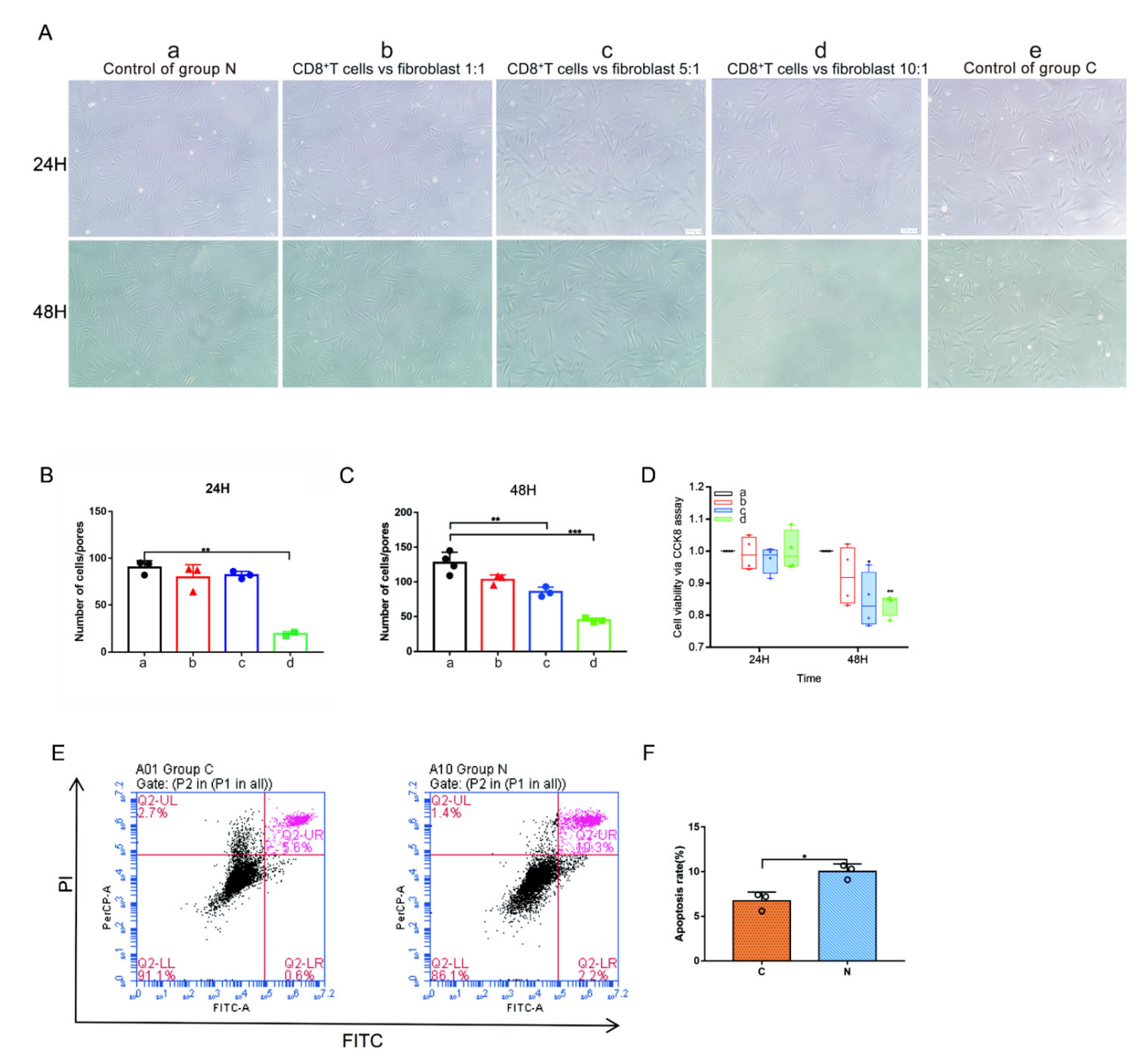
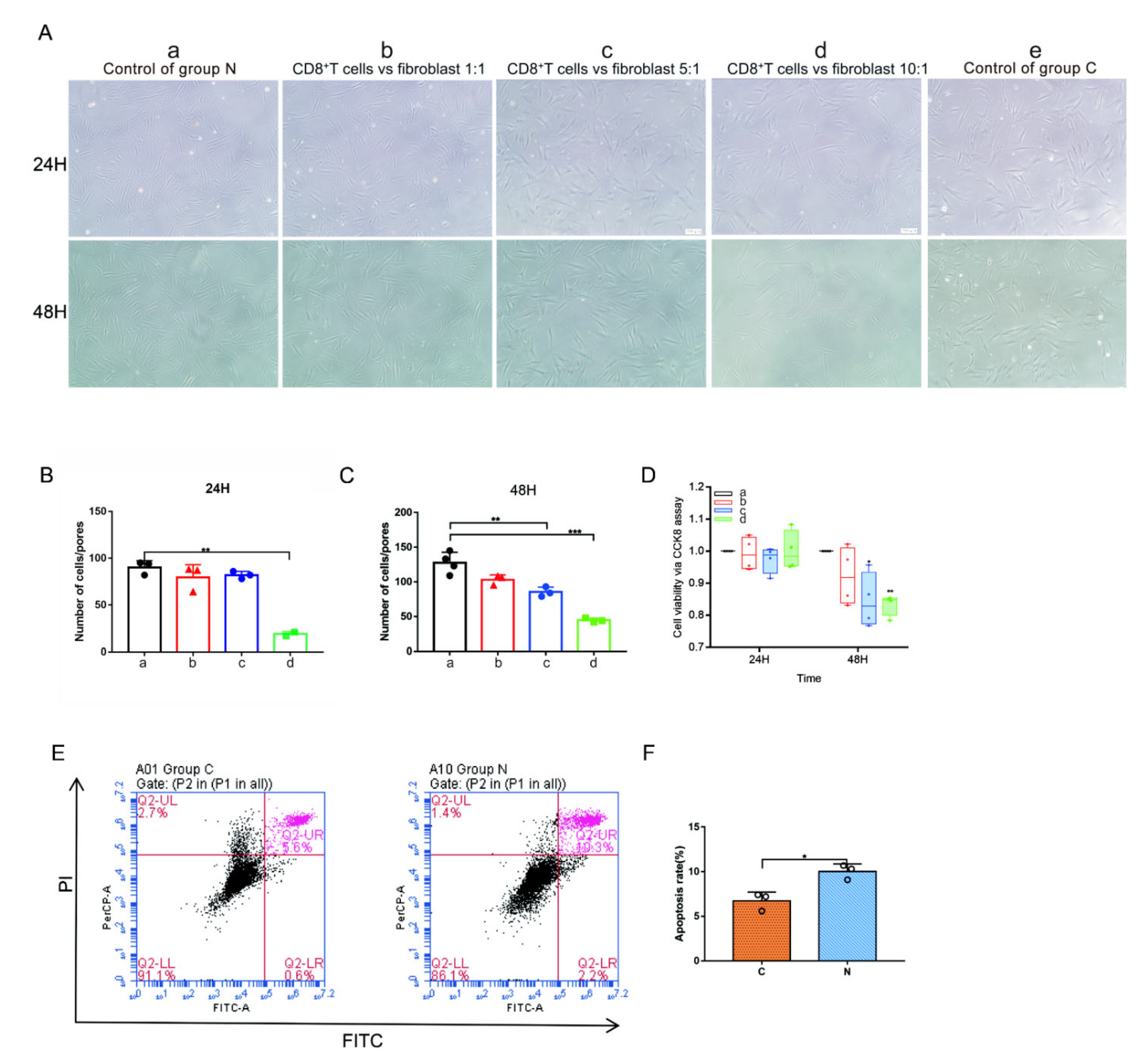
2.7. CD8+ T Cells and Keloid-Associated Fibroblast Coculture
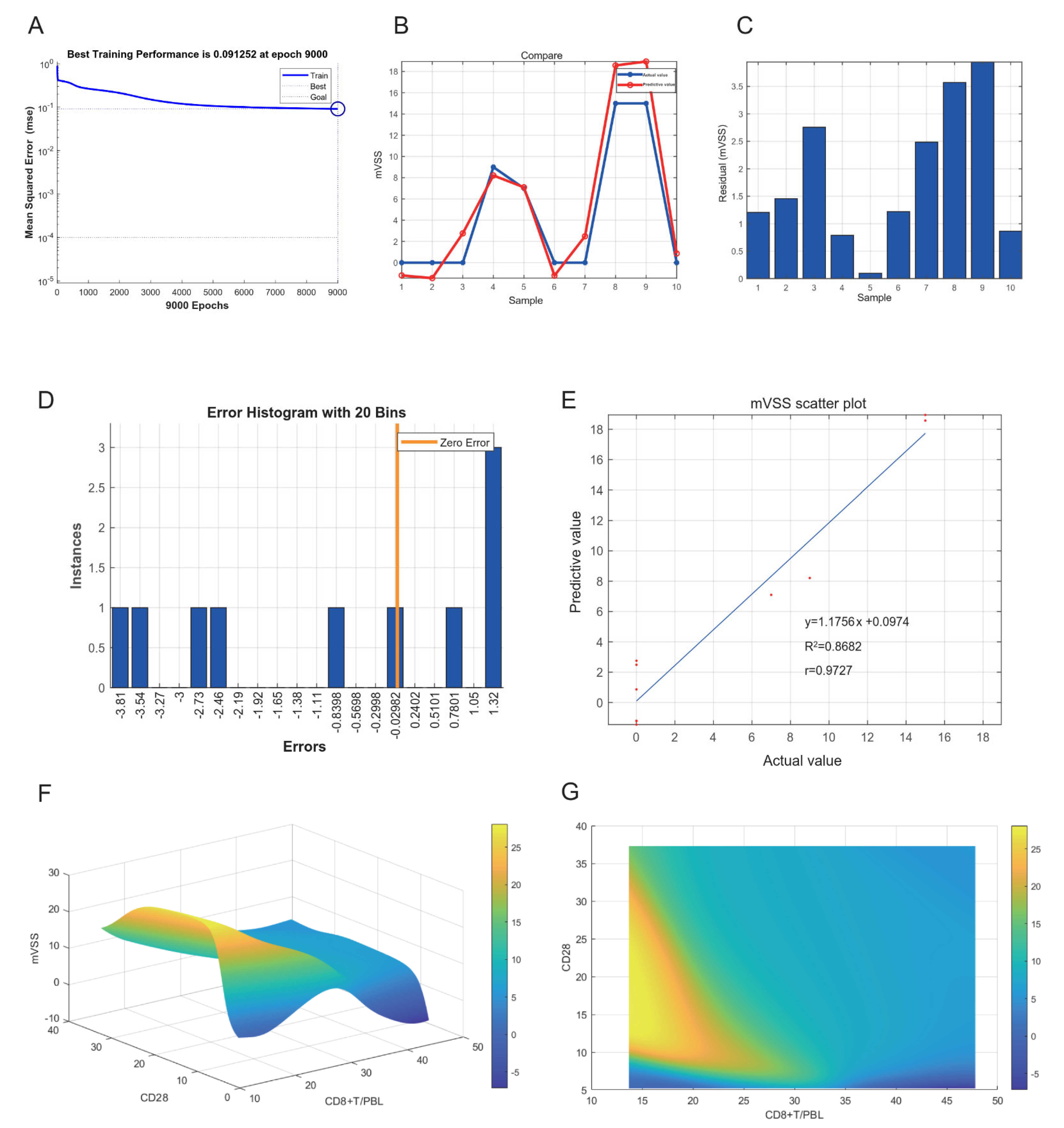
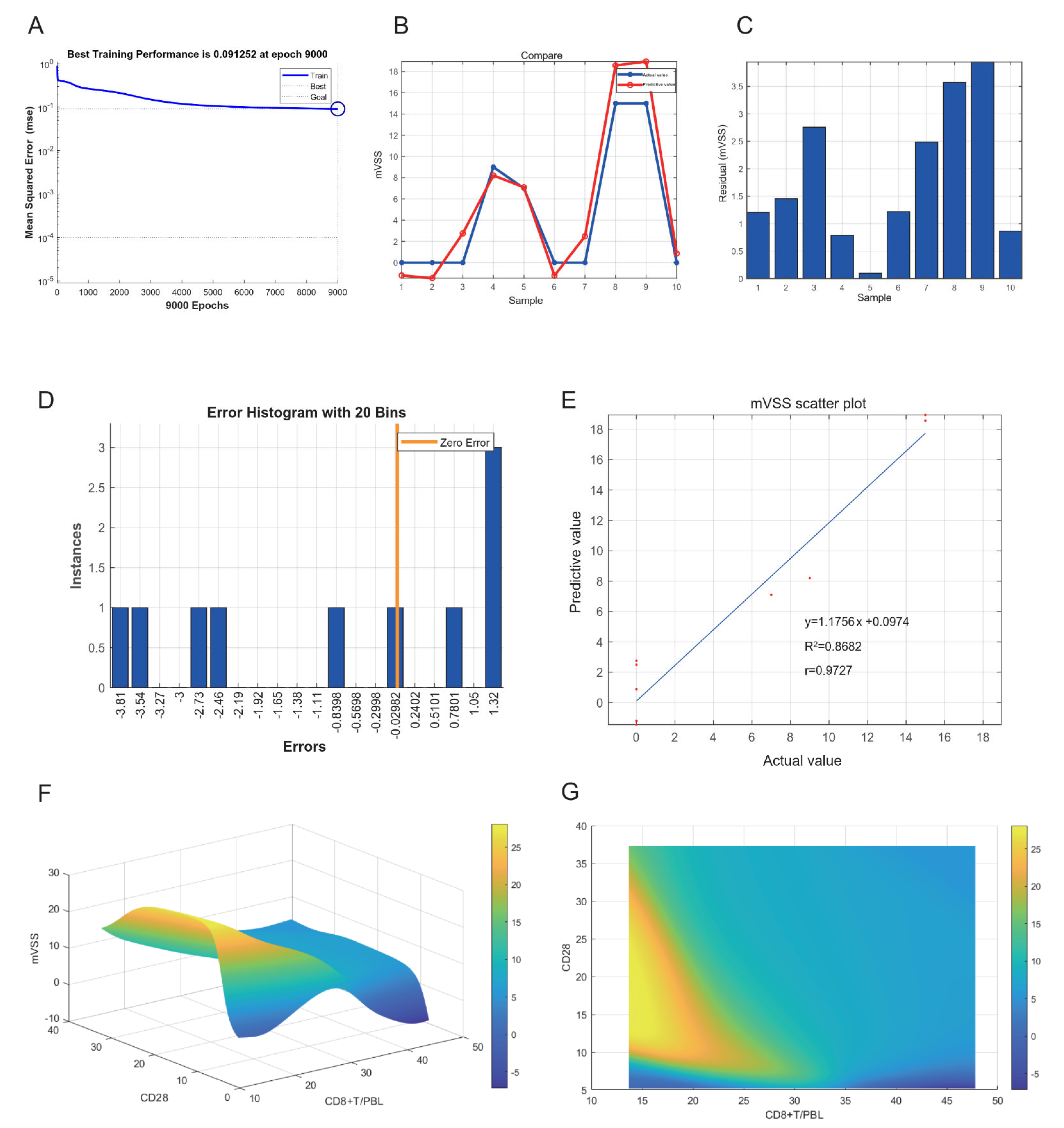
2.8. Neural Network Model of Keloids
3. Discussion
4. Methods
4.1. Patients
4.2. Fibroblasts from Patients with Keloids Cocultured with CD8+ T Cells
4.3. Neural Network Model
4.4. Exploration of Rabbit Ear Model Construction
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andrews, J.P.; Marttala, J.; Macarak, E.; Rosenbloom, J.; Uitto, J. Keloids: The Paradigm of Skin Fibrosis—Pathomechanisms and Treatment. Matrix Biol. 2016, 51, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R. Keloid and Hypertrophic Scars Are the Result of Chronic Inflammation in the Reticular Dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Ao, J.Y.J. Recent Understandings of Biology, Prophylaxis and Treatment Strategies for Hypertrophic Scars and Keloids. Int. J. Mol. Sci. 2018, 19, 711. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.; Maderal, A.; Raphael, B. Keloids and Hypertrophic Scars: Pathophysiology, Classification, and Treatment. Dermatol. Surg. 2017, 43 (Suppl. 1), S3–S18. [Google Scholar] [CrossRef]
- Jaloux, C.; Bertrand, B.; Degardin, N.; Casanova, D.; Kerfant, N.; Philandrianos, C. Keloid Scars (Part Ii): Treatment and Prevention. Ann. Chir. Plast. Esthet. 2017, 62, 87–96. [Google Scholar] [CrossRef]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast Proliferation And Heterogeneity Are Supported By Macrophages During Skin Repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef]
