
Perineuronal Nets in the Prefrontal Cortex of a Schizophrenia Mouse Model: Assessment of Neuroanatomical, Electrophysiological, and Behavioral Contributions

 and
and 
Abstract
1. Introduction
2. Results
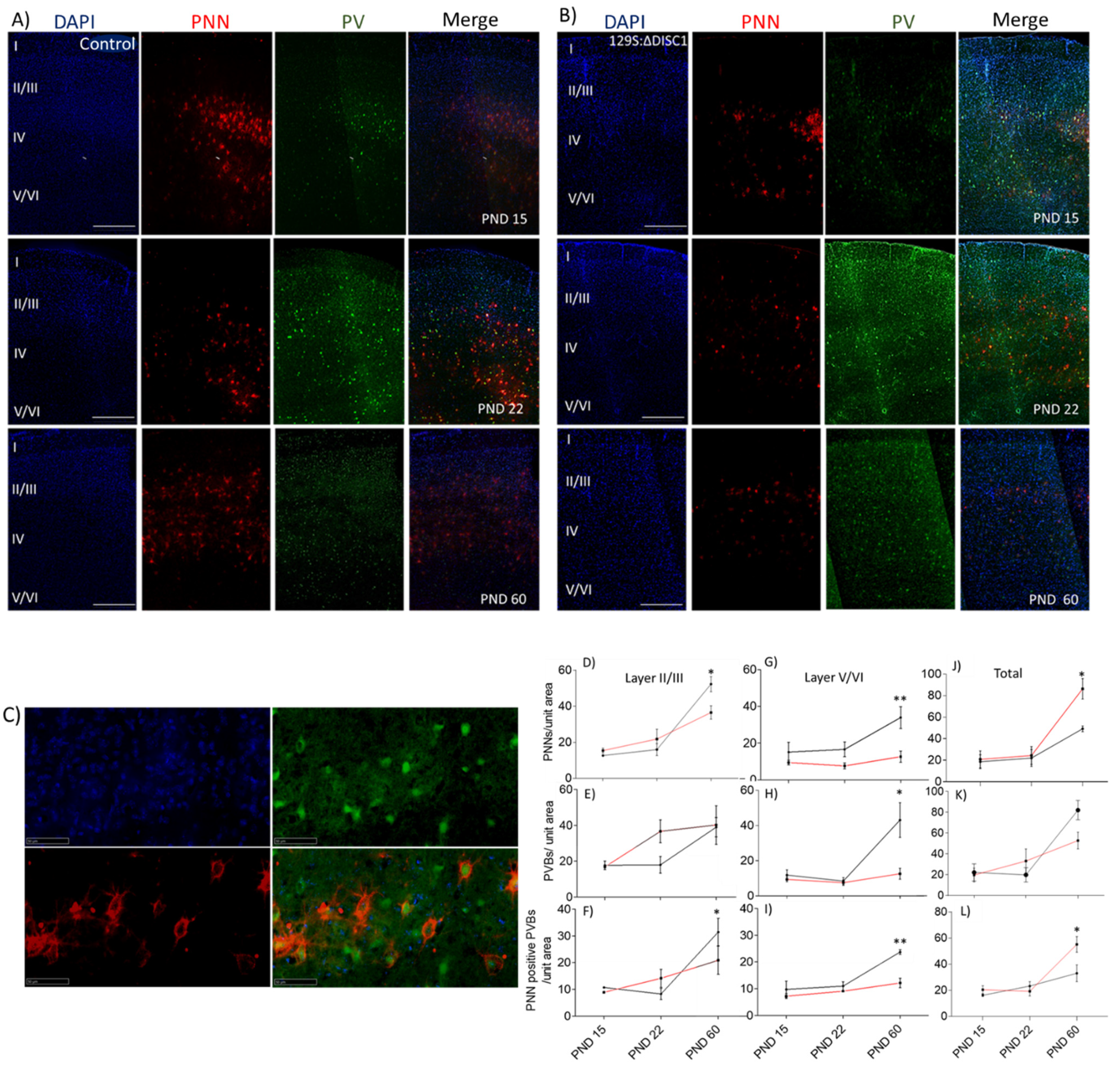
2.1. Developmental Expression of PNNs and PVs in the PFC
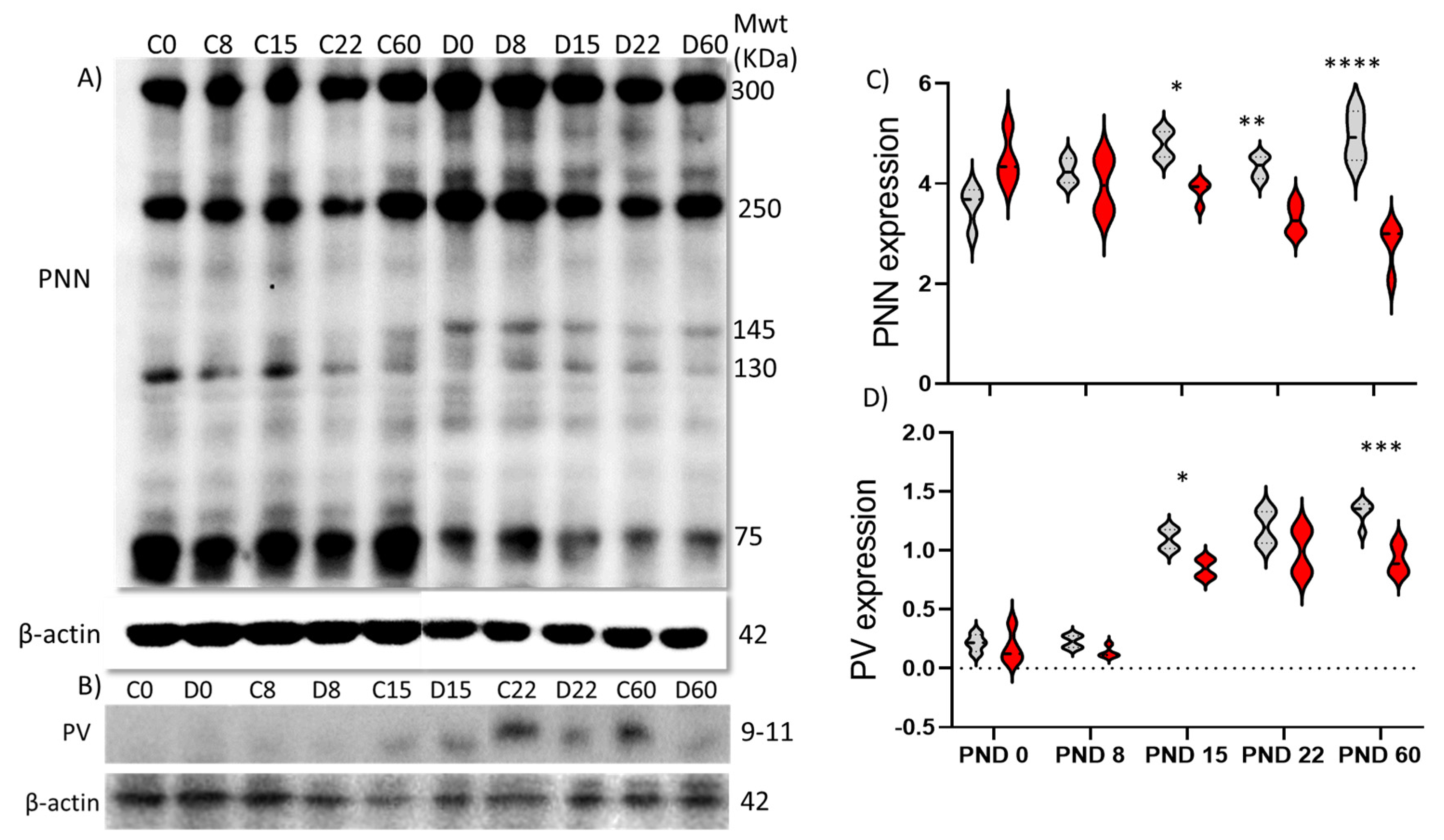
2.2. Protein Expression
2.3. Chondroitinase Injections in PFC
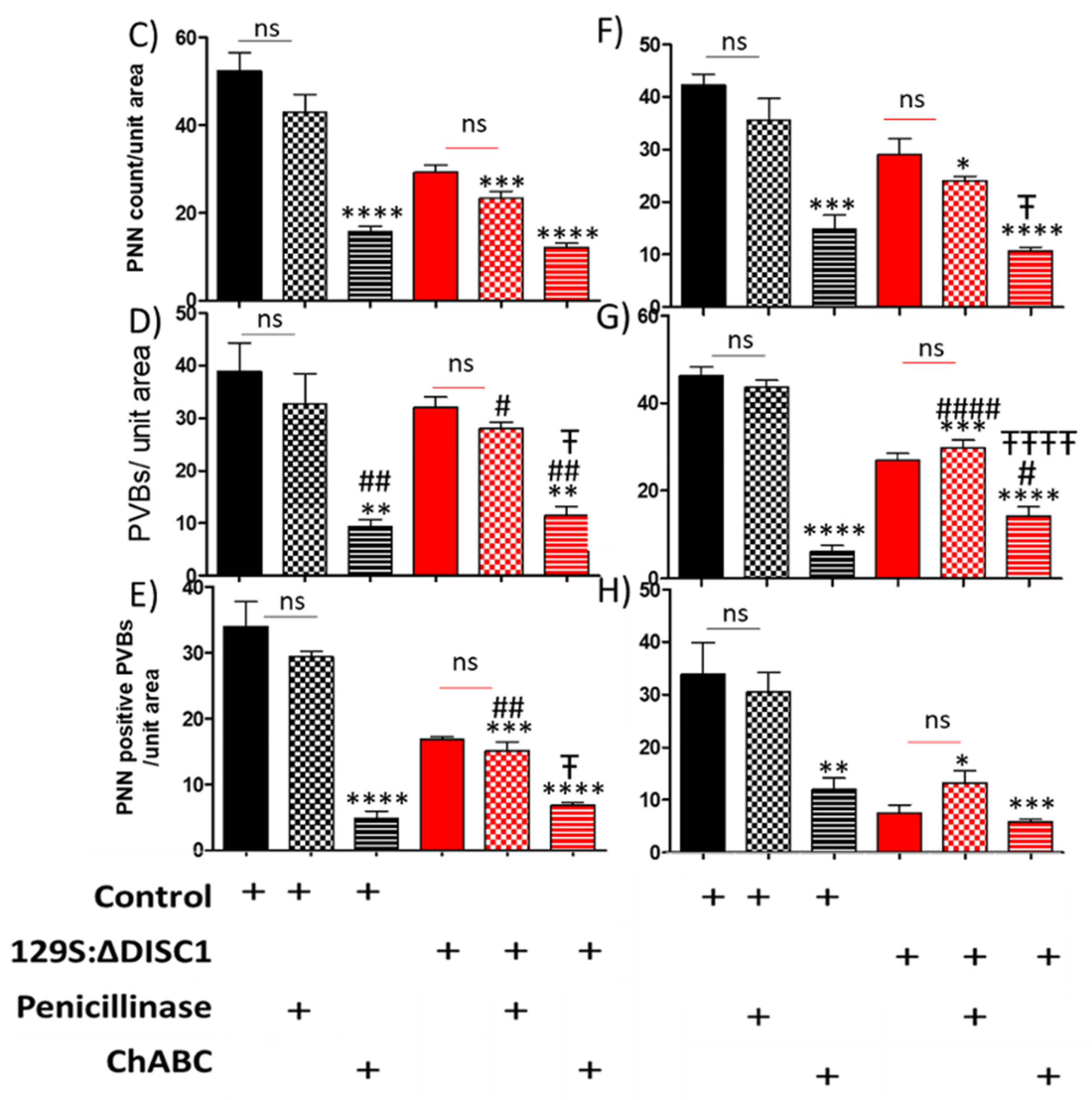
2.3.1. Chondroitinase Injections Reduce the Number of Well-Formed PNNs
2.3.2. Chondroitinase Treated Animals Show Aberrant PFC Associated Behaviors
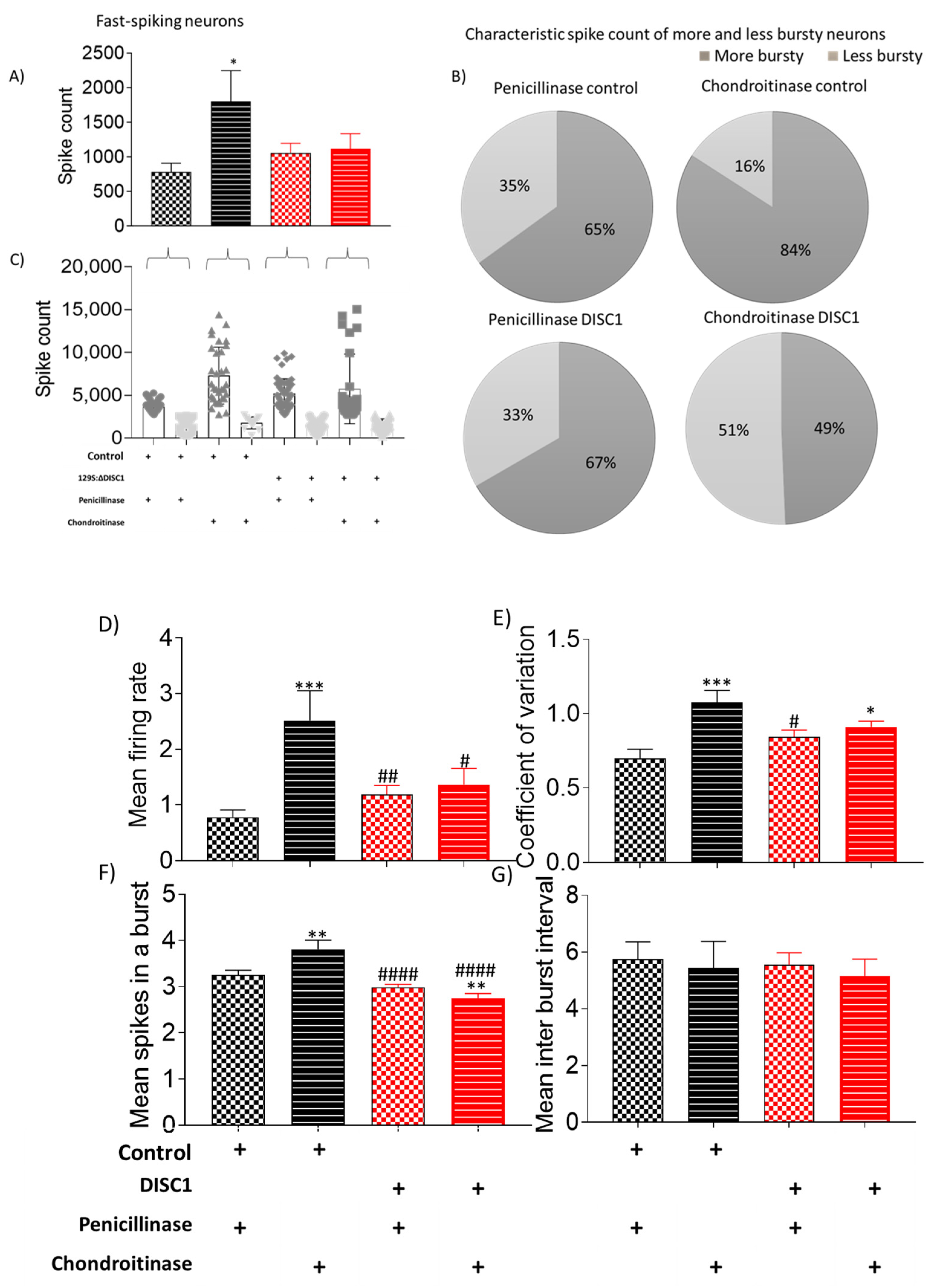
2.3.3. Chondroitinase Treated Animals Exhibit Electrophysiological Changes in PFC
3. Discussion
3.1. Development of PNNs and PVs
3.2. Chondroitinase Treatment of PNNs in PFC
4. Materials and Methods
4.1. Histological Staining and Imaging
4.2. Western Blotting for Protein Expression
4.3. Chondroitinase ABC (chABC) Treatment of PFC (Adult Animals)
4.4. Behavioral Testing
4.5. In Vivo Electrophysiology in PFC of Chondroitinase (ChABC) and Penicillinase (p) Treated Animals
4.6. Neural Spike Processing and Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Immonen, J.; Jääskeläinen, E.; Korpela, H.; Miettunen, J. Age at onset and the outcomes of schizophrenia: A systematic review and meta-analysis. Early Interv. Psychiatry 2017, 11, 453–460. [Google Scholar] [CrossRef]
- McGorry, P.D.; McFarlane, C.; Patton, G.; Bell, R.; Hibbert, M.E.; Jackson, H.; Bowes, G. The prevalence of prodromal features of schizophrenia in adolescence: A preliminary survey. Acta Psychiatr. Scand. 1995, 92, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Gejman, P.V.; Sanders, A.R.; Duan, J. The Role of Genetics in the Etiology of Schizophrenia. Psychiatr. Clin. N. Am. 2010, 33, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, O.; Niwa, S.; Hiramatsu, K.; Kameyama, T.; Rymar, K.; Itoh, K. Abnormalities in late positive components of event-related potentials may reflect a genetic predisposition to schizophrenia. Biol. Psychiatry 1984, 19, 293–303. [Google Scholar] [PubMed]
- Sobell, J.L.; Heston, L.L.; Sommer, S.S. Novel association approach for determining the genetic predisposition to schizophrenia: Case-control resource and testing of a candidate gene. Am. J. Med. Genet. 1993, 48, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.; Lawrie, S.M.; Hodges, A.; Clafferty, R.; Cosway, R.; Johnstone, E.C. Genetic liability, illicit drug use, life stress and psychotic symptoms: Preliminary findings from the Edinburgh study of people at high risk for schizophrenia. Soc. Psychiatry Psychiatr. Epidemiol. 2001, 36, 338–342. [Google Scholar] [CrossRef]
