
Impact of Microbial Metabolites on Microbiota–Gut–Brain Axis in Inflammatory Bowel Disease

 ,
,  , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. The Gut Microbiota and IBD
3. The Microbiota–Gut–Brain Axis and IBD
3.1. The Gut Immune System
3.2. The ENS
3.3. The Vagus Nerve
3.4. Spinal Pathways and Visceral Pain
3.5. Hormonal Connections: The HPA Axis and Stress Response
4. Microbial Metabolites and IBD
4.1. Short-Chain Fatty Acids
4.2. Bile Acids
4.3. Trp Metabolism: Focus on Kynurenine and AhR Ligands in IBD
4.3.1. Kynurenine
4.3.2. AhR Ligands
5. Microbiota-Based Approaches in IBD Therapy
5.1. Antibiotics
5.2. Probiotics and Prebiotics
5.3. Fecal Microbiota Transplantation
6. Future Perspectives and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Tontini, G.E.; Vecchi, M.; Pastorelli, L.; Neurath, M.F.; Neumann, H. Differential diagnosis in inflammatory bowel disease colitis: State of the art and future perspectives. World J. Gastroenterol. 2015, 21, 21–46. [Google Scholar] [CrossRef]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Caetano M Antunes, L.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Lomax, A.E.; Fernández, E.; Sharkey, K.A. Plasticity of the enteric nervous system during intestinal inflammation. Neurogastroenterol. Motil. 2005, 17, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Filpa, V.; Bistoletti, M.; Caon, I.; Moro, E.; Grimaldi, A.; Moretto, P.; Baj, A.; Giron, M.C.; Karousou, E.; Viola, M.; et al. Changes in hyaluronan deposition in the rat myenteric plexus after experimentally-induced colitis. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Brierley, S.M.; Linden, D.R. Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 611–627. [Google Scholar] [CrossRef]
- Bistoletti, M.; Micheloni, G.; Baranzini, N.; Bosi, A.; Conti, A.; Filpa, V.; Pirrone, C.; Millefanti, G.; Moro, E.; Grimaldi, A.; et al. Homeoprotein OTX1 and OTX2 involvement in rat myenteric neuron adaptation after DNBS-induced colitis. Peer J. 2020, 8, e8442. [Google Scholar] [CrossRef]
- Gkouskou, K.K.; Deligianni, C.; Tsatsanis, C.; Eliopoulos, A.G. The gut microbiota in mouse models of inflammatory bowel disease. Front. Cell. Infect. Microbiol. 2014, 4, 28. [Google Scholar] [CrossRef]
- Sinagra, E.; Utzeri, E.; Morreale, G.C.; Fabbri, C.; Pace, F.; Anderloni, A. Microbiota-gut-brain axis and its affect inflammatory bowel disease: Pathophysiological concepts and insights for clinicians. World J. Clin. Cases 2020, 8, 1013–1025. [Google Scholar] [CrossRef]
- Narula, N.; Pinto-Sanchez, M.I.; Calo, N.C.; Ford, A.C.; Bercik, P.; Reinisch, W.; Moayyedi, P. Anxiety But Not Depression Predicts Poor Outcomes in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Gracie, D.J.; Guthrie, E.A.; Hamlin, P.J.; Ford, A.C. Bi-directionality of Brain–Gut Interactions in Patients With Inflammatory Bowel Disease. Gastroenterology 2018, 154, 1635–1646.e3. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA. 2014, 111, 18321–18326. [Google Scholar] [CrossRef]
- Mikocka-Walus, A.; Knowles, S.R.; Keefer, L.; Graff, L. Controversies Revisited: A Systematic Review of the Comorbidity of Depression and Anxiety with Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2015, 22, 752–762. [Google Scholar] [CrossRef]
- Cámara, R.J.A.; Ziegler, R.; Begré, S.; Schoepfer, A.M.; Von Känel, R. The role of psychological stress in inflammatory bowel disease: Quality assessment of methods of 18 prospective studies and suggestions for future research. Digestion 2009, 80, 129–139. [Google Scholar]
- Walker, J.R.; Ediger, J.P.; Graff, L.A.; Greenfeld, J.M.; Clara, I.; Lix, L.; Rawsthorne, P.; Miller, N.; Rogala, L.; McPhail, C.M.; et al. The Manitoba IBD cohort study: A population-based study of the prevalence of lifetime and 12-month anxiety and mood disorders. Am. J. Gastroenterol. 2008, 103, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.G.; Srinath, A.I.; Youk, A.O.; Kirshner, M.A.; Nicole McCarthy, F.; Keljo, D.J.; Bousvaros, A.; Demaso, D.R.; Szigethy, E.M. Predictors of depression in youth with crohn disease. J. Pediatric Gastroenterol. Nutr. 2014, 58, 569–573. [Google Scholar] [CrossRef]
- Szigethy, E.; Levy-Warren, A.; Whitton, S.; Bousvaros, A.; Gauvreau, K.; Leichtner, A.M.; Beardslee, W.R. Depressive Symptoms and Inflammatory Bowel Disease in Children and Adolescents: A Cross-Sectional Study. J. Pediatric Gastroenterol. Nutr. 2004, 39, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Engström, I. Mental Health and Psychological Functioning in Children and Adolescents with Inflammatory Bowel Disease: A Comparison with Children having Other Chronic Illnesses and with Healthy Children. J. Child. Psychol. Psychiatry 1992, 33, 563–582. [Google Scholar] [CrossRef] [PubMed]
- Gracie, D.J.; Hamlin, P.J.; Ford, A.C. The influence of the brain–gut axis in inflammatory bowel disease and possible implications for treatment. Lancet Gastroenterol. Hepatol. 2019, 4, 632–642. [Google Scholar] [CrossRef]
- Labanski, A.; Langhorst, J.; Engler, H.; Elsenbruch, S. Stress and the brain-gut axis in functional and chronic-inflammatory gastrointestinal diseases: A transdisciplinary challenge. Psychoneuroendocrinology 2020, 111, 104501. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Savidge, T.; Shulman, R.J. Brain Gut Microbiome Interactions and Functional Bowel Disorders. Gastroenterology 2014, 146, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.L.; Bernstein, C.N. Brain-gut interactions in inflammatory bowel disease. Gastroenterology 2013, 144, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Bistoletti, M.; Bosi, A.; Banfi, D.; Cristina, G.; Baj, A. The microbiota-gut-brain axis: Focus on the fundamental communication pathways. In Progress in Molecular Biology and Translational Science; Elsevier B.V: Amsterdam, The Netherlands, 2020; Volume 176, pp. 43–110. [Google Scholar]
- Baj, A.; Moro, E.; Bistoletti, M.; Orlandi, V.; Crema, F.; Giaroni, C. Glutamatergic signaling along the microbiota-gut-brain axis. Int. J. Mol. Sci. 2019, 20, 1482. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The microbiota-gut-brain axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Oligschlaeger, Y.; Yadati, T.; Houben, T.; Condello Oliván, C.M.; Shiri-Sverdlov, R. Inflammatory Bowel Disease: A Stressed “Gut/Feeling”. Cells 2019, 8, 659. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Swanson, K.S. Gut microbiota of humans, dogs and cats: Current knowledge and future opportunities and challenges. Br. J. Nutr. 2015, 113, S6–S17. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. Plos Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Groussin, M.; Mazel, F.; Alm, E.J. Co-evolution and Co-speciation of Host-Gut Bacteria Systems. Cell Host Microbe 2020, 28, 12–22. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA. 2011, 108, 4554–4561. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.C.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef]
- Zuo, T.; Kamm, M.A.; Colombel, J.F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory bowel disease. Frontiers in Microbiology 2018, 9, 2247. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8836–8847. [Google Scholar] [CrossRef]
- Koboziev, I.; Reinoso Webb, C.; Furr, K.L.; Grisham, M.B. Role of the enteric microbiota in intestinal homeostasis and inflammation. Free Radic. Biol. Med. 2014, 68, 122–133. [Google Scholar] [CrossRef]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Casén, C.; Vebø, H.C.; Sekelja, M.; Hegge, F.T.; Karlsson, M.K.; Ciemniejewska, E.; Dzankovic, S.; Frøyland, C.; Nestestog, R.; Engstrand, L.; et al. Deviations in human gut microbiota: A novel diagnostic test for determining dysbiosis in patients with IBS or IBD. Aliment. Pharmacol. Ther. 2015, 42, 71–83. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Schultsz, C.; Van Den Berg, F.M.; Ten Kate, F.W.; Tytgat, G.N.J.; Dankert, J. The Intestinal Mucus Layer From Patients With Inflammatory Bowel Disease Harbors High. Numbers of Bacteria Compared With Controls. Gastroenterology 1999, 117, 1089–1097. [Google Scholar] [CrossRef]
- Chow, J.; Mazmanian, S.K. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe 2010, 7, 265–276. [Google Scholar] [CrossRef]
- Alam, M.T.; Amos, G.C.A.; Murphy, A.R.J.; Murch, S.; Wellington, E.M.H.; Arasaradnam, R.P. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Carvalho, F.A.; Ley, R.E.; Gewirtz, A.T. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 2014, 63, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.P.; Longhi, C.; Marazzato, M.; Conte, A.L.; Aleandri, M.; Lepanto, M.S.; Zagaglia, C.; Nicoletti, M.; Aloi, M.; Totino, V.; et al. Adherent-invasive Escherichia coli (AIEC) in pediatric Crohn’s disease patients: Phenotypic and genetic pathogenic features. BMC Res. Notes 2014, 7, 748. [Google Scholar] [CrossRef]
- Sokol, H.; Lepage, P.; Seksik, P.; Doré, J.; Marteau, P. Temperature Gradient Gel Electrophoresis of Fecal 16S rRNA Reveals Active Escherichia coli in the Microbiota of Patients with Ulcerative Colitis. J. Clin. Microbiol. 2006, 44, 3172–3177. [Google Scholar] [CrossRef]
- Ohkusa, T.; Okayasu, I.; Ogihara, T.; Morita, K.; Ogawa, M.; Sato, N. Induction of experimental ulcerative colitis by Fusobacterium varium isolated from colonic mucosa of patients with ulcerative colitis. Gut 2003, 52, 79–83. [Google Scholar] [CrossRef]
- Willing, B.; Halfvarson, J.; Dicksved, J.; Rosenquist, M.; Järnerot, G.; Engstrand, L.; Tysk, C.; Jansson, J.K. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohnʼs disease. Inflamm. Bowel Dis. 2009, 15, 653–660. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Varela, E.; Manichanh, C.; Gallart, M.; Torrejón, A.; Borruel, N.; Casellas, F.; Guarner, F.; Antolin, M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013, 38, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Livanos, A.E.; Snider, E.J.; Whittier, S.; Chong, D.H.; Wang, T.C.; Abrams, J.A.; Freedberg, D.E. Rapid gastrointestinal loss of Clostridial Clusters IV and XIVa in the ICU associates with an expansion of gut pathogens. PLoS ONE 2018, 13, e0200322. [Google Scholar] [CrossRef]
- Hamilton, A.L.; Kamm, M.A.; De Cruz, P.; Wright, E.K.; Feng, H.; Wagner, J.; Sung, J.J.Y.; Kirkwood, C.D.; Inouye, M.; Teo, S.-M. Luminal microbiota related to Crohn’s disease recurrence after surgery. Gut Microbes 2020, 11, 1713–1728. [Google Scholar] [CrossRef] [PubMed]
