
Identification of an IL-22-Dependent Gene Signature as a Pharmacodynamic Biomarker


Abstract
:1. Introduction
2. Results
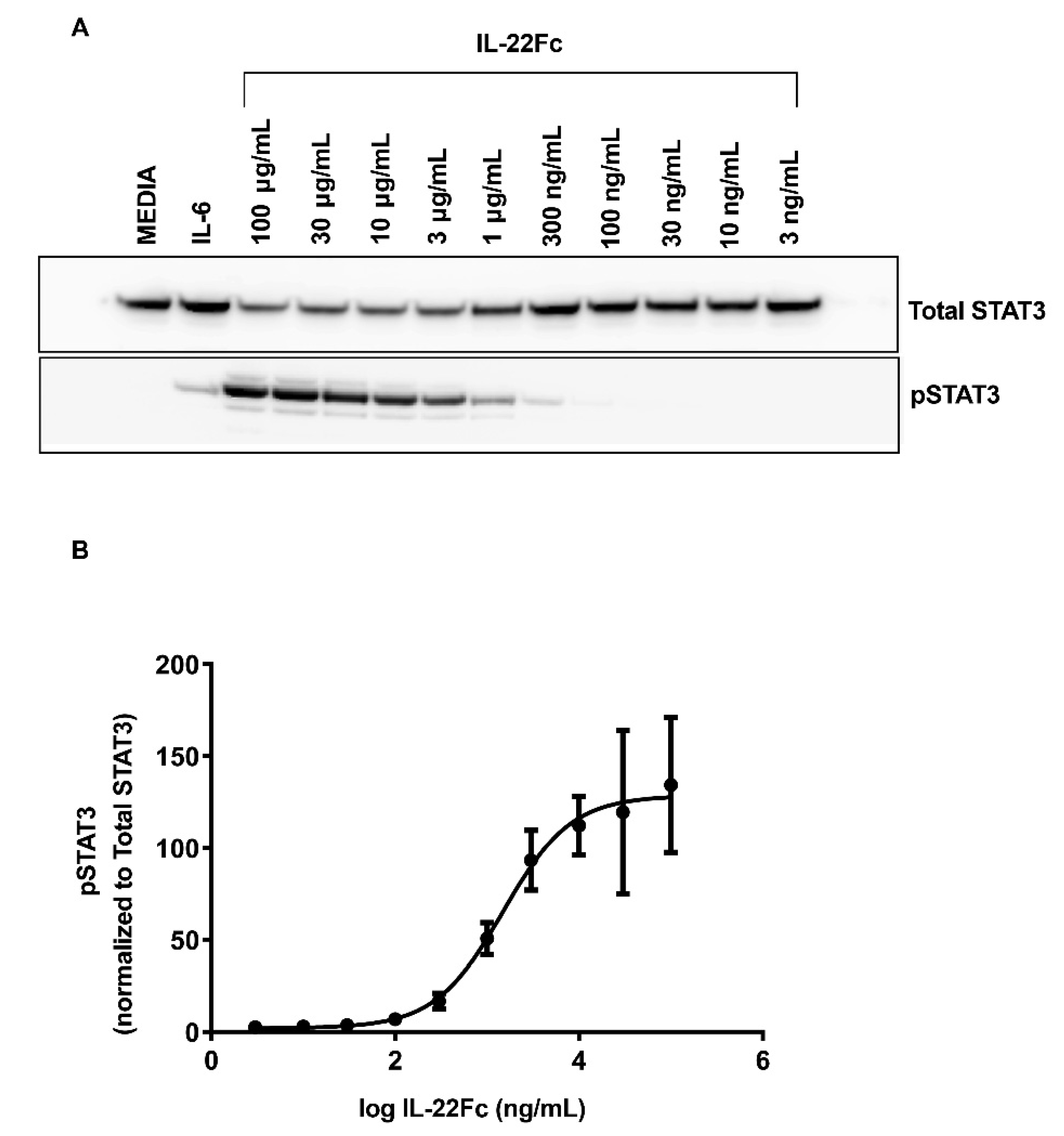
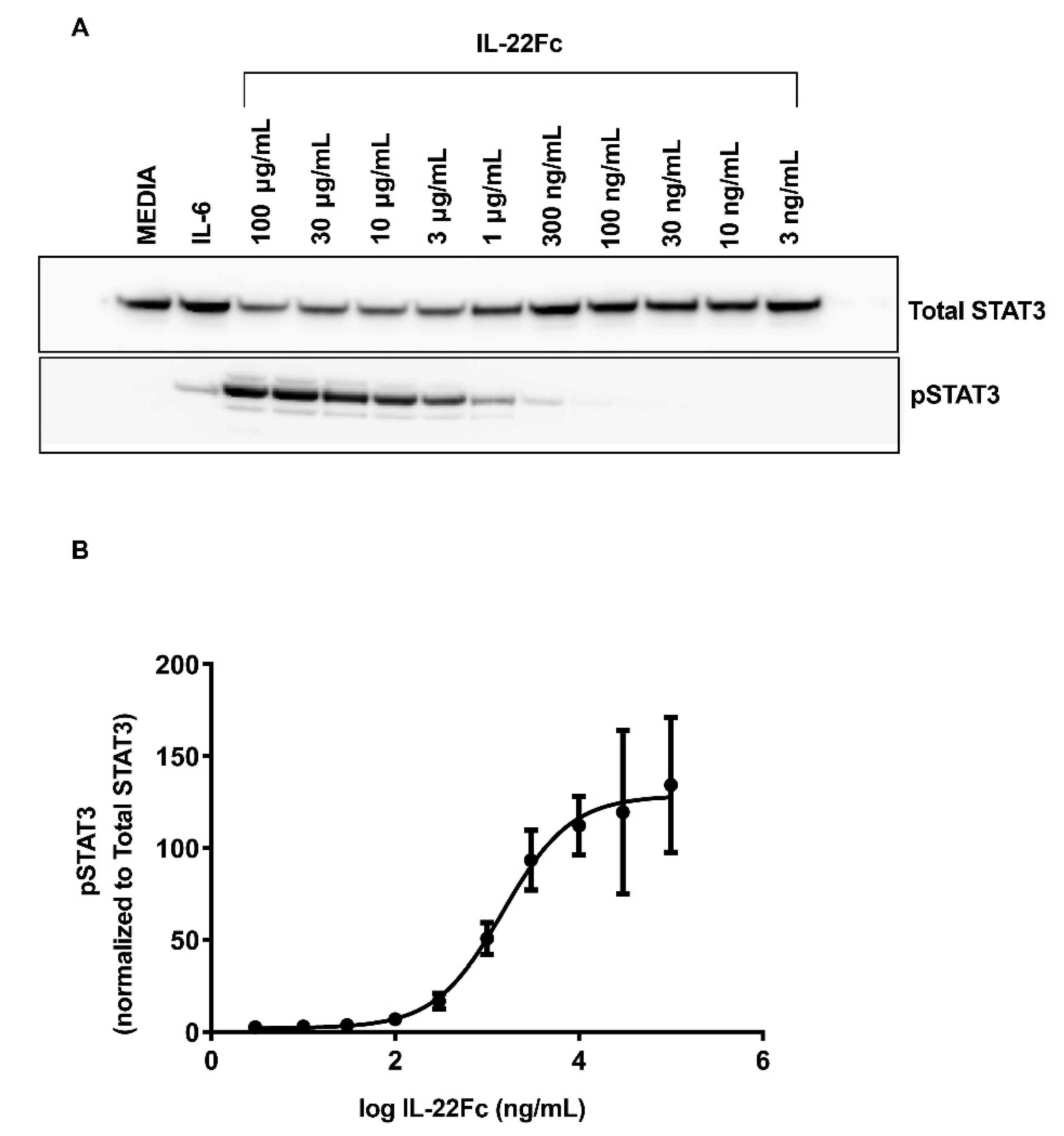
2.1. Characterizing HT-29 Cells
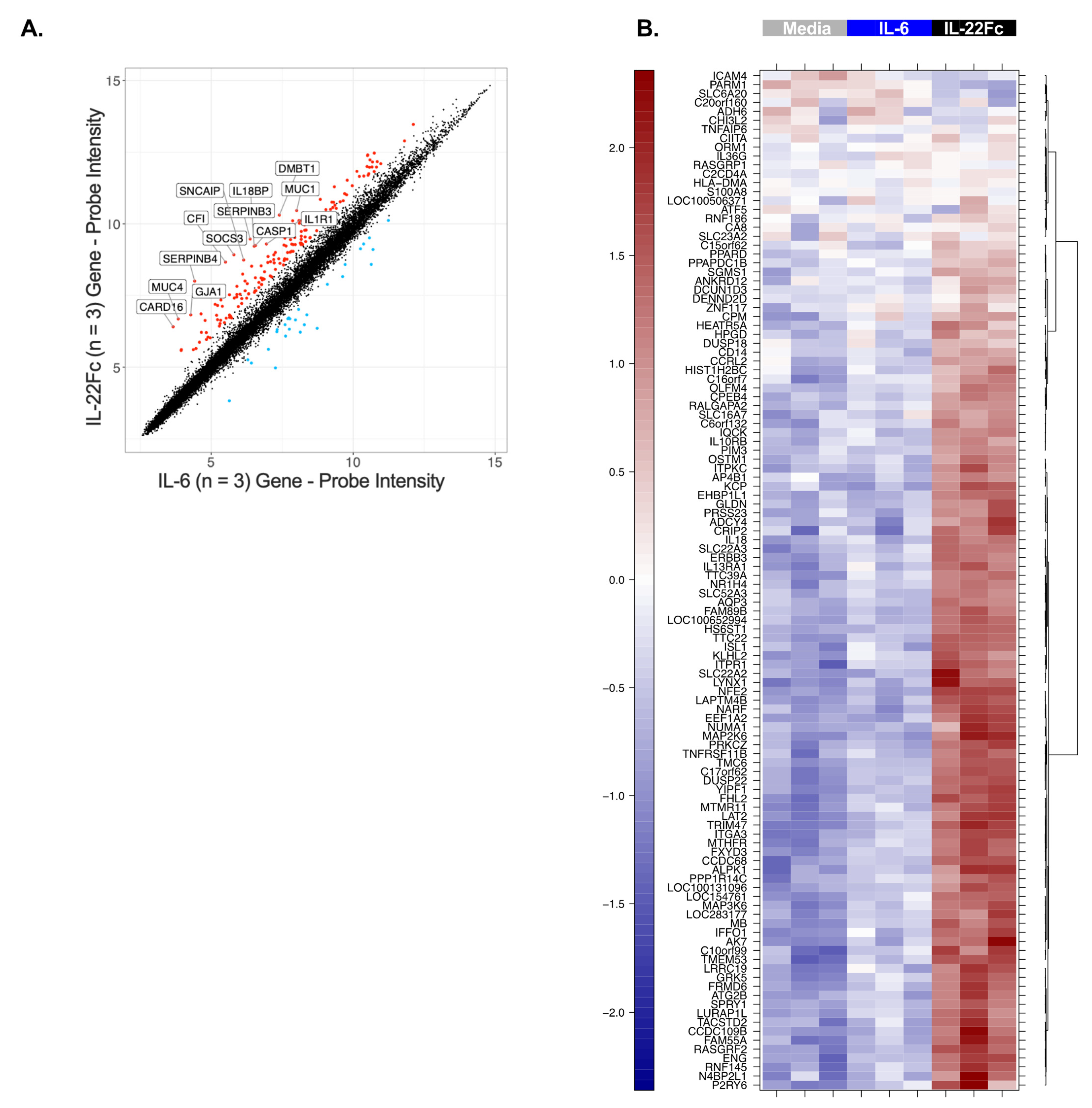
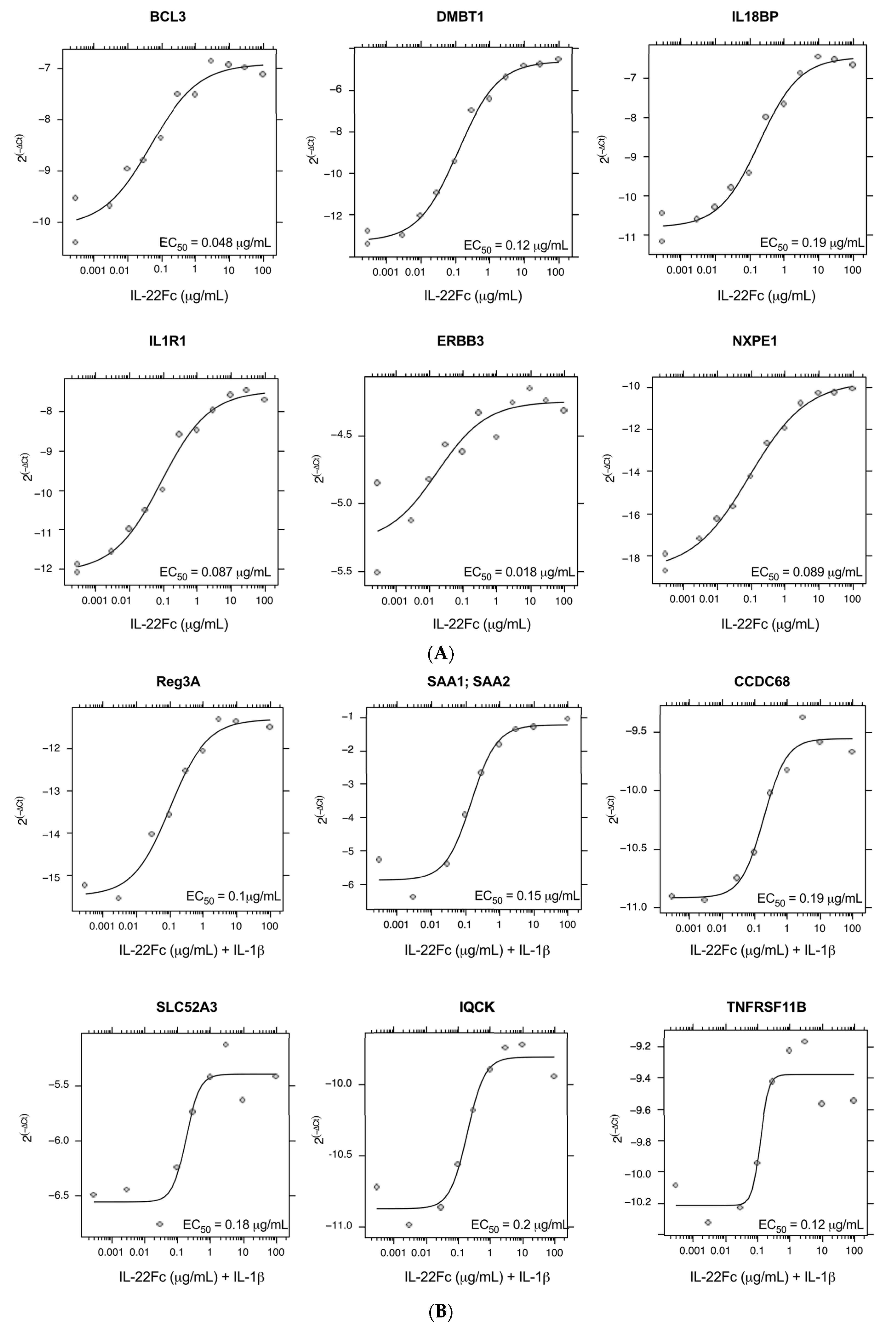
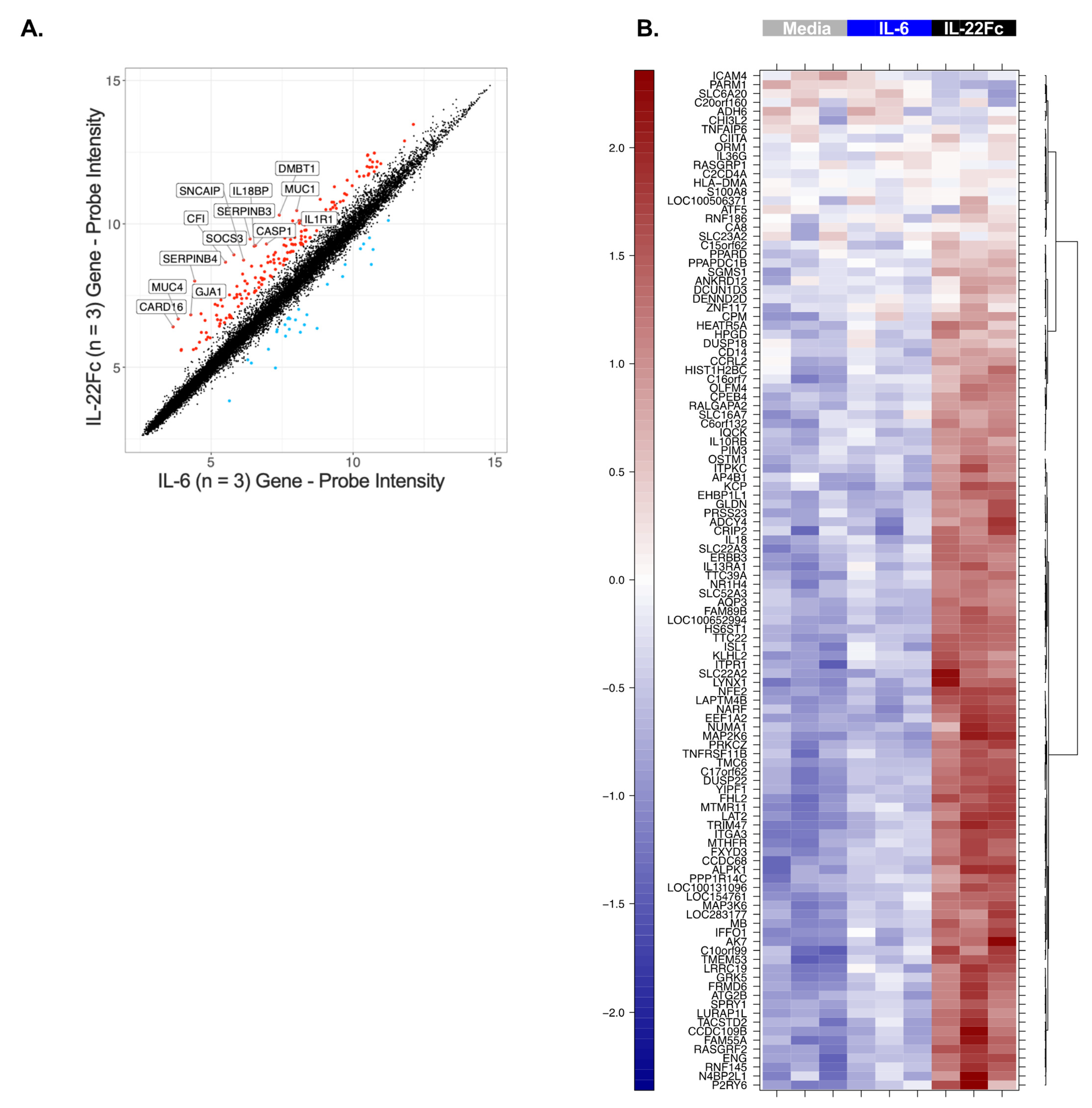
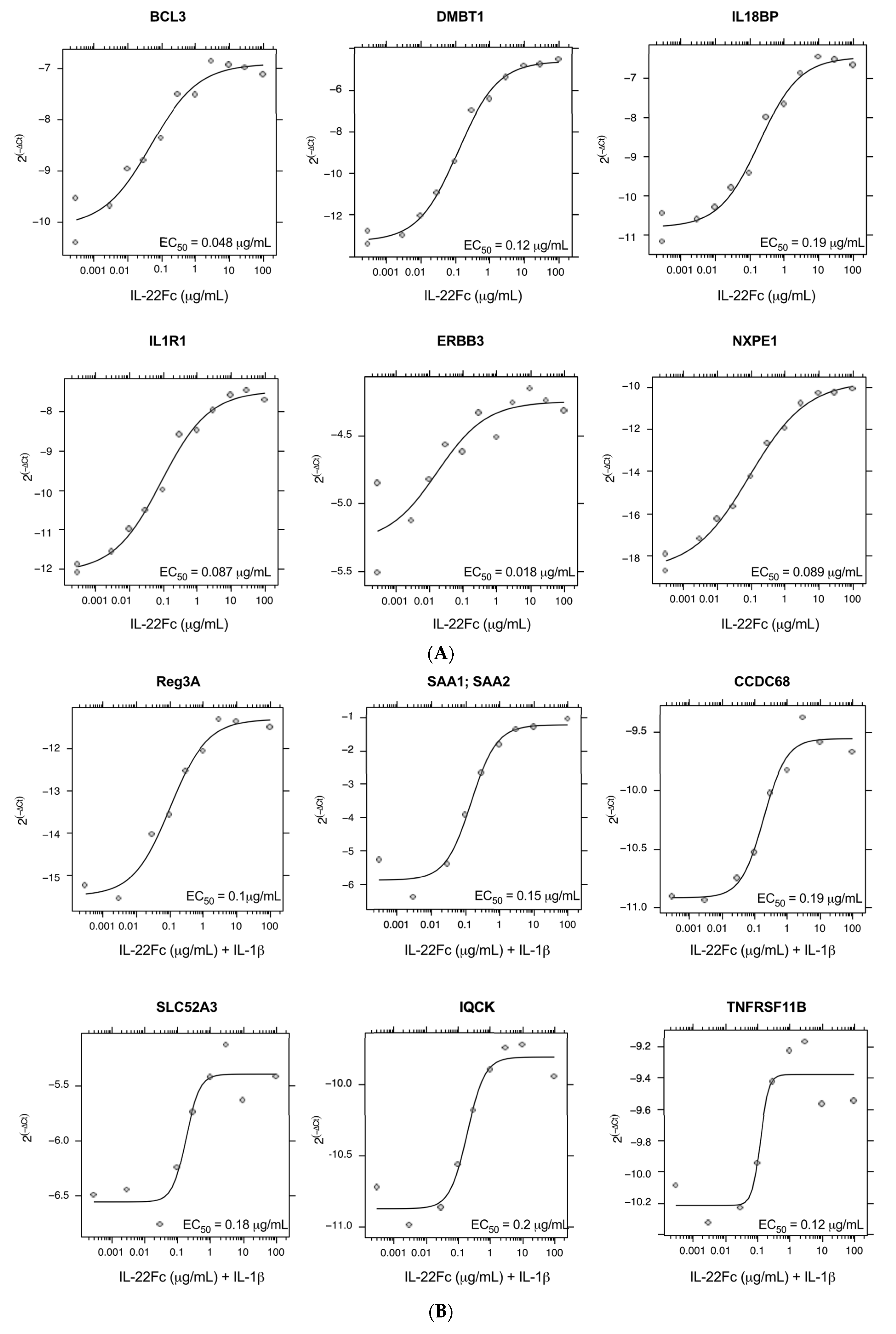
2.2. Development of a Specific, Sensitive and Dose-Dependent IL-22 Responsive Gene Signature in HT-29 Cells
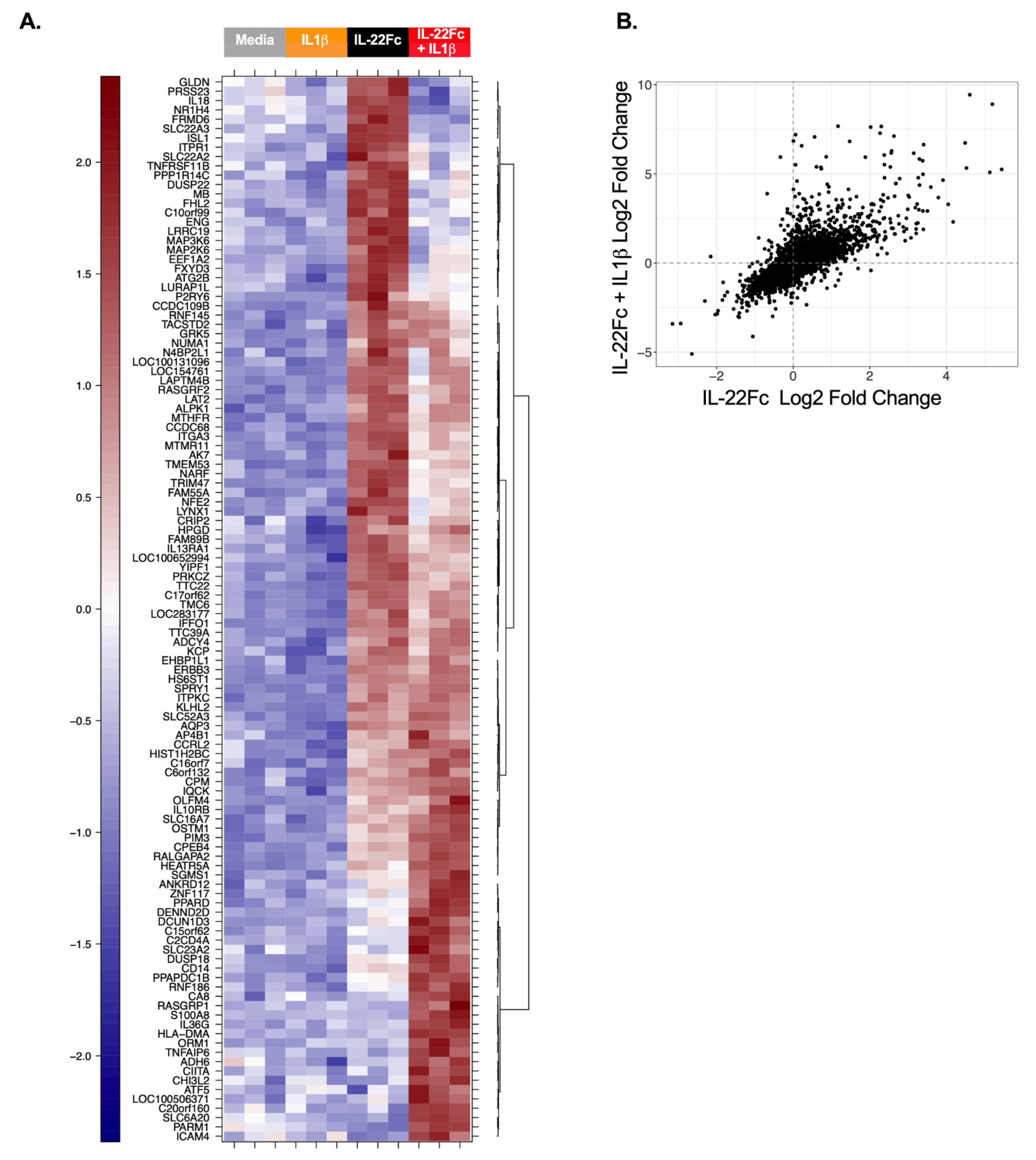
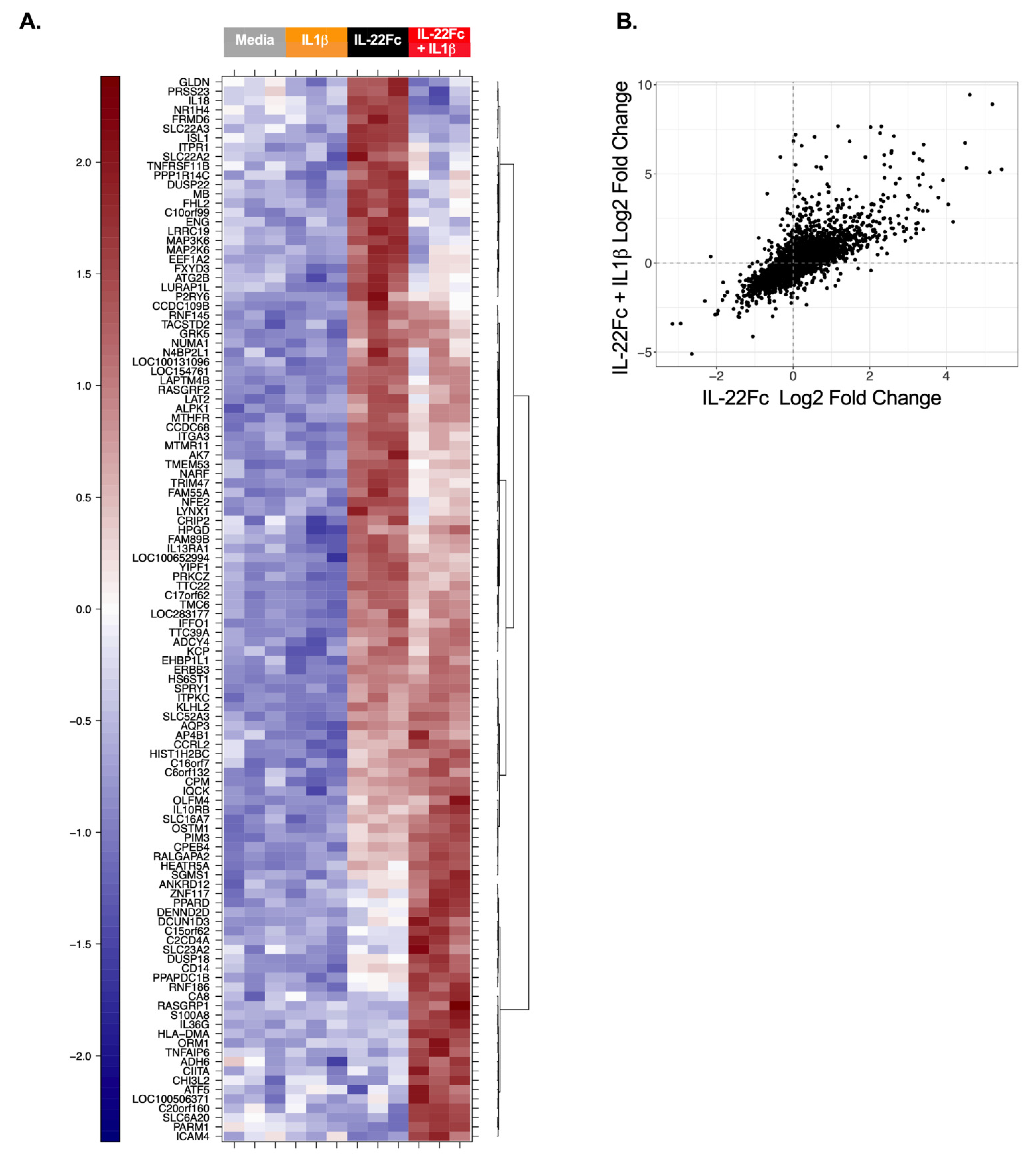
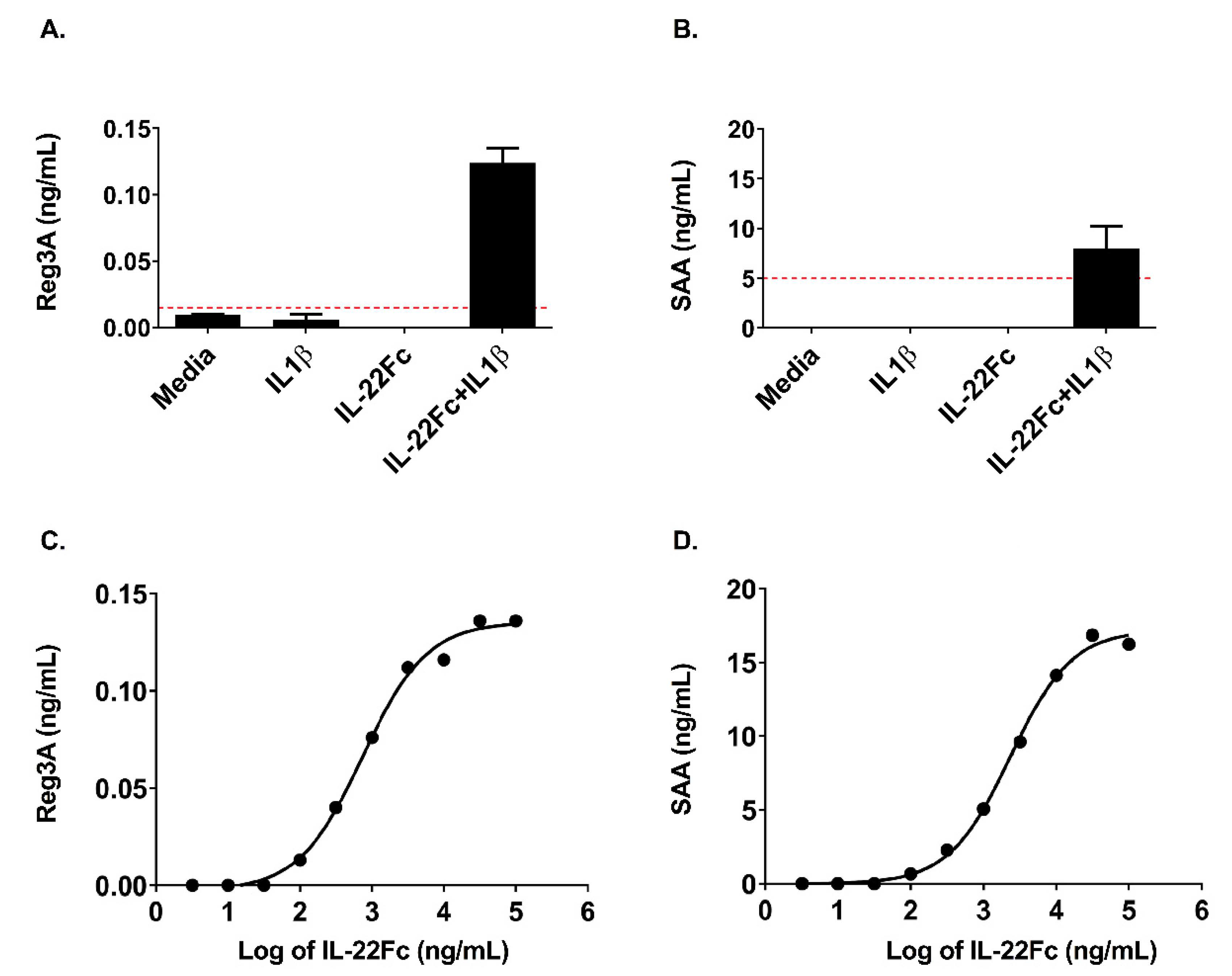
2.3. HT-29 Cell Secretion of Antimicrobial Peptides REG3A and SAA Confirms the IL-22/IL-1 Gene Expression Pattern
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. In Vitro Stimulation of HT-29 Cells and the Analysis of the Phospho-STAT3
4.3. In Vitro Stimulation of the HT-29 Cells, mRNA Sample Preparation and Microarray Analysis
4.4. Quantitative Real-Time PCR Analysis
4.5. Microarray Analyses
- up-regulated in IL-22Fc v. Media, and
- up-regulated in IL-22Fc v. IL-6, and
- up-regulated in IL-22Fc v. IL-1β
- not up-regulated in IL-6 v. Media, or
- not up-regulated in IL-1β v. Media, or
- not up-regulated in IL-6 + IL-1β v Media
- up-regulated in IL-22Fc + IL-1β v. Media, and
- up-regulated in IL-22Fc + IL-1β v. IL-6 + IL-1β, and
- up-regulated in IL-22Fc + IL-1β v. IL-1β
- not up-regulated in IL-6 v. Media, or
- not up-regulated in IL-1β v. Media, or