- Tsuang, M. Schizophrenia: Genes and environment. Biol. Psychiatry 2000, 47, 210–220. [Google Scholar] [CrossRef]
- Whalley, H.C.; Simonotto, E.; Marshall, I.; Owens, D.G.C.; Goddard, N.H.; Johnstone, E.C.; Lawrie, S.M. Functional disconnectivity in subjects at high genetic risk of schizophrenia. Brain 2005, 128, 2097–2108. [Google Scholar] [CrossRef]
- Van Os, J.; Kenis, G.; Rutten, B.P.F. The environment and schizophrenia. Nature 2010, 468, 203–212. [Google Scholar] [CrossRef]
- van Os, J.; Rutten, B.P.; Poulton, R. Gene-Environment Interactions in Schizophrenia: Review of Epidemiological Findings and Future Directions. Schizophr. Bull. 2008, 34, 1066–1082. [Google Scholar] [CrossRef]
- Morgan, C.; Fisher, H. Environment and Schizophrenia: Environmental Factors in Schizophrenia: Childhood Trauma--A Critical Review. Schizophr. Bull. 2006, 33, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Pogue-Geile, M.F.; Zubin, J. Negative Symptomatology and Schizophrenia: A Conceptual and Empirical Review. Int. J. Ment. HealTH 1987, 16, 3–45. [Google Scholar] [CrossRef]
- Heinrichs, R.W.; Zakzanis, K.K. Neurocognitive deficit in schizophrenia: A quantitative review of the evidence. Neuropsychology 1998, 12, 426–445. [Google Scholar] [CrossRef] [PubMed]
- Brune, M. "Theory of Mind" in Schizophrenia: A Review of the Literature. Schizophr. Bull. 2005, 31, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Cunill, R.; Castells, X.; Simeon, D. Relationships Between Obsessive-Compulsive Symptomatology and Severity of Psychosis in Schizophrenia. J. Clin. Psychiatry 2008, 70, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Bellak, L. Schizophrenia: A Review of the Syndrome; Logos Press: New York, NY, USA, 1958. [Google Scholar]
- McClellan, J.; Werry, J. Practice Parameter for the Assessment and Treatment of Children and Adolescents with Schizophrenia. J. Am. Acad. Child. Adolesc. Psychiatry 2001, 40, 4S–23S. [Google Scholar] [CrossRef]
- Inoue, K.; Nakajima, T.; Kato, N. A Longitudinal Study of Schizophrenia in Adolescence: I. The One- to Three-Year Outcome. Psychiatry Clin. Neurosci. 1986, 40, 143–151. [Google Scholar] [CrossRef]
- Fleischhaker, C.; Schulz, E.; Tepper, K.; Martin, M.; Hennighausen, K.; Remschmidt, H. Long-Term Course of Adolescent Schizophrenia. Schizophr. Bull. 2005, 31, 769–780. [Google Scholar] [CrossRef]
- Maynard, T.; Sikich, L.; Lieberman, J.A.; LaMantia, A.-S. Neural Development, Cell-Cell Signaling, and the "Two-Hit" Hypothesis of Schizophrenia. Schizophr. Bull. 2001, 27, 457–476. [Google Scholar] [CrossRef]
- Weickert, C.S.; Weinberger, D.R. A candidate molecule approach to defining developmental pathology in schizophrenia. Schizophr. Bull. 1998, 24, 303–316. [Google Scholar] [CrossRef][Green Version]