- Louw, L. Keloids in Rural Black South Africans. Part 1: General Overview and Essential Fatty Acid Hypotheses for Keloid Formation and Prevention. Prostaglandins Leukot Essent Fat. Acids 2000, 63, 237–245. [Google Scholar] [CrossRef]
- Shan, M.; Liu, H.; Hao, Y.; Meng, T.; Feng, C.; Song, K.; Wang, Y. Il-4 And Ccr7 Play An Important Role In The Development Of Keloids In Patients With A Family History. Am. J. Transl. Res. 2022, 14, 3381–3394. [Google Scholar]
- Wang, C.H.; Shan, M.J.; Liu, H.; Hao, Y.; Song, K.X.; Wu, H.W.; Meng, T.; Feng, C.; Qi, Z.; Wang, Z.; et al. Hyperbaric oxygen treatment on keloid tumor immune gene expression. Chin. Med. J. 2021, 134, 2205–2213. [Google Scholar] [CrossRef]
- Shan, M.; Liu, H.; Song, K.; Liu, S.; Hao, Y.; Wang, Y. Immune-related gene expression in skin, inflamed and keloid tissue from patients with keloids. Oncol. Lett. 2022, 23, 72. [Google Scholar] [CrossRef]
- Shan, M.; Liu, H.; Hao, Y.; Song, K.; Meng, T.; Feng, C.; Wang, Y.; Huang, Y. Metabolomic Profiling Reveals That 5-Hydroxylysine and 1-Methylnicotinamide Are Metabolic Indicators of Keloid Severity. Front. Genet. 2021, 12, 804248. [Google Scholar] [CrossRef]
- Shan, M.; Wang, Y. Viewing keloids within the immune microenvironment. Am. J. Transl. Res. 2022, 14, 718–727. [Google Scholar]
- Dong, X.; Zhang, M.; Chen, Y.; Li, C.; Wang, Y.; Jin, X. A comparison expression analysis of CXCR4, CXCL9 and Caspase-9 in dermal vascular endothelial cells between keloids and normal skin on chemotaxis and apoptosis. J. Plast. Surg. Hand Surg. 2021, 56, 93–102. [Google Scholar] [CrossRef]
