
Fibroblast Growth Factor 21 Response in a Preclinical Alcohol Model of Acute-on-Chronic Liver Injury

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. ACLI Model