- Pittayanon, R.; Lau, J.T.; Leontiadis, G.I.; Tse, F.; Yuan, Y.; Surette, M.; Moayyedi, P. Differences in Gut Microbiota in Patients With vs Without Inflammatory Bowel Diseases: A Systematic Review. Gastroenterology 2020, 158, 930–946.e1. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Yassour, M.; Sauk, J.; Garner, A.; Jiang, X.; Arthur, T.; Lagoudas, G.K.; Vatanen, T.; Fornelos, N.; Wilson, R.; et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017, 9, 1–12. [Google Scholar] [CrossRef]
- Winter, S.E.; Lopez, C.A.; Bäumler, A.J. The dynamics of gut-associated microbial communities during inflammation. Embo Rep. 2013, 14, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Chen, E.Z.; Baldassano, R.N.; Otley, A.R.; Griffiths, A.M.; Lee, D.; Bittinger, K.; Bailey, A.; Friedman, E.S.; Hoffmann, C.; et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 2015, 18, 489–500. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 1–11. [Google Scholar] [CrossRef]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Ward, T.L.; McKinlay, C.E.; Miller, H.; Gonzalez, A.; McDonald, D.; Knight, R. Rethinking enterotypes. Cell Host Microbe 2014, 16, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Örtqvist, A.K.; Cao, Y.; Simon, T.G.; Roelstraete, B.; Song, M.; Joshi, A.D.; Staller, K.; Chan, A.T.; Khalili, H.; et al. Antibiotic use and the development of inflammatory bowel disease: A national case-control study in Sweden. Lancet Gastroenterol. Hepatol. 2020, 5, 986–995. [Google Scholar] [CrossRef]
- Bamola, V.D.; Ghosh, A.; Kapardar, R.K.; Lal, B.; Cheema, S.; Sarma, P.; Chaudhry, R. Gut microbial diversity in health and disease: Experience of healthy Indian subjects, and colon carcinoma and inflammatory bowel disease patients. Microb. Ecol. Health Dis. 2017, 28, 1322447. [Google Scholar] [CrossRef] [PubMed]
- Mondot, S.; Kang, S.; Furet, J.P.; Aguirre De Carcer, D.; McSweeney, C.; Morrison, M.; Marteau, P.; Doré, J.; Leclerc, M. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm. Bowel Dis. 2011, 17, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Hedin, C.R.; McCarthy, N.E.; Louis, P.; Farquharson, F.M.; McCartney, S.; Taylor, K.; Prescott, N.J.; Murrells, T.; Stagg, A.J.; Whelan, K.; et al. Altered intestinal microbiota and blood T cell phenotype are shared by patients with Crohn’s disease and their unaffected siblings. Gut 2014, 63, 1578–1586. [Google Scholar] [CrossRef]
- Frank, D.N.; St. Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef]
- Fujimoto, T.; Imaeda, H.; Takahashi, K.; Kasumi, E.; Bamba, S.; Fujiyama, Y.; Andoh, A. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroenterol. Hepatol. 2013, 28, 613–619. [Google Scholar] [CrossRef]
- Jacobs, J.P.; Goudarzi, M.; Singh, N.; Tong, M.; McHardy, I.H.; Ruegger, P.; Asadourian, M.; Moon, B.H.; Ayson, A.; Borneman, J.; et al. A Disease-Associated Microbial and Metabolomics State in Relatives of Pediatric Inflammatory Bowel Disease Patients. CMGH 2016, 2, 750–766. [Google Scholar] [CrossRef]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, H.; Kataoka, K.; Ishikawa, H.; Ikata, K.; Arimochi, H.; Iwasaki, T.; Ohnishi, Y.; Kuwahara, T.; Yasutomo, K. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig. Dis. Sci. 2012, 57, 2955–2964. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.; Xu, B.; Wang, X.; Zhang, Y.; Wang, H.; Kong, X.; Zhu, H.; Wu, K. The biodiversity and composition of the dominant fecal microbiota in patients with inflammatory bowel disease. Diagn. Microbiol. Infect. Dis. 2013, 75, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Verma, A.K.; Ahuja, V.; Paul, J. Real-time analysis of mucosal flora in patients with inflammatory bowel disease in India. J. Clin. Microbiol. 2010, 48, 4279–4282. [Google Scholar] [CrossRef]
- Favier, C.; Neut, C.; Mizon, C.; Cortot, A.; Colombel, J.F.; Mizon, J. Fecal β-D-galactosidase production and Bifidobacteria are decreased in Crohn’s disease. Dig. Dis. Sci. 1997, 42, 817–822. [Google Scholar] [CrossRef]
- Nishida, A.; Imaeda, H.; Ohno, M.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Andoh, A. Efficacy and safety of single fecal microbiota transplantation for Japanese patients with mild to moderately active ulcerative colitis. J. Gastroenterol. 2017, 52, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Ohkusa, T.; Sato, N.; Ogihara, T.; Morita, K.; Ogawa, M.; Okayasu, I. Fusobacterium varium localized in the colonic mucosa of patients with ulcerative colitis stimulates species-specific antibody. J. Gastroenterol. Hepatol. 2002, 17, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, H.; He, H.; Du, Y.; Hu, J.; Li, Y.; Li, Y.; Zhou, Y.; Wang, H.; Chen, Y.; et al. Increased Enterococcus faecalis infection is associated with clinically active Crohn disease. Medicine 2016, 95, e5019. [Google Scholar] [CrossRef] [PubMed]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species roseburia hominis and faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhu, C.; Quan, Y.; Yang, J.; Yuan, W.; Yang, Z.; Wu, S.; Luo, W.; Tan, B.; Wang, X. Insights into Roseburia intestinalis which alleviates experimental colitis pathology by inducing anti-inflammatory responses. J. Gastroenterol. Hepatol. 2018, 33, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Martinez-Medina, M.; Aldeguer, X.; Lopez-Siles, M.; González-Huix, F.; López-Oliu, C.; Dahbi, G.; Bianco, J.E.; Blanco, J.; Garcia-Gil, L.J.; Darfeuille-Michaud, A. Molecular diversity of Escherichia coli in the human gut: New ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 872–882. [Google Scholar] [CrossRef]
- Pierce, E.S. Ulcerative colitis and Crohn’s disease: Is Mycobacterium avium subspecies paratuberculosis the common villain? Gut Pathog. 2010, 2, 1–11. [Google Scholar] [CrossRef]
- Zamani, S.; Zali, M.R.; Aghdaei, H.A.; Sechi, L.A.; Niegowska, M.; Caggiu, E.; Keshavarz, R.; Mosavari, N. Mycobacterium avium subsp. paratuberculosis and associated risk factors for inflammatory bowel disease in Iranian patients. Gut Pathog. 2017, 1–10. [Google Scholar] [CrossRef]
- Mulak, A.; Bonaz, B. Irritable bowel syndrome: A model of the brain-gut interactions. Medical Science Monitor 2004, 10, RA55–RA62. [Google Scholar] [PubMed]
- Baj, A.; Bistoletti, M.; Bosi, A.; Moro, E.; Giaroni, C.; Crema, F. Marine Toxins and Nociception: Potential Therapeutic Use in the Treatment of Visceral Pain Associated with Gastrointestinal Disorders. Toxins 2019, 11, 449. [Google Scholar] [CrossRef]
- Filpa, V.; Moro, E.; Protasoni, M.; Crema, F.; Frigo, G.; Giaroni, C. Role of glutamatergic neurotransmission in the enteric nervous system and brain-gut axis in health and disease. Neuropharmacology 2016, 111, 14–33. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kibe, R.; Ooga, T.; Aiba, Y.; Sawaki, E.; Koga, Y.; Benno, Y. Cerebral low-molecular metabolites influenced by intestinal microbiota: A pilot study. Front. Syst. Neurosci. 2013, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Bazin, T.; Pellissier, S. The vagus nerve at the interface of the microbiota-gut-brain axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef]
- Mishima, Y.; Sartor, R.B. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J. Gastroenterol. 2020, 55, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.; Ning, M.X.; Chen, D.K.; Ma, W.T. Interactions between the gut microbiota and the host innate immune response against pathogens. Front. Immunol. 2019, 10, 607. [Google Scholar] [CrossRef]
- Roy, U.; Gálvez, E.J.C.; Iljazovic, A.; Lesker, T.R.; Błażejewski, A.J.; Pils, M.C.; Heise, U.; Huber, S.; Flavell, R.A.; Strowig, T. Distinct Microbial Communities Trigger Colitis Development upon Intestinal Barrier Damage via Innate or Adaptive Immune Cells. Cell Rep. 2017, 21, 994–1008. [Google Scholar] [CrossRef]
- Kullberg, M.C.; Andersen, J.F.; Gorelick, P.L.; Caspar, P.; Suerbaum, S.; Fox, J.G.; Cheever, A.W.; Jankovic, D.; Sher, A. Induction of colitis by a CD4+ T cell clone specific for a bacterial epitope. Proc. Natl. Acad. Sci. USA 2003, 100, 15830–15835. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S.; Lord, G.M.; Punit, S.; Lugo-Villarino, G.; Mazmanian, S.K.K.; Ito, S.; Glickman, J.N.; Glimcher, L.H. Communicable Ulcerative Colitis Induced by T-bet Deficiency in the Innate Immune System. Cell 2007, 131, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867. [Google Scholar] [CrossRef]
- McGovern, D.; Powrie, F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut 2007, 56, 1333–1336. [Google Scholar] [CrossRef]
- Argollo, M.C.; Allocca, M.; Furfaro, F.; Peyrin-Biroulet, L.; Danese, S. Interleukin-23 Blockers: Born to be First-line Biologic Agents in Inflammatory Bowel Disease? Curr. Pharm. Des. 2019, 25, 25–31. [Google Scholar] [CrossRef]
- Chandran, P.; Satthaporn, S.; Robins, A.; Eremin, O. Inflammatory bowel disease: Dysfunction of GALT and gut bacterial flora (II). Surg. J. R. Coll. Surg. Edinb. Irel. 2003, 1, 125–136. [Google Scholar] [CrossRef]
- Gasche, C.; Bakos, S.; Dejaco, C.; Tillinger, W.; Zakeri, S.; Reinisch, W. IL-10 secretion and sensitivity in normal human intestine and inflammatory bowel disease. J. Clin. Immunol. 2000, 20, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Levast, B.; Li, Z.; Madrenas, J. The role of IL-10 in microbiome-associated immune modulation and disease tolerance. Cytokine 2015, 75, 291–301. [Google Scholar] [CrossRef]
- Moran, C.J.; Walters, T.D.; Guo, C.H.; Kugathasan, S.; Klein, C.; Turner, D.; Wolters, V.M.; Bandsma, R.H.; Mouzaki, M.; Zachos, M.; et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm. Bowel Dis. 2013, 19, 115–123. [Google Scholar] [CrossRef]
- Hoshi, N.; Schenten, D.; Nish, S.A.; Walther, Z.; Gagliani, N.; Flavell, R.A.; Reizis, B.; Shen, Z.; Fox, J.G.; Iwasaki, A.; et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat. Commun. 2012, 3, 1–10. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Malvin, N.P.; Seno, H.; Stappenbeck, T.S. Colonic epithelial response to injury requires Myd88 signaling in myeloid cells. Mucosal Immunol. 2012, 5, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-Like Receptor 2 Controls Mucosal Inflammation by Regulating Epithelial Barrier Function. Gastroenterology 2007, 132, 1359–1374. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Allen, I.C.; Tekippe, E.M.E.; Woodford, R.M.T.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P.Y. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; Van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Wu, S.; Yan, C.; Zhao, C.; Jin, H.; Yan, N.; Xu, J.; Wu, Y.; Li, C.; Shao, Q.; et al. Butyrate upregulates the TLR4 expression and the phosphorylation of MAPKs and NK-κB in colon cancer cell in vitro. Oncol. Lett. 2018, 16, 4439–4447. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like receptors and inflammatory bowel disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A. Neuroinflammation in inflammatory bowel disease. J. Neuroinflammation 2010, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Giron, M.C.; Qesari, M.; Porzionato, A.; Caputi, V.; Zoppellaro, C.; Banzato, S.; Grillo, A.R.; Spagnol, L.; De Caro, R.; et al. Toll-like receptor 2 regulates intestinal inflammation by controlling integrity of the enteric nervous system. Gastroenterology 2013, 145, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Shea-Donohue, T.; Urban, J.F. Neuroimmune modulation of gut function. Handb. Exp. Pharmacol. 2017, 239, 247–267. [Google Scholar] [PubMed]