- Bagabir, R.; Byers, R.J.; Chaudhry, I.H.; Müller, W.; Paus, R.; Bayat, A. Site-Specific Immunophenotyping of Keloid Disease Demonstrates Immune Upregulation and the Presence of Lymphoid Aggregates. Br. J. Dermatol. 2012, 167, 1053–1066. [Google Scholar] [CrossRef]
- Kim, H.; Wang, S.Y.; Kwak, G.; Yang, Y.; Kwon, I.C.; Kim, S.H. Exosome-Guided Phenotypic Switch of M1 to M2 Macrophages for Cutaneous Wound Healing. Adv. Sci. 2019, 6, 1900513. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T Cells in Cancer Immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef]
- Jin, Q.; Gui, L.; Niu, F.; Yu, B.; Lauda, N.; Liu, J.; Mao, X.; Chen, Y. Macrophages in Keloid Are Potent at Promoting the Differentiation and Function of Regulatory T Cells. Exp. Cell Res. 2018, 362, 472–476. [Google Scholar] [CrossRef]
- Wu, J.; Del Duca, E.; Espino, M.; Gontzes, A.; Cueto, I.; Zhang, N.; Estrada, Y.D.; Pavel, A.B.; Krueger, J.G.; Guttman-Yassky, E. RNA Sequencing Keloid Transcriptome Associates Keloids with Th2, Th1, Th17/Th22, and Jak3-Skewing. Front. Immunol. 2020, 11, 597741. [Google Scholar] [CrossRef]
- Seoudy, W.M.; Mohy ELDien, S.M.; Abdel Reheem, T.A.; Elfangary, M.M.; Erfan, M.A. Macrophages of the M1 and M2 Types Play A Role in Keloids Pathogenesis. Int. Wound J. 2022. [Google Scholar] [CrossRef]
- Murao, N.; Seino, K.; Hayashi, T.; Ikeda, M.; Funayama, E.; Furukawa, H.; Yamamoto, Y.; Oyama, A. Treg-Enriched Cd4+ T Cells Attenuate Collagen Synthesis in Keloid Fibroblasts. Exp. Dermatol. 2014, 23, 266–271. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the Antitumour Response of Cd8(+) T Cells by Modulating Cholesterol Metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef]