- O’Donnell, P. Cortical disinhibition in the neonatal ventral hippocampal lesion model of schizophrenia: New vistas on possible therapeutic approaches. Pharmacol. Ther. 2012, 133, 19–25. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Cho, R.Y.; Lewis, D.A. Alterations in Cortical Network Oscillations and Parvalbumin Neurons in Schizophrenia. Biol. Psychiatry 2015, 77, 1031–1040. [Google Scholar] [CrossRef]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef]
- Jadi, M.P.; Behrens, M.M.; Sejnowski, T.J. Abnormal Gamma Oscillations in N-Methyl-D-Aspartate Receptor Hypofunction Models of Schizophrenia. Biol. Psychiatry 2016, 79, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Rogasch, N.C.; Daskalakis, Z.J.; Fitzgerald, P.B. Cortical Inhibition, Excitation, and Connectivity in Schizophrenia: A Review of Insights from Transcranial Magnetic Stimulation. Schizophr. Bull. 2014, 40, 685–696. [Google Scholar] [CrossRef]
- Vogels, T.P.; Abbott, L.F. Gating Deficits in Model Networks: A Path to Schizophrenia? Pharmacopsychiatry 2007, 40, S73–S77. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Singer, W. Oscillations and Neuronal Dynamics in Schizophrenia: The Search for Basic Symptoms and Translational Opportunities. Biol. Psychiatry 2015, 77, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Selten, M.; Van Bokhoven, H.; Kasri, N.N. Inhibitory control of the excitatory/inhibitory balance in psychiatric disorders. F1000Research 2018, 7, 23. [Google Scholar] [CrossRef]
- Jardri, R.; Hugdahl, K.; Hughes, M.; Brunelin, J.; Waters, F.; Alderson-Day, B.; Smailes, D.; Sterzer, P.; Corlett, P.R.; Leptourgos, P.; et al. Are Hallucinations Due to an Imbalance Between Excitatory and Inhibitory Influences on the Brain? Schizophr. Bull. 2016, 42, 1124–1134. [Google Scholar] [CrossRef]
- Adhikari, A.; Topiwala, M.A.; Gordon, J.A. Synchronized Activity between the Ventral Hippocampus and the Medial Prefrontal Cortex during Anxiety. Neuron 2010, 65, 257–269. [Google Scholar] [CrossRef]
- Amilhon, B.; Huh, C.Y.L.; Manseau, F.; Ducharme, G.; Nichol, H.; Adamantidis, A.; Williams, S. Parvalbumin Interneurons of Hippocampus Tune Population Activity at Theta Frequency. Neuron 2015, 86, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Baeg, E.; Kim, Y.; Huh, K.; Mook-Jung, I.; Kim, H.; Jung, M. Dynamics of Population Code for Working Memory in the Prefrontal Cortex. Neuron 2003, 40, 177–188. [Google Scholar] [CrossRef]
- Ueno, H.; Suemitsu, S.; Okamoto, M.; Matsumoto, Y.; Ishihara, T. Parvalbumin neurons and perineuronal nets in the mouse prefrontal cortex. Neurosci. 2017, 343, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Miyamae, T.; Chen, K.; Lewis, D.A.; Gonzalez-Burgos, G. Distinct Physiological Maturation of Parvalbumin-Positive Neuron Subtypes in Mouse Prefrontal Cortex. J. Neurosci. 2017, 37, 4883–4902. [Google Scholar] [CrossRef]