- not up-regulated in IL-6 + IL-1β v Media
4.6. SAA and REG3A ELISA Assays
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perusina Lanfranca, M.; Lin, Y.; Fang, J.; Zou, W.; Frankel, T. Biological and pathological activities of interleukin-22. J. Mol. Med. 2016, 94, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013, 252, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, K.; Dimartino, V.; Cavani, A. IL-17 and IL-22 in immunity: Driving protection and pathology. Eur. J. Immunol. 2017, 47, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, S.; Beigel, F.; Olszak, T.; Zitzmann, K.; Eichhorst, S.T.; Otte, J.M.; Diepolder, H.; Marquardt, A.; Jagla, W.; Popp, A.; et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G827–G838. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, A.; Yano, A.; Himuro, H.; Ezaki, Y.; Sadanaga, T.; Mizoguchi, E. Clinical importance of IL-22 cascade in IBD. J. Gastroenterol. 2018, 53, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhl, H.; Scheiermann, P.; Bachmann, M.; Hardle, L.; Heinrichs, A.; Pfeilschifter, J. IL-22 in tissue-protective therapy. Br. J. Pharmacol. 2013, 169, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Parks, O.B.; Pociask, D.A.; Hodzic, Z.; Kolls, J.K.; Good, M. Interleukin-22 Signaling in the Regulation of Intestinal Health and Disease. Front. Cell Dev. Biol. 2015, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.H.; Aggarwal, S.; Ho, W.H.; Foster, J.; Zhang, Z.; Stinson, J.; Wood, W.I.; Goddard, A.D.; Gurney, A.L. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J. Biol. Chem. 2000, 275, 31335–31339. [Google Scholar] [CrossRef] [Green Version]