- Olivo Pimentel, V.; Yaromina, A.; Marcus, D.; Dubois, L.J.; Lambin, P. A novel co-culture assay to assess anti-tumor CD8(+) T cell cytotoxicity via luminescence and multicolor flow cytometry. J. Immunol. Methods 2020, 487, 112899. [Google Scholar] [CrossRef]
- Kato, T.; Noma, K.; Ohara, T.; Kashima, H.; Katsura, Y.; Sato, H.; Komoto, S.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-Associated Fibroblasts Affect Intratumoral CD8(+) and FoxP3(+) T Cells Via IL6 in the Tumor Microenvironment. Clin. Cancer Res. 2018, 24, 4820–4833. [Google Scholar] [CrossRef]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Martin-Orozco, N.; Dong, C. Inhibitory costimulation and anti-tumor immunity. Semin. Cancer Biol. 2007, 17, 288–298. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Topp, M.S.; Riddell, S.R.; Akatsuka, Y.; Jensen, M.C.; Blattman, J.N.; Greenberg, P.D. Restoration of CD28 expression in CD28- CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. J. Exp. Med. 2003, 198, 947–955. [Google Scholar] [CrossRef]
- Shaker, S.A.; Ayuob, N.N.; Hajrah, N.H. Cell talk: A phenomenon observed in the keloid scar by immunohistochemical study. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 153–159. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Huang, H.; Hao, S.; Li, F.; Ye, Z.; Yang, J.; Xiang, J. CD4+ Th1 cells promote CD8+ Tc1 cell survival, memory response, tumor localization and therapy by targeted delivery of interleukin 2 via acquired pMHC I