
Withholding of M-CSF Supplement Reprograms Macrophages to M2-Like via Endogenous CSF-1 Activation


Abstract
1. Introduction
2. Results
2.1. Withholding of M-CSF Supplement Still Directs M2 Macrophages Polarization for Long Time
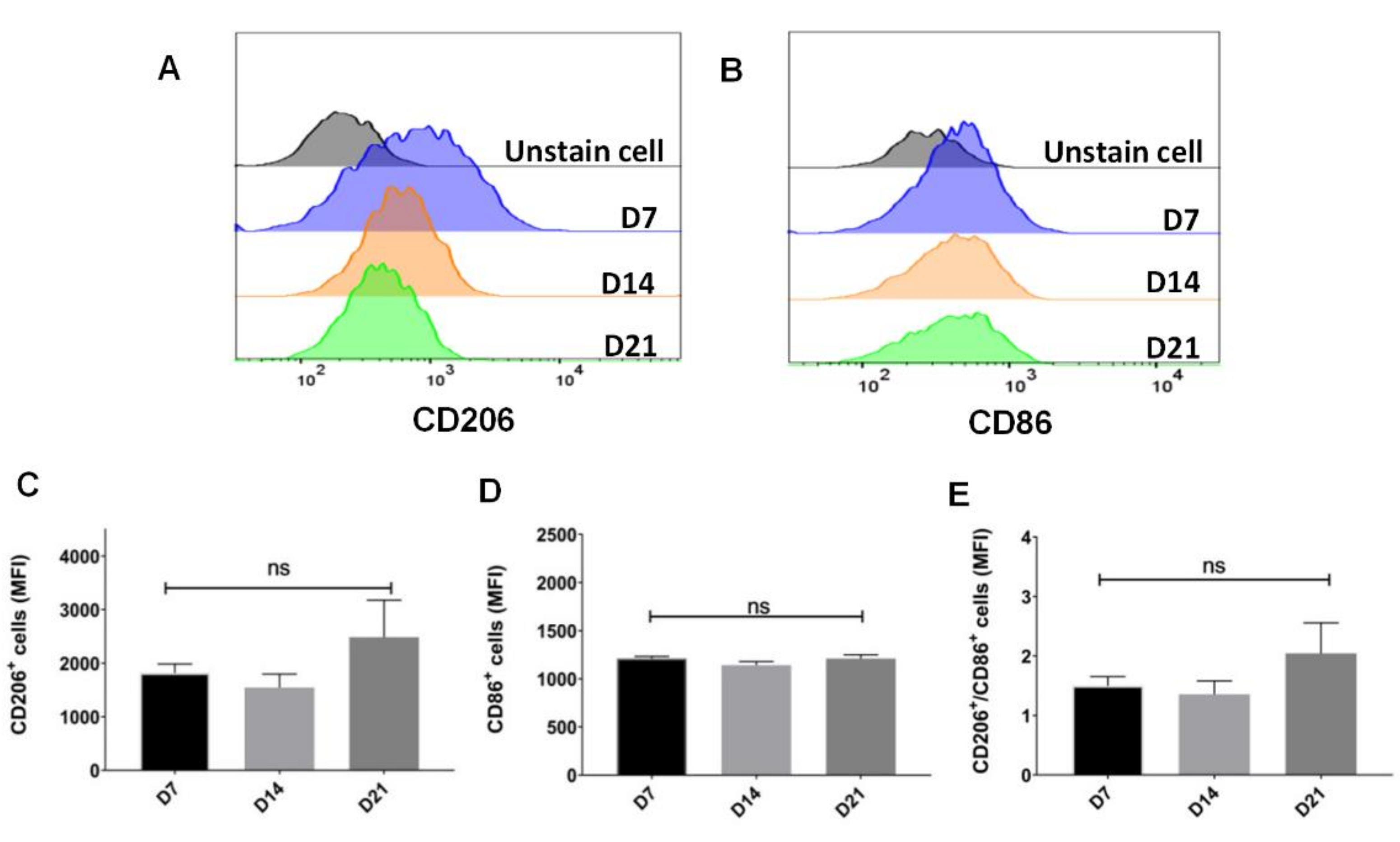
2.2. Sustainable Expression of CD206 on Single Cell after Withholding of M-CSF Supplement
2.3. A Selective Pressure for Cell Survival Was Initiated after Withholding of M-CSF Supplement
2.4. Trend to Polarization into M2, Instead of M1 Subtype, after Withholding of M-CSF Supplement
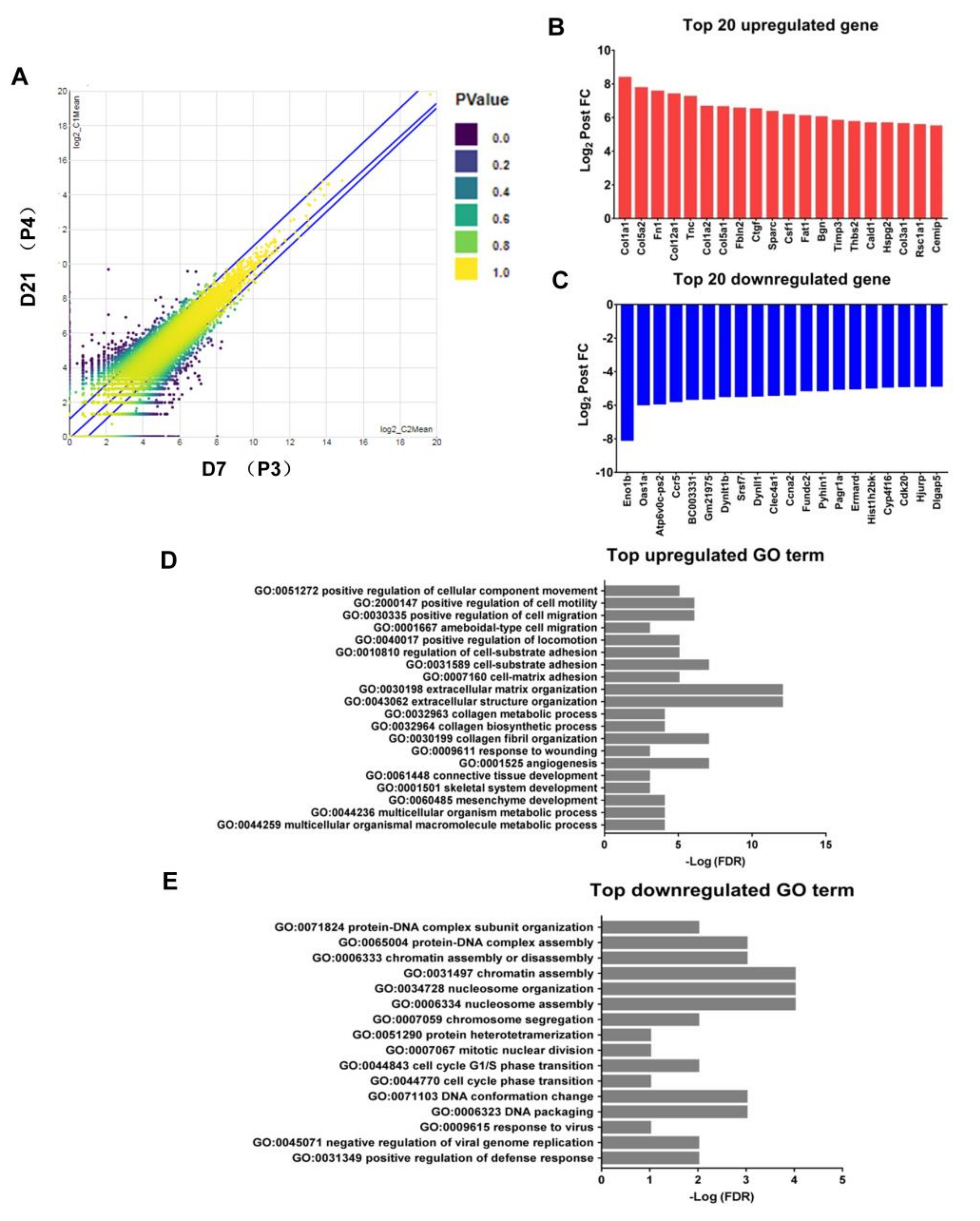
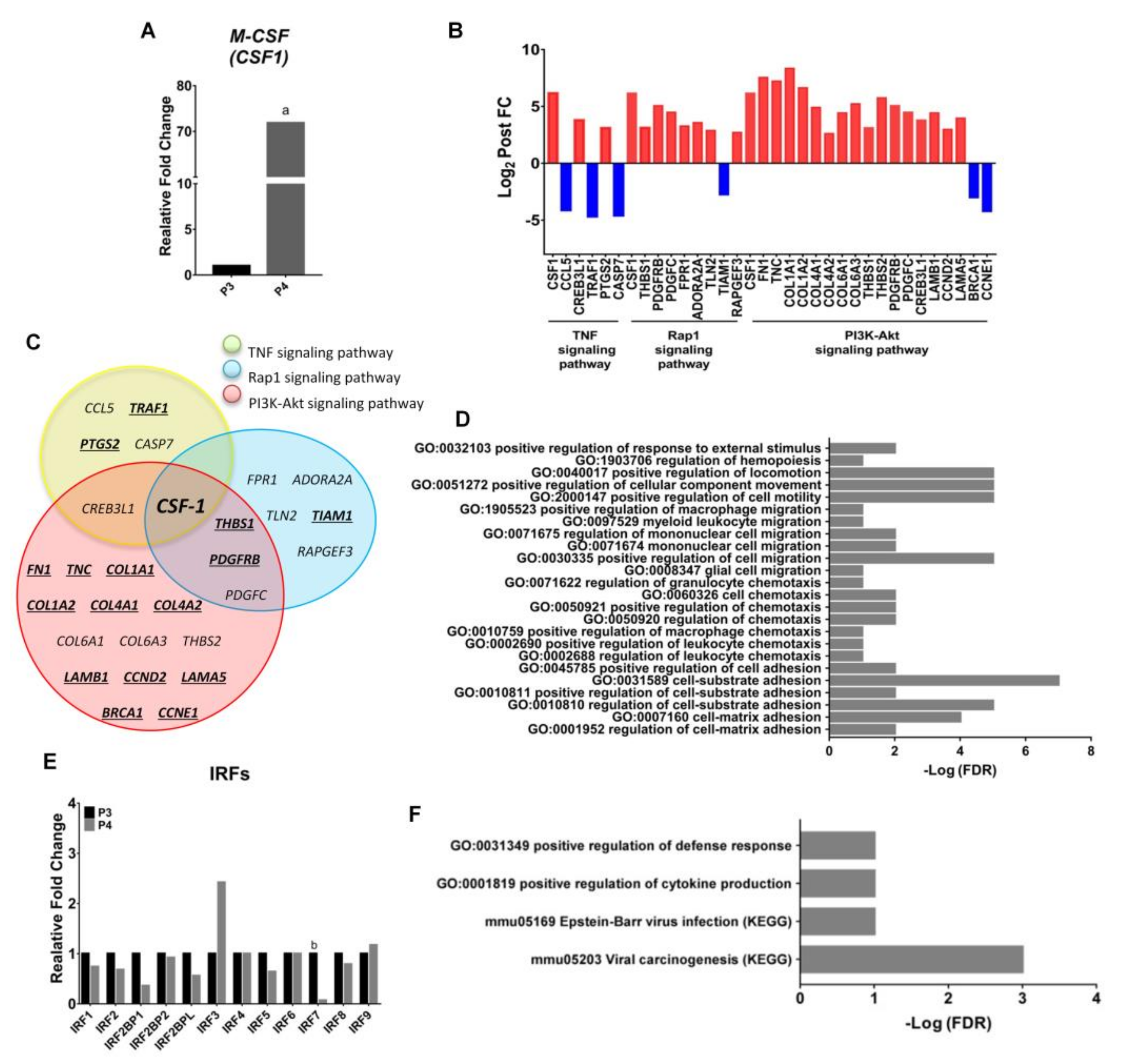
2.5. Endogenous Expression of CSF-1 Was Augmented in M2 Macrophages after Withholding of M-CSF Supplement
2.6. Upregulation of the Genes Related to Pro-Tumor Activity in M2-Like Macrophages after Withholding of M-CSF
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Differentiation of Bone Marrow-Derived Macrophages (BMDMs)
4.3. Macrophage Subtype Polarization
4.4. Immunocytochemistry
4.5. Flow Cytometry Analysis
4.6. Apoptosis Assay
4.7. Real-Time PCR
4.8. Library Preparation and Sequencing
4.9. Bioinformatics Analysis
4.10. MicroRNA Expression Analysis
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron. 2011, 4, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. New mechanisms of tumor-associated macrophages on promoting tumor progression: Recent research advances and potential targets for tumor immunotherapy. J. Immunol. Res. 2016, 2016, 9720912. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Locati, M. Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization: Lessons and open questions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1478–1483. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Pei, B.X.; Sun, B.S.; Zhang, Z.F.; Wang, A.L.; Ren, P. Interstitial tumor-associated macrophages combined with tumor-derived colony-stimulating factor-1 and interleukin-6, a novel prognostic biomarker in non–small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2014, 148, 1208–1216. [Google Scholar] [CrossRef]