- Wolk, K.; Kunz, S.; Asadullah, K.; Sabat, R. Cutting edge: Immune cells as sources and targets of the IL-10 family members? J. Immunol. 2002, 168, 5397–5402. [Google Scholar] [CrossRef]
- Andoh, A.; Zhang, Z.; Inatomi, O.; Fujino, S.; Deguchi, Y.; Araki, Y.; Tsujikawa, T.; Kitoh, K.; Kim-Mitsuyama, S.; Takayanagi, A.; et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 2005, 129, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, H.; Kuroiwa, T.; Hiramatsu, N.; Kaneko, Y.; Hiromura, K.; Ueki, K.; Nojima, Y. Expression of interleukin-22 in rheumatoid arthritis: Potential role as a proinflammatory cytokine. Arthritis Rheum. 2005, 52, 1037–1046. [Google Scholar] [CrossRef]
- Lejeune, D.; Dumoutier, L.; Constantinescu, S.; Kruijer, W.; Schuringa, J.J.; Renauld, J.C. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J. Biol. Chem. 2002, 277, 33676–33682. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Li, Z.; Pan, W.; Qin, L.; Zhu, G.; Ke, Y.; Wu, J.; Bo, P.; Meng, S. Participation of Gab1 and Gab2 in IL-22-mediated keratinocyte proliferation, migration, and differentiation. Mol. Cell. Biochem. 2012, 369, 255–266. [Google Scholar] [CrossRef]