- del Rio, J.; de Lecea, L.; Ferrer, I.; Soriano, E. The development of parvalbumin-immunoreactivity in the neocortex of the mouse. Dev. Brain Res. 1994, 81, 247–259. [Google Scholar] [CrossRef]
- Fader, S.M.; Imaizumi, K.; Yanagawa, Y.; Lee, C.C. Wisteria Floribunda Agglutinin-Labeled Perineuronal Nets in the Mouse Inferior Colliculus, Thalamic Reticular Nucleus and Auditory Cortex. Brain Sci. 2016, 6, 13. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.-H.; Bukhari, S.A.; Ardelt, M.I.; Pantazopoulos, H.; Hamati, F.; Salt, T.E.; Cuenod, M.; Do, K.Q.; Berretta, S. The thalamic reticular nucleus in schizophrenia and bipolar disorder: Role of parvalbumin-expressing neuron networks and oxidative stress. Mol. Psychiatry 2018, 23, 2057–2065. [Google Scholar] [CrossRef]
- Barros, C.S.; Franco, S.J.; Müller, U. Extracellular Matrix: Functions in the Nervous System. Cold Spring Harb. Perspect. Biol. 2011, 3, a005108. [Google Scholar] [CrossRef] [PubMed]
- Brückner, G.; Brauer, K.; Härtig, W.; Wolff, J.R.; Rickmann, M.J.; Derouiche, A.; Delpech, B.; Girard, N.; Oertel, W.H.; Reichenbach, A. Perineuronal nets provide a polyanionic, glia-associated form of microenvironment around certain neurons in many parts of the rat brain. Glia 1993, 8, 183–200. [Google Scholar] [CrossRef]
- Balmer, T.S.; Carels, V.; Frisch, J.L.; Nick, T.A. Modulation of Perineuronal Nets and Parvalbumin with Developmental Song Learning. J. Neurosci. 2009, 29, 12878–12885. [Google Scholar] [CrossRef]
- Hensch, T.K. Critical Period Mechanisms in Developing Visual Cortex. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 69, pp. 215–237. [Google Scholar]
- Hensch, T.K.; Bilimoria, P.M. Re-opening Windows: Manipulating Critical Periods for Brain Development. Cerebrum Dana Forum Brain Sci. 2012, 2012, 11. [Google Scholar]
- Testa, D.; Prochiantz, A.; Di Nardo, A.A. Perineuronal nets in brain physiology and disease. Semin. Cell Dev. Biol. 2019, 89, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Carulli, D.; de Winter, F.; Verhaagen, J. Semaphorins in Adult Nervous System Plasticity and Disease. Front. Synaptic Neurosci. 2021, 13, 672891. [Google Scholar] [CrossRef] [PubMed]
- Bernard, C.; Prochiantz, A. Otx2-PNN Interaction to Regulate Cortical Plasticity. Neural Plast. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Berretta, S.; Pantazopoulos, H.; Chelini, G. The Tetrapartite Synapse in Schizophrenia: Role of the Extracellular Matrix and Glial Cell in PSD Pathology. Biol. Psychiatry 2018, 83, S6. [Google Scholar] [CrossRef]
