
Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development


Abstract
1. Introduction
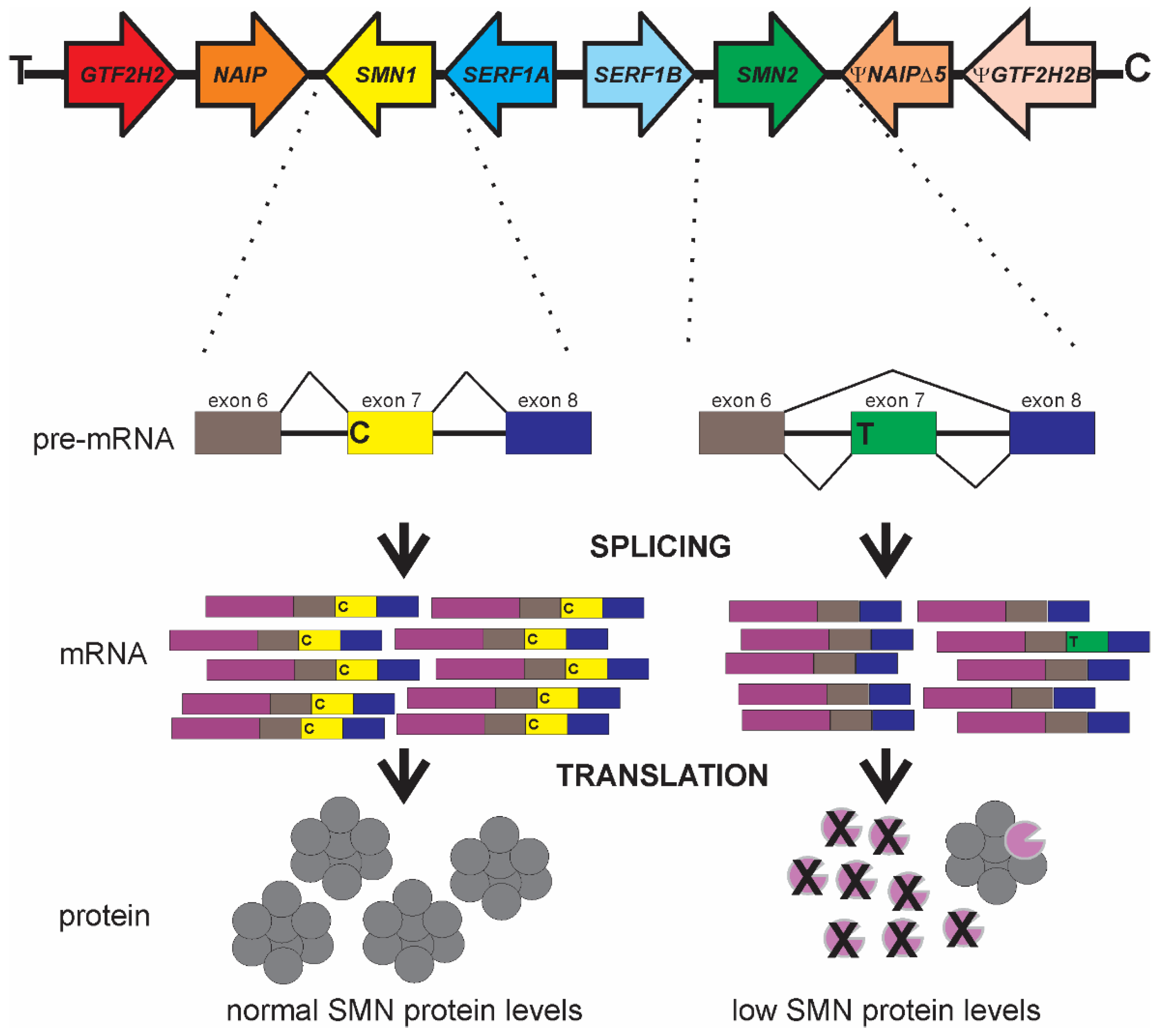
2. Genetics and Biology of SMA
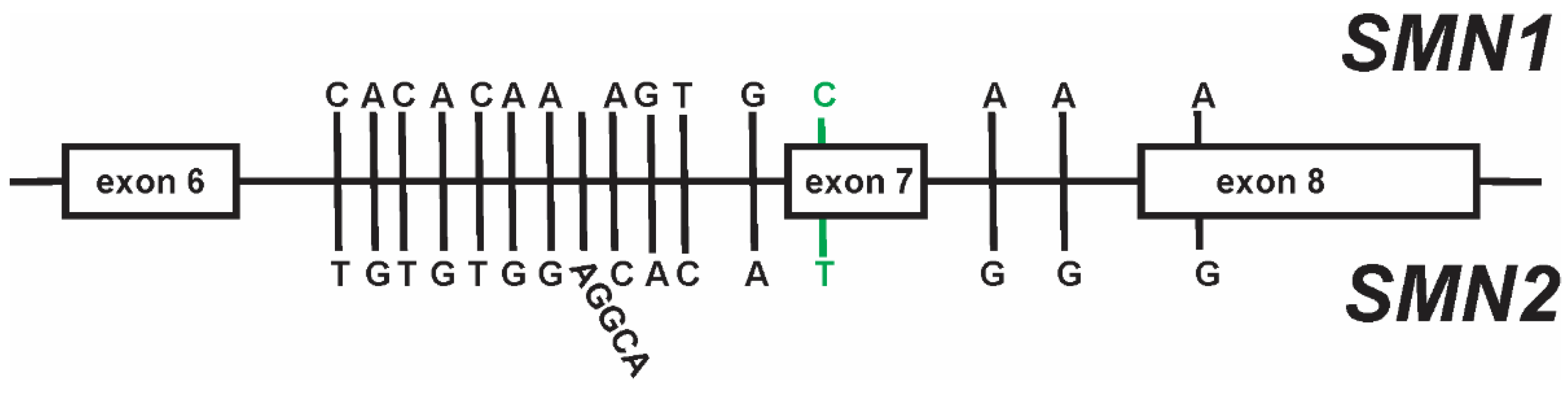
3. SMN2 as a Disease Modifier for SMA
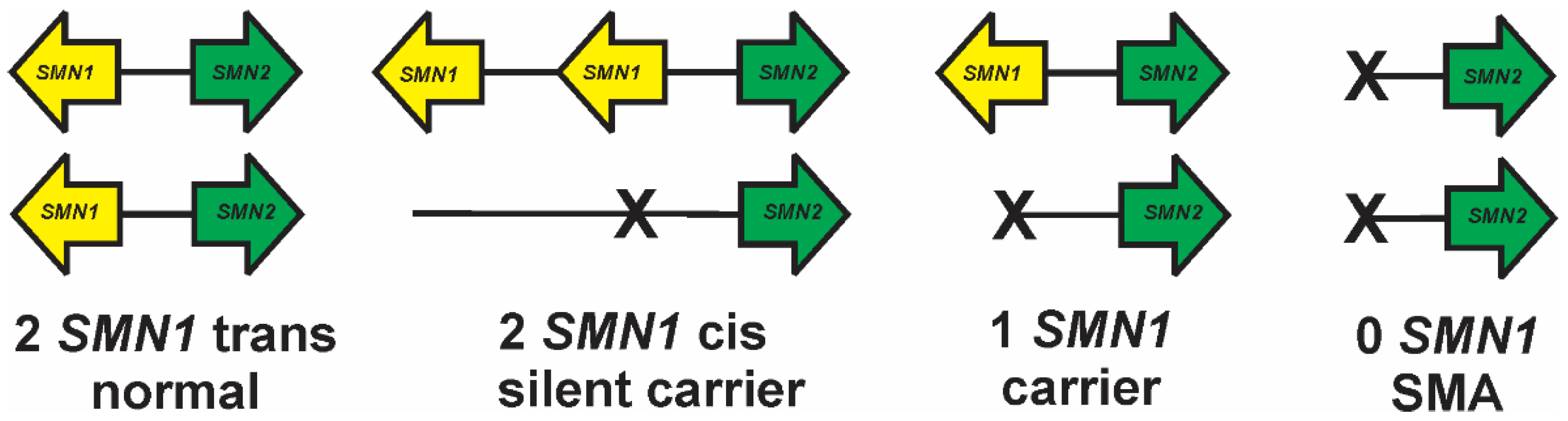
4. Measurement of SMN1 and SMN2 CNV
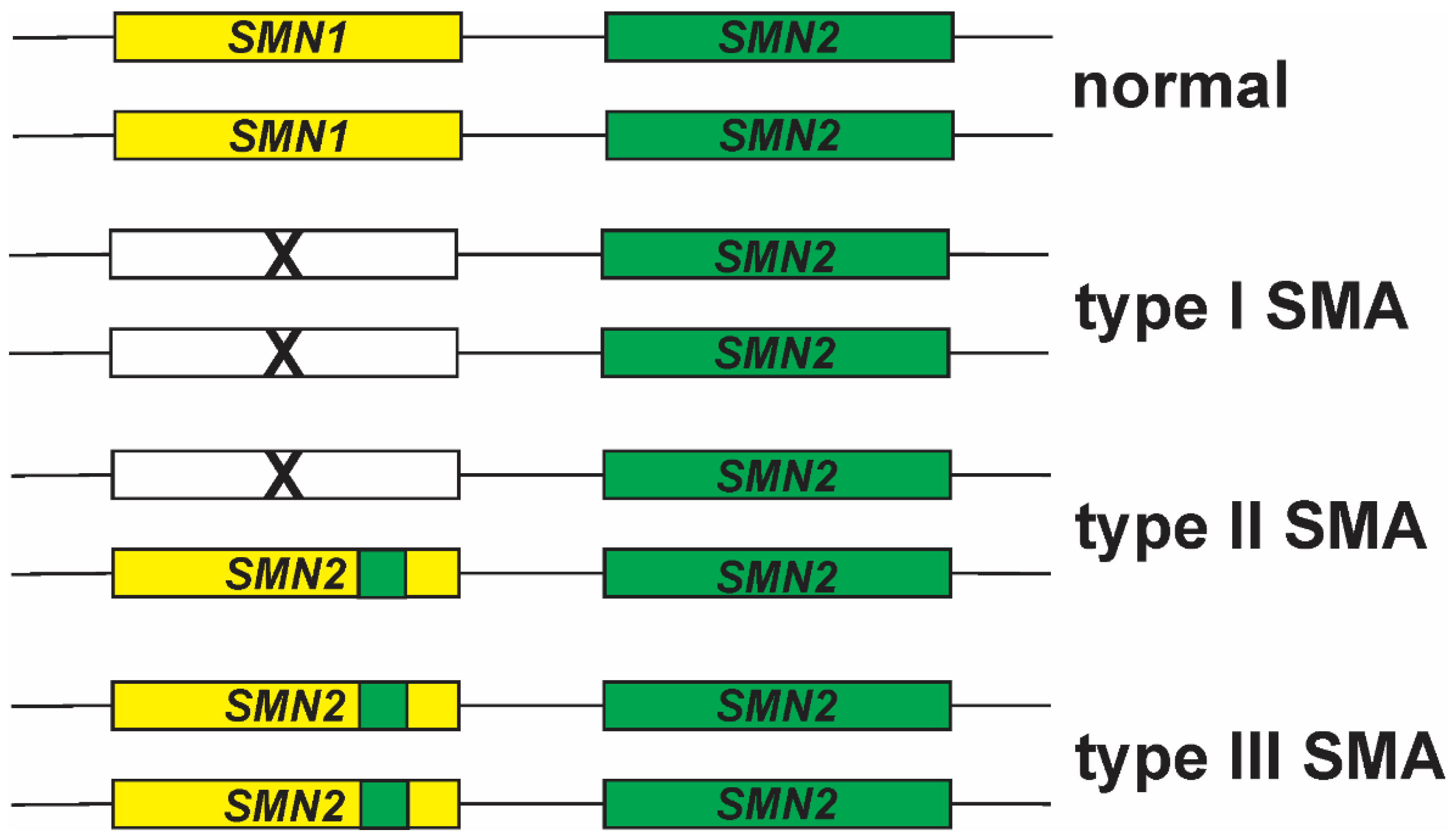
5. SMN1 to SMN2 Gene Conversions and Partial Deletions
6. SMN2 Copy Number and Therapeutic Efficacy
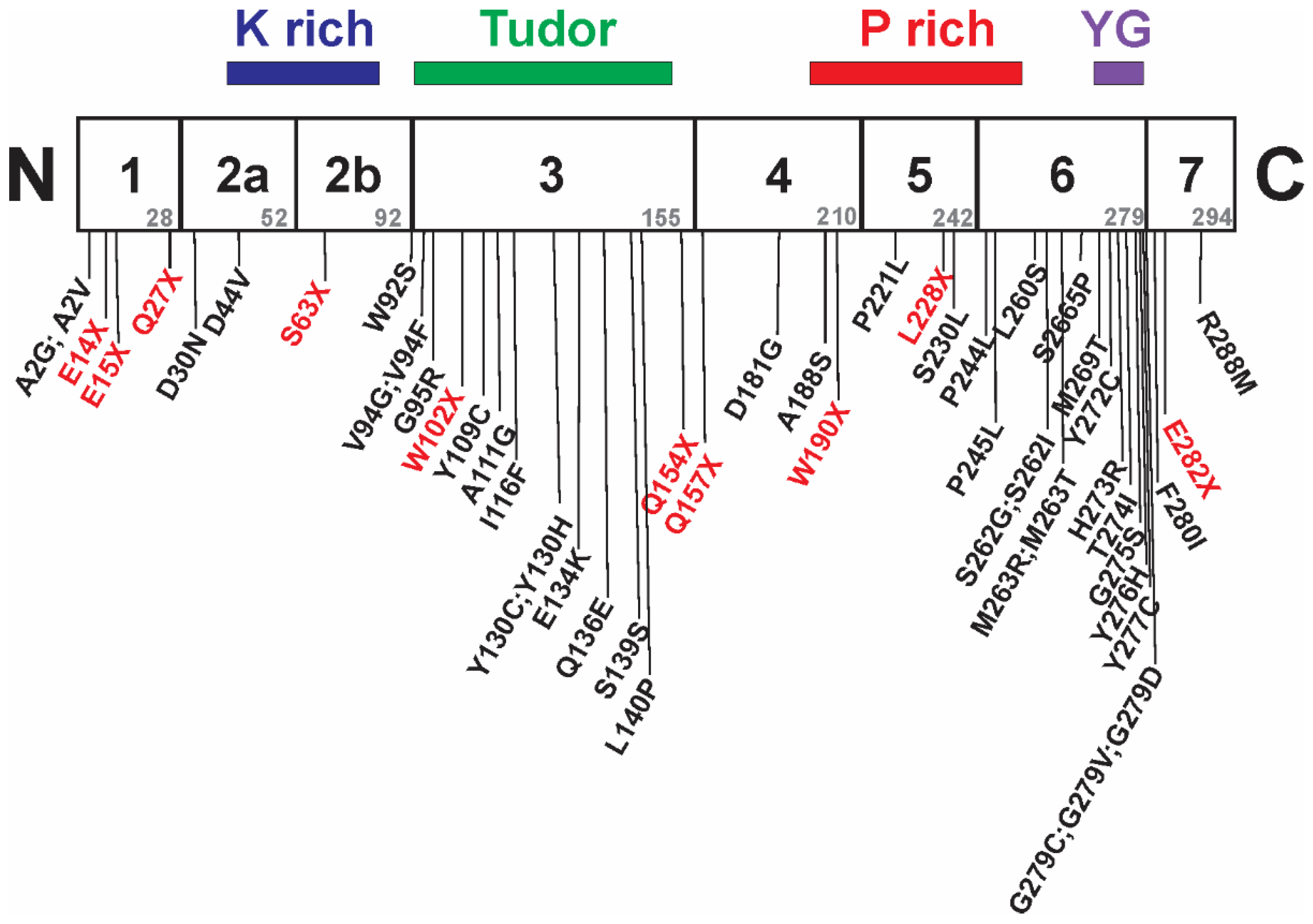
7. Intragenic Mutations in SMN1 and SMA
8. Silent Carriers and Compound Heterozygotes in SMA
9. Intrafamilial Variation in SMA Clinical Presentation
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Crawford, T.O.; Pardo, C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, I.; Barceló, M.J.; Soler, C.; Parra, J.; Baiget, M.; Tizzano, E. Prenatal diagnosis for risk of spinal muscular atrophy. Br. J. Obstet. Gynaecol. 2002, 109, 1244–1249. [Google Scholar] [CrossRef]
- Pearn, J. Incidence, prevalence and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 1978, 15, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, E.A.; Nagan, N.; Zhu, H.; Akmaev, V.R.; Zhou, Z.; Rohlfs, A.M.; Flynn, K.; Hendrickson, B.C.; Scholl, T.; Sirko-Osadsa, D.A.; et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of > 72400 specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, S.; Orr-Urtreger, A.; Bardugo, E.; Shomrat, R.; Yaron, Y. Large-scale population screening for spinal muscular atrophy: Clinical implications. Genet. Med. 2011, 13, 110–114. [Google Scholar] [CrossRef]
- Su, Y.N.; Hung, C.C.; Lin, S.Y.; Chen, F.Y.; Chern, J.P.S.; Tsai, C.; Chang, T.S.; Yang, C.C.; Li, H.; Ho, H.N.; et al. Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005–2009: A prospective population-based cohort study. PLoS ONE 2011, 6, e17067. [Google Scholar] [CrossRef]
- Lyahyai, J.; Sbiti, A.; Barkat, A.; Ratbi, I.; Sefiani, A. Spinal muscular atrophy carrier frequency and estimated prevalence of the disease in Moroccan newborns. Genet. Test. Mol. Biomark. 2012, 16, 215–218. [Google Scholar] [CrossRef]
- Sangaré, M.; Hendrickson, B.; Sango, H.A.; Chen, K.; Nofziger, J.; Amara, A.; Dutra, A.; Schindler, A.B.; Guindo, A.; Traoré, M.; et al. Genetics of low spinal muscular atrophy carrier frequency in sub-Saharan Africa. Ann. Neurol. 2014, 75, 525–532. [Google Scholar] [CrossRef]
- Zaldívar, T.; Montejo, Y.; Acevedo, A.M.; Guerra, R.; Vargas, J.; Garofalo, N.; Alvarez, R.; Alvarez, M.A.; Hardiman, O. Evidence of reduced frequency of spinal muscular atrophy type I in the Cuban population. Neurology 2005, 65, 636–638. [Google Scholar] [CrossRef]
- Labrum, R.; Rodda, J.; Krause, A. The molecular basis of spinal muscular atrophy (SMA) in South African black patients. Neuromuscul. Disord. 2007, 17, 684–692. [Google Scholar] [CrossRef]
- Hendrickson, B.C.; Donohoe, C.; Akmaev, V.R.; Sugarman, E.A.; Labrousse, P.; Boguslavskiy, L.; Flynn, K.; Rohlfs, E.M.; Walker, A.; Allitto, B.; et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet. 2009, 46, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, K.J.; Prior, T.W.; Scott, C.B.; McNaught, T.P.; Wride, M.C.; Reyna, S.P.; Bromberg, M.B. Natural history of denervation in SMA: Relation to age, SMN2 copy number and function. Ann. Neurol. 2005, 57, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Munsat, T.L.; Davies, K.E. International SMA Consortium meeting. Neuromuscul. Disord. 1992, 2, 423–428. [Google Scholar] [CrossRef]
- Russman, B.S. Spinal muscular atrophy: Clinical classification and disease heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Shababi, M.; Lorson, C.L.; Rudnik-Schöneborn, S. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2012, 19, 40–50. [Google Scholar] [CrossRef]
- Arnold, A.S.; Gueye, M.; Guettier-Sigrist, S.; Courdier-Fruh, I.; Coupin, G.; Poindron, P.; Gies, J.P. Reduced expression of nicotinic AChRs in myotubes from spinal muscular atrophy I patients. Lab. Investig. 2004, 84, 1271–1278. [Google Scholar] [CrossRef]
- Martínez-Hernández, R.; Soler-Botija, C.; Also, E.; Alias, L.; Caselles, L.; Gich, I.; Bernal, S.; Tizzano, E.F. The developmental pattern of myotubes in spinal muscular atrophy indicates prenatal delay of muscle maturation. J. Neuropathol. Exp. Neurol. 2009, 68, 474–481. [Google Scholar] [CrossRef]
- Rudnik-Schöneborn, S.; Goebel, H.H.; Schlote, W.; Molaian, S.; Omran, H.; Ketelsen, U.; Korinthenberg, R.; Wenzel, D.; Lauffer, H.; Kreiβ-Nachtsheim, M.; et al. Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy. Neurology 2003, 60, 983–987. [Google Scholar] [CrossRef]
- Yonekawa, T.; Komaki, H.; Saito, Y.; Sugai, K.; Sasaki, M. Peripheral nerve abnormalities in pediatric patients with spinal muscular atrophy. Brain Dev. 2013, 35, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Shishikura, K.; Hara, M.; Sasaki, Y.; Misugi, K. A neuropathological study of Werdnig-Hoffmann disease with special reference to the thalamus and posterior roots. Acta Neuropathol. 1983, 60, 99–106. [Google Scholar] [CrossRef]
- Ito, Y.; Kumada, S.; Uchiyama, A.; Saito, K.; Osawa, M.; Yagishita, A.; Kurata, K.; Hayashi, M. Thalamic lesions in a long-surviving child with spinal muscular atrophy type I: MRI and EEG findings. Brain Dev. 2003, 26, 53–56. [Google Scholar] [CrossRef]
- Rudnik-Schöneborn, S.; Vogelgesang, S.; Armbrust, S.; Graul-Neumann, L.; Fusch, C.; Zerres, K. Digital necroses and vascular thrombosis in severe spinal muscular atrophy. Muscle Nerve 2010, 42, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Palladino, A.; Passamano, L.; Taglia, A.; D’Ambrosio, P.; Scutifero, M.; Cecio, M.R.; Picillo, E.; Viggiano, E.; Torre, V.; De Luca, F.; et al. Cardiac involvement in patients with spinal muscular atrophies. Acta Myol. 2011, 30, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.Q.C.; Araujo, M.; Swoboda, K.J. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J. Pediatr. 2009, 155, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Swoboda, K.J.; Michalski, J.P.; Wang, G.S.; Reeks, C.; Beauvais, A.; Murphy, K.; Woulfe, J.; Screaton, R.A.; Scott, F.W.; et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 2012, 72, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I.; Sladky, J.T. Dicarboxylic aciduria in an infant with spinal muscular atrophy. Ann. Neurol. 1986, 20, 734–736. [Google Scholar] [CrossRef]
- Tein, I.; Sloane, A.E.; Donner, E.J.; Lehotay, D.C.; Millington, D.S.; Kelley, R.I. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: Primary or secondary defect(s)? Pediatr. Neurol. 1995, 12, 21–30. [Google Scholar] [CrossRef]
- Crawford, T.O.; Sladky, J.T.; Hurko, O.; Besner-Johnston, A.; Kelley, R.I. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann. Neurol. 1999, 45, 337–343. [Google Scholar] [CrossRef]
- Kölbel, H.; Hauffa, B.P.; Wudy, S.A.; Bouikidis, A.; Della Marina, A.; Schara, U. Hyperleptinemia in children with autosomal recessive spinal muscular atrophy type I-III. PLoS ONE 2017, 12, e0173144. [Google Scholar]
- Brandt, S. Hereditary factors in infantile progressive muscular atrophy; study of 112 cases in 70 families. Am. J. Dis. Child. 1949, 78, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.E. Advances in SMA research: Review of gene deletions. Neuromuscul. Disord. 1996, 6, 397–408. [Google Scholar] [CrossRef]
- Schmutz, J.; Martin, J.; Terry, A.; Couronne, O.; Grimwood, J.; Lowry, S.; Gordon, L.A.; Scott, D.; Xie, G.; Huang, W.; et al. The DNA sequence and comparative analysis of human chromosome 5. Nature 2004, 431, 268–274. [Google Scholar] [CrossRef]