- Ding, P.; Wang, W.; Wang, J.; Yang, Z.; Xue, L. Expression of tumor-associated macrophage in progression of human glioma. Cell Biochem. Biophys. 2014, 70, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.N.; Wang, C.J.; Chao, Y.J.; Lai, M.D.; Shan, Y.S. The significance of the co-existence of osteopontin and tumor-associated macrophages in gastric cancer progression. BMC Cancer 2015, 15, 128. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Yuan, A.; Hsiao, Y.J.; Chen, H.Y.; Chen, H.W.; Ho, C.C.; Chen, Y.Y.; Liu, Y.C.; Hong, T.H.; Yu, S.L.; Chen, J.J.; et al. Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci. Rep. 2015, 5, 14273. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Greenland, E.L.; Pixley, F.J. Promotion of tumor invasion by tumor-associated macrophages: The role of CSF-1-activated phosphatidylinositol 3 kinase and Src family kinase motility signaling. Cancers 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Mescher, A.L. Macrophages and fibroblasts during inflammation and tissue repair in models of organ regeneration. Regeneration 2017, 4, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Paietta, E.; Racevskis, J.; Stanley, E.R.; Andreeff, M.; Papenhausen, P.; Wiernik, P.H. Expression of the macrophage growth factor, CSF-1 and its receptor c-fms by a Hodgkin’s disease-derived cell line and its variants. Cancer Res. 1990, 50, 2049–2055. [Google Scholar]
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef] [PubMed]
- Von Tresckow, B.; Morschhauser, F.; Ribrag, V.; Topp, M.S.; Chien, C.; Seetharam, S.; Aquino, R.; Kotoulek, S.; de Boer, C.J.; Engert, A. An open-label, multicenter, phase I/II study of JNJ-40346527, a CSF-1R inhibitor, in patients with relapsed or refractory Hodgkin lymphoma. Clin. Cancer Res. 2015, 21, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Kubota, Y.; Takubo, K.; Shimizu, T.; Ohno, H.; Kishi, K.; Shibuya, M.; Saya, H.; Suda, T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 2009, 206, 1089–1102. [Google Scholar] [CrossRef]
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology 2013, 2, e26968. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264. [Google Scholar] [CrossRef]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J. Immunother. Cancer 2019, 7, 140. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef]