- Sorg, B.A.; Berretta, S.; Blacktop, J.M.; Fawcett, J.W.; Kitagawa, H.; Kwok, J.C.; Miquel, M. Casting a Wide Net: Role of Perineuronal Nets in Neural Plasticity. J. Neurosci. 2016, 36, 11459–11468. [Google Scholar] [CrossRef]
- Pantazopoulos, H.; Berretta, S. In Sickness and in Health: Perineuronal Nets and Synaptic Plasticity in Psychiatric Disorders. Neural Plast. 2016, 2016, 1–23. [Google Scholar] [CrossRef]
- Reichelt, A.C.; Hare, D.; Bussey, T.J.; Saksida, L.M. Perineuronal Nets: Plasticity, Protection, and Therapeutic Potential. Trends Neurosci. 2019, 42, 458–470. [Google Scholar] [CrossRef]
- Ingalhalikar, M.; Parker, D.; Ghanbari, Y.; Smith, A.; Hua, K.; Mori, S.; Abel, T.; Davatzikos, C.; Verma, R. Connectome and Maturation Profiles of the Developing Mouse Brain Using Diffusion Tensor Imaging. Cereb. Cortex 2015, 25, 2696–2706. [Google Scholar] [CrossRef]
- de Lecea, L.; Soriano, E. Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Mol. Brain Res. 1995, 32, 1–13. [Google Scholar] [CrossRef]
- Frysztak, R.J.; Neafsey, E.J. The Effect of Medial Frontal Cortex Lesions on Respiration, “Freezing,” and Ultrasonic Vocalizations during Conditioned Emotional Responses in Rats. Cereb. Cortex 1991, 1, 418–425. [Google Scholar] [CrossRef]
- Sultana, R.; Ogundele, O.M.; Lee, C. Contrasting characteristic behaviours among common laboratory mouse strains. R. Soc. Open Sci. 2019, 6, 190574. [Google Scholar] [CrossRef]
- Grosche, J.; Schmidt, S.; Margolis, R.U.; Delpech, B.; Seidenbecher, C.I.; Czaniera, R.; Schachner, M. Postnatal development of perineuronal nets in wild-type mice and in a mutant deficient in tenascin-R. J. Comp. Neurol. 2000, 428, 616–629. [Google Scholar] [CrossRef]
- Ueno, H.; Fujii, K.; Suemitsu, S.; Murakami, S.; Kitamura, N.; Wani, K.; Aoki, S.; Okamoto, M.; Ishihara, T.; Takao, K. Expression of aggrecan components in perineuronal nets in the mouse cerebral cortex. IBRO Rep. 2018, 4, 22–37. [Google Scholar] [CrossRef]
- Ueno, H.; Fujii, K.; Takao, K.; Suemitsu, S.; Murakami, S.; Kitamura, N.; Wani, K.; Matsumoto, Y.; Okamoto, M.; Ishihara, T. Alteration of parvalbumin expression and perineuronal nets formation in the cerebral cortex of aged mice. Mol. Cell. Neurosci. 2019, 95, 31–42. [Google Scholar] [CrossRef]
- Gottschling, C.; Wegrzyn, D.; Denecke, B.; Faissner, A. Elimination of the four extracellular matrix molecules tenascin-C, tenascin-R, brevican and neurocan alters the ratio of excitatory and inhibitory synapses. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]