- Courseaux, A.; Richard, F.; Grosgeorge, J.; Ortola, C.; Viale, A.; Turc-Carel, C.; Dutrillaux, B.; Gaudray, P.; Nahon, J.L. Segmental duplications in euchromatic regions of human chromosome 5: A source of evolutionary instability and transcriptional innovation. Genome Res. 2003, 13, 369–381. [Google Scholar] [CrossRef]
- Rochette, C.F.; Gilbert, N.; Simard, L.R. SMN gene duplication and emergence of the SMN2 gene occured in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 2001, 108, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Roy, N.; Mahadevan, M.S.; McLean, M.; Shutler, G.; Yaraghi, Z.; Farahani, R.; Baird, S.; Besner-Johnston, A.; Lefebvre, C.; Kang, X.; et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995, 80, 167–178. [Google Scholar] [CrossRef]
- Carter, T.A.; Bönnemann, C.G.; Wang, C.H.; Obici, S.; Parano, E.; De Fatima Bonaldo, M.; Ross, B.A.; Penchaszadeh, G.K.; MacKenzie, A.; Soares, M.B.; et al. A multicopy transcription-repair gene, BTF2p44, maps to the SMA region and demonstrates SMA associated deletions. Hum. Mol. Genet. 1997, 6, 229–236. [Google Scholar] [CrossRef][Green Version]
- Bürglen, L.; Seroz, T.; Miniou, P.; Lefebvre, S.; Burlet, P.; Munnich, A.; Pequignot, E.V.; Egly, J.M.; Melki, J. The gene encoding p44, a subunit of the transcription factor TFIIH, is involved in large-scale deletions associated with Werdnig-Hoffmann disease. Am. J. Hum. Genet. 1997, 60, 72–79. [Google Scholar]
- Scharf, J.M.; Endrizzi, M.G.; Wetter, A.; Huang, S.; Thompson, T.G.; Zerres, K.; Dietrich, W.F.; Wirth, B.; Kunkel, L.M. Identification of a candidate modifying gene for spinal muscular atrophy by comparative genomics. Nat. Genet. 1998, 20, 83–86. [Google Scholar] [CrossRef]
- Butchbach, M.E.R.; Burghes, A.H.M. Perspectives on models of spinal muscular atrophy for drug discovery. Drug Discov. Today Dis. Models 2004, 1, 151–156. [Google Scholar] [CrossRef]
- Butchbach, M.E.R. Copy number variations in the Survival Motor Neuron genes: Implications for spinal muscular atrophy and other neurodegenerative diseases. Front. Mol. Biosci. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.R.; Owen, N.; Talbot, K.; Patel, S.; Muntoni, F.; Ignatius, J.; Dubowitz, V.; Davies, K.E. Gene deletions in spinal muscular atrophy. J. Med. Genet. 1996, 33, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Burlet, P.; Bürglen, L.; Clermont, O.; Lefebvre, S.; Viollet, L.; Munnich, A.; Melki, J. Large scale deletions of the 5q13 region are specific to Werdnig-Hoffmann disease. J. Med. Genet. 1996, 33, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Hahnen, E.; Morgan, K.; DiDonato, C.J.; Dadze, A.; Rudnik-Schöneborn, S.; Simard, L.R.; Zerres, K.; Burghes, A.H.M. Allelic association and deletions in autosomal recessive proximal spinal muscular atrophy: Association of marker genotype with disease severity and candidate cDNAs. Hum. Mol. Genet. 1995, 4, 1273–1284. [Google Scholar] [CrossRef]
- Velasco, E.; Valero, C.; Valero, A.; Moreno, F.; Hernández-Chico, C. Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies of c BCD541 and SMA phenotype. Hum. Mol. Genet. 1996, 5, 257–263. [Google Scholar] [CrossRef]
- Burghes, A.H.M.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef]
- Coovert, D.D.; Le, T.T.; McAndrew, P.E.; Strasswimmer, J.; Crawford, T.O.; Mendell, J.R.; Coulson, S.E.; Androphy, E.J.; Prior, T.W.; Burghes, A.H.M. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997, 6, 1205–1214. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Kolb, S.J.; Gubitz, A.K.; Olszewski, R.F., Jr.; Ottinger, E.; Sumner, C.J.; Fischbeck, K.H.; Dreyfuss, G. A novel cell immunoassay to measure survival of motor neurons protein in blood cells. BMC Neurol. 2006, 6, 6. [Google Scholar] [CrossRef]
- Simard, L.R.; Bélanger, M.C.; Morissette, S.; Wride, M.; Prior, T.W.; Swoboda, K.J. Preclinical validation of a multiplex real-time assay to quantify SMN mRNA in patients with SMA. Neurology 2007, 68, 451–456. [Google Scholar] [CrossRef]
- Sumner, C.J.; Kolb, S.J.; Harmison, G.G.; Jeffries, N.O.; Schadt, K.; Finkel, R.S.; Dreyfuss, G.; Fischbeck, K.H. SMN mRNA and protein levels in peripheral blood. Biomarkers for SMA clinical trials. Neurology 2006, 66, 1067–1073. [Google Scholar] [CrossRef]
- Crawford, T.O.; Paushkin, S.; Kobayashi, D.T.; Forrest, S.J.; Joyce, C.L.; Finkel, R.S.; Kaufmann, P.; Swoboda, K.J.; Tiziano, F.; Lomastro, R.; et al. Evaluation of SMN protein, transcript and copy number in the Biomarkers for Spinal Muscular Atrophy (BforSMA) clinical study. PLoS ONE 2012, 7, e33572. [Google Scholar] [CrossRef]
- Vezain, M.; Saugier-Veber, P.; Melki, J.; Toutain, A.; Bieth, E.; Husson, M.; Pedespan, J.M.; Viollet, L.; Pénisson-Besnier, I.; Fehrenbach, S.; et al. A sensitive assay for measuring SMN mRNA levels in peripheral blood and in muscle samples of patients affected with spinal muscular atrophy. Eur. J. Hum. Genet. 2007, 15, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Tiziano, F.D.; Pinto, A.M.; Fiori, S.; Lomastro, R.; Messina, S.; Bruno, C.; Pini, A.; Pane, M.; D’Amico, A.; Ghezzo, A.; et al. SMN transcript levels in leukocytes of SMA patients determined by absolute real-time PCR. Eur. J. Hum. Genet. 2010, 18, 52–58. [Google Scholar] [CrossRef]
- Pellizzoni, L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007, 8, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta 2017, 1860, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.M.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Lorson, C.L.; Androphy, E.J. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 2000, 9, 259–265. [Google Scholar] [CrossRef]
- Cho, S.; Dreyfuss, G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010, 24, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.G.; Muñoz, E.; Tandon, A.; Kwon, D.Y.; Sumner, C.J.; Fischbeck, K.H. Regulation of SMN protein stability. Mol. Cell. Biol. 2009, 29, 1107–1115. [Google Scholar] [CrossRef]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.R.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H.M. SMNΔ7, the major product of the centromeric survival motor neuron gene (SMN2), extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef]
- McAndrew, P.E.; Parsons, D.W.; Simard, L.R.; Rochette, C.; Ray, P.N.; Mendell, J.R.; Prior, T.W.; Burghes, A.H.M. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997, 60, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Swoboda, K.J.; Scott, H.D.; Hejmanowski, A.Q. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet. 2005, 130A, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Brichta, L.; Schrank, B.; Lochmüller, H.; Blick, S.; Baasner, A.; Heller, R. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet. 2006, 119, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, B.; Prior, T.; Zhang, X.; Miller, R.; Kolb, S.J.; Moore, D.; Bradley, W.; Barohn, R.; Bryan, W.; Gelinas, D.; et al. An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve 2009, 40, 652–656. [Google Scholar] [CrossRef]
- Tiziano, F.D.; Bertini, E.; Messina, S.; Angelozzi, C.; Pane, M.; D’Amico, A.; Alfieri, P.; Fiori, S.; Battini, R.; Berardinelli, A.; et al. The Hammersmith functional score correlates with the SMN2 copy number: A multicentric study. Neuromuscul. Disord. 2007, 17, 400–403. [Google Scholar] [CrossRef]
- Stabley, D.L.; Harris, A.W.; Holbrook, J.; Chubbs, N.J.; Lozo, K.W.; Crawford, T.O.; Swoboda, K.J.; Funanage, V.L.; Wang, W.; Mackenzie, W.; et al. SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol. Genet. Genomic Med. 2015, 3, 248–257. [Google Scholar] [CrossRef]
- Mailman, M.D.; Heinz, J.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Wirth, B.; Burghes, A.H.M.; Prior, T.W. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002, 4, 20–26. [Google Scholar] [CrossRef]