- Mitra, A.; Raychaudhuri, S.K.; Raychaudhuri, S.P. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine 2012, 60, 38–42. [Google Scholar] [CrossRef]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Aujla, S.J.; Chan, Y.R.; Zheng, M.; Fei, M.; Askew, D.J.; Pociask, D.A.; Reinhart, T.A.; McAllister, F.; Edeal, J.; Gaus, K.; et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008, 14, 275–281. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Karow, M.; Flavell, R.A. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007, 27, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Kolumam, G.; Wu, X.; Lee, W.P.; Hackney, J.A.; Zavala-Solorio, J.; Gandham, V.; Danilenko, D.M.; Arora, P.; Wang, X.; Ouyang, W. IL-22R Ligands IL-20, IL-22, and IL-24 Promote Wound Healing in Diabetic db/db Mice. PLoS ONE 2017, 12, e0170639. [Google Scholar] [CrossRef] [PubMed]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanich, E.G.; Rae, J.; Sukumaran, S.; Lutman, J.; Lekkerkerker, A.; Ouyang, W.; Wang, X.; Lee, D.; Danilenko, D.M.; Diehl, L.; et al. Pre-clinical and translational pharmacology of a human interleukin-22 IgG fusion protein for potential treatment of infectious or inflammatory diseases. Biochem. Pharmacol. 2018, 152, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Eidenschenk, C.; Rutz, S.; Liesenfeld, O.; Ouyang, W. Role of IL-22 in microbial host defense. Curr. Top. Microbiol. Immunol. 2014, 380, 213–236. [Google Scholar] [PubMed]
- Fukui, H.; Sekikawa, A.; Tanaka, H.; Fujimori, Y.; Katake, Y.; Fujii, S.; Ichikawa, K.; Tomita, S.; Imura, J.; Chiba, T.; et al. DMBT1 is a novel gene induced by IL-22 in ulcerative colitis. Inflamm. Bowel Dis. 2011, 17, 1177–1188. [Google Scholar] [CrossRef]