complexes. Immunology 2007, 120, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. IFNγ-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J. Leukoc. Biol. 2021, 110, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Guerder, S.; Flavell, R.A. T-cell activation. Two for T. Curr. Biol. 1995, 5, 866–868. [Google Scholar] [CrossRef]
- Rundqvist, H.; Veliça, P.; Barbieri, L.; Gameiro, P.A.; Bargiela, D.; Gojkovic, M.; Mijwel, S.; Reitzner, S.M.; Wulliman, D.; Ahlstedt, E.; et al. Cytotoxic T-cells mediate exercise-induced reductions in tumor growth. eLife 2020, 9, e59996. [Google Scholar] [CrossRef] [PubMed]
- Shahinian, A.; Pfeffer, K.; Lee, K.P.; Kündig, T.M.; Kishihara, K.; Wakeham, A.; Kawai, K.; Ohashi, P.S.; Thompson, C.B.; Mak, T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993, 261, 609–612. [Google Scholar] [CrossRef]
- Nurieva, R.; Thomas, S.; Nguyen, T.; Martin-Orozco, N.; Wang, Y.; Kaja, M.K.; Yu, X.Z.; Dong, C. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 2006, 25, 2623–2633. [Google Scholar] [CrossRef]
- Edner, N.M.; Carlesso, G.; Rush, J.S.; Walker, L. Targeting co-stimulatory molecules in autoimmune disease. Nat. Rev. Drug Discov. 2020, 19, 860–883. [Google Scholar] [CrossRef]
- Borthwick, L.A.; Wynn, T.A.; Fisher, A.J. Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta 2013, 1832, 1049–1060. [Google Scholar] [CrossRef]
- Nguyen, J.K.; Austin, E.; Huang, A.; Mamalis, A.; Jagdeo, J. The IL-4/IL-13 axis in skin fibrosis and scarring: Mechanistic concepts and therapeutic targets. Arch. Dermatol. Res. 2020, 312, 81–92. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jameson, S.C.; Hogquist, K.A. Alternative memory in the CD8 T cell lineage. Trends Immunol. 2011, 32, 50–56. [Google Scholar] [CrossRef]