- Bosi, A.; Banfi, D.; Bistoletti, M.; Giaroni, C.; Baj, A. Tryptophan Metabolites Along the Microbiota-Gut-Brain Axis: An Interkingdom Communication System Influencing the Gut in Health and Disease. Int. J. Tryptophan Res. 2020, 13, 13. [Google Scholar] [CrossRef]
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Giaroni, C.; Zanetti, E.; Pascale, A.; Oldrini, R.; Canciani, L.; Giuliani, D.; Amadio, M.; Chiaravalli, A.M.; Lecchini, S.; Frigo, G.M. Involvement of Ca2+-dependent PKCs in the adaptive changes of μ-opioid pathways to sympathetic denervation in the guinea pig colon. Biochem. Pharmacol. 2009, 78, 1233–1241. [Google Scholar] [CrossRef]
- Furness, J.B.; Jones, C.; Nurgali, K.; Clerc, N. Intrinsic primary afferent neurons and nerve circuits within the intestine. Prog. Neurobiol. 2004, 72, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Bistoletti, M.; Bosi, A.; Caon, I.; Chiaravalli, A.M.; Moretto, P.; Genoni, A.; Moro, E.; Karousou, E.; Viola, M.; Crema, F.; et al. Involvement of hyaluronan in the adaptive changes of the rat small intestine neuromuscular function after ischemia/reperfusion injury. Sci. Rep. 2020, 10, 11521. [Google Scholar] [CrossRef] [PubMed]
- Bistoletti, M.; Caputi, V.; Baranzini, N.; Marchesi, N.; Filpa, V.; Marsilio, I.; Cerantola, S.; Terova, G.; Baj, A.; Grimaldi, A.; et al. Antibiotic treatment-induced dysbiosis differently affects BDNF and TrkB expression in the brain and in the gut of juvenile mice. PLoS ONE 2019, 14, e0212856. [Google Scholar] [CrossRef]
- Giaroni, C. Purinergic signalling and development of the autonomic nervous system. Auton. Neurosci. Basic Clin. 2015, 191, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Alverdy, J.; Holbrook, C.; Rocha, F.; Seiden, L.; Licheng, R.; Wu; Musch, M.; Chang, E.; Ohman, D.; Suh, S. Gut-derived sepsis occurs when the right pathogen with the right virulence genes meets the right host: Evidence for in vivo virulence expression in Pseudomonas aeruginosa. Ann. Surg. 2000, 232, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Bernstein, C.N.; Gearry, R.; Hviid, A.; Kolho, K.L.; Kronman, M.P.; Shaw, S.; Van Kruiningen, H.; Colombel, J.F.; Atreja, A. Antibiotics associated with increased risk of New-Onset Crohn’s disease but not ulcerative colitis: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Rolig, A.S.; Mittge, E.K.; Ganz, J.; Troll, J.V.; Melancon, E.; Wiles, T.J.; Alligood, K.; Stephens, W.Z.; Eisen, J.S.; Guillemin, K. The enteric nervous system promotes intestinal health by constraining microbiota composition. PLoS Biol. 2017, 15, e2000689. [Google Scholar] [CrossRef]
- Collins, J.; Borojevic, R.; Verdu, E.F.; Huizinga, J.D.; Ratcliffe, E.M. Intestinal microbiota influence the early postnatal development of the enteric nervous system. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2014, 26, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Mcvey Neufeld, K.A.; Perez-Burgos, A.; Mao, Y.K.; Bienenstock, J.; Kunze, W.A. The gut microbiome restores intrinsic and extrinsic nerve function in germ-free mice accompanied by changes in calbindin. Neurogastroenterol. Motil. 2015, 27, 627–636. [Google Scholar] [CrossRef]
- Caputi, V.; Marsilio, I.; Filpa, V.; Cerantola, S.; Orso, G.; Bistoletti, M.; Paccagnella, N.; De Martin, S.; Montopoli, M.; Dall’Acqua, S.; et al. Antibiotic-induced dysbiosis of the microbiota impairs gut neuromuscular function in juvenile mice. Br. J. Pharmacol. 2017, 174, 3623–3639. [Google Scholar] [CrossRef]
- Caputi, V.; Marsilio, I.; Cerantola, S.; Roozfarakh, M.; Lante, I.; Galuppini, F.; Rugge, M.; Napoli, E.; Giulivi, C.; Orso, G.; et al. Toll-like receptor 4 modulates small intestine neuromuscular function through nitrergic and purinergic pathways. Front. Pharmacol. 2017, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Marsilio, I.; Caputi, V.; Latorre, E.; Cerantola, S.; Paquola, A.; Alcalde, A.I.; Mesonero, J.E.; O’Mahony, S.M.; Bertazzo, A.; Giaroni, C.; et al. Oxidized phospholipids affect small intestine neuromuscular transmission and serotonergic pathways in juvenile mice. Neurogastroenterol. Motil. 2020, e14036. [Google Scholar] [CrossRef]
- Cerantola, S.; Caputi, V.; Marsilio, I.; Ridolfi, M.; Faggin, S.; Bistoletti, M.; Giaroni, C.; Giron, M.C. Involvement of Enteric Glia in Small Intestine Neuromuscular Dysfunction of Toll-Like Receptor 4-Deficient Mice. Cells 2020, 9, 838. [Google Scholar] [CrossRef]
- Kunze, W.A.; Mao, Y.K.; Wang, B.; Huizinga, J.D.; Ma, X.; Forsythe, P.; Bienenstock, J. Lactobacillus reuteri enhances excitability of colonic AH neurons by inhibiting calcium-dependent potassium channel opening. J. Cell. Mol. Med. 2009, 13, 2261–2270. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.K.; Kasper, D.L.; Wang, B.; Forsythe, P.; Bienenstock, J.; Kunze, W.A. Bacteroides fragilis polysaccharide A is necessary and sufficient for acute activation of intestinal sensory neurons. Nat. Commun. 2013, 4, 1465. [Google Scholar] [CrossRef] [PubMed]
- Husebye, E.; Hellström, P.M.; Sundler, F.; Chen, J.; Midtvedt, T. Influence of microbial species on small intestinal myoelectric activity and transit in germ-free rats. Am. J. Physiol.—Gastrointest. Liver Physiol. 2001, 280, G368–G380. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Neuhuber, W.L. Functional and chemical anatomy of the afferent vagal system. Auton. Neurosci. 2000, 85, 1–17. [Google Scholar] [CrossRef]
- Wang, F.-B.; Powley, T.L. Vagal innervation of intestines: Afferent pathways mapped with new en bloc horseradish peroxidase adaptation. Cell Tissue Res. 2007, 329, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.B.; Callaghan, B.P.; Rivera, L.R.; Cho, H.-J. The Enteric Nervous System and Gastrointestinal Innervation: Integrated Local and Central Control. Adv. Exp. Med. Biol. 2014, 817, 39–71. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, J.R.; Ho, W.; Linden, D.R.; Mawe, G.M.; Sharkey, K.A. Enteroendocrine cells and 5-HT availability are altered in mucosa of guinea pigs with TNBS ileitis. Am. J. Physiol.—Gastrointest. Liver Physiol. 2004, 287, G998–G1007. [Google Scholar]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Plata-Salaman, C.R.; Langhans, W. Subdiaphragmatic vagal deafferentation fails to block feeding- suppressive effects of LPS and IL-1β in rats. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 1997, 273, R1193–R1198. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Okuma, Y.; Matsuda, T.; Nomura, Y. Novel pathway for LPS-induced afferent vagus nerve activation: Possible role of nodose ganglion. Auton. Neurosci. Basic Clin. 2005, 120, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Van felius, I.D.; akkermans, L.M.A.; bosscha, K.; Verheem, A.; Harmsen, W.; Visser, M.R.; Gooszen, H.G. Interdigestive small bowel motility and duodenal bacterial overgrowth in experimental acute pancreatitis. Neurogastroenterol. Motil. 2003, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M.; Varcoe, J.J.; Bailey, M.T. Anxiogenic effect of subclinical bacterial infection in mice in the absence of overt immune activation. Physiol. Behav. 1998, 65, 63–68. [Google Scholar] [CrossRef]
- Goehler, L.E.; Gaykema, R.P.A.; Opitz, N.; Reddaway, R.; Badr, N.; Lyte, M. Activation in vagal afferents and central autonomic pathways: Early responses to intestinal infection with Campylobacter jejuni. Brain Behav. Immun. 2005, 19, 334–344. [Google Scholar] [CrossRef]
- Klarer, M.; Krieger, J.P.; Richetto, J.; Weber-Stadlbauer, U.; Günther, L.; Winter, C.; Arnold, M.; Langhans, W.; Meyer, U. Abdominal vagal afferents modulate the brain transcriptome and behaviors relevant to schizophrenia. J. Neurosci. 2018, 38, 1634–1647. [Google Scholar] [CrossRef] [PubMed]
- Desbonnet, L.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Microbiota is essential for social development in the mouse. Mol. Psychiatry 2014, 19, 146–148. [Google Scholar] [CrossRef]
- Perez-Burgos, A.; Wang, B.; Mao, Y.-K.; Mistry, B.; Neufeld, K.-A.M.; Bienenstock, J.; Kunze, W. Psychoactive bacteria Lactobacillus rhamnosus (JB-1) elicits rapid frequency facilitation in vagal afferents. Am. J. Physiol.—Gastrointest. Liver Physiol. 2013, 304, G211–G220. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Bercik, P.; Park, A.J.; Sinclair, D.; Khoshdel, A.; Lu, J.; Huang, X.; Deng, Y.; Blennerhassett, P.A.; Fahnestock, M.; Moine, D.; et al. The anxiolytic effect of Bifidobacterium longum NCC3001 involves vagal pathways for gut-brain communication. Neurogastroenterol. Motil. 2011, 23, 1132–1139. [Google Scholar] [CrossRef]
- Van Der Kleij, H.; O’Mahony, C.; Shanahan, F.; O’Mahony, L.; Bienenstock, J. Protective effects of Lactobacillus reuteri and Bifidobacterium infantis in murine models for colitis do not involve the vagus nerve. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2008, 295, R1131–R1137. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Sinniger, V.; Pellissier, S. The vagus nerve in the neuro-immune axis: Implications in the pathology of the gastrointestinal tract. Front. Immunol. 2017, 8, 1452. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, W.J.; van der Zanden, E.P.; The, F.O.; Bijlsma, M.F.; van Westerloo, D.J.; Bennink, R.J.; Berthoud, H.R.; Uematsu, S.; Akira, S.; van den Wijngaard, R.M.; et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 2005, 6, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Nourian, M.; Chaleshi, V.; Pishkar, L.; Azimzadeh, P.; Baradaran Ghavami, S.; Balaii, H.; Alinaghi, S.; Shahrokh, S.; Asadzadeh Aghdaei, H.; Zali, M.R. Evaluation of tumor necrosis factor (TNF)-α mRNA expression level and the rs1799964 polymorphism of the TNF-α gene in peripheral mononuclear cells of patients with inflammatory bowel diseases. Biomed. Rep. 2017, 6, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.Q.; Taché, Y. Abdominal surgery induced gastric ileus and activation of M1-like macrophages in the gastric myenteric plexus: Prevention by central vagal activation in rats. Am. J. Physiol.—Gastrointest. Liver Physiol. 2017, 313, G320–G329. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdés-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Meregnani, J.; Clarençon, D.; Vivier, M.; Peinnequin, A.; Mouret, C.; Sinniger, V.; Picq, C.; Job, A.; Canini, F.; Jacquier-Sarlin, M.; et al. Anti-inflammatory effect of vagus nerve stimulation in a rat model of inflammatory bowel disease. Auton. Neurosci. Basic Clin. [CrossRef] [PubMed]