- Diegelmann, J.; Czamara, D.; Le Bras, E.; Zimmermann, E.; Olszak, T.; Bedynek, A.; Goke, B.; Franke, A.; Glas, J.; Brand, S. Intestinal DMBT1 expression is modulated by Crohn’s disease-associated IL23R variants and by a DMBT1 variant which influences binding of the transcription factors CREB1 and ATF-2. PLoS ONE 2013, 8, e77773. [Google Scholar] [CrossRef]
- Kolls, J.K.; McCray, P.B., Jr.; Chan, Y.R. Cytokine-mediated regulation of antimicrobial proteins. Nat. Rev. Immunol. 2008, 8, 829–835. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Zhong, S.; Pai, R.; Rae, J.; Sukumaran, S.; Stefanich, E.G.; Lutman, J.; Doudement, E.; Wang, X.; Harder, B.; et al. Nonclinical safety assessment of a human interleukin-22FC IG fusion protein demonstrates in vitro to in vivo and cross-species translatability. Pharmacol. Res. Perspect. 2018, 6, e00434. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Chowdhry, S.; Pizarro, T.T. Opposing Functions of Classic and Novel IL-1 Family Members in Gut Health and Disease. Front. Immunol. 2013, 4, 181. [Google Scholar] [CrossRef] [Green Version]
- Keir, M.E.; Ichikawa, R.; Acres, M.; Hackney, J.; Hulme, G.; Fuh, F.; Carey, C.D.; Palmer, J.; Jones, C.J.; Long, A.K.; et al. Regulation and role of alphaE integrin and gut homing integrins in migration and retention of intestinal lymphocytes during inflammatory bowel disease. Med. Res. Rev. 2020, 40, 245–262. [Google Scholar]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Fuller, C.K.; Song, Y.; Meng, Q.; Zhang, B.; Yang, X.; Li, H. Sherlock: Detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am. J. Hum. Genet. 