- Qu, Y.; Ge, X.; Bai, J.; Wang, L.; Cao, Y.; Lu, Y.; Jin, Y.; Wang, H.; Song, F. Association of copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein gene with the natural history in a Chinese spinal muscular atrophy cohort. J. Child Neurol. 2014, 30, 429–436. [Google Scholar] [CrossRef]
- Amara, A.; Adala, L.; Ben Charfeddine, I.; Mamaï, O.; Mili, A.; Ben Lazreg, T.; H-Mida, D.; Amri, F.; Salem, N.; Boughammura, L.; et al. Correlation of SMN2, NAIP, p44, H4F5 and Occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur. J. Paediatr. Neurol. 2012, 16, 167–174. [Google Scholar] [CrossRef]
- Brkusanin, M.; Kosac, A.; Jovanovic, V.; Pesovic, J.; Brajuskovic, G.; Dimitrijevic, N.; Todorovic, S.; Romac, S.; Milic Rasic, V.; Savic-Pavicevic, D. Joint effect of the SMN2 and SERF1A genes on childhood-onset types of spinal muscular atrophy in Serbian patients. J. Hum. Genet. 2015, 60, 723–728. [Google Scholar] [CrossRef]
- Van der Steege, G.; Grootscholten, P.M.; van der Vlies, P.; Draaijers, T.G.; Osinga, J.; Cobben, J.M.; Scheffer, H.; Buys, C.H.C.M. PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995, 345, 985–986. [Google Scholar] [CrossRef]
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef]
- Anhuf, D.; Eggermann, T.; Rudnik-Schöneborn, S.; Zerres, K. Determination of SMN1 and SMN2 copy number using TaqMan technology. Hum. Mutat. 2003, 22, 74–78. [Google Scholar] [CrossRef]
- Gómez-Curet, I.; Robinson, K.G.; Funanage, V.L.; Crawford, T.O.; Scavina, M.; Wang, W. Robust quantification of the SMN gene copy number by real-time TaqMan PCR. Neurogenetics 2007, 8, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.E.; Thomas, N.H.; Lewis, C.M.; Abbs, S.J.; Rodrigues, N.R.; Davies, K.E.; Mathew, C.G. Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur. J. Hum. Genet. 1998, 6, 467–474. [Google Scholar] [CrossRef]
- Gérard, B.; Ginet, N.; Matthijs, G.; Evrard, P.; Baumann, C.; Da Silva, F.; Gérard-Blanleut, M.; Mayer, M.; Grandchamp, B.; Elion, J. Genotype determination at the survival motor neuron locus in a normal population and SMA carriers using competitive PCR and primer extension. Hum. Mutat. 2004, 16, 253–263. [Google Scholar] [CrossRef]
- Su, Y.N.; Hung, C.C.; Li, H.; Lee, C.N.; Cheng, W.F.; Tsao, P.N.; Chang, M.C.; Yu, C.L.; Hsieh, W.S.; Lin, W.L.; et al. Quantitative analysis of SMN1 and SMN2 genes based on DHPLC: A highly efficient and reliable carrier-screening test. Hum. Mutat. 2005, 25, 460–467. [Google Scholar] [CrossRef]
- Scarciolla, O.; Stuppia, L.; De Angelis, M.V.; Murru, S.; Palka, C.; Giuliani, R.; Pace, M.; Di Muzio, A.; Torrente, I.; Morella, A.; et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 2006, 7, 269–276. [Google Scholar] [CrossRef]
- Arkblad, E.L.; Darin, N.; Berg, K.; Kimber, E.; Brandberg, G.; Lindberg, C.; Holmberg, E.; Tulinius, M.; Nordling, M. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul. Disord. 2006, 16, 830–838. [Google Scholar] [CrossRef]
- Huang, C.H.; Chang, Y.Y.; Chen, C.H.; Kuo, Y.S.; Hwu, W.L.; Gerdes, T.; Ko, T.M. Copy number analysis of survival motor neuron genes by multiplex ligation-dependent probe amplification. Genet. Med. 2007, 9, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Alías, L.; Bernal, S.; Barceló, M.J.; Also-Rallo, E.; Martínez-Hernández, R.; Rodríguez-Alvarez, F.J.; Hernández-Chico, C.; Baiget, M.; Tizzano, E.F. Accuracy of marker analysis, quantitative real-time polymerase chain reaction and multiple ligation-dependent probe amplification to determine SMN2 copy number in patients with spinal muscular atrophy. Genet. Test. Mol. Biomark. 2011, 15, 587–594. [Google Scholar] [CrossRef]
- Fang, P.; Li, L.; Zhou, W.J.; Wu, W.Q.; Zhong, Z.Y.; Yan, T.Z.; Xie, J.S.; Huang, J.; Lin, L.; Zhao, Y.; et al. Molecular characterization and copy number of SMN1, SMN2 and NAIP in Chinese patients with spinal muscular atrophy and unrelated healthy controls. BMC Musculoskelet. Disord. 2015, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Kirwin, S.M.; Vinette, K.M.B.; Gonzalez, I.L.; Al Abdulwahed, H.; Al-Sannaa, N.; Funanage, V.L. A homozygous double mutation in SMN1: A complicated genetic diagnosis of SMA. Mol. Genet. Genomic Med. 2013, 1, 113–117. [Google Scholar] [CrossRef]
- Dobrowolski, S.F.; Pham, H.T.; Pouch-Downes, F.; Prior, T.W.; Naylor, E.W.; Swoboda, K.J. Newborn screening for spinal muscular atrophy by calibrated short-amplicon melt profiling. Clin. Chem. 2012, 58, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Jong, Y.J.; Chang, J.G.; Chen, Y.L.; Wu, S.M. Universal fluorescent multiplex PCR and capillary electrophoresis for evaluation of gene conversion between SMN1 and SMN2 in spinal muscular atrophy. Anal. Bioanal. Chem. 2010, 397, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Chang, J.G.; Chen, Y.L.; Jong, Y.J.; Wu, S.M. Multi-exon genotyping of SMN gene in spinal muscular atrophy by universal fluorescent PCR and capillary electrophoresis. Electrophoresis 2010, 31, 2396–2404. [Google Scholar] [CrossRef]
- Wang, C.C.; Shih, C.J.; Jong, Y.J.; Wu, S.M. Universal fluorescent tri-probe ligation equipped with capillary electrophoresis for targeting SMN1 and SMN2 genes in diagnosis of spinal muscular atrophy. Anal. Chim. Acta 2014, 833, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, J.; Zhang, Y.; Li, L.; Tang, X.; Wang, L.; Guo, J.; Jin, C.; Tighe, S.; Zhang, Y.; et al. The analysis of the association between the copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein genes and the clinical phenotypes in 40 patients with spinal muscular atrophy: Observational study. Medicine 2020, 99, e18809. [Google Scholar] [CrossRef] [PubMed]
- Stabley, D.L.; Holbrook, J.; Scavina, M.; Crawford, T.O.; Swoboda, K.J.; Robbins, K.M.; Butchbach, M.E.R. Detection of SMN1 and SMN2 gene conversion events and partial SMN1 deletions using array digital PCR. Neurogenetics 2021, 22, 53–64. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Müller-Felber, W.; Vill, K.; Schwartz, O.; Gläser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Röschinger, W.; Becker, M.; Durner, J.; et al. Infants diagnosed with spinal muscular atrophy and 4 SMN2 copies through newborn screening—Opportunity or burden? J. Neuromuscul. Dis. 2020, 7, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, I.; Bernal, S.; Blasco-Pérez, L.; Calucho, M.; Alias, L.; Fuentes-Prior, P.; Tizzano, E.F. Practical guidelines to manage discordant situations of SMN2 copy number in patients with spinal muscular atrophy. Neurol. Genet. 2020, 6, e530. [Google Scholar] [CrossRef]
- DiDonato, C.J.; Chen, X.N.; Noya, D.; Korenberg, J.R.; Nadeau, J.H.; Simard, L.R. Cloning, characterization and copy number of the murine survival motor neuron gene: Homolog of the spinal muscular atrophy-determining gene. Genome Res. 1997, 7, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Viollet, L.; Bertrandy, S.; Beuno Brunialti, A.L.; Lefebvre, S.; Burlet, P.; Clermont, O.; Cruaud, C.; Guénet, J.L.; Munnich, A.; Melki, J. cDNA isolation, expression and chromosomal localization of the mouse survival motor neuron gene (Smn). Genomics 1997, 40, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.; Götz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Diaz, C.; Nicole, S.; Velasco, M.E.; Borra-Cebrian, C.; Panozzo, C.; Frugier, T.; Millet, G.; Roblot, N.; Joshi, V.; Melki, J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum. Mol. Genet. 2002, 11, 1439–1447. [Google Scholar] [CrossRef]
- Nicole, S.; Desforges, B.; Millet, G.; Lesbordes, J.; Cifuentes-Diaz, C.; Vertes, D.; Cao, M.L.; De Backer, F.; Languille, L.; Roblot, N.; et al. Intact satellite cells lead to remarkable protection against Smn gene defect in differentiation skeletal muscle. J. Cell Biol. 2003, 161, 571–582. [Google Scholar] [CrossRef]