- Carstens, K.E.; Phillips, M.; Pozzo-Miller, L.; Weinberg, R.; Dudek, S.M. Perineuronal Nets Suppress Plasticity of Excitatory Synapses on CA2 Pyramidal Neurons. J. Neurosci. 2016, 36, 6312–6320. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, K.P.; Velez, D.X.F.; Habeeb, M.; Gandhi, S.P. Precocious deposition of perineuronal nets on Parvalbumin inhibitory neurons transplanted into adult visual cortex. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Miyata, S.; Nadanaka, S.; Igarashi, M.; Kitagawa, H. Structural Variation of Chondroitin Sulfate Chains Contributes to the Molecular Heterogeneity of Perineuronal Nets. Front. Integr. Neurosci. 2018, 12, 3. [Google Scholar] [CrossRef]
- Sigal, Y.M.; Bae, H.; Bogart, L.J.; Hensch, T.K.; Zhuang, X. Structural maturation of cortical perineuronal nets and their perforating synapses revealed by superresolution imaging. Proc. Natl. Acad. Sci. USA 2019, 116, 7071–7076. [Google Scholar] [CrossRef]
- Atallah, B.V.; Bruns, W.; Carandini, M.; Scanziani, M. Parvalbumin-Expressing Interneurons Linearly Transform Cortical Responses to Visual Stimuli. Neuron 2012, 73, 159–170. [Google Scholar] [CrossRef]
- Cho, K.; Hoch, R.; Lee, A.T.; Patel, T.; Rubenstein, J.L.; Sohal, V.S. Gamma Rhythms Link Prefrontal Interneuron Dysfunction with Cognitive Inflexibility in Dlx5/6+/− Mice. Neuron 2015, 85, 1332–1343. [Google Scholar] [CrossRef]
- Kim, H.; Ährlund-Richter, S.; Wang, X.; Deisseroth, K.; Carlén, M. Prefrontal Parvalbumin Neurons in Control of Attention. Cell 2016, 164, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Lagler, M.; Ozdemir, A.T.; Lagoun, S.; Malagon-Vina, H.; Borhegyi, Z.; Hauer, R.; Jelem, A.; Klausberger, T. Divisions of Identified Parvalbumin-Expressing Basket Cells during Working Memory-Guided Decision Making. Neuron 2016, 91, 1390–1401. [Google Scholar] [CrossRef] [PubMed]
- Slaker, M.; Churchill, L.; Todd, R.P.; Blacktop, J.M.; Zuloaga, D.G.; Raber, J.; Darling, R.A.; Brown, T.E.; Sorg, B.A. Removal of Perineuronal Nets in the Medial Prefrontal Cortex Impairs the Acquisition and Reconsolidation of a Cocaine-Induced Conditioned Place Preference Memory. J. Neurosci. 2015, 35, 4190–4202. [Google Scholar] [CrossRef]
- Roach, B.; Ford, J.; Hoffman, R.; Mathalon, D. Converging evidence for gamma synchrony deficits in schizophrenia. Advances in Clinical Neurophysiology. In Proceedings of the 27th International Congress of Clinical Neurophysiology, AAEM 50th Anniversary and 57th Annual Meeting of the ACNS Joint Meeting, San Francisco, CA, USA, 15–20 September 2003; Volume 62, pp. 163–180. [Google Scholar] [CrossRef]
- Bygrave, A.M.; Masiulis, S.; Nicholson, E.; Berkemann, M.; Barkus, C.; Sprengel, R.; Harrison, P.J.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Knockout of NMDA-receptors from parvalbumin interneurons sensitizes to schizophrenia-related deficits induced by MK-801. Transl. Psychiatry 2016, 6, e778. [Google Scholar] [CrossRef]
- Sultana, R.; Shrestha, A.; Lee, C.; Ogundele, O.M. Disc1 Carrier Mice Exhibit Alterations in Neural pIGF-1Rβ and Related Kinase Expression. Front. Cell. Neurosci. 2020, 14, 14. [Google Scholar] [CrossRef]