2013, 92, 667–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Hulur, I.; Gamazon, E.R.; Skol, A.D.; Xicola, R.M.; Llor, X.; Onel, K.; Ellis, N.A.; Kupfer, S.S. Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci. BMC Genom. 2015, 16, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Rybaczyk, L.; Rozmiarek, A.; Circle, K.; Grants, I.; Needleman, B.; Wunderlich, J.E.; Huang, K.; Christofi, F.L. New bioinformatics approach to analyze gene expressions and signaling pathways reveals unique purine gene dysregulation profiles that distinguish between CD and UC. Inflamm. Bowel Dis. 2009, 15, 971–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Fang, M.; Jostins, L.; Umicevic Mirkov, M.; Boucher, G.; Anderson, C.A.; Andersen, V.; Cleynen, I.; Cortes, A.; Crins, F.; et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.S.; Wiles, B.M.; McGee, D.W. IL-22 Enhances TNF-alpha- and IL-1-Induced CXCL8 Responses by Intestinal Epithelial Cell Lines. Inflammation 2017, 40, 1726–1734. [Google Scholar] [CrossRef]
- Madsen, J.; Mollenhauer, J.; Holmskov, U. Review: Gp-340/DMBT1 in mucosal innate immunity. Innate Immun. 2010, 16, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, K.; Modrusan, Z.; Cornelius, J.; Chavali, A.; Kasman, I.; Komuves, L.; Mo, L.; Diehl, L. Intestinal Epithelial Cell Up-Regulation of LY6 Molecules during Colitis Results in Enhanced Chemokine Secretion. J. Immunol. 2008, 180, 3874–3881. [Google Scholar] [CrossRef] [Green Version]