- Vitte, J.M.; Davoult, B.; Roblot, N.; Mayer, M.; Joshi, V.; Courageot, S.; Tronche, F.; Vadrot, J.; Moreau, M.H.; Kemeny, F.; et al. Deletion of murine Smn exon 7 directed to liver leads to severe defect of liver development associated with iron overload. Am. J. Pathol. 2004, 165, 1731–1741. [Google Scholar] [CrossRef]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossoll, W.; Prior, T.W.; et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn-/- mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Arnoux, T.; Bielli, S.; Durand, E.; Rotrou, Y.; Jablonka, S.; Robert, F.; Giraudon-Paoli, M.; Riessland, M.; Mattei, M.G.; et al. Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol. Dis. 2010, 38, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.L.; Xiao, S.; McWhorter, M.L.; Donn, T.; Wolf-Saxon, E.; Bohnsack, M.T.; Moens, C.B.; Beattie, C.E. Zebrafish survival motor neuron mutants exhibit presynaptic neuromuscular junction defects. Hum. Mol. Genet. 2009, 18, 3615–3625. [Google Scholar] [CrossRef] [PubMed]
- Hao Le, t.h.i.; Burghes, A.H.M.; Beattie, C.E. Generation and characterization of a genetic zebrafish model of SMA carrying the human SMN2 gene. Mol. Neurodegener. 2011, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; Jansen, M.D.; Stam, M.; Wijngaarde, C.A.; Curial, C.A.D.; Medic, J.; Sodaar, P.; Schouten, J.; Vijzelaar, R.; Lemmink, H.H.; et al. Intragenic and structural variation in the SMN locus and clinical variability in spinal muscular atrophy. Brain Commun. 2020, 2, fcaa075. [Google Scholar] [CrossRef]
- Prior, T.W.; Nagan, N.; Sugarman, E.A.; Batish, S.D.; Braastad, C. Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med. 2011, 13, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Vijzelaar, R.; Senetsalaar, R.; Clausen, M.; Mason, A.G.; Rinsma, M.; Zegers, M.; Molleman, N.; Boschloo, R.; Yilmaz, R.; Kuilboer, R.; et al. The frequency of SMN gene variants lacking exon 7 and 8 is highly population dependent. PLoS ONE 2019, 14, e0220211. [Google Scholar] [CrossRef]
- Zhong, Q.; Bhattacharya, S.; Kotsopoulos, S.; Olson, J.; Taly, V.; Griffiths, A.D.; Link, D.R.; Larson, J.W. Multiplex digital PCR: Breaking the one target per color barrier of quantitative PCR. Lab Chip 2011, 11, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Stabley, D.L.; Holbrook, J.; Harris, A.W.; Swoboda, K.J.; Crawford, T.O.; Sol-Church, K.; Butchbach, M.E.R. Establishing a reference dataset for the authentication of spinal muscular atrophy cell lines using STR profiling and digital PCR. Neuromuscul. Disord. 2017, 27, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Folch, N.; Gavrilov, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S.; Matern, D.; Oglesbee, D. Multiplex droplet digital PCR method applicable to newborn screening, carrier status and assessment of spinal muscular atrophy. Clin. Chem. 2018, 64, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, H.; Shin, S.; Lee, S.T.; Lee, K.A.; Choi, J.R. Analytical validation of the droplet digital PCR assay for the diagnosis of spinal muscular atrophy. Clin. Chim. Acta 2020, 510, 787–789. [Google Scholar] [CrossRef]
- Jiang, L.; Lin, R.; Gallagher, S.; Zayac, A.; Butchbach, M.E.R.; Hung, P. Development and validation of a 4-color multiplexing spinal muscular atrophy (SMA) genotyping assay on a novel integrated digital PCR instrument. Sci. Rep. 2020, 10, 19892. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.L.; Silver, A.J.; Chan, D.; Borroto, C.; Spurrier, B.; Silver, L.M. Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Med. Genet. 2015, 16, 100. [Google Scholar] [CrossRef]
- Feng, Y.; Ge, X.; Meng, L.; Scull, J.; Li, J.; Tian, X.; Zhang, T.; Jin, W.; Cheng, H.; Wang, X.; et al. The next generation of population-based spinal muscular atrophy carrier screening: Comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet. Med. 2017, 19, 936–994. [Google Scholar] [CrossRef]
- Lopez-Lopez, D.; Loucera, C.; Carmona, R.; Aquino, V.; Salgado, J.; Pasalodos, S.; Miranda, M.; Alonso, Á.; Dopazo, J. SMN1 copy-number and sequence variant analysis from next-generation sequencing data. Hum. Mutat. 2020, 41, 2073–2077. [Google Scholar] [CrossRef]
- Tan, C.A.; Westbrook, M.J.; Truty, R.; Kvitek, D.J.; Kennemer, M.; Winder, T.L.; Shieh, P.B. Incorporating spinal muscular atrophy analysis by next-generation sequencing into a comprehensive multigene panel for neuromuscular diseases. Genet. Test. Mol. Biomark. 2020, 24, 616–624. [Google Scholar] [CrossRef]
- Chen, X.; Sanchis-Juan, A.; French, C.E.; Connell, A.J.; Delon, I.; Kingsbury, Z.; Chawla, A.; Halpern, A.L.; Taft, R.J.; BioResource, N.I.H.R.; et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet. Med. 2020, 22, 945–953. [Google Scholar] [CrossRef]
- Burghes, A.H.M. When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 1997, 61, 9–15. [Google Scholar] [CrossRef]
- Campbell, L.; Potter, A.; Ignatius, J.; Dubowitz, V.; Davies, K. Genomic variation and gene conversion in spinal muscular atrophy: Implications for disease process and clinical phenotype. Am. J. Hum. Genet. 1997, 61, 40–50. [Google Scholar] [CrossRef]
- DiDonato, C.J.; Ingraham, S.E.; Mendell, J.R.; Prior, T.W.; Lenard, S.; Moxley, R.T., III; Florence, J.; Burghes, A.H.M. Deletion and conversion in spinal muscular atrophy: Is there a relationship to severity? Ann. Neurol. 1997, 41, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Hahnen, E.; Schönling, J.; Rudnik-Schöneborn, S.; Zerres, K.; Wirth, B. Hybrid survival motor neuron genes in patients with autosomal recessive spinal muscular atrophy: New insights into molecular mechanisms responsible for the disease. Am. J. Hum. Genet. 1996, 59, 1057–1065. [Google Scholar] [PubMed]
- Van der Steege, G.; Grootscholten, P.M.; Cobben, J.M.; Zappata, S.; Scheffer, H.; den Dunnen, J.T.; van Ommen, G.J.B.; Brahe, C.; Buys, C.H.C.M. Apparent gene conversions involving the SMN gene in the region of the spinal muscular atrophy locus at chromosome 5. Am. J. Hum. Genet. 1996, 59, 834–838. [Google Scholar] [PubMed]
- Devriendt, K.; Lammens, M.; Schollen, E.; Van Hole, C.; Dom, R.; Devlieger, H.; Cassiman, J.J.; Fryns, J.P.; Matthijs, G. Clinical and molecular genetic features of congenital spinal muscular atrophy. Ann. Neurol. 1996, 40, 731–738. [Google Scholar] [CrossRef]
- Qu, Y.J.; Bai, J.L.; Cao, Y.Y.; Wang, H.; Jin, Y.W.; Du, J.; Ge, X.S.; Zhang, W.H.; Li, Y.; He, S.X.; et al. Mutation spectrum of the survival of motor neuron 1 and functional analysis of variants in Chinese spinal muscular atrophy. J. Mol. Diagn. 2016, 18, 741–752. [Google Scholar] [CrossRef]
- Alías, L.; Bernal, S.; Calucho, M.; Martínez, E.; March, F.; Gallano, P.; Fuentes-Prior, P.; Abuli, A.; Serra-Juhe, C.; Tizzano, E.F. Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling. Eur. J. Hum. Genet. 2018, 26, 1554–1557. [Google Scholar] [CrossRef]
- Kubo, Y.; Nishio, H.; Saito, K. A new method for SMN1 and hybrid SMN gene analysis in spinal muscular atrophy using long-range PCR followed by sequencing. J. Hum. Genet. 2015, 60, 233–239. [Google Scholar] [CrossRef]
- Chen, T.H.; Tzeng, C.C.; Wang, C.C.; Wu, S.M.; Chang, J.G.; Yang, S.N.; Hung, C.H.; Jong, Y.J. Identification of bidirectional gene conversion between SMN1 and SMN2 by simultaneous analysis of SMN dosage and hybrid genes in a Chinese population. J. Neurol. Sci. 2011, 308, 83–87. [Google Scholar] [CrossRef]
- Ogino, S.; Gao, S.; Leonard, D.G.B.; Paessler, M.; Wilson, R.B. Inverse correlation between SMN1 and SMN2 copy numbers: Evidence for gene conversion from SMN2 to SMN1. Eur. J. Hum. Genet. 2003, 11, 275–277. [Google Scholar] [CrossRef][Green Version]