- Sahraei, M.; Chaube, B.; Liu, Y.; Sun, J.; Kaplan, A.; Price, N.L.; Ding, W.; Oyaghire, S.; García-Milian, R.; Mehta, S.; et al. Suppressing miR-21 activity in tumor-associated macrophages promotes an antitumor immune response. J. Clin. Investig. 2019, 129, 5518–5536. [Google Scholar] [CrossRef]
- De Nardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Ao, J.Y.; Zhu, X.D.; Chai, Z.T.; Cai, H.; Zhang, Y.Y.; Zhang, K.Z.; Kong, L.Q.; Zhang, N.; Ye, B.G.; Ma, D.N.; et al. Colony-stimulating factor 1 receptor blockade inhibits tumor growth by altering the polarization of tumor-associated macrophages in hepatocellular carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Tolcher, A.W.; Jones, S.F.; Beeram, M.; Infante, J.R.; Larsen, P.; Rasor, K.; Garrus, J.E.; Li, J.; Cable, P.L.; et al. A phase 1 study of ARRY-382, an oral inhibitor of colony-stimulating factor-1 receptor (CSF1R), in patients with advanced or metastatic cancers. Mol. Cancer Ther. 2013, 12, A252. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An ivy foundation early phase clinical trials consortium phase II study. Neuro-oncology 2015, 18, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Gluck, L.; Martin, L.P.; Olszanski, A.J.; Tolcher, A.W.; Ngarmchamnanrith, G.; Rasmussen, E.; Amore, B.M.; Nagorsen, D.; Hill, J.S.; et al. First-in-human study of AMG 820, a monoclonal anti-colony-stimulating factor 1 receptor antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 5703–5710. [Google Scholar] [CrossRef]
- O’Brien, S.A.; Orf, J.; Skrzypczynska, K.M.; Tan, H.; Kim, J.; De Voss, J.; Belmontes, B.; Egen, J.G. Activity of tumor-associated macrophage depletion by CSF1R blockade is highly dependent on the tumor model and timing of treatment. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.; Blazek, K.; Byrne, A.J.; Perocheau, D.P.; Udalova, I.A. IRF5 is a specific marker of inflammatory macrophages in vivo. Mediat. Inflamm. 2013, 2013, 245804. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017, 47, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado, J.D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef]
- Xuan, W.; Qu, Q.; Zheng, B.; Xiong, S.; Fan, G.H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2015, 97, 61–69. [Google Scholar] [CrossRef]