- Ghia, J.E.; Blennerhassett, P.; Kumar-Ondiveeran, H.; Verdu, E.F.; Collins, S.M. The Vagus Nerve: A Tonic Inhibitory Influence Associated With Inflammatory Bowel Disease in a Murine Model. Gastroenterology 2006, 131, 1122–1130. [Google Scholar] [CrossRef]
- O’Mahony, C.; Van Der Kleij, H.; Bienenstock, J.; Shanahan, F.; O’Mahony, L. Loss of vagal anti-inflammatory effect: In vivo visualization and adoptive transfer. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2009, 297, R1118–R1126. [Google Scholar]
- Ghia, J.E.; Blennerhassett, P.; Collins, S.M. Impaired parasympathetic function increases susceptibility to inflammatory bowel disease in a mouse model of depression. J. Clin. Investig. 2008, 118, 2209–2218. [Google Scholar] [CrossRef]
- Pellissier, S.; Dantzer, C.; Canini, F.; Mathieu, N.; Bonaz, B. Psychological adjustment and autonomic disturbances in inflammatory bowel diseases and irritable bowel syndrome. Psychoneuroendocrinology 2010, 35, 653–662. [Google Scholar] [CrossRef]
- Rubio, A.; Pellissier, S.; Picot, A.; Dantzer, C.; Bonaz, B. The link between negative affect, vagal tone, and visceral sensitivity in quiescent Crohn’s disease. Neurogastroenterol. Motil. 2014, 26, 1200–1203. [Google Scholar] [CrossRef]
- Defaye, M.; Gervason, S.; Altier, C.; Berthon, J.Y.; Ardid, D.; Filaire, E.; Carvalho, F.A. Microbiota: A novel regulator of pain. J. Neural Transm. 2020, 127, 445–465. [Google Scholar] [CrossRef]
- Moloney, R.D.; O’Mahony, S.M.; Dinan, T.G.; Cryan, J.F. Stress-induced visceral pain: Toward animal models of irritable-bowel syndrome and associated comorbidities. Front. Psychiatry 2015, 6, 15. [Google Scholar] [CrossRef]
- Bielefeldt, K.; Davis, B.; Binion, D.G. Pain and inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory Bowel Disease in Children and Adolescents HHS Public Access. JAMA Pediatrics 2015, 169, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Minderhoud, I.M.; Oldenburg, B.; Wismeijer, J.A.; Van Berge Henegouwen, G.P.; Smout, A.J.P.M. IBS-like symptoms in patients with inflammatory bowel disease in remission; relationships with quality of life and coping behavior. Dig. Dis. Sci. 2004, 49, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Morales-Soto, W.; Gulbransen, B.D. Enteric Glia: A New Player in Abdominal Pain. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, J.N. Visceral pain: The neurophysiological mechanism. Handb. Exp. Pharmacol. 2009, 194, 31–74. [Google Scholar]
- Azpiroz, F.; Bouin, M.C.; Mayer, E.A.; Poitras, P.; Serra, J.; Spiller, R.C. Mechanisms of hypersensitivity in IBS and functional disorders. Neurogastroenterol. Motil. 2007, 19, 62–88. [Google Scholar] [CrossRef]
- Enck, P.; Aziz, Q.; Barbara, G.; Farmer, A.D.; Fukudo, S.; Mayer, E.A.; Niesler, B.; Quigley, E.M.M.; Rajilić-Stojanović, M.; Schemann, M.; et al. Irritable bowel syndrome. Nat. Rev. Dis. Primers 2016, 2, 1–24. [Google Scholar] [CrossRef]
- Guo, R.; Chen, L.H.; Xing, C.; Liu, T. Pain regulation by gut microbiota: Molecular mechanisms and therapeutic potential. Br. J. Anaesth. 2019, 123, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Mcvey Neufeld, K.A.; Mao, Y.K.; Bienenstock, J.; Foster, J.A.; Kunze, W.A. The microbiome is essential for normal gut intrinsic primary afferent neuron excitability in the mouse. Neurogastroenterol. Motil. 2013, 25, 183-e88. [Google Scholar] [CrossRef]
- Perez-Burgos, A.; Wang, L.; Mcvey Neufeld, K.-A.; Mao, Y.-K.; Ahmadzai, M.; Janssen, L.J.; Stanisz, A.M.; Bienenstock, J.; Kunze, W.A. The TRPV1 channel in rodents is a major target for antinociceptive effect of the probiotic Lactobacillus reuteri DSM 17938. J. Physiol. 2015, 593, 3943–3957. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Felice, V.D.; Nally, K.; Savignac, H.M.; Claesson, M.J.; Scully, P.; Woznicki, J.; Hyland, N.P.; Shanahan, F.; Quigley, E.M.; et al. Disturbance of the gut microbiota in early-life selectively affects visceral pain in adulthood without impacting cognitive or anxiety-related behaviors in male rats. Neuroscience 2014, 277, 885–901. [Google Scholar] [CrossRef] [PubMed]
- McKernan, D.P.; Fitzgerald, P.; Dinan, T.G.; Cryan, J.F. The probiotic Bifidobacterium infantis 35624 displays visceral antinociceptive effects in the rat. Neurogastroenterol. Motil. 2010, 22, 1029-e268. [Google Scholar] [CrossRef] [PubMed]
- Moloney, R.D.; Johnson, A.C.; O’Mahony, S.M.; Dinan, T.G.; Greenwood-Van Meerveld, B.; Cryan, J.F. Stress and the Microbiota-Gut-Brain Axis in Visceral PaRelevance to Irritable Bowel Syndrome. Cns Neurosci. Ther. 2016, 22, 102–117. [Google Scholar] [CrossRef]
- Johnson, A.C.; Greenwood-Van Meerveld, B.; McRorie, J. Effects of Bifidobacterium infantis 35624 on post-inflammatory visceral hypersensitivity in the rat. Dig. Dis. Sci. 2011, 56, 3179–3186. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Wang, L.; Forsythe, P.; Goettsche, G.; Mao, Y.; Wang, Y.; Tougas, G.; Bienenstock, J. Inhibitory effects of Lactobocillus reuteri on visceral pain induced by colorectal distension in Sprague-Dawley rats. Gut 2006, 55, 191–196. [Google Scholar] [CrossRef]
- Verdú, E.F.; Bercik, P.; Verma-Gandhu, M.; Huang, X.-X.; Blennerhassett, P.; Jackson, W.; Mao, Y.; Wang, L.; Rochat, F.; Collins, S.M. Specific probiotic therapy attenuates antibiotic induced visceral hypersensitivity in mice. Gut 2006, 55, 182–190. [Google Scholar] [CrossRef]
- Smith, P.; Willemsen, D.; Popkes, M.; Metge, F.; Gandiwa, E.; Reichard, M.; Valenzano, D.R. Regulation of life span by the gut microbiota in the short-lived african turquoise killifish. eLife 2017, 6, e27014. [Google Scholar] [CrossRef]
- O’ Mahony, S.M.; Clarke, G.; McKernan, D.P.; Bravo, J.A.; Dinan, T.G.; Cryan, J.F. Differential visceral nociceptive, behavioural and neurochemical responses to an immune challenge in the stress-sensitive Wistar Kyoto rat strain. Behav. Brain Res. 2013, 253, 310–317. [Google Scholar] [CrossRef]
- Keszthelyi, D.; Troost, F.J.; Simrén, M.; Ludidi, S.; Kruimel, J.W.; Conchillo, J.M.; Masclee, A.A. Revisiting concepts of visceral nociception in irritable bowel syndrome. Eur. J. Pain 2012, 16, 1444–1454. [Google Scholar] [CrossRef]
- Ibeakanma, C.; Vanner, S. TNFα is a key mediator of the pronociceptive effects of mucosal supernatant from human ulcerative colitis on colonic DRG neurons. Gut 2010, 59, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Targownik, L.E.; Sexton, K.A.; Bernstein, M.T.; Beatie, B.; Sargent, M.; Walker, J.R.; Graff, L.A. The relationship among perceived stress, symptoms, and inflammation in persons with inflammatory bowel disease. Am. J. Gastroenterol. 2015, 110, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Reber, S.O. Stress and animal models of inflammatory bowel disease-An update on the role of the hypothalamo-pituitary-adrenal axis. Psychoneuroendocrinology 2012, 37, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mawdsley, J.E.; Rampton, D.S. Psychological stress in IBD: New insights into pathogenic and therapeutic implications. Gut 2005, 54, 1481–1491. [Google Scholar] [CrossRef]
- Lasconi, C.; Pahl, M.C.; Cousminer, D.L.; Doege, C.A.; Chesi, A.; Hodge, K.M.; Leonard, M.E.; Lu, S.; Johnson, M.E.; Su, C.; et al. Variant-to-Gene-Mapping Analyses Reveal a Role for the Hypothalamus in Genetic Susceptibility to Inflammatory Bowel Disease. Cell. Mol. Gastroenterol. Hepatol. 2020, 11, 667–682. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Gareau, M.G.; Jury, J.; MacQueen, G. Probiotic treatment of rat pups normalises corticosterone release and ameliorates colonic dysfunction induced by maternal separation (Gut (2007) 56, (1522-1528)). Gut 2008, 57, 560. [Google Scholar]
- Shanks, N.; Windle, R.J.; Perks, P.A.; Harbuz, M.S.; Jessop, D.S.; Ingram, C.D.; Lightman, S.L. Early-life exposure to endotoxin alters hypothalamic-pituitary-adrenal function and predisposition to inflammation. Proc. Natl. Acad. Sci. USA 2000, 97, 5645–5650. [Google Scholar] [CrossRef] [PubMed]
- Ait-Belgnaoui, A.; Durand, H.; Cartier, C.; Chaumaz, G.; Eutamene, H.; Ferrier, L.; Houdeau, E.; Fioramonti, J.; Bueno, L.; Theodorou, V. Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology 2012, 37, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; Rinaman, L.; Cryan, J.F. Stress & the gut-brain axis: Regulation by the microbiome. Neurobiol. Stress 2017, 7, 124–136. [Google Scholar] [PubMed]
- Scott, L.V.; Clarke, G.; Dinan, T.G. The brain-gut axis: A target for treating stress-related disorders. Mod. Trends Pharmacopsychiatry 2013, 28, 90–99. [Google Scholar]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Hyland, N.P.; Dinan, T.G.; Cryan, J.F. Maternal separation as a model of brain-gut axis dysfunction. Psychopharmacology 2011, 214, 71–88. [Google Scholar] [CrossRef]
- Deng, Y.; Li, M.; Mei, L.; Cong, L.M.; Liu, Y.; Zhang, B.B.; He, C.Y.; Zheng, P.Y.; Yuan, J.L. Manipulation of intestinal dysbiosis by a bacterial mixture ameliorates loperamide-induced constipation in rats. Benef. Microbes 2018, 9, 453–464. [Google Scholar] [CrossRef]
- Gareau, M.G.; Silva, M.A.; Perdue, M.H. Pathophysiological mechanisms of stress-induced intestinal damage. Curr. Mol. Med. 2008, 8, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Lafuse, W.P.; Gearinger, R.; Fisher, S.; Nealer, C.; Mackos, A.R.; Bailey, M.T. Exposure to a social stressor induces translocation of commensal lactobacilli to the spleen and priming of the innate immune system. J. Immunol. 2017, 198, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Campos-Rodríguez, R.; Godínez-Victoria, M.; Abarca-Rojano, E.; Pacheco-Yépez, J.; Reyna-Garfias, H.; Barbosa-Cabrera, R.E.; Drago-Serrano, M.E. Stress modulates intestinal secretory immunoglobulin A. Front. Integr. Neurosci. 2013, 7, 86. [Google Scholar]
- Lyte, M.; Vulchanova, L.; Brown, D.R. Stress at the intestinal surface: Catecholamines and mucosa-bacteria interactions. Cell Tissue Res. 2011, 343, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gárate, I.; Garcia-Bueno, B.; Madrigal, J.L.M.; Caso, J.R.; Alou, L.; Gomez-Lus, M.L.; Micó, J.A.; Leza, J.C. Stress-induced neuroinflammation: Role of the toll-like receptor-4 pathway. Biol. Psychiatry 2013, 73, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Bastiaanssen, T.F.S.; Cowan, C.S.M.; Claesson, M.J.; Dinan, T.G.; Cryan, J.F. Making Sense of … the Microbiome in Psychiatry. Int. J. Neuropsychopharmacol. 2019, 22, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Emge, J.R.; Huynh, K.; Miller, E.N.; Kaur, M.; Reardon, C.; Barrett, K.E.; Gareau, M.G. Modulation of the microbiota-gut-brain axis by probiotics in a murine model of inflammatory bowel disease. Am. J. Physiol.—Gastrointest. Liver Physiol. 2016, 310, G989–G998. [Google Scholar] [CrossRef]