- Mazzei, R.; Gambardella, A.; Conforti, F.L.; Magariello, A.; Patitucci, A.; Gabiele, A.L.; Sprovieri, T.; Labate, A.; Valentino, P.; Bono, F.; et al. Gene conversion events in adult-onset spinal muscular atrophy. Acta Neurol. Scand. 2004, 109, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, I.; Barceló, M.J.; del Rio, E.; Martín, Y.; Hernández-Chico, C.; Bussaglia, E.; Baiget, M.; Tizzano, E.F. Characterization of SMN hybrid genes in Spanish SMA patients: De novo, homozygous and compound heterozygous cases. Hum. Genet. 2001, 108, 222–229. [Google Scholar] [PubMed]
- Wirth, B. Spinal muscular atrophy: In the challenge lies a solution. Trends Neurosci. 2021, 44, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Ruhno, C.; McGovern, V.L.; Avenarius, M.R.; Snyder, P.J.; Prior, T.W.; Nery, F.C.; Muhtaseb, A.; Roggenbuck, J.S.; Kissel, J.T.; Sansone, V.A.; et al. Complete sequencing of the SMN2 gene in SMA patients detect SMN gene deletion junctions and variants in SMN that modify the SMA phenotype. Hum. Genet. 2019, 138, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Pérez, L.; Paramonov, I.; Leno, J.; Bernal, S.; Alías, L.; Fuentes-Prior, P.; Cuscó, I.; Tizzano, E.F. Beyond copy number: A new, rapid and versatile methods for sequencing the entire SMN2 gene in SMA patients. Hum. Mutat. 2021, 42, 787–795. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.H.; Sun, J.; Krainer, A.R.; Hua, Y.; Prior, T.W. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 2768–2780. [Google Scholar] [CrossRef]
- Kashima, T.; Rao, N.; Manley, J.L. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 3426–3431. [Google Scholar] [CrossRef]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.M.; Kissel, J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Vezain, M.; Saukkonen, A.M.; Goina, E.; Touraine, R.; Manel, V.; Toutain, A.; Fehrenbach, S.; Frébourg, T.; Pagani, F.; Tosi, M.; et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010, 31, E1110–E1125. [Google Scholar] [CrossRef]
- Bernal, S.; Alías, L.; Barceló, M.J.; Also-Rallo, E.; Martínez-Hernández, R.; Gámez, J.; Guillén-Navarro, E.; Rosell, J.; Hernando, I.; Rodríguez-Alvarez, F.J.; et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J. Med. Genet. 2010, 47, 640–642. [Google Scholar] [CrossRef]
- Gambardella, A.; Mazzei, R.; Toscano, A.; Annesi, G.; Pasqua, A.; Annesi, F.; Quattrone, F.; Oliveri, R.L.; Valentino, P.; Bono, F.; et al. Spinal muscular atrophy due to an isolated deletion of exon 8 of the telomeric survival motor neuron gene. Ann. Neurol. 1998, 44, 836–839. [Google Scholar] [CrossRef]
- Thauvin-Robinet, C.; Drunat, S.; Saugier Veber, P.; Cantereau, D.; Cossée, M.; Cassini, C.; Soichot, P.; Masurel-Paulet, A.; De Monléon, J.V.; Sagot, P.; et al. Homozygous SMN1 exons 1-6 deletion: Pitfalls in genetic counseling and general recommendations for spinal muscular atrophy molecular diagnosis. Am. J. Med. Genet. 2012, 158, 1735–1741. [Google Scholar] [CrossRef]
- Wirth, B.; Herz, M.; Wetter, A.; Moskau, S.; Hahnen, E.; Rudnik-Schöneborn, S.; Wienker, T.; Zerres, K. Quantitative analysis of survival motor neuron copies: Identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation and implications for genetic counseling. Am. J. Hum. Genet. 1999, 64, 1340–1356. [Google Scholar] [CrossRef]
- Jedličková, I.; Přistoupilová, A.; Nosková, L.; Majer, F.; Stránecký, V.; Hartmannová, H.; Hodaňová, K.; Trešlová, H.; Hýblová, M.; Solár, P.; et al. Spinal muscular atrophy caused by a novel Alu-mediated deletion of exons 2a-5 in SMN1 undetectable with routine genetic testing. Mol. Genet. Genomic Med. 2020, 8, e1238. [Google Scholar] [CrossRef]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef]
- Ottesen, E.W.; Seo, J.; Singh, N.N.; Singh, R.N. A multilayered control of the human Survival Motor Neuron gene expression by Alu elements. Front. Microbiol. 2017, 8, 2252. [Google Scholar] [CrossRef]
- Sen, S.K.; Han, K.S.; Wang, J.; Lee, J.; Wang, H.; Callinan, P.A.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, M.A. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006, 79, 41–53. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in type 1 spinal muscular atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef]
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Finkel, R.; Howell, R.R.; Klinger, K.W.; Kuntz, N.; et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef]
- Glascock, J.; Sampson, J.; Connolly, A.M.; Darras, B.T.; Day, J.W.; Finkel, R.; Howell, R.R.; Klinger, K.W.; Kuntz, N.; Prior, T.; et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J. Neuromuscul. Dis. 2020, 7, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.L.; Qu, Y.J.; Cao, Y.Y.; Li, E.Z.; Wang, L.W.; Li, Y.; Zhu, Y.L.; Zhang, W.H.; Jin, Y.W.; Wang, H.; et al. Subtle mutation detection of SMN1 gene in Chinese spinal muscular atrophy patients: Implications of molecular diagnostic procedure for SMN1 gene mutations. Genet. Test. Mol. Biomark. 2014, 18, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Grimmler, M.; Schwarzer, V.; Schoenen, F.; Fischer, U.; Wirth, B. Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum. Mutat. 2005, 25, 64–71. [Google Scholar] [CrossRef]
- Wijaya, Y.O.S.; Ar Rochmah, M.; Niba, E.T.E.; Morisada, N.; Noguchi, Y.; Hidaka, Y.; Ozasa, S.; Inoue, T.; Shimazu, T.; Takahashi, Y.; et al. Phenotypes of SMA patients retaining SMN1 with intragenic mutation. Brain Dev. 2021, 43, 745–758. [Google Scholar] [CrossRef]
- Prior, T.W. Spinal muscular atrophy diagnostics. J. Child Neurol. 2007, 22, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Sossi, V.; Giuli, A.; Vitali, T.; Tiziano, F.; Mirabella, M.; Antonelli, A.; Neri, G.; Brahe, C. Premature termination mutations in exon 3 of the SMN1 gene are associated with exon skipping and a relatively mild SMA phenotype. Eur. J. Hum. Genet. 2001, 9, 113–120. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mendonça, R.H.; Matsui Jr, C.; Polido, G.J.; Silva, A.M.S.; Kulikowski, L.; Torchio Dias, A.; Zanardo, E.A.; Solla, D.J.F.; Gurgel-Giannetti, J.; de Moura, A.C.M.L.; et al. Intragenic variants in the SMN1 gene determine the clinical phenotype in 5q spinal muscular atrophy. Neurol. Genet. 2020, 6, e505. [Google Scholar] [CrossRef] [PubMed]
- Brichta, L.; Garbes, L.; Jedrzejowska, M.; Grellscheid, S.N.; Holker, I.; Zimmermann, K.; Wirth, B. Nonsense-mediated messenger RNA decay of survival motor neuron 1 causes spinal muscular atrophy. Hum. Genet. 2008, 123, 141–153. [Google Scholar] [CrossRef]
- Alías, L.; Bernal, S.; Fuentes-Prior, P.; Barceló, M.J.; Also, E.; Martínez-Hernández, R.; Rodríguez-Alvarez, F.J.; Martín, Y.; Aller, E.; Grau, E.; et al. Mutation update of spinal muscular atrophy in Spain: Molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum. Genet. 2009, 125, 29–39. [Google Scholar] [CrossRef]
- Tsai, C.H.; Jong, Y.J.; Hu, C.J.; Chen, C.M.; Shih, M.C.; Chang, C.P.; Chang, J.G. Molecular analysis of SMN, NAIP and P44 genes of SMA patients and their families. J. Neurol. Sci. 2001, 190, 35–40. [Google Scholar] [CrossRef]
- Xu, Y.; Xiao, B.; Liu, Y.; Qu, X.X.; Dai, M.Y.; Ying, X.M.; Jiang, W.T.; Zhang, J.M.; Liu, X.Q.; Chen, Y.W.; et al. Identification of novel SMN1 subtle mutations using an allele-specific RT-PCR. Neuromuscul. Disord. 2020, 30, 219–226. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, Q.J.; Lin, Q.F.; Chen, Y.F.; Lin, X.Z.; Lin, M.T.; Murong, S.X.; Wang, N.; Chen, W.J. Molecular analysis of SMN1, SMN2, NAIP, GTF2H2 and H4F5 genes in 157 Chinese patients with spinal muscular atrophy. Gene 2013, 518, 325–329. [Google Scholar] [CrossRef]