- Gill, S.E.; Gharib, S.A.; Bench, E.M.; Sussman, S.W.; Wang, R.T.; Rims, C.; Birkland, T.P.; Wang, Y.; Manicone, A.M.; McGuire, J.K.; et al. Tissue inhibitor of metalloproteinases-3 moderates the proinflammatory status of macrophages. Am. J. Respir. Cell Mol. Biol. 2013, 49, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in M1 (LPS+) vs. classically and M2 (LPS–) vs. alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Liao, X.; Bu, Y.; Xu, Z.; Jia, F.; Chang, F.; Liang, J.; Jia, Q.; Lv, Y. WISP1 predicts clinical prognosis and is associated with tumor purity, immunocyte infiltration, and macrophage M2 polarization in pan-cancer. Front. Genet. 2020, 11, 502. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, W.; Dai, X.; Zhang, C.; Zhang, Q.; Lu, J. Identification of the collagen family as prognostic biomarkers and immune-associated targets in gastric cancer. Int. Immunopharmacol. 2020, 87, 106798. [Google Scholar] [CrossRef]
- Mouton, A.J.; De Leon-Pennell, K.Y.; Gonzalez, O.J.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef]
- Murrey, M.W.; Steer, J.H.; Greenland, E.L.; Proudfoot, J.M.; Joyce, D.A.; Pixley, F.J. Adhesion, motility and matrix-degrading gene expression changes in CSF-1-induced mouse macrophage differentiation. J. Cell Sci. 2020, 133, jcs232405. [Google Scholar] [CrossRef]
- Tanaka, T.; Murakami, K.; Bando, Y.; Yoshida, S. Interferon regulatory factor 7 participates in the M 1-like microglial polarization switch. Glia 2015, 63, 595–610. [Google Scholar] [CrossRef]
- Caescu, C.I.; Guo, X.; Tesfa, L.; Bhagat, T.D.; Verma, A.; Zheng, D.; Stanley, E.R. Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood 2015, 125, e1–e13. [Google Scholar] [CrossRef]
- Hume, D.A.; Gordon, S. Regulation of bone marrow macrophage proliferation. Adv. Exp. Med. Biol. 1982, 155, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, X.; Cao, Q.; Wang, Y.; Zheng, G.; Tan, T.K.; Zhao, H.; Zhao, Y.; Wang, Y.; Harris, D.C. Characterizatio n of murine macrophages from bone marrow, spleen and peritoneum. BMC Immunol. 2013, 14, 6. [Google Scholar] [CrossRef]
- Wajant, H. The role of TNF in cancer. In Death Receptors and Cognate Ligands in Cancer; Kalthoff, H., Ed.; Springer: Berlin, Germany, 2009; pp. 1–15. [Google Scholar]