- Salvo, E.; Stokes, P.; Keogh, C.E.; Brust-Mascher, I.; Hennessey, C.; Knotts, T.A.; Sladek, J.A.; Rude, K.M.; Swedek, M.; Rabasa, G.; et al. A murine model of pediatric inflammatory bowel disease causes microbiota-gut-brain axis deficits in adulthood. Am. J. Physiol.—Gastrointest. Liver Physiol. 2020, 319, G361–G374. [Google Scholar] [CrossRef]
- Jang, H.M.; Lee, H.J.; Jang, S.E.; Han, M.J.; Kim, D.H. Evidence for interplay among antibacterial-induced gut microbiota disturbance, neuro-inflammation, and anxiety in mice. Mucosal Immunol. 2018, 11, 1386–1397. [Google Scholar] [CrossRef]
- Takahashi, K.; Nakagawasai, O.; Nemoto, W.; Odaira, T.; Sakuma, W.; Onogi, H.; Nishijima, H.; Furihata, R.; Nemoto, Y.; Iwasa, H.; et al. Effect of Enterococcus faecalis 2001 on colitis and depressive-like behavior in dextran sulfate sodium-treated mice: Involvement of the brain-gut axis. J. Neuroinflammation 2019, 16, 1–6. [Google Scholar] [CrossRef]
- Bercik, P.; Verdu, E.F.; Foster, J.A.; MacRi, J.; Potter, M.; Huang, X.; Malinowski, P.; Jackson, W.; Blennerhassett, P.; Neufeld, K.A.; et al. Chronic gastrointestinal inflammation induces anxiety-like behavior and alters central nervous system biochemistry in mice. Gastroenterology 2010, 139, 2102–2112.e1. [Google Scholar] [CrossRef]
- Sumi, Y.; Miyakawa, M.; Kanzaki, M.; Kotake, Y. Vitamin B-6 Deficiency in Germfree Rats. J. Nutr. 1977, 107, 1707–1714. [Google Scholar] [CrossRef]
- Wostmann, B.S.; Larkin, C.; Moriarty, A.; Bruckner-Kardoss, E. Dietary intake, energy metabolism, and excretory losses of adult male germfree Wistar rats. Lab. Anim. Sci. 1983, 33, 46–50. [Google Scholar] [PubMed]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA. 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Høverstad, T.; Midtvedt, T. Short-chain fatty acids in germfree mice and rats. J. Nutr. 1986, 116, 1772–1776. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Cherbut, C.; Ferrier, L.; Rozé, C.; Anini, Y.; Blottière, H.; Lecannu, G.; Galmiche, J.P. Short-chain fatty acids modify colonic motility through nerves and polypeptide YY release in the rat. Am. J. Physiol. 1998, 275, G1415–G1422. [Google Scholar] [CrossRef]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef]
- Arnoldussen, I.A.C.; Wiesmann, M.; Pelgrim, C.E.; Wielemaker, E.M.; van Duyvenvoorde, W.; Amaral-Santos, P.L.; Verschuren, L.; Keijser, B.J.F.; Heerschap, A.; Kleemann, R.; et al. Butyrate restores HFD-induced adaptations in brain function and metabolism in mid-adult obese mice. Int. J. Obes. 2017, 41, 935–944. [Google Scholar] [CrossRef]
- Val-Laillet, D.; Guérin, S.; Coquery, N.; Nogret, I.; Formal, M.; Romé, V.; Le Normand, L.; Meurice, P.; Randuineau, G.; Guilloteau, P.; et al. Oral sodium butyrate impacts brain metabolism and hippocampal neurogenesis, with limited effects on gut anatomy and function in pigs. FASEB J. 2018, 32, 2160–2171. [Google Scholar] [CrossRef]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Nilsson, N.E.; Kotarsky, K.; Owman, C.; Olde, B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem. Biophys. Res. Commun. 2003, 303, 1047–1052. [Google Scholar] [CrossRef]
- Docampo, M.D.; Stein-Thoeringer, C.K.; Lazrak, A.; Burgos da Silva, M.D.; Cross, J.; van den Brink, M.R.M. Expression of the Butyrate/Niacin Receptor, GPR109a on T Cells Plays an Important Role in a Mouse Model of Graft Versus Host Disease. Blood 2018, 132, 61. [Google Scholar] [CrossRef]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y., M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic T reg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Gurav, A.; Sivaprakasam, S.; Bhutia, Y.D.; Boettger, T.; Singh, N.; Ganapathy, V. Slc5a8, a Na+-coupled high-affinity transporter for short-chain fatty acids, is a conditional tumour suppressor in colon that protects against colitis and colon cancer under low-fibre dietary conditions. Biochem. J. 2015, 469, 267–278. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huda-Faujan, N.; Abdulamir, A.S.; Fatimah, A.B.; Anas, O.M.; Shuhaimi, M.; Yazid, A.M.; Loong, Y.Y. The Impact of the Level of the Intestinal Short Chain Fatty Acids in Inflammatory Bowel Disease Patients Versus Healthy Subjects. Open Biochem. J. 2010, 4, 53–58. [Google Scholar] [CrossRef]
- De Preter, V.; Arijs, I.; Windey, K.; Vanhove, W.; Vermeire, S.; Schuit, F.; Rutgeerts, P.; Verbeke, K. Impaired butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway*. Inflamm. Bowel Dis. 2012, 18, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Thibault, R.; De Coppet, P.; Daly, K.; Bourreille, A.; Cuff, M.; Bonnet, C.; Mosnier, J.F.; Galmiche, J.P.; Shirazi-Beechey, S.; Segain, J.P. Down-Regulation of the Monocarboxylate Transporter 1 Is Involved in Butyrate Deficiency During Intestinal Inflammation. Gastroenterology 2007, 133, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, O.; Creanza, T.M.; Bossa, F.; Palumbo, O.; Maglietta, R.; Ancona, N.; Corritore, G.; Latiano, T.; Martino, G.; Biscaglia, G.; et al. Genome-wide Pathway Analysis Using Gene Expression Data of Colonic Mucosa in Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1260–1268. [Google Scholar] [CrossRef]
- Barichello, T.; Generoso, J.S.; Simões, L.R.; Faller, C.J.; Ceretta, R.A.; Petronilho, F.; Lopes-Borges, J.; Valvassori, S.S.; Quevedo, J. Sodium Butyrate Prevents Memory Impairment by Re-establishing BDNF and GDNF Expression in Experimental Pneumococcal Meningitis. Mol. Neurobiol. 2015, 52, 734–740. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef]
- Nagatsu, T. Tyrosine hydroxylase: Human isoforms, structure and regulation in physiology and pathology. Essays Biochem. 1995, 30, 15–35. [Google Scholar]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.E.; Akbarian, S. Antidepressant-Like Effects of the Histone Deacetylase Inhibitor, Sodium Butyrate, in the Mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef]
- During, M.J.; Cao, L.; Zuzga, D.S.; Francis, J.S.; Fitzsimons, H.L.; Jiao, X.; Bland, R.J.; Klugmann, M.; Banks, W.A.; Drucker, D.J.; et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 2003, 9, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef]
- Maltz, R.M.; Keirsey, J.; Kim, S.C.; Mackos, A.R.; Gharaibeh, R.Z.; Moore, C.C.; Xu, J.; Bakthavatchalu, V.; Somogyi, A.; Bailey, M.T. Prolonged restraint stressor exposure in outbred CD-1 mice impacts microbiota, colonic inflammation, and short chain fatty acids. PLoS ONE 2018, 13, e0196961. [Google Scholar] [CrossRef] [PubMed]
- van de Wouw, M.; Boehme, M.; Lyte, J.M.; Wiley, N.; Strain, C.; O’Sullivan, O.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 2018, 596, 4923–4944. [Google Scholar] [CrossRef]
- Rea, K.; Dinan, T.G.; Cryan, J.F. The Brain-Gut Axis Contributes to Neuroprogression in Stress-Related Disorders. Mod. Trends Pharm. 2017, 31, 152–161. [Google Scholar]
- Dalile, B.; Vervliet, B.; Bergonzelli, G.; Verbeke, K.; Van Oudenhove, L. Colon-delivered short-chain fatty acids attenuate the cortisol response to psychosocial stress in healthy men: A randomized, placebo-controlled trial. Neuropsychopharmacology 2020, 45, 2257–2266. [Google Scholar] [CrossRef]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Maruyama, T.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 2006, 191, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.-Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Gahan, C.G.M.; Hill, C. The interaction between bacteria and bile. Fems Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef]
- Joyce, S.A.; MacSharry, J.; Casey, P.G.; Kinsella, M.; Murphy, E.F.; Shanahan, F.; Hill, C.; Gahan, C.G.M. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. USA 2014, 111, 7421–7426. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and Gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.L.; Renooij, W.; Murzilli, S.; Klomp, L.W.J.; Siersema, P.D.; Schipper, M.E.I.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Attinkara, R.; Mwinyi, J.; Truninger, K.; Regula, J.; Gaj, P.; Rogler, G.; Kullak-Ublick, G.A.; Eloranta, J.J. Association of genetic variation in the NR1H4 gene, encoding the nuclear bile acid receptor FXR, with inflammatory bowel disease. BMC Res. Notes 2012, 5, 461. [Google Scholar] [CrossRef]
- Nijmeijer, R.M.; Gadaleta, R.M.; van Mil, S.W.C.; van Bodegraven, A.A.; Crusius, J.B.A.; Dijkstra, G.; Hommes, D.W.; de Jong, D.J.; Stokkers, P.C.F.; Verspaget, H.W.; et al. Farnesoid X Receptor (FXR) Activation and FXR Genetic Variation in Inflammatory Bowel Disease. PLoS ONE 2011, 6, e23745. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Wang, Q.; Almousa, A.A.; Jansen, L.E.; Choi, Y.-H.; Schwarz, U.I.; Kim, R.B. Genetic variation in the farnesoid X-receptor predicts Crohn’s disease severity in female patients. Sci. Rep. 2020, 10, 11725. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.; Detloff, S.J.; Ao, M.; Khan, N.; French, S.; Sirajuddin, H.; Nair, T.; Rao, M.C. The Yin and Yang of bile acid action on tight junctions in a model colonic epithelium. Physiol. Rep. 2017, 5, e13294. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchianò, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunuol. 2017, 199, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, T.; Inoue, T.; Yorifuji, N.; Iguchi, M.; Fujiwara, K.; Narabayashi, K.; Kakimoto, K.; Nouda, S.; Okada, T.; Kuramoto, T.; et al. The effects of a TGR5 agonist and a dipeptidyl peptidase IV inhibitor on dextran sulfate sodium-induced colitis in mice. J. Gastroenterol. Hepatol. 2015, 30 (Suppl. 1), 60–65. [Google Scholar] [CrossRef]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef]
- Van den Bossche, L.; Hindryckx, P.; Devisscher, L.; Devriese, S.; Van Welden, S.; Holvoet, T.; Vilchez-Vargas, R.; Vital, M.; Pieper, D.H.; Bussche, J.V.; et al. Ursodeoxycholic acid and its taurine- or glycine-conjugated species reduce colitogenic dysbiosis and equally suppress experimental colitis in mice. Appl. Environ. Microbiol. 2017, 83, e02766-16. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Corlianò, M.; Singaraja, R.R. Bile Acids: A Communication Channel in the Gut-Brain Axis. Neuromolecular Med. 2020, 1–19. [Google Scholar] [CrossRef]