- Ganji, H.; Nouri, N.; Salehi, M.; Aryani, O.; Houshmand, M.; Basiri, K.; Fazel-Najafabadi, E.; Sedghi, M. Deletion of intragenic SMN1 mutations in spinal muscular atrophy patients with a single copy of SMN1. J. Child Neurol. 2015, 30, 558–562. [Google Scholar] [CrossRef]
- Zeng, J.; Lin, Y.; Yan, A.; Ke, L.; Zhu, Z.; Lan, F. Establishment of a molecular diagnostic system for spinal muscular atrophy. Experience from a clinical laboratory in China. J. Mol. Diagn. 2011, 13, 41–47. [Google Scholar] [CrossRef]
- Qu, Y.; Du, J.; Li, E.; Bai, J.; Jin, Y.; Hong, W.; Wang, H.; Song, F. Subtle mutations in the SMN1 gene in Chinese patients with SMA: p.Arg288Met mutation causing SMN1 transcript exclusion of exon7. BMC Med. Genet. 2012, 13, 86. [Google Scholar]
- Skordis, L.A.; Dunckley, M.G.; Burglen, L.; Campbell, L.; Talbot, K.; Patel, S.; Melki, J.; Davies, J.E.; Dubowitz, V.; Muntoni, F. Characterisation of novel point mutations in the survival motor neuron gene SMN, in three patients with SMA. Hum. Genet. 2001, 108, 356–357. [Google Scholar] [CrossRef]
- Zapletalová, E.; Hedvicáková, P.; Kozák, L.; Vondrácek, P.; Gaillyová, R.; Maríková, T.; Kalina, Z.; Jüttnerová, V.; Fajkus, J.; Fajkusová, L. Analysis of point mutations in the SMN1 gene in SMA patients bearing a single SMN1 copy. Neuromuscul. Disord. 2007, 17, 476–481. [Google Scholar] [CrossRef]
- Jedrzejowska, M.; Gos, M.; Zimowski, J.G.; Kostera-Pruszczyk, A.; Ryniewicz, B.; Hausmanowa-Petrusewicz, I. Novel point mutations in survival motor neuron 1 gene expand the spectrum of phenotypes observed in spinal muscular atrophy patients. Neuromuscul. Disord. 2014, 24, 617–623. [Google Scholar] [CrossRef]
- Clermont, O.; Burlet, P.; Benit, P.; Chanterau, S.; Saugier-Veber, P.; Munnich, A.; Cusin, V. Molecular analysis of SMA patients without homozygous SMN1 deletions using a new strategy for identification of SMN1 subtle mutations. Hum. Mutat. 2004, 24, 417–427. [Google Scholar] [CrossRef]
- Bussaglia, E.; Clermont, O.; Tizzano, E.; Lefebvre, S.; Bürglen, L.; Cruaud, C.; Urtizberea, J.A.; Colomer, J.; Munnich, A.; Baiget, M.; et al. A frame-shift deletion in the survival motor neuron gene in Spanish spinal muscular atrophy patients. Nat. Genet. 2004, 11, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Brahe, C.; Clermont, O.; Zappata, S.; Tiziano, F.; Melki, J.; Neri, G. Frameshift mutation in the survival motor neuron gene in a severe case of SMA type I. Hum. Mol. Genet. 1996, 5, 1971–1976. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gonçalves-Rocha, M.; Oliveira, J.R.M.; Rodrigues, L.; Santos, R. New approaches in molecular diagnosis and population carrier screening for spinal muscular atrophy. Genet. Test. Mol. Biomark. 2011, 15, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; McAndrew, P.E.; Iannaccone, S.T.; Mendell, J.R.; Burghes, A.H.M.; Prior, T.W. Intragenic telSMN mutations: Frequency, distribution, evidence of a founder effect and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am. J. Hum. Genet. 1998, 63, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; McAndrew, P.E.; Allinson, P.S.; Parker, W.D., Jr.; Burghes, A.H.M.; Prior, T.W. Diagnosis of spinal muscular atrophy in an SMN non-deletion patient with quantitative PCR screen and mutation analysis. J. Med. Genet. 1998, 35, 674–676. [Google Scholar] [CrossRef]
- Sharifi, Z.; Taheri, M.; Fallah, M.S.; Abiri, M.; Golnabi, F.; Begherian, H.; Zeinali, R.; Farahzadi, H.; Alborji, M.; Tehrani, P.G.; et al. Comprehensive mutation analysis and report of 12 novel mutations in a cohort of patients with spinal muscular atrophy in Iran. J. Mol. Neurosci. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Martín, Y.; Valero, A.; del Castillo, E.; Pascual, S.I.; Hernández-Chico, C. Genetic study of SMA patients without homozygous SMN1 deletions: Identification of compound heterozygotes and characterisation of novel intragenic SMN1 mutations. Hum. Genet. 2002, 110, 257–263. [Google Scholar] [CrossRef]
- Parsons, D.W.; McAndrew, P.E.; Monani, U.R.; Mendell, J.R.; Burghes, A.H.M.; Prior, T.W. An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: Further evidence for SMN as the primary SMA-determining gene. Hum. Mol. Genet. 1996, 5, 1727–1732. [Google Scholar] [CrossRef]
- Takarada, T.; Ar Rochmah, M.; Harahap, N.I.F.; Shinohara, M.; Saito, T.; Saito, K.; Lai, P.S.; Bouike, Y.; Takeshima, Y.; Awano, H.; et al. SMA mutations in SMN Tudor and C-terminal domains destabilize the protein. Brain Dev. 2017, 39, 606–612. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sato, H.; Lai, P.S.; Nurputra, D.K.; Harahap, N.I.F.; Morikawa, S.; Nishimura, N.; Kurashige, T.; Ohshita, T.; Nakajima, H.; et al. Intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some spinal muscular atrophy (SMA) patients. Brain Dev. 2014, 36, 914–920. [Google Scholar] [CrossRef]
- Kotani, T.; Sutomo, R.; Sasongko, T.H.; Sadewa Gunadi, A.H.; Minato, T.; Fujii, E.; Endo, S.; Lee, M.J.; Ayaki, M.; Harada, Y.; et al. A novel mutation at the N-terminal of SMN Tudor domain inhibits its interaction with target proteins. J. Neurol. 2007, 254, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, Y.; Mei, S.; Chen, C.H.; Liu, L.; Wang, C.; Zhao, G.; Kong, X. Identification of two novel SMN1 point mutations associated with a very severe SMA-I phenotype. Eur. J. Med. Genet. 2020, 63, 104006. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, I.; Barceló, M.J.; del Río, E.; Baiget, M.; Tizzano, E.F. Detection of novel mutations in the SMN Tudor domain in type I SMA patients. Neurology 2004, 63, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Fraidakis, M.J.; Drunat, S.; Maisonobe, T.; Gerard, B.; Pradat, P.F.; Meininger, V.; Salachas, F. Genotype-phenotype relationship in 2 SMA III patients with novel mutations in the Tudor domain. Neurology 2012, 78, 551–556. [Google Scholar] [CrossRef]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Wadman, R.I.; Stam, M.; Gijzen, M.; Lemmink, H.H.; Snoeck, I.N.; Wijngaarde, C.A.; Braun, K.P.J.; Schoenmakers, M.A.G.C.; van den Berg, L.H.; Dooijes, D.; et al. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0-4. J. Neurol. Neurosurg. Psychiatry 2017, 88, 365–367. [Google Scholar] [CrossRef]
- Rochette, C.F.; Surh, L.C.; Ray, R.N.; McAndrew, P.E.; Prior, T.W.; Burghes, A.H.M.; Vanasse, M.; Simard, L.R. Molecular diagnosis of non-deletion SMA patients using quantitative PCR of SMN exon 7. Neurogenetics 1997, 1, 141–147. [Google Scholar] [CrossRef]
- Hahnen, E.; Schönling, J.; Rudnik-Schöneborn, S.; Raschke, H.; Zerres, K.; Wirth, B. Missense mutations in exon 6 of the survival motor neuron gene in patients with spinal muscular atrophy (SMA). Hum. Mol. Genet. 1997, 6, 821–825. [Google Scholar] [CrossRef]
- Sumner, C.J.; Huynh, T.N.; Markowitz, J.A.; Perhac, J.S.; Hill, B.; Coovert, D.D.; Schussler, K.; Chen, X.; Jarecki, J.; Burghes, A.H.M.; et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 2003, 54, 647–654. [Google Scholar] [CrossRef]
- Ar Rochmah, M.; Awano, H.; Awaya, T.; Harahap, N.I.F.; Morisada, N.; Bouike, Y.; Saito, A.; Kubo, Y.; Saito, K.; Lai, P.S.; et al. Spinal muscular atrophy carriers with two SMN1 copies. Brain Dev. 2017, 39, 851–860. [Google Scholar] [CrossRef]
- Wang, C.H.; Papendick, B.D.; Bruinsma, P.; Day, J.K. Identification of a novel missense mutation of the SMNT gene in two siblings with spinal muscular atrophy. Neurogenetics 1998, 1, 273–276. [Google Scholar] [PubMed]
- Talbot, K.; Ponting, C.P.; Theodosiou, A.M.; Rodrigues, N.R.; Surtees, R.; Mountford, R.; Davies, K.E. Missense mutation clustering in the survival motor neuron gene: A role for conserved tyrosine and glycine rich region of the protein in RNA metabolism? Hum. Mol. Genet. 1997, 6, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Cho, S.I.; Chae, J.H.; Chung, K.N.; Ra, E.K.; Kim, S.Y.; Seong, M.W.; Kim, J.Y.; Park, S.S. False homozygous deletions of SMN1 exon 7 using Dra I PCR-RFLP caused by a novel mutation in spinal muscular atrophy. Genet. Test. Mol. Biomark. 2009, 13, 511–513. [Google Scholar] [CrossRef]