- Ardizzoia, A.; Lissoni, P.; Brivio, F.; Tisi, E.; Perego, M.S.; Grassi, M.G.; Pittalis, S.; Crispino, S.; Barni, S.; Tancini, G. Tumor necrosis factor in solid tumors: Increased blood levels in the metastatic disease. J. Biol. Regul. Homeost. Agents 1992, 6, 103–107. [Google Scholar] [PubMed]
- Rivas, M.A.; Carnevale, R.P.; Proietti, C.J.; Rosemblit, C.; Beguelin, W.; Salatino, M.; Charreau, E.H.; Frahm, I.; Sapia, S.; Brouckaert, P.; et al. TNFα acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-kappa B-dependent pathways. Exp. Cell Res. 2008, 314, 509–529. [Google Scholar] [CrossRef]
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J. Transl Med. 2018, 16, 242. [Google Scholar] [CrossRef] [PubMed]
- Guilbert, L.J.; Winkler-Lowen, B.; Smith, A.; Branch, D.R.; Garcia-Lloret, M. Analysis of the synergistic stimulation of mouse macrophage proliferation by macrophage colony-stimulating factor (CSF-1) and tumor necrosis factor alpha (TNF-α). J. Leukoc. Biol. 1993, 54, 65–72. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Cai, Y.; Sukhova, G.K.; Wong, H.K.; Xu, A.; Tergaonkar, V.; Vanhoutte, P.M.; Tang, E.H.C. Rap1 induces cytokine production in pro-inflammatory macrophages through NFκB signaling and is highly expressed in human atherosclerotic lesions. Cell Cycle 2015, 14, 3580–3592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chenwei, L.; Mahmood, R.; Van Golen, K.; Greenson, J.; Li, G.; D’Silva, N.J.; Li, X.; Burant, C.F.; Logsdon, C.D.; et al. Identification of a putative tumor suppressor gene Rap1GAP in pancreatic cancer. Cancer Res. 2006, 66, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Wang, R.C.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol. Med. 2017, 14, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Arthur, W.T.; Quilliam, L.A.; Cooper, J.A. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J. Cell Biol. 2004, 167, 111–122. [Google Scholar] [CrossRef]