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Pérez, A.; Peterson, V.; et al. Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 2017, 24, 166–178. [Google Scholar] [PubMed]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms, Tryptophan Metabolism, and Kynurenine Pathway: A Complex Interconnected Loop Influencing Human Health Status. Int. J. Tryptophan Res. 2019, 12, 275. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Tryptophan availability for kynurenine pathway metabolism across the life span: Control mechanisms and focus on aging, exercise, diet and nutritional supplements. Neuropharmacology 2017, 112, 248–263. [Google Scholar] [CrossRef]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. Ijtr 2009, 2, 45–60. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased Tryptophan Metabolism Is Associated With Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef] [PubMed]
- Hisamatsu, T.; Okamoto, S.; Hashimoto, M.; Muramatsu, T.; Andou, A.; Uo, M.; Kitazume, M.T.; Matsuoka, K.; Yajima, T.; Inoue, N.; et al. Novel, objective, multivariate biomarkers composed of plasma amino acid profiles for the diagnosis and assessment of inflammatory bowel disease. PLoS ONE 2012, 7, e31131. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.K.; Thaker, A.I.; Kanuri, N.; Riehl, T.E.; Rowley, C.W.; Stenson, W.F.; Ciorba, M.A. Serum analysis of tryptophan catabolism pathway: Correlation with Crohn’s disease activity. Inflamm. Bowel Dis. 2012, 18, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Ciorba, M.A. Indoleamine 2,3 dioxygenase in intestinal disease. Curr. Opin. Gastroenterol. 2013, 29, 146–152. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, K.; Harkin, A. Stress-related regulation of the kynurenine pathway: Relevance to neuropsychiatric and degenerative disorders. Neuropharmacology 2017, 112, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Strasser, B.; Becker, K.; Fuchs, D.; Gostner, J.M. Kynurenine pathway metabolism and immune activation: Peripheral measurements in psychiatric and co-morbid conditions. Neuropharmacology 2017, 112, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Shoaie, S.; Bergentall, M.; Ghaffari, P.; Zhang, C.; Larsson, E.; Bäckhed, F.; Nielsen, J. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 2015, 11, 834. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Desbonnet, L.; Garrett, L.; Clarke, G.; Bienenstock, J.; Dinan, T.G. The probiotic Bifidobacteria infantis: An assessment of potential antidepressant properties in the rat. J. Psychiatr. Res. 2008, 43, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Orhan, F.; Bhat, M.; Sandberg, K.; Ståhl, S.; Piehl, F.; Svensson, C.; Erhardt, S.; Schwieler, L.; Farde, L.; Flyckt, L.; et al. Tryptophan Metabolism Along the Kynurenine Pathway Downstream of Toll-like Receptor Stimulation in Peripheral Monocytes. Scand. J. Immunol. 2016, 84, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-De La Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxidative Med. Cell. Longev. 2013, 2013, 1–14. [Google Scholar] [CrossRef]
- Giuliani, D.; Giaroni, C.; Zanetti, E.; Canciani, L.; Borroni, P.; Lecchini, S.; Frigo, G. Involvement of glutamate receptors of the NMDA type in the modulation of acetylcholine and glutamate overflow from the guinea pig ileum during in vitro hypoxia and hypoglycaemia. Neurochem. Int. 2006, 48, 191–200. [Google Scholar] [CrossRef]
- Carpanese, E.; Moretto, P.; Filpa, V.; Marchet, S.; Moro, E.; Crema, F.; Frigo, G.; Giaroni, C. Antagonism of ionotropic glutamate receptors attenuates chemical ischemia-induced injury in rat primary cultured myenteric ganglia. PLoS ONE 2014, 9, e113613. [Google Scholar] [CrossRef]
- Giaroni, C.; Zanetti, E.; Giuliani, D.; Oldrini, R.; Marchet, S.; Moro, E.; Borroni, P.; Trinchera, M.; Crema, F.; Lecchini, S.; et al. Protein kinase c modulates NMDA receptors in the myenteric plexus of the guinea pig ileum during in vitro ischemia and reperfusion. Neurogastroenterol. Motil. 2011, 23, e91–e103. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, D.; Troost, F.J.; Masclee, A.A.M. Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol. Motil. 2009, 21, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Jürgens, B.; Hainz, U.; Fuchs, D.; Felzmann, T.; Heitger, A. Interferon-γ-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood 2009, 114, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.W.S.; Terentis, A.C.; King, N.J.C.; Thomas, S.R. Role of indoleamine 2,3-dioxygenase in health and disease. Clin. Sci. 2015, 129, 601–672. [Google Scholar] [CrossRef]
- Brooks, A.K.; Lawson, M.A.; Smith, R.A.; Janda, T.M.; Kelley, K.W.; McCusker, R.H. Interactions between inflammatory mediators and corticosteroids regulate transcription of genes within the Kynurenine Pathway in the mouse hippocampus. J. Neuroinflammation 2016, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, H.; Wen, Q.; Zhang, Y. Indoleamine 2,3-dioxygenase expression in human inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2012, 24, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.A.; Ciorba, M.A.; Meckel, K.; Lim, C.K.; Guillemin, G.J.; Weber, C.R.; Bissonnette, M.; Pekow, J.R. Tryptophan Metabolism through the Kynurenine Pathway is Associated with Endoscopic Inflammation in Ulcerative Colitis. Inflamm. Bowel Dis. 2018, 24, 1471. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.; Szabó, E.; Vécsei, L. molecules Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Scarpellini, E.; Colica, C.; Boccuto, L.; Salehi, B.; Sharifi-Rad, J.; Aiello, V.; Romano, B.; De Lorenzo, A.; Izzo, A.A.; et al. Gut microbiota and obesity: A role for probiotics. Nutrients 2019, 11, 2690. [Google Scholar] [CrossRef]
- Tiszlavicz, Z.; Németh, B.; Fülöp, F.; Vécsei, L.; Tápai, K.; Ocsovszky, I.; Mándi, Y. Different inhibitory effects of kynurenic acid and a novel kynurenic acid analogue on tumour necrosis factor-α (TNF-α) production by mononuclear cells, HMGB1 production by monocytes and HNP1-3 secretion by neutrophils. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 382, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Mándi, Y.; Endrész, V.; Mosolygó, T.; Burián, K.; Lantos, I.; Fülöp, F.; Szatmári, I.; Lorinczi, B.; Balog, A.; Vécsei, L. The opposite effects of kynurenic acid and different kynurenic acid analogs on tumor necrosis factor-α (TNF-α) production and tumor necrosis factor-stimulated gene-6 (TSG-6) expression. Front. Immunol. 2019, 10, 1406. [Google Scholar] [CrossRef]
- Kiank, C.; Zeden, J.P.; Drude, S.; Domanska, G.; Fusch, G.; Otten, W.; Schuett, C. Psychological stress-induced, IDO1-dependent tryptophan catabolism: Implications on immunosuppression in mice and humans. PLoS ONE 2010, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kaszaki, J.; Palásthy, Z.; Érczes, D.; Rácz, A.; Torday, C.; Varga, G.; Vécsei, L.; Boros, M. Kynurenic acid inhibits intestinal hypermotility and xanthine oxidase activity during experimental colon obstruction in dogs. Neurogastroenterol. Motil. 2008, 20, 53–62. [Google Scholar] [CrossRef]
- Kaszaki, J.; Érces, D.; Varga, G.; Szabó, A.; Vécsei, L.; Boros, M. Kynurenines and intestinal neurotransmission: The role of N-methyl-d-aspartate receptors. J. Neural Transm. 2012, 119, 211–223. [Google Scholar] [CrossRef]
- Érces, D.; Varga, G.; Fazekas, B.; Kovács, T.; Tkés, T.; Tiszlavicz, L.; Fülöp, F.; Vécsei, L.; Boros, M.; Kaszaki, J. N-methyl-d-aspartate receptor antagonist therapy suppresses colon motility and inflammatory activation six days after the onset of experimental colitis in rats. Eur. J. Pharmacol. 2012, 691, 225–234. [Google Scholar] [CrossRef]
- Kelly, J.R.; Borre, Y.; O’ Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Wichers, M.C.; Koek, G.H.; Robaeys, G.; Verkerk, R.; Scharpé, S.; Maes, M. IDO and interferon-alpha-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatry 2005, 10, 538–544. [Google Scholar] [CrossRef]
- Waclawiková, B.; El Aidy, S. Role of microbiota and tryptophan metabolites in the remote effect of intestinal inflammation on brain and depression. Pharmaceuticals 2018, 11, 63. [Google Scholar] [CrossRef]
- Savitz, J.; Dantzer, R.; Meier, T.B.; Wurfel, B.E.; Victor, T.A.; McIntosh, S.A.; Ford, B.N.; Morris, H.M.; Bodurka, J.; Teague, T.K.; et al. Activation of the kynurenine pathway is associated with striatal volume in major depressive disorder. Psychoneuroendocrinology 2015, 62, 54–58. [Google Scholar] [CrossRef]
- Clarke, G.; McKernan, D.P.; Gaszner, G.; Quigley, E.M.; Cryan, J.F.; Dinan, T.G. A Distinct Profile of Tryptophan Metabolism along the Kynurenine Pathway Downstream of Toll-Like Receptor Activation in Irritable Bowel Syndrome. Front. Pharmacol. 2012, 3, 90. [Google Scholar] [CrossRef]
- McRoberts, J.A.; Coutinho, S.V.; Marvizón, J.C.G.; Grady, E.F.; Tognetto, M.; Sengupta, J.N.; Ennes, H.S.; Chaban, V.V.; Amadesi, S.; Creminon, C.; et al. Role of peripheral N-methyl-D-aspartate (NMDA) receptors in visceral nociception in rats. Gastroenterology 2001, 120, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, W.; De Man, J.G.; Pelckmans, P.A.; De Winter, B.Y. Neuroanatomy of lower gastrointestinal pain disorders. World J. Gastroenterol. 2014, 20, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Majláth, Z.; Török, N.; Toldi, J.; Vécsei, L. Memantine and Kynurenic Acid: Current Neuropharmacological Aspects. Curr. Neuropharmacol. 2016, 14, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Grócz, G.; Zádor, F.; Dvorácskó, S.; Bohár, Z.; Benyhe, S.; Tömböly, C.; Párdutz, Á.; Vécsei, L. Interactions between the kynurenine and the endocannabinoid system with special emphasis on migraine. Int. J. Mol. Sci. 2017, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Darkoh, C.; Chappell, C.; Gonzales, C.; Okhuysen, P. A rapid and specific method for the detection of indole in complex biological samples. Appl. Environ. Microbiol. 2015, 81, 8093–8097. [Google Scholar] [CrossRef]
- Pavlova, T.; Vidova, V.; Bienertova-Vasku, J.; Janku, P.; Almasi, M.; Klanova, J.; Spacil, Z. Urinary intermediates of tryptophan as indicators of the gut microbial metabolism. Anal. Chim. Acta 2017, 987, 72–80. [Google Scholar] [CrossRef]
- Yanofsky, C.; Horn, V.; Gollnick, P. Physiological studies of tryptophan transport and tryptophanase operon induction in Escherichia coli. J. Bacteriol. 1991, 173, 6009–6017. [Google Scholar] [CrossRef]
- Aoki, R.; Aoki-Yoshida, A.; Suzuki, C.; Takayama, Y. Indole-3-Pyruvic Acid, an Aryl Hydrocarbon Receptor Activator, Suppresses Experimental Colitis in Mice. J. Immunol. 2018, 201, 3683–3693. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The Aryl Hydrocarbon Receptor: Multitasking in the Immune System. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Omenetti, S. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef]