- Qu, Y.J.; Song, F.; Yang, Y.L.; Jin, Y.W.; Bai, J.L. Compound heterozygous mutation in two unrelated cases of Chinese spinal muscular atrophy patients. Chin. Med. J. 2011, 124, 385–389. [Google Scholar] [PubMed]
- Zhu, S.Y.; Xiong, F.; Chen, Y.J.; Yan, T.Z.; Zeng, J.; Li, L.; Zhang, Y.N.; Chen, W.Q.; Bao, X.H.; Zhang, C.; et al. Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population. Eur. J. Hum. Genet. 2010, 18, 978–984. [Google Scholar]
- Eggermann, T.; Eggermann, K.; Elbracht, M.; Zerres, K.; Rudnik-Schöneborn, S. A new splice site mutation in the SMN1 gene causes discrepant results in SMN1 deletion screening approaches. Neuromuscul. Disord. 2008, 18, 146–149. [Google Scholar] [CrossRef]
- Monani, U.R.; Pastore, M.T.; Gavrilina, T.O.; Jablonka, S.; Le, T.T.; Andreassi, C.; DiCocco, J.M.; Lorson, C.; Androphy, E.J.; Sendtner, M.; et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell Biol. 2003, 160, 41–52. [Google Scholar] [CrossRef]
- Workman, E.; Saieva, L.; Carrel, T.L.; Crawford, T.O.; Liu, D.; Lutz, C.; Beattie, C.E.; Pellizzoni, L.; Burghes, A.H.M. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum. Mol. Genet. 2009, 18, 2115–2229. [Google Scholar] [CrossRef]
- Iyer, C.C.; Corlett, K.M.; Massoni-Laporte, A.; Duque, S.I.; Madabusi, N.; Tisdale, S.; McGovern, V.L.; Le, T.T.; Zaworksi, P.G.; Arnold, W.D.; et al. Mild SMN missense alleles are only functional in the presence of SMN2 in mammals. Hum. Mol. Genet. 2018, 27, 3404–3416. [Google Scholar] [CrossRef] [PubMed]
- Carrel, T.L.; McWhorter, M.L.; Workman, E.; Zhang, H.; Wolstencroft, E.C.; Lorson, C.; Bassell, G.J.; Burghes, A.H.M.; Beattie, C.E. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J. Neurosci. 2006, 26, 11014–11022. [Google Scholar] [CrossRef] [PubMed]
- McGovern, V.L.; Kray, K.M.; Arnold, W.D.; Duque, S.I.; Iyer, C.C.; Massoni-Laporte, A.; Workman, E.; Patel, A.; Battle, D.J.; Burghes, A.H.M. Intragenic complementation of amino and carboxy terminal SMN missense mutations can rescue Smn null mice. Hum. Mol. Genet. 2020, 29, 3493–3503. [Google Scholar] [CrossRef]
- Mailman, M.D.; Hemingway, T.; Darsey, R.L.; Glasure, C.E.; Huang, Y.; Chadwick, R.B.; Heinz, J.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; et al. Hybrids monosomal for human chromosome 5 reveal the presence of a spinal muscular atrophy (SMA) carrier with two SMN1 copies on one chromosome. Hum. Genet. 2001, 108, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Alías, L.; Barceló, M.J.; Bernal, S.; Martínez-Hernández, R.; Also-Rallo, E.; Vázquez, C.; Santana, A.; Millán, J.M.; Baiget, M.; Tizzano, E.F. Improving detection and genetic counseling in carriers of spinal muscular atrophy with two copies of the SMN1 gene. Clin. Genet. 2014, 85, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, L.; Peter, I.; Zhu, J.; Scott, S.A.; Zhao, G.; Eversley, C.; Kornreich, R.; Desnick, R.J.; Edelmann, L. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet. Med. 2014, 16, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Bayrak-Toydemir, P.; Lynnes, T.C.; Mao, R.; Metcalf, J.D.; Muralidharan, K.; Iwata-Otsubo, A.; Pham, H.T.; Pratt, V.M.; Qureshi, S.A.; et al. Characterization of reference materials for spinal muscular atrophy genetic testing: A Genetic Testing Reference Materials Coordination Program collaborative project. J. Mol. Diagn. 2021, 23, 103–110. [Google Scholar] [CrossRef]
- Ceylan, A.C.; Erdem, H.B.; Şahin, İ.; Agarwal, M. SMN1 gene copy number analysis for spinal muscular atrophy (SMA) in a Turkish cohort by CODE-SEQ technology, an integrated solution for detection of SMN1 and SMN2 copy numbers and the “2+0” genotype. Neurol. Sci. 2020, 41, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.K.; Huang, C.K.; Jin, H.; Zou, H.; Yanakakis, L.; Du, J.; Fiddler, M.; Naeem, R.; Goldstein, Y. Enhanced carrier screening for spinal muscular atrophy: Detection of silent (SMN1:2+0) carriers utilizing a novel TaqMan genotyping method. Lab. Med. 2020, 51, 408–415. [Google Scholar] [CrossRef]
- Talbot, J.; Rodrigues, N.; Bernert, G.; Bittner, R.; Davies, K. Evidence for compound heterozygositiy causing mild and severe forms of autosomal recessive spinal muscular atrophy. J. Med. Genet. 1996, 33, 1019–1021. [Google Scholar] [CrossRef][Green Version]
- Muqit, M.M.K.; Moss, J.; Sewry, C.; Lane, R.J.M. Phenotypic variability in siblings with type III spinal muscular atrophy. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1762–1764. [Google Scholar] [CrossRef]
- Arkblad, E.; Tulinius, M.; Kroksmark, A.K.; Henricsson, M.; Darin, N. A population-based study of genotypic and phenotypic variability in children with spinal muscular atrophy. Acta Paediatr. 2009, 98, 865–872. [Google Scholar] [CrossRef]
- Jones, C.C.; Cook, S.F.; Jarecki, J.; Belter, L.; Reyna, S.P.; Staropoli, J.; Farwell, W.; Hobby, K. Spinal muscular atrophy (SMA) subtype concordance in siblings: Findings from the Cure SMA cohort. J. Neuromuscul. Dis. 2020, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Burghes, A.H.M.; Ingraham, S.E.; Kóte-Jarai, Z.; Rosenfeld, S.; Herta, N.; Nadkarni, N.; DiDonato, C.J.; Carpten, J.; Hurko, O.; Florence, J.; et al. Linkage mapping of the spinal muscular atrophy gene. Hum. Genet. 1994, 93, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Hahnen, E.; Forkert, R.; Marke, C.; Rudnik-Schöneborn, S.; Schönling, J.; Zerres, K.; Wirth, B. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum. Mol. Genet. 1995, 4, 1927–1933. [Google Scholar] [CrossRef]
- Cobben, J.M.; van der Steege, G.; Grootscholten, P.; de Visser, M.; Scheffer, H.; Buys, C.H.C.M. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am. J. Hum. Genet. 1995, 57, 805–808. [Google Scholar] [PubMed]
- Cuscó, I.; Barceló, M.J.; Rojas-García, R.; Illa, I.; Gámez, J.; Cervera, C.; Pou, A.; Izquierdo, G.; Baiget, M.; Tizzano, E.F. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J. Neurol. 2006, 253, 21–25. [Google Scholar] [CrossRef]
- Wirth, B.; Garbes, L.; Riessland, M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic applications. Curr. Opin. Genet. Dev. 2013, 23, 330–338. [Google Scholar] [CrossRef]
- Oprea, G.E.; Kröber, S.; McWhorter, M.L.; Rossoll, W.; Müller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef]
- Stratigopoulos, G.; Lanzano, P.; Deng, L.; Guo, J.; Kaufmann, P.; Darras, B.; Finkel, R.; Tawil, R.; McDermott, M.P.; Martens, W.; et al. Association of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Arch. Neurol. 2010, 67, 1252–1256. [Google Scholar] [CrossRef]
- Yanyan, C.; Yujin, Q.; Jinli, B.; Yuwei, J.; Hong, W.; Fang, S. Correlation of PLS3 expression with disease severity in children with spinal muscular atrophy. J. Hum. Genet. 2014, 59, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.; Also-Rallo, E.; Martínez-Hernández, R.; Alías, L.; Rodríguez-Alvarez, F.J.; Millán, J.M.; Hernández-Chico, C.; Baiget, M.; Tizzano, E.F. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul. Disord. 2011, 21, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Yener, I.H.; Topaloglu, H.; Erdem-Özdamar, S.; Dayangac-Erdem, D. Transcript levels of plastin 3 and neuritin 1 modifer genes in spinal muscular atrophy siblings. Pediatr. Int. 2017, 59, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; Jansen, M.D.; Curial, C.A.D.; groen, E.J.N.; Stam, M.; Wijngaarde, C.A.; Medic, J.; Sodaar, P.; van Eijk, K.R.; Huibers, M.M.H.; et al. Analysis of FUS, PFN2, TDP-43 and PLS3 as potential disease severity modifiers in spinal muscular atrophy. Neurol. Genet. 2020, 6, e386. [Google Scholar] [CrossRef] [PubMed]