- Fransen, K.; Visschedijk, M.C.; van Sommeren, S.; Fu, J.Y.; Franke, L.; Festen, E.A.M.; Stokkers, P.C.F.; van Bodegraven, A.A.; Crusius, J.B.A.; Hommes, D.W.; et al. Analysis of SNPs with an effect on gene expression identifies UBE2L3 and BCL3 as potential new risk genes for Crohn’s disease. Hum. Mol. Genet. 2010, 19, 3482–3488. [Google Scholar] [CrossRef] [Green Version]
- Consortium, U.I.G.; Barrett, J.C.; Lee, J.C.; Lees, C.W.; Prescott, N.J.; Anderson, C.A.; Phillips, A.; Wesley, E.; Parnell, K.; Zhang, H.; et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 2009, 41, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.I.; Lees, C.W. Genetics of ulcerative colitis. Inflamm. Bowel Dis. 2011, 17, 831–848. [Google Scholar] [CrossRef] [PubMed]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Loci * | Gene | Loci * |
|---|---|---|---|
| ADH6 | ITPKC | ||
| AK7 | ITPR1 | ||
| ALPK1 | KCP | ||
| ANKRD12 | LAPTM4B | ||
| AP4B1 | LAT2 | ||
| ATF5 | LURAP1L | ||
| ATG2B | LYNX1 | CD [31] | |
| BCL3† | MAP2K6 | ||
| CA8 | MB | ||
| CCDC68 | MTHFR | ||
| CCM2L | NARF | ||
| CCRL2 | IBD [32] | NR1H4 | |
| CD14 | NUMA1 | ||
| CDH1# | UC [32] | NXPE1 | UC [33] |
| CHI3L2 | ORM1 | ||
| CIITA | OSTM1 | ||
| CPEB4 | CD [34] | P2RY6 | UC [35] |
| CPM | PARM1 | ||
| DCUN1D3 | PPAPDC1B | ||
| DENND2D | PPARD | ||
| DMBT1 | IBD [31,34] | PPP1R14C | |
| EEF1A2 | PRKCZ | ||
| EPCAM# | PRSS23 | ||
| ERBB3 | RALGAPA2 | ||