- Christian, S.L.; Lee, R.L.; McLeod, S.J.; Burgess, A.E.; Li, A.H.; Dang-Lawson, M.; Lin, K.B.; Gold, M.R. Activation of the Rap GTPases in B lymphocytes modulates B cell antigen receptor-induced activation of Akt but has no effect on MAPK activation. J. Biol. Chem. 2003, 278, 41756–41767. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, N.G.; Yu, W.; Cox, D.; Wyckoff, J.; Condeelis, J.; Stanley, E.R.; Pixley, F.J. Phosphorylation of CSF-1R Y721 mediates its association with PI3K to regulate macrophage motility and enhancement of tumor cell invasion. J. Cell Sci. 2011, 124, 2021–2031. [Google Scholar] [CrossRef]
- Yang, F.; Gao, J.Y.; Chen, H.; Du, Z.H.; Zhang, X.Q.; Gao, W. Targeted inhibition of the phosphoinositide 3-kinase impairs cell proliferation, survival, and invasion in colon cancer. Onco. Targets Ther. 2017, 10, 4413–4422. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, J.B.; Ding, Y.; Huang, D.S.; Liang, A.J.; Zeng, W.K.; Zeng, Z.P.; Qin, C.Q.; Barden, B. Inhibition of the PI3K/akt pathway reduces tumor necrosis factor-alpha production in the cellular response to wear particles in Vitro. Artif. Organs. 2013, 37, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef]
- Sachdeva, M.; Zhu, S.; Mo, Y.Y. MicroRNA-21 as a Novel Therapeutic Target. Curr. Canc. Ther. Rev. 2010, 6, 41–50. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, J.; Jia, Q.; Ren, Y.; Wang, Y.; Shi, L.; Liu, N.; Wang, G.; Pu, P.; You, Y.; et al. Reduction of miR-21 induces glioma cell apoptosis via activating caspase 9 and 3. Oncol. Rep. 2010, 24, 195–201. [Google Scholar] [CrossRef]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Si, M.L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.Y. miR-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar] [CrossRef] [PubMed]
- Ruan, F.; Wang, Y.F.; Chai, Y. Diagnostic values of miR-21, miR-124, and M-CSF in patients with early cervical cancer. Technol. Cancer Res. Treat. 2020, 19, 1533033820914983. [Google Scholar] [CrossRef]
- An, Y.; Yang, Q. MiR-21 modulates the polarization of macrophages and increases the effects of M2 macrophages on promoting the chemoresistance of ovarian cancer. Life Sci. 2020, 242, 117162. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA- seq experiments. Bioinformatics 2013, 29, 2073. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Chai, Z.T.; Zhu, X.D.; Ao, J.Y.; Wang, W.Q.; Gao, D.M.; Kong, J.; Zhang, N.; Zhang, Y.Y.; Ye, B.G.; Ma, D.N.; et al. microRNA-26a suppresses recruitment of macrophages by down-regulating macrophage colony-stimulating factor expression through the PI3K/Akt pathway in hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Semerad, C.L.; Christopher, M.J.; Liu, F.; Short, B.; Simmons, P.J.; Winkler, I.; Levesque, J.P.; Chappel, J.; Ross, F.P.; Link, D.C. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood 2005, 106, 3020–3027. [Google Scholar] [CrossRef]
- Wu, D.; Pan, P.; Su, X.; Zhang, L.; Qin, Q.; Tan, H.; Huang, L.; Li, Y. Interferon regulatory factor-1 mediates alveolar macrophage pyroptosis during LPS-induced acute lung injury in mice. Shock 2016, 46, 329–338. [Google Scholar] [CrossRef]
- Sun, K.; He, S.B.; Qu, J.G.; Dang, S.C.; Chen, J.X.; Gong, A.H.; Xie, R.; Zhang, J.X. IRF5 regulates lung macrophages M2 polarization during severe acute pancreatitis in vitro. World J. Gastroenterol. 2016, 22, 9368–9377. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Bao, R.; Fan, X.; Tao, T.; Zhu, J.; Wang, J.; Li, J.; Bo, L.; Deng, X. PD-L1 blockade attenuated sepsis-induced liver injury in a mouse cecal ligation and puncture model. Mediat. Inflamm. 2013, 2013, 361501. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-C.; Lai, Y.-S.; Hsuuw, Y.-D.; Chang, K.-T. Withholding of M-CSF Supplement Reprograms Macrophages to M2-Like via Endogenous CSF-1 Activation. Int. J. Mol. Sci. 2021, 22, 3532. https://doi.org/10.3390/ijms22073532
Chen Y-C, Lai Y-S, Hsuuw Y-D, Chang K-T. Withholding of M-CSF Supplement Reprograms Macrophages to M2-Like via Endogenous CSF-1 Activation. International Journal of Molecular Sciences. 2021; 22(7):3532. https://doi.org/10.3390/ijms22073532
Chicago/Turabian StyleChen, Yu-Chih, Yin-Siew Lai, Yan-Der Hsuuw, and Ko-Tung Chang. 2021. "Withholding of M-CSF Supplement Reprograms Macrophages to M2-Like via Endogenous CSF-1 Activation" International Journal of Molecular Sciences 22, no. 7: 3532. https://doi.org/10.3390/ijms22073532
APA StyleChen, Y.-C., Lai, Y.-S., Hsuuw, Y.-D., & Chang, K.-T. (2021). Withholding of M-CSF Supplement Reprograms Macrophages to M2-Like via Endogenous CSF-1 Activation. International Journal of Molecular Sciences, 22(7), 3532. https://doi.org/10.3390/ijms22073532