- Hosseinibarkooie, S.; Peters, M.; Torres-Benito, L.; Rastetter, R.H.; Hupperich, K.; Hoffmann, A.; Mendoza-Ferreira, N.; Kaczmarek, A.; Janzen, E.; Milbradt, J.; et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet. 2016, 99, 647–665. [Google Scholar] [CrossRef] [PubMed]
- Janzen, E.; Mendoza-Ferreira, N.; Hosseinibarkooie, S.; Schneider, S.; Hupperich, K.; Tschanz, T.; Grysko, V.; Riessland, M.; Hammerschmidt, M.; Rigo, F.; et al. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 2018, 141, 2343–2361. [Google Scholar] [CrossRef]
- Eshraghi, M.; McFall, E.; Gibeault, S.; Kothary, R. Effect of genetic background on the phenotype of the Smn2B/- mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 4494–4506. [Google Scholar]
- Ackermann, B.; Kröber, S.; Torres-Benito, L.; Borgmann, A.; Peters, M.; Barkooie, S.M.H.; Tejero, R.; Jakubik, M.; Schreml, J.; Milbradt, J.; et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 2013, 22, 1328–1347. [Google Scholar] [CrossRef]
- McGovern, V.L.; Massoni-Laporte, A.; Wang, X.; Le, T.T.; Le, H.T.; Beattie, C.E.; Rich, M.M.; Burghes, A.H.M. Plastin 3 expression does not modify spinal muscular atrophy severity in the Δ7 SMA mouse. PLoS ONE 2015, 10, e0132364. [Google Scholar] [CrossRef]
- Kaifer, K.A.; Villalón, E.; Osman, E.Y.; Glascock, J.J.; Arnold, L.L.; Cornelison, D.D.W.; Lorson, C.L. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2017, 2, e89970. [Google Scholar] [CrossRef]
- Alrafiah, A.; Karyka, E.; Coldicott, I.; Iremonger, K.; Lewis, K.E.; Ning, K.; Azzouz, M. Plastin 3 promotes motor axonal growth and extends survival in a mouse model of spinal muscular atrophy. Mol. Ther. Methods Clin. Dev. 2018, 9, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Löhr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta suppression protects against spinal muscular atrophy in humans and acress species by restoring impaired endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315. [Google Scholar] [CrossRef]
- Jiang, J.; Huang, J.; Gu, J.; Cai, X.; Zhao, H.; Lu, H. Genomic analysis of a spinal muscular atrophy (SMA) discordant family identifies a novel mutation in TLL2, an activator of growth differentiation factor 8 (myostatin): A case report. BMC Med. Genet. 2019, 20, 204. [Google Scholar] [CrossRef]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Pauschkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Akten, B.; Kye, M.J.; Hao Le, t.h.i.; Wertz, M.H.; Singh, S.; Nie, D.; Huang, J.; Merianda, T.T.; Twiss, J.L.; Beattie, C.E.; et al. Interaction of survival of motor neuron (SMN) with HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc. Natl. Acad. Sci. USA 2011, 108, 10337–10342. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.J.; Kobayashi, D.T.; Lynes, M.M.; Naryshkin, N.N.; Tiziano, F.D.; Zaworski, P.G.; Rubin, L.L.; Jarecki, J. Assays for the identification and prioritization of drug candidates for spinal muscular atrophy. Assay Drug Dev. Technol. 2014, 12, 315–341. [Google Scholar] [CrossRef]
- Arnold, W.D.; Burghes, A.H.M. Spinal muscular atrophy: Development and implementation of potential therapeutics. Ann. Neurol. 2013, 74, 348–362. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Mutation | Mutation | Phenotype | Reference(s) |
|---|---|---|---|
| Nonsense mutations | p.E14X | I | [127,155] |
| p.Q15X | I, II, III | [127,144,155,156] | |
| p.Q27X | I | [157] | |
| p.S63X | I | [157] | |
| p.W102X | II, III | [158,159] | |
| p.Q154X | III | [160] | |
| p.Q157X | II | [160,161] | |
| p.W190X | I | [162] | |
| p.L228X | I | [127,155,163,164] | |
| p.Q282X | I | [165] | |
| p.R288X | II | [166] | |
| Frameshift mutations | c.-7-9del | III | [127] |
| c.19delG | I | [164] | |
| c.22dupA | I, II, III | [127,155,163,164,167,168] | |
| c.48_55dupGGATTCCG | I | [87] | |
| c.56delT | II | [127,155,168] | |
| c.81+1dupG | II | [169,170] | |
| c.90_91insT | I, II | [144,162] | |
| c.98delT | I | [170] | |
| c.100delT | N/A | [171] | |
| c.109dupA | N/A | [158] | |
| c.124insT | I | [144] | |
| c.198_214del | N/A | [172] | |
| c.208_209ins4 | III | [144] | |
| c.241-242in4 | III | [144] | |
| c.286delG | I | [166] | |
| c.312dupA | III | [162] | |
| c.314_317dup | III | [172] | |
| c.401_402delAG | I | [164] | |
| c.411delT | I | [162] | |
| c.429_435del | I | [171] | |
| c.430del4 | I, II, III | [173] | |
| c.431delC | I | [171] | |
| c.439_443del | I | [159,174] | |
| c.472del5 | I | [174] | |
| c.509_510delGT | N/A | [158] | |
| c.524delC | N/A | [175] | |
| c.542delGT | II | [176,177] | |
| c.549delC | N/A | [178] | |
| c.551_552insA | I | [164] | |
| c.585dupT | I | [172] | |
| c.591delA | II | [144] | |
| c.627_628ins65 | I | [161] | |
| c.722delC | I | [171] | |
| c.734_735insC | I | [160,175] | |
| c.735_736insA | N/A | [178] | |
| c.740dupC | N/A | [179] | |
| c.744delC | I | [127] | |
| c.770-780dup11 | I | [158,160,162,172,175] | |
| c.773insC | III | [179] | |
| c.811_814dupGGCT | II | [127] | |
| c.813ins/dup11 | I, II | [176,179,180] | |
| c.819_820insT | I | [157,181,182] | |
| Missense mutations | p.A2G | II, III | [127,155,158,160,176] |
| p.A2V | III | [129,157,182] | |
| p.D30N | II | [156] | |
| p.D44V | III | [156] | |
| p.W92S | I | [157,181,182,183] | |
| p.V94F | I | [184] | |
| p.V94G | II | [172] | |
| p.G95R | III | [156,178] | |
| p.Y109C | III | [127] | |
| p.A111G | I | [156] | |
| p.I116F | I | [160,175,185] | |
| p.Y130C | III | [158,186] | |
| p.Y130H | III | [186] | |
| p.E134K | I, II | [127,156,168,187] | |
| p.Q136E | I | [185] | |
| p.S139S | N/A | [158,159] | |
| p.L141P | I | [157] | |
| p.D181G | N/A | [188] | |
| p.A188S | I | [170] | |
| p.P221L | I | [87] | |
| p.S230L | II, III | [127,165,168,171] | |
| p.P244L | III | [171] | |
| p.P245L | III | [189] | |
| p.L260S | II | [172] | |
| p.S262G | III | [156] | |
| p.S262I | III | [65,158,190] | |
| p.M263R | I | [172] | |
| p.M263T | II | [162] | |
| p.S266P | II | [158] | |
| p.M269T | III | [160] | |
| p.Y272C | I, II, III | [144,156,162,164,170,172,189,191] | |
| p.H273R | II | [158] | |
| p.T274I | II, III | [71,144,170,190] | |
| p.G275S | III | [172] | |
| p.Y276H | I | [157,192] | |
| p.Y277C | II, III | [127,129,168,182] | |
| p.G279C | II, III | [178,193] | |
| p.G279V | I | [194] | |
| p.G279D | N/A | [143] | |
| p.F280I | N/A | [178] | |
| p.R288M | I, II | [168,195,196] | |
| Splice-site mutations | c.*3+3A>T | I | [184] |
| c.628-140A>G | N/A | [161] | |
| c.834+2T>G | I | [162] | |
| c.835-1G>A | III | [197] | |
| c.835-2A>G | I | [157,198] | |
| c.835-3C>A | I | [157] | |
| c.835-5T>G | I | [127] | |
| c.867+2T>G | I | [179] | |
| c.868-11del7 | I | [37] | |
| c.888+3delAGAG | I | [37,172] | |
| c.922+3del4 | I | [97] | |
| c.922+6T>G | III | [144] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butchbach, M.E.R. Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. Int. J. Mol. Sci. 2021, 22, 7896. https://doi.org/10.3390/ijms22157896
Butchbach MER. Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. International Journal of Molecular Sciences. 2021; 22(15):7896. https://doi.org/10.3390/ijms22157896
Chicago/Turabian StyleButchbach, Matthew E. R. 2021. "Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development" International Journal of Molecular Sciences 22, no. 15: 7896. https://doi.org/10.3390/ijms22157896
APA StyleButchbach, M. E. R. (2021). Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. International Journal of Molecular Sciences, 22(15), 7896. https://doi.org/10.3390/ijms22157896