- Morris, S.C.; Heidorn, S.M.; Herbert, D.R.; Perkins, C.; Hildeman, D.A.; Khodoun, M.V.; Finkelman, F.D. Endogenously produced IL-4 nonredundantly stimulates CD8+ T cell proliferation. J. Immunol. 2009, 182, 1429–1438. [Google Scholar] [CrossRef]
- Seneviratne, S.L.; Black, A.P.; Jones, L.; di Gleria, K.; Bailey, A.S.; Ogg, G.S. Interleukin-4 promotes human CD8 T cell expression of CCR7. Immunology 2007, 120, 66–72. [Google Scholar] [CrossRef]
- Korbecki, J.; Grochans, S.; Gutowska, I.; Barczak, K.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of Receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 Ligands. Int. J. Mol. Sci. 2020, 21, 7619. [Google Scholar] [CrossRef]
- Wilgus, T.A. Vascular Endothelial Growth Factor and Cutaneous Scarring. Adv. Wound Care 2019, 8, 671–678. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, K.; Zhou, H.; Peng, L.; You, W.; Fu, Z. Profiles of immune infiltration in colorectal cancer and their clinical significant: A gene expression-based study. Cancer Med. 2018, 7, 4496–4508. [Google Scholar] [CrossRef]
- Zhou, R.; Zhang, J.; Zeng, D.; Sun, H.; Rong, X.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Immune cell infiltration as a biomarker for the diagnosis and prognosis of stage I-III colon cancer. Cancer Immunol. Immunother. 2019, 68, 433–442. [Google Scholar] [CrossRef]
- Novak, M.L.; Koh, T.J. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013, 93, 875–881. [Google Scholar] [CrossRef]