| FHL2 | RASGRP1 | CD [36] | |
| FRMD6 | REG3A# | ||
| FXYD3 | REG3G# | ||
| GLDN | RNF145 | ||
| HIST1H2BC | RNF186 | UC [37] | |
| HLA-DMA | S100A8 | ||
| HS6ST1 | S100A9# | ||
| ICAM4 | SAA1;SAA2# | ||
| IFFO1 | SAA2# | ||
| IL10RB | SBNO2† | ||
| IL13RA1 | SLC16A7 | ||
| IL18 | SLC22A2 | ||
| IL18BP | SLC22A3 | ||
| IL1R1 | SLC23A2 | ||
| IL1RAP | SLC52A3 | ||
| IL22RA1# | SPRY1 | ||
| IL22RA2# | TMEM53 | ||
| IL36G | TNFAIP6 | ||
| IQCK | TNFRSF11B | ||
| ISL1 | TTC39A | ||
| ITGA3 | YIPF1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rae, J.; Hackney, J.; Huang, K.; Keir, M.; Herman, A. Identification of an IL-22-Dependent Gene Signature as a Pharmacodynamic Biomarker. Int. J. Mol. Sci. 2021, 22, 8205. https://doi.org/10.3390/ijms22158205
Rae J, Hackney J, Huang K, Keir M, Herman A. Identification of an IL-22-Dependent Gene Signature as a Pharmacodynamic Biomarker. International Journal of Molecular Sciences. 2021; 22(15):8205. https://doi.org/10.3390/ijms22158205
Chicago/Turabian StyleRae, Julie, Jason Hackney, Kevin Huang, Mary Keir, and Ann Herman. 2021. "Identification of an IL-22-Dependent Gene Signature as a Pharmacodynamic Biomarker" International Journal of Molecular Sciences 22, no. 15: 8205. https://doi.org/10.3390/ijms22158205
APA StyleRae, J., Hackney, J., Huang, K., Keir, M., & Herman, A. (2021). Identification of an IL-22-Dependent Gene Signature as a Pharmacodynamic Biomarker. International Journal of Molecular Sciences, 22(15), 8205. https://doi.org/10.3390/ijms22158205