- Yang, S.; Cacquevel, M.; Saksida, L.; Bussey, T.; Schneider, B.; Aebischer, P.; Melani, R.; Pizzorusso, T.; Fawcett, J.; Spillantini, M.G. Perineuronal net digestion with chondroitinase restores memory in mice with tau pathology. Exp. Neurol. 2015, 265, 48–58. [Google Scholar] [CrossRef]
- Bozzelli, P.L.; Alaiyed, S.; Kim, E.; Villapol, S.; Conant, K. Proteolytic Remodeling of Perineuronal Nets: Effects on Synaptic Plasticity and Neuronal Population Dynamics. Neural Plast. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Choleris, E. The effect of demonstrator age and number on duration of socially-induced food preferences in house mouse (Mus domesticus). Behav. Process. 1997, 41, 69–77. [Google Scholar] [CrossRef]
- Bicks, L.K.; Ekoike, H.; Eakbarian, S.; Emorishita, H. Prefrontal Cortex and Social Cognition in Mouse and Man. Front. Psychol. 2015, 6, 1805. [Google Scholar] [CrossRef] [PubMed]
- Dalley, J.W.; Cardinal, R.; Robbins, T. Prefrontal executive and cognitive functions in rodents: Neural and neurochemical substrates. Neurosci. Biobehav. Rev. 2004, 28, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.R.; Gao, W.-J. PV Interneurons: Critical Regulators of E/I Balance for Prefrontal Cortex-Dependent Behavior and Psychiatric Disorders. Front. Neural Circuits 2018, 12, 37. [Google Scholar] [CrossRef]
- Prabhakar, V.; Raman, R.; Capila, I.; Bosques, C.J.; Pojasek, K.; Sasisekharan, R. Biochemical characterization of the chondroitinase ABC I active site. Biochem. J. 2005, 390, 395–405. [Google Scholar] [CrossRef]
- Fawcett, J.W. The extracellular matrix in plasticity and regeneration after CNS injury and neurodegenerative disease. Prog. Brain Res. 2015, 218, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Lee, C. Expression of Behavioral Phenotypes in Genetic and Environmental Mouse Models of Schizophrenia. Front. Behav. Neurosci. 2020, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Franklin, K.B.J.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: New York, NY, USA, 2001. [Google Scholar]
- Funahashi, S. Working Memory in the Prefrontal Cortex. Brain Sci. 2017, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Maren, S. Prefrontal-Hippocampal Interactions in Memory and Emotion. Front. Syst. Neurosci. 2015, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; A Zilli, E.; Paley, A.M.; E Hasselmo, M. Working memory performance correlates with prefrontal-hippocampal theta interactions but not with prefrontal neuron firing rates. Front. Integr. Neurosci. 2010, 4, 2. [Google Scholar] [CrossRef]