- Gankande, T.U.; Wood, F.M.; Edgar, D.W.; Duke, J.M.; DeJong, H.M.; Henderson, A.E.; Wallace, H.J. A modified Vancouver Scar Scale linked with TBSA (mVSS-TBSA): Inter-rater reliability of an innovative burn scar assessment method. Burns 2013, 39, 1142–1149. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Esmaelpoor, J.; Moradi, M.H.; Kadkhodamohammadi, A. A multistage deep neural network model for blood pressure estimation using photoplethysmogram signals. Comput. Biol. Med. 2020, 120, 103719. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D. Cytoscape: A Software Environment For Integrated Models Of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Canzler, S.; Hackermüller, J. Multigsea: A Gsea-Based Pathway Enrichment Analysis for Multi-Omics Data. BMC Bioinform. 2020, 21, 561. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.; Tanaseichuk, O.; Benner, C.; Metascape, S.K.C. Provides A Biologist-Oriented Resource For The Analysis Of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells With Cibersort. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Age of Onset (Years) | Gender | mVSS a |
|---|---|---|---|
| C6 | 27 | male | 0 |
| C10 | 54 | female | 0 |
| C15 | 31 | female | 0 |
| C16 | 25 | male | 0 |
| C19 | 32 | female | 0 |
| N7 | 24 | female | 11 |
| N8 | 32 | male | 10 |
| N9 | 37 | female | 10 |
| N14 | 21 | male | 9 |
| N24 | 26 | female | 11 |
| N25 | 24 | female | 9 |
| N36 | 38 | male | 12 |
| Gene Set Name | SIZE | ES | NES | Rank at Max |
|---|---|---|---|---|
| Upregulated | ||||