- Lee, J.S.; Cella, M.; McDonald, K.G.; Garlanda, C.; Kennedy, G.D.; Nukaya, M.; Mantovani, A.; Kopan, R.; Bradfield, C.A.; Newberry, R.D.; et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2012, 13, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Kimura, A.; Naka, T.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med. 2009, 206, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA. 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Murugaiyan, G.; Farez, M.F.; Mitsdoerffer, M.; Tukpah, A.M.; Burns, E.J.; Weiner, H.L. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 20768–20773. [Google Scholar] [CrossRef]
- Zindl, C.L.; Lai, J.F.; Lee, Y.K.; Maynard, C.L.; Harbour, S.N.; Ouyang, W.; Chaplin, D.D.; Weaver, C.T. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc. Natl. Acad. Sci. USA. 2013, 110, 12768–12773. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus reuteri induces gut intraepithelial CD4+CD8αα+ T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; DeLuca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, M.; Yin, L.; Ren, W.; Bin, P.; Xia, Y.; Liu, G.; Yang, H.; Tan, B.; Yin, Y. Effects of dietary tryptophan supplementation in the acetic acid-induced colitis mouse model. Food Funct. 2018, 9, 4143–4152. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248.e1. [Google Scholar] [CrossRef]
- Goettel, J.A.; Gandhi, R.; Kenison, J.E.; Yeste, A.; Murugaiyan, G.; Sambanthamoorthy, S.; Griffith, A.E.; Patel, B.; Shouval, D.S.; Weiner, H.L.; et al. AHR Activation Is Protective against Colitis Driven by T Cells in Humanized Mice. Cell Rep. 2016, 17, 1318–1329. [Google Scholar] [CrossRef]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Whitfield-Cargile, C.M.; Cohen, N.D.; Chapkin, R.S.; Weeks, B.R.; Davidson, L.A.; Goldsby, J.S.; Hunt, C.L.; Steinmeyer, S.H.; Menon, R.; Suchodolski, J.S.; et al. The microbiota-derived metabolite indole decreases mucosal inflammation and injury in a murine model of NSAID enteropathy. Gut Microbes 2016, 7, 246–261. [Google Scholar] [CrossRef]
- Takamura, T.; Harama, D.; Fukumoto, S.; Nakamura, Y.; Shimokawa, N.; Ishimaru, K.; Ikegami, S.; Makino, S.; Kitamura, M.; Nakao, A. Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunol. Cell Biol. 2011, 89, 817–822. [Google Scholar] [CrossRef]
- Wlodarska, M.; Luo, C.; Kolde, R.; d’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, K.; Han, B.; Sheng, B.; Yin, J.; Pu, A.; Li, L.; Sun, L.; Yu, M.; Qiu, Y.; et al. Aryl hydrocarbon receptor inhibits inflammation in DSS-induced colitis via the MK2/p-MK2/TTP pathway. Int. J. Mol. Med. 2018, 41, 868–876. [Google Scholar] [CrossRef]
- Mu, C.; Zhu, W. Antibiotic effects on gut microbiota, metabolism, and beyond. Appl. Microbiol. Biotechnol. 2019, 103, 9277–9285. [Google Scholar] [CrossRef] [PubMed]
- Ledder, O.; Turner, D. Antibiotics in IBD: Still a Role in the Biological Era? Inflamm. Bowel Dis. 2018, 24, 1676–1688. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 153, 327–339.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Wang, Z.R.; Yang, C.Q. Meta-analysis of broad-spectrum antibiotic therapy in patients with active inflammatory bowel disease. Exp. Ther. Med. 2012, 4, 1051–1056. [Google Scholar] [CrossRef]
- West, R.L.; Van Der Woude, C.J.; Hansen, B.E.; Felt-Bersma, R.J.F.; Van Tilburg, A.J.P.; Drapers, J.A.G.; Kuipers, E.J. Clinical and endosonographic effect of ciprofloxacin on the treatment of perianal fistulae in Crohn’s disease with infliximab: A double-blind placebo-controlled study. Aliment. Pharmacol. Ther. 2004, 20, 1329–1336. [Google Scholar] [CrossRef]
- Doherty, G.; Bennett, G.; Patil, S.; Cheifetz, A.; Moss, A.C. Interventions for prevention of post-operative recurrence of Crohn’s disease. Cochrane Database Syst. Rev. 2009, CD006873. [Google Scholar] [CrossRef] [PubMed]
- Lev-Tzion, R.; Ledder, O.; Shteyer, E.; Tan, M.L.N.; Uhlig, H.H.; Turner, D. Oral Vancomycin and Gentamicin for Treatment of Very Early Onset Inflammatory Bowel Disease. Digestion 2017, 95, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.C.; Moayyedi, P.; Lacy, B.E.; Lembo, A.J.; Saito, Y.A.; Schiller, L.R.; Soffer, E.E.; Spiegel, B.M.R.; Quigley, E.M.M. American college of gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am. J. Gastroenterol. 2014, 106, S2–S26. [Google Scholar] [CrossRef] [PubMed]
- Zommiti, M.; Feuilloley, M.G.J.; Connil, N. Update of Probiotics in Human World: A Nonstop Source of Benefactions till the End of Time. Microorganisms 2020, 8, 1907. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, T.; Sequoia, J.; Jennings, W.; Dorn, B. Probiotics for Gastrointestinal Conditions: A Summary of the Evidence. Am. Fam. Physician. 2017, 96, 170–178. [Google Scholar]
- Ancona, A.; Petito, C.; Iavarone, I.; Petito, V.; Galasso, L.; Leonetti, A.; Turchini, L.; Belella, D.; Ferrarrese, D.; Addolorato, G.; et al. The gut–brain axis in irritable bowel syndrome and inflammatory bowel disease. Dig. Liver Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.M.; Plosker, G.L.; Figgitt, D.P. VSL#3 probiotic mixture: A review of its use in chronic inflammatory bowel diseases. Drugs 2006, 66, 1371–1387. [Google Scholar]
- Silva, N.O.; de Brito, B.B.; da Silva, F.A.F.; Santos, M.L.C.; Melo, F.F. de Probiotics in inflammatory bowel disease: Does it work? World J. Meta-Anal. 2020, 8, 54–66. [Google Scholar] [CrossRef]
- Corridoni, D.; Pastorelli, L.; Mattioli, B.; Locovei, S.; Ishikawa, D.; Arseneau, K.O.; Chieppa, M.; Cominelli, F.; Pizarro, T.T. Probiotic Bacteria Regulate Intestinal Epithelial Permeability in Experimental Ileitis by a TNF-Dependent Mechanism. PLoS ONE 2012, 7, e42067. [Google Scholar] [CrossRef]
- Liu, Y.; Tran, D.Q.; Rhoads, J.M. Probiotics in Disease Prevention and Treatment. J. Clin. Pharmacol. 2018, 58, S164–S179. [Google Scholar] [CrossRef]
- Rousseaux, C.; Thuru, X.; Gelot, A.; Barnich, N.; Neut, C.; Dubuquoy, L.; Dubuquoy, C.; Merour, E.; Geboes, K.; Chamaillard, M.; et al. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat. Med. 2007, 13, 35–37. [Google Scholar] [CrossRef]
- Umbrello, G.; Esposito, S. Microbiota and neurologic diseases: Potential effects of probiotics. J. Transl. Med. 2016, 14, 298. [Google Scholar] [CrossRef] [PubMed]
- Ng, Q.X.; Peters, C.; Ho, C.Y.X.; Lim, D.Y.; Yeo, W.S. A meta-analysis of the use of probiotics to alleviate depressive symptoms. J. Affect. Disord. 2018, 228, 13–19. [Google Scholar] [CrossRef]
- Wasilewski, A.; Zielińska, M.; Storr, M.; Fichna, J. Beneficial Effects of Probiotics, Prebiotics, Synbiotics, and Psychobiotics in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Major, G.; Pritchard, S.; Murray, K.; Alappadan, J.P.; Hoad, C.L.; Marciani, L.; Gowland, P.; Spiller, R. Colon Hypersensitivity to Distension, Rather Than Excessive Gas Production, Produces Carbohydrate-Related Symptoms in Individuals With Irritable Bowel Syndrome. Gastroenterology 2017, 152, 124–133.e2. [Google Scholar] [CrossRef]
- Cox, S.R.; Prince, A.C.; Myers, C.E.; Irving, P.M.; Lindsay, J.O.; Lomer, M.C.; Whelan, K. Fermentable carbohydrates [FODMAPs] exacerbate functional gastrointestinal symptoms in patients with inflammatory bowel disease: A randomised, double-blind, placebo-controlled, cross-over, re-challenge trial. J. Crohn’s Colitis 2017, 11, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Prince, A.C.; Myers, C.E.; Joyce, T.; Irving, P.; Lomer, M.; Whelan, K. Fermentable carbohydrate restriction (Low FODMAP Diet) in clinical practice improves functional gastrointestinal symptoms in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2016, 22, 1129–1136. [Google Scholar] [CrossRef]
- Pedersen, N.; Ankersen, D.V.; Felding, M.; Wachmann, H.; Végh, Z.; Molzen, L.; Burisch, J.; Andersen, J.R.; Munkholm, P. Low-FODMAP diet reduces irritable bowel symptoms in patients with inflammatory bowel disease. World J. Gastroenterol. 2017, 23, 3356. [Google Scholar] [CrossRef]
- Cox, S.R.; Lindsay, J.O.; Fromentin, S.; Stagg, A.J.; McCarthy, N.E.; Galleron, N.; Ibraim, S.B.; Roume, H.; Levenez, F.; Pons, N.; et al. Effects of Low FODMAP Diet on Symptoms, Fecal Microbiome, and Markers of Inflammation in Patients With Quiescent Inflammatory Bowel Disease in a Randomized Trial. Gastroenterology 2020, 158, 176–188.e7. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.A.; Goertz, J.E.; Ren, T.; Rich, S.S.; Onengut-Gumuscu, S.; Farber, E.; Wu, M.; Overall, C.C.; Kipnis, J.; Gaultier, A. Microbiota alteration is associated with the development of stress-induced despair behavior. Sci. Rep. 2017, 7, 43859. [Google Scholar] [CrossRef]
- Martín, R.; Miquel, S.; Benevides, L.; Bridonneau, C.; Robert, V.; Hudault, S.; Chain, F.; Berteau, O.; Azevedo, V.; Chatel, J.M.; et al. Functional characterization of novel Faecalibacterium prausnitzii strains isolated from healthy volunteers: A step forward in the use of F. prausnitzii as a next-generation probiotic. Front. Microbiol. 2017, 8, 1226. [Google Scholar]
- Wong, A.C.; Levy, M. New Approaches to Microbiome-Based Therapies. mSystems 2019, 4, e00122-19. [Google Scholar] [CrossRef]
- Gagliardi, A.; Totino, V.; Cacciotti, F.; Iebba, V.; Neroni, B.; Bonfiglio, G.; Trancassini, M.; Passariello, C.; Pantanella, F.; Schippa, S. Rebuilding the gut microbiota ecosystem. Int. J. Environ. Res. Public Health 2018, 15, 1679. [Google Scholar] [CrossRef]
- Colman, R.J.; Rubin, D.T. Fecal microbiota transplantation as therapy for inflammatory bowel disease: A systematic review and meta-analysis. J. Crohn’s Colitis 2014, 8, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Costello, S.P.; Hughes, P.A.; Waters, O.; Bryant, R.V.; Vincent, A.D.; Blatchford, P.; Katsikeros, R.; Makanyanga, J.; Campaniello, M.A.; Mavrangelos, C.; et al. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients with Ulcerative Colitis: A Randomized Clinical Trial. JAMA 2019, 321, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Nusbaum, D.J.; Sun, F.; Ren, J.; Zhu, Z.; Ramsy, N.; Pervolarakis, N.; Kunde, S.; England, W.; Gao, B.; Fiehn, O.; et al. Gut microbial and metabolomic profiles after fecal microbiota transplantation in pediatric ulcerative colitis patients. Fems Microbiol. Ecol. 2018, 94, 133. [Google Scholar] [CrossRef] [PubMed]
- Angelberger, S.; Reinisch, W.; Makristathis, A.; Lichtenberger, C.; Dejaco, C.; Papay, P.; Novacek, G.; Trauner, M.; Loy, A.; Berry, D. Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am. J. Gastroenterol. 2013, 108, 1620–1630. [Google Scholar] [CrossRef]