- Newman, J.D. Neural circuits underlying crying and cry responding in mammals. Behav. Brain Res. 2007, 182, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Ghandi, T.; Davila, A.M.; Lee, C.C.; Ogundele, O.M. Upregulated SK2 Expression and Impaired CaMKII Phosphorylation Are Shared Synaptic Defects Between 16p11.2del and 129S:Δdisc1 Mutant Mice. ASN Neuro 2018, 10, 1759091418817641. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Sultana, R.; Lee, C.; Ogundele, O.M. SK Channel Modulates Synaptic Plasticity by Tuning CaMKIIα/β Dynamics. Front. Synaptic Neurosci. 2019, 11, 18. [Google Scholar] [CrossRef]
- Madisen, L.; Mao, T.; Koch, H.; Zhuo, J.-M.; Berenyi, A.; Fujisawa, S.; Hsu, Y.-W.; Garcia, A.J.; Gu, X.; Zanella, S.; et al. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 2012, 15, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.G.; Boyden, E.S. Optogenetic tools for analyzing the neural circuits of behavior. Trends Cogn. Sci. 2011, 15, 592–600. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
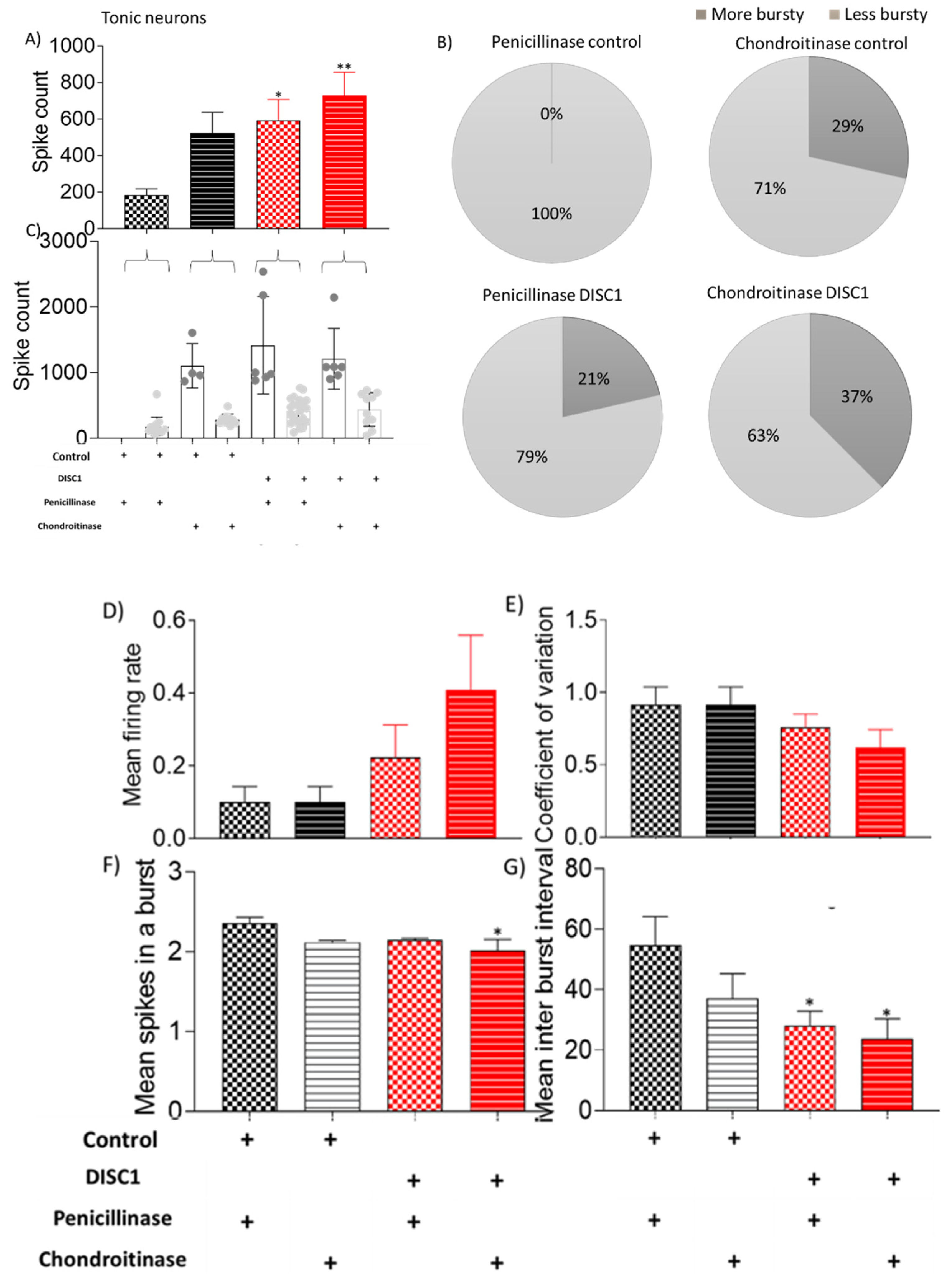{kind=link}
{kind=link}
| S. No. | Behavioral Test | Vs. ChABC Control | Vs. ChABC 129s:ΔDISC1 |
|---|---|---|---|
| 1 | Sociability | Yes (p = 0.0356) | Yes (p = 0.0001) |
| 2 | Novelty | Yes (p = 0.0106) | Yes (p = 0.0001) |
| 3 | Y maze | No | Yes (p = 0.05) |
| 4 | Tail Suspension test | No | Yes (p = 0.03) |
| 5 | Stress calls | Yes (p = 0.0002) | Yes (p = 0.0001) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultana, R.; Brooks, C.B.; Shrestha, A.; Ogundele, O.M.; Lee, C.C. Perineuronal Nets in the Prefrontal Cortex of a Schizophrenia Mouse Model: Assessment of Neuroanatomical, Electrophysiological, and Behavioral Contributions. Int. J. Mol. Sci. 2021, 22, 11140. https://doi.org/10.3390/ijms222011140
Sultana R, Brooks CB, Shrestha A, Ogundele OM, Lee CC. Perineuronal Nets in the Prefrontal Cortex of a Schizophrenia Mouse Model: Assessment of Neuroanatomical, Electrophysiological, and Behavioral Contributions. International Journal of Molecular Sciences. 2021; 22(20):11140. https://doi.org/10.3390/ijms222011140
Chicago/Turabian StyleSultana, Razia, Charles Brady Brooks, Amita Shrestha, Olalekan Michael Ogundele, and Charles Chulsoo Lee. 2021. "Perineuronal Nets in the Prefrontal Cortex of a Schizophrenia Mouse Model: Assessment of Neuroanatomical, Electrophysiological, and Behavioral Contributions" International Journal of Molecular Sciences 22, no. 20: 11140. https://doi.org/10.3390/ijms222011140
APA StyleSultana, R., Brooks, C. B., Shrestha, A., Ogundele, O. M., & Lee, C. C. (2021). Perineuronal Nets in the Prefrontal Cortex of a Schizophrenia Mouse Model: Assessment of Neuroanatomical, Electrophysiological, and Behavioral Contributions. International Journal of Molecular Sciences, 22(20), 11140. https://doi.org/10.3390/ijms222011140