| KEGG_EPITHELIAL_CELL_SIGNALING_IN_HELICOBACTER_PYLORI_INFECTION | 61 | 0.563 | 1.525 | 3156 |
| KEGG_BASE_EXCISION_REPAIR | 31 | 0.459 | 1.522 | 6082 |
| KEGG_ANTIGEN_PROCESSING_AND_PRESENTATION | 57 | 0.653 | 1.519 | 2554 |
| KEGG_T_CELL_RECEPTOR_SIGNALING_PATHWAY | 101 | 0.605 | 1.510 | 3648 |
| KEGG_TOLL_LIKE_RECEPTOR_SIGNALING_PATHWAY | 88 | 0.702 | 1.508 | 2235 |
| KEGG_PATHOGENIC_ESCHERICHIA_COLI_INFECTION | 45 | 0.499 | 1.505 | 4454 |
| Downregulated | ||||
| KEGG_DILATED_CARDIOMYOPATHY | 86 | −0.524 | −1.434 | 1865 |
| KEGG_HYPERTROPHIC_CARDIOMYOPATHY_HCM | 80 | −0.499 | −1.407 | 2006 |
| KEGG_LONG_TERM_POTENTIATION | 65 | −0.390 | −1.300 | 1772 |
| KEGG_ADHERENS_JUNCTION | 72 | −0.425 | −1.272 | 2780 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, M.; Liu, H.; Hao, Y.; Song, K.; Feng, C.; Wang, Y. The Role of CD28 and CD8+ T Cells in Keloid Development. Int. J. Mol. Sci. 2022, 23, 8862. https://doi.org/10.3390/ijms23168862
Shan M, Liu H, Hao Y, Song K, Feng C, Wang Y. The Role of CD28 and CD8+ T Cells in Keloid Development. International Journal of Molecular Sciences. 2022; 23(16):8862. https://doi.org/10.3390/ijms23168862
Chicago/Turabian StyleShan, Mengjie, Hao Liu, Yan Hao, Kexin Song, Cheng Feng, and Youbin Wang. 2022. "The Role of CD28 and CD8+ T Cells in Keloid Development" International Journal of Molecular Sciences 23, no. 16: 8862. https://doi.org/10.3390/ijms23168862
APA StyleShan, M., Liu, H., Hao, Y., Song, K., Feng, C., & Wang, Y. (2022). The Role of CD28 and CD8+ T Cells in Keloid Development. International Journal of Molecular Sciences, 23(16), 8862. https://doi.org/10.3390/ijms23168862