- De Leon, L.M.; Watson, J.B.; Kelly, C.R. Transient flare of ulcerative colitis after fecal microbiota transplantation for recurrent clostridium difficile infection. Clin. Gastroenterol. Hepatol. 2013, 11, 1036–1038. [Google Scholar] [CrossRef]
- Kelly, C.R.; Kahn, S.; Kashyap, P.; Laine, L.; Rubin, D.; Atreja, A.; Moore, T.; Wu, G. AGA SECTION Update on Fecal Microbiota Transplantation 2015: Indications, Methodologies, Mechanisms, and Outlook. Gastroenterology 2015, 149, 223–237. [Google Scholar] [CrossRef]
- Ramai, D.; Zakhia, K.; Ofosu, A.; Ofori, E.; Reddy, M. Fecal microbiota transplantation: Donor relation, fresh or frozen, delivery methods, cost-effectiveness. Ann. Gastroenterol. 2019, 32, 30–38. [Google Scholar] [CrossRef] [PubMed]



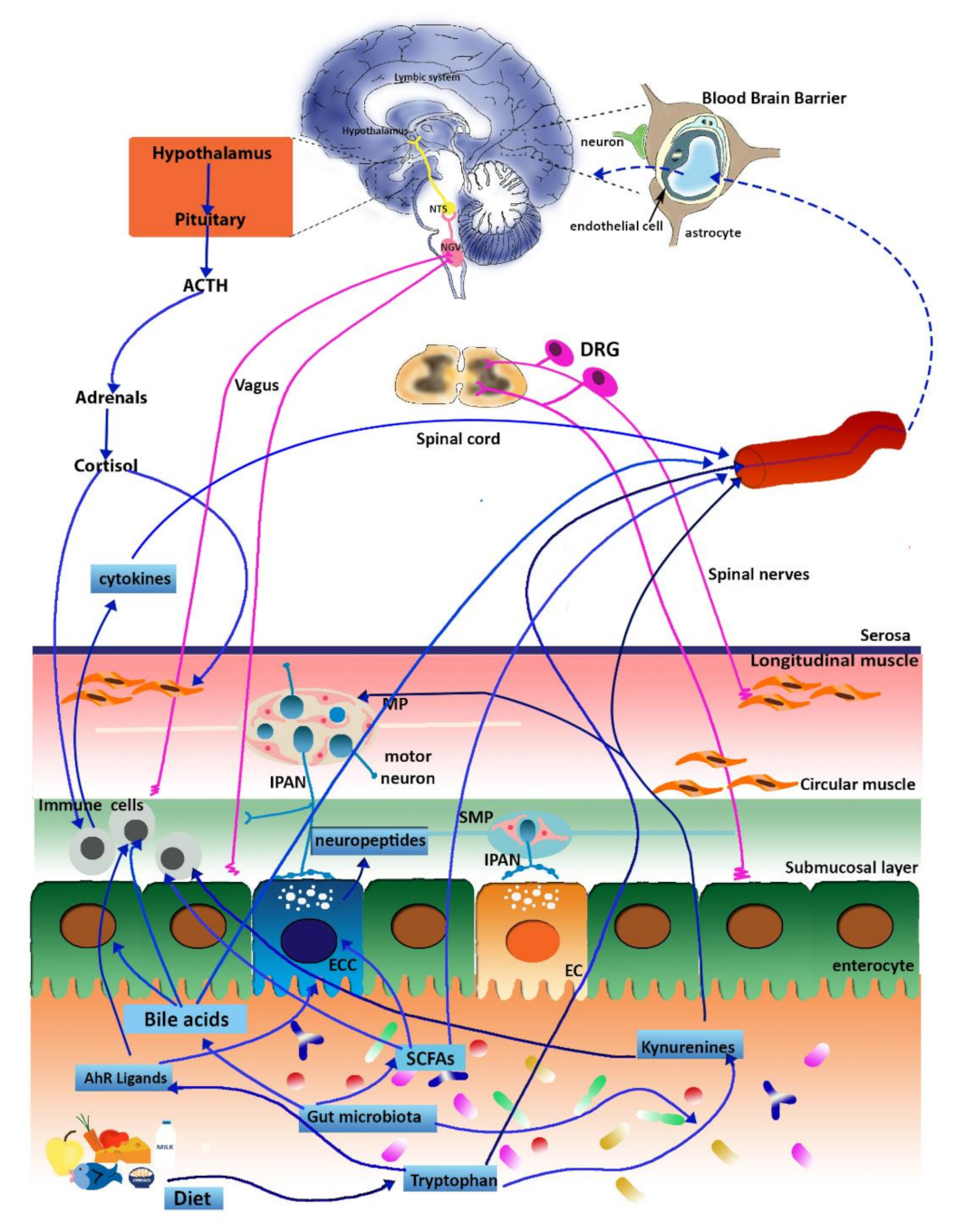{kind=link}
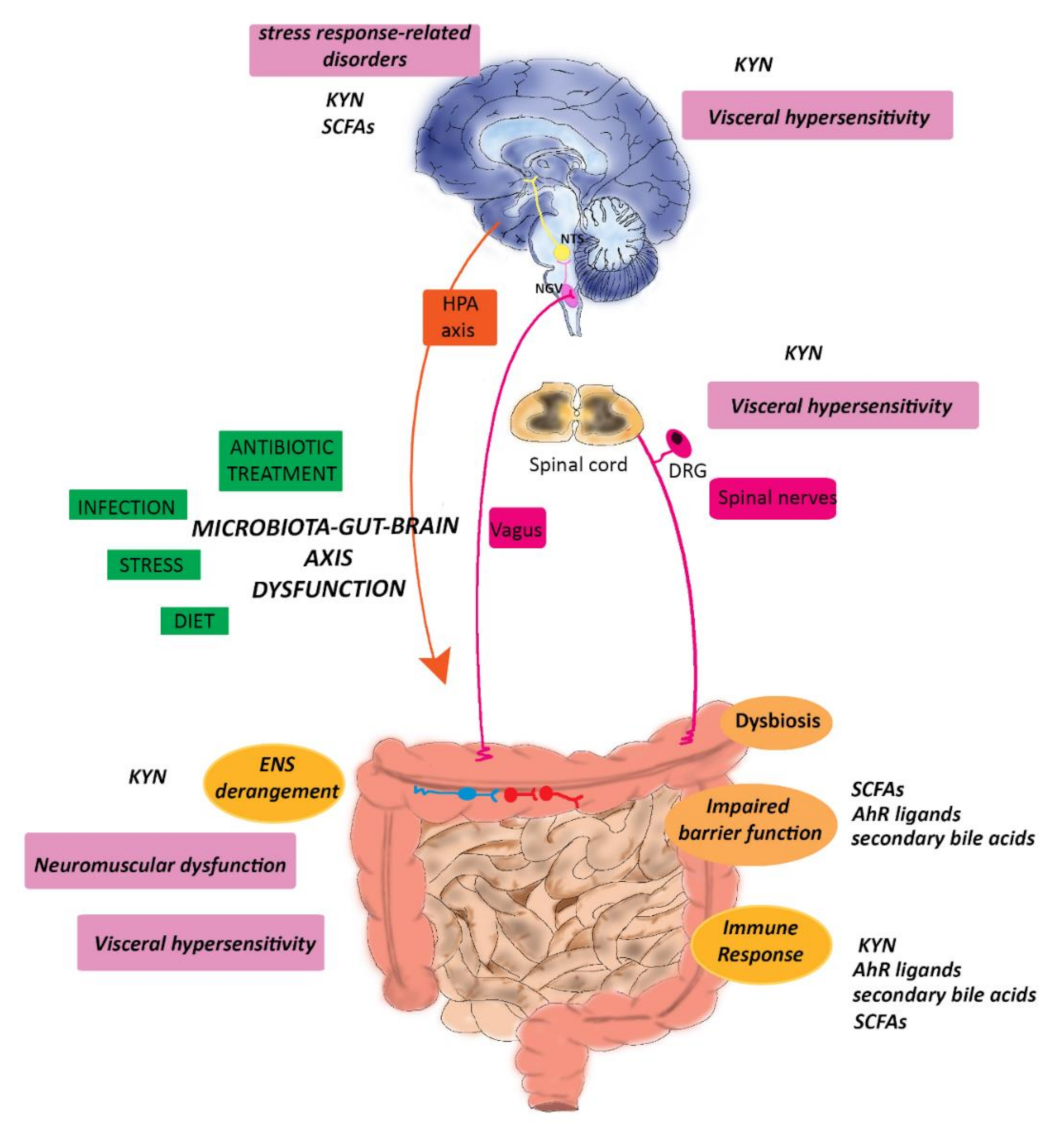{kind=link}
| Level | Bacteria | Variation | References |
|---|---|---|---|
| Phylum | Firmicutes | decrease | [41,65,66,67,68,69,70,71] |
| Bacteroidetes | decrease | [41,65,66,67,68,69,70,71] | |
| Proteobacteria(Enterobacteriaceae) | increase | [41,65,66,67,68,69,70,71,72] | |
| Actinobacteria | increase | [41,65,68] | |
| Genus | Bacteroides | decrease | [73,74,75,76] |
| Lactobacillus | decrease | [73,74,75,76,77] | |
| Eubacterium | decrease | [73,74] | |
| Clostridium spp. | decrease | [55,56,78,79] | |
| Fusobacterium spp. | increase | [80,81,82] | |
| Species | Faecalibacterium prausnitzii | decrease | [51,52,54,79,83] |
| Roseburia hominis | decrease | [83,84] | |
| Bifidobacterium adolescentis | decrease | [79] | |
| Dialister invisus | decrease | [79] | |
| Escherichia coli AIEC | increase | [48,51,85,86] | |
| Ruminococcus gnavus | increase | [58,79] | |
| Mycobacterium avium subsp. Para-tuberculosis | increase | [87,88] |
| Metabolite/ Teatment | Cell Type/Tissue | Findings | Reference |
|---|---|---|---|
| SCFAs | |||
| Administration of a SCFA smixture (os) | Mouse colon mucosal Tregs | Regulation of cell function and size with protective effect against colitis via FFAR-2 | [227] |
| Administration of n-butyrate (os) | Mouse colon mucosal macrophages | Downregulation of LPS-induced proinflammatory mediators, including nitric oxide, IL-6, and IL-12, via HDAC inhibition | [228] |
| High-fiber feeding | Mouse colon epithelial cells | Promotion of NLRP3 inflammasome-mediated IL-18 release contributing to gut homeostasis and protection from colitis via FFAR-2 and GPR109A | [230] |
| Administration of n-butyrate (ip) | Mouse hippocampus, prefrontal cortex | Anti-depressant effect associated with short-term HDAC inhibition and increased BDNF levels (only in prefrontal cortex) | [238] |
| Endogenous SCFA levels | Mouse colon mucosa, stools | Reduction of SCFAs and Lactobacillus spp fecal content, increase in GPR109A colonic expression after stressor exposure during infection with Citrobacter Rodentium underlying gut inflammation | [241] |
| Administration of a SCFAs mixture (os) | Mouse hypothalamus, hippocampus, colon mucosa | -Decreased stress-induced CRF expression in the hypothalamus, and CRF-receptor 1 in the hippocampus -Amelioration of psychosocial stress-induced responses -Anti-depressant and anxiolytic effects -Rescue of stress-induced enhancement of colonic mucosa permeability | [242] |
| Bile acids | |||
| Administration of the FXR agonist, INT-747, (i.p.) | Mouse colon mucosal macrophages | -Reduced activation and release of IL-1β, IL-2, IL-6, IFN-γ, and TNF-α -Amelioration of TNBS- and DSS-colitis | [253] |
| -Administration of INT-747 (os, in mice) -INT effect in vitro on LMPCs cultures | -Mouse colon epithelial cells - IBD patient’s LMPCs | -Amelioration of TNBS- and DSS-colitis, rescue of the mouse epithelial barrier function and reduced proinflammatory cytokine release in the mouse colon -Decrease TNF-α secretion in LMPCs from patients with IBD, | [254] |
| Administration of ciprofloxacin (i.p.) or oleanolic acid (os) to activate TGR5 | Mouse and human colon mucosa | -Increased levels of TGR5 in the mouse colon after TNBS- and DSS-colitis, and in the colon of IBD patients -reduced colonic TLR4-mediated TNF-α secretion in TNBS-treated mice | [258] |
| -Administration of TGR5 agonists -BAR501, (os) (ref 352) -BTA (os) (ref 253) -Several TGR5 agonist in vitro (ref 254) | -Mouse colon mucosa macrophages -LMPCs from IBD patient’s intestinal mucosa | -Amelioration of the severity of colitis in TNBS, DSS and oxazolone-treated mice -Shift of intestinal macrophages from a classically activated proinflammatory M1 to an anti-inflammatory M2 phenotype in mice -reduced colonic expression of pro-inflammatory genes (TNF-α, IFN-γ, IL-1β, IL-6, and CCL2 mRNAs) and increased expression of IL-10 and TGF-β mRNAs in mice -reduced TNF-α release from proinflammatory macrophages isolated from IBD patient’s LMPCs | [260,261,262] |
| Administration of UDCA, TUDCA, GUDCA (os) | Mouse colon | -Amelioration of the severity of colitis in DSS-treated mice -Normalization of Firmicutes to Bacteroidetes ratio after colitis-induced dysbiosis -Prevention of Clostridium cluster XIVa loss and increased abundance of the protective species, Akkermansia muciniphila | [263] |
| Tryptophan metabolites | |||
| Kynurenines | |||
| IDO1 | -Mouse colon crypt epithelial cells -Mouse colon lamina propria infiltrating APCs | - IDO1 levels of expression are fundamental to reduce Treg immune suppression and the severity of the inflammatory injury after TNBS-, DSS- and T-cell transfer-colitis | reviewed in ref [291] |
| Endogenous IDO1 and KynA | Epithelial cells and LMPCs from IBD patients’ biopsies | Increased IDO1 expression in epithelial and mononuclear cells positively related to the severity of inflamed region | [293] |
| UC patients’s serum and mucosa | Increased IDO mucosal expression and increased KynA serum levels, positively correlated with endoscopic inflammation | [294] | |
| Exposure in vitro to KynA and synthetic analogues | Human monocytic isolated cells | Reduced production of proinflammatory TNF-α induced by Staphylococcus aureus and Chlamydia pneumoniae, via TSG-6 expression | [297,298] |
| Endogenous IDO1, TRP, KynA | Mouse brain, spleen, Peyer’s patches | -Psychological stress-induced IDO1 activity, blocked by anti-TNFα and IFNγ treatment, and increased plasmatic KynA/TRP ratio | [299] |
| Administration of KynA (i.v.) | Dog colon ENS | -KynA antagonized hypermotility and inflammatory changes (increased XOR and MPO activity) associated with transitory colonic obstruction, via enteric NMDA receptors | [300] |
| Administration of KynA and its analogue, SRZ-72 (i.v.) | Rat colon ENS | -Reduced proinflammatory cytokine release, nitrite/nitrate and nitrotyrosine formation after TNBS-induced colitis -Normalization of microcirculation and of the rate of bowel movements after TNBS-induced colitis NMDA receptors located on enteric neurons | [302] |
| Endogenous Kyn/TRP | Rat intestine and CNS | FMT from depressed patients to bacterial microbiota-depleted animals induced: -Increased anxiety-like and anhedonic behavior -Increased Kyn/TRP plasma ratio -Increased bowel movements -Reduced microbiota richness | [303] |
| Kyn/TRP and Kyn | Mouse colon and hippocampus | Kyn and Kyn/TRP and cytokine plasma levels increased after Trichuris muris-induced mild to moderate colonic inflammation associated with -anxiety-like behaviors and reduced hippocampal BDNF mRNA levels | [210] |
| AhR ligands | |||
| -Endogenous AhR -administration of AhR agonist FICZ in vitro and ip | -UC and CD biopsies -Mouse colon | -Reduced AhR levels in DSS-treated mice colon and IBD biopsies -Reduced iIFNγ levels and IL-22 upregulation in patients’ LPMCs - Reduction of the severity of TNBS-, DSS and T-cell transfer-induced colitis in mice by Ficz -Downregulation of proinflammatory cytokines and upregulation of IL-22 in TNBS-treated mice which was reversed by FICZ antagonism -AhR activation by FICZ ameliorates DSS-induced colitis in mice via the MK2/p-MK2/TTP pathway | [329,336] |
| Administration of AhR agonist, ITE (ip) | Mouse colon | -Reduction of colitis severity in a humanized murine model of human CD4+ T cells transfer after TNBS administration -Increased CD39, Granzyme B, and IL-10-secreting human Treg cells isolated from colonic LPMCs. | [330] |
| Endogenous IAA | -Mouse colon and stools -IBD patients’ stools | -Susceptibility to colitis and altered microbiota in Card9 −/− mice associated with significant reduction of IAA fecal levels Reduced IAA and TRP levels and increased Kyn levels in fecal samples from IBD patients, particularly in those with CARD9 risk alleles associated with IBD | [331] |
| Endogenous and exogenous IPA | -IBD patient’s serum -Mouse colon and serum | -Reduced serum indole and IPA levels -Reduced serum indole and IPA levels after DSS colitis in mice -Improvement of DSS-colitis by exogenously administered IPA via epithelial IL-10 signaling | [332] |
| Endogenous IA | -Mouse colon goblet cells -Stools from CD and UC patients | -Administration of IA producing Peptostreptococcus russellii carrying the phenyllactate gene cluster (fldAIBC) to mice, reduced susceptibility to DSS, improved mucin producing goblet cell differentiation, promoted immune tolerance -reduced abundance of phenyllactate gene cluster in UC by metagenomic analysis | [335] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banfi, D.; Moro, E.; Bosi, A.; Bistoletti, M.; Cerantola, S.; Crema, F.; Maggi, F.; Giron, M.C.; Giaroni, C.; Baj, A. Impact of Microbial Metabolites on Microbiota–Gut–Brain Axis in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 1623. https://doi.org/10.3390/ijms22041623
Banfi D, Moro E, Bosi A, Bistoletti M, Cerantola S, Crema F, Maggi F, Giron MC, Giaroni C, Baj A. Impact of Microbial Metabolites on Microbiota–Gut–Brain Axis in Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2021; 22(4):1623. https://doi.org/10.3390/ijms22041623
Chicago/Turabian StyleBanfi, Davide, Elisabetta Moro, Annalisa Bosi, Michela Bistoletti, Silvia Cerantola, Francesca Crema, Fabrizio Maggi, Maria Cecilia Giron, Cristina Giaroni, and Andreina Baj. 2021. "Impact of Microbial Metabolites on Microbiota–Gut–Brain Axis in Inflammatory Bowel Disease" International Journal of Molecular Sciences 22, no. 4: 1623. https://doi.org/10.3390/ijms22041623
APA StyleBanfi, D., Moro, E., Bosi, A., Bistoletti, M., Cerantola, S., Crema, F., Maggi, F., Giron, M. C., Giaroni, C., & Baj, A. (2021). Impact of Microbial Metabolites on Microbiota–Gut–Brain Axis in Inflammatory Bowel Disease. International Journal of Molecular Sciences, 22(4), 1623. https://doi.org/10.3390/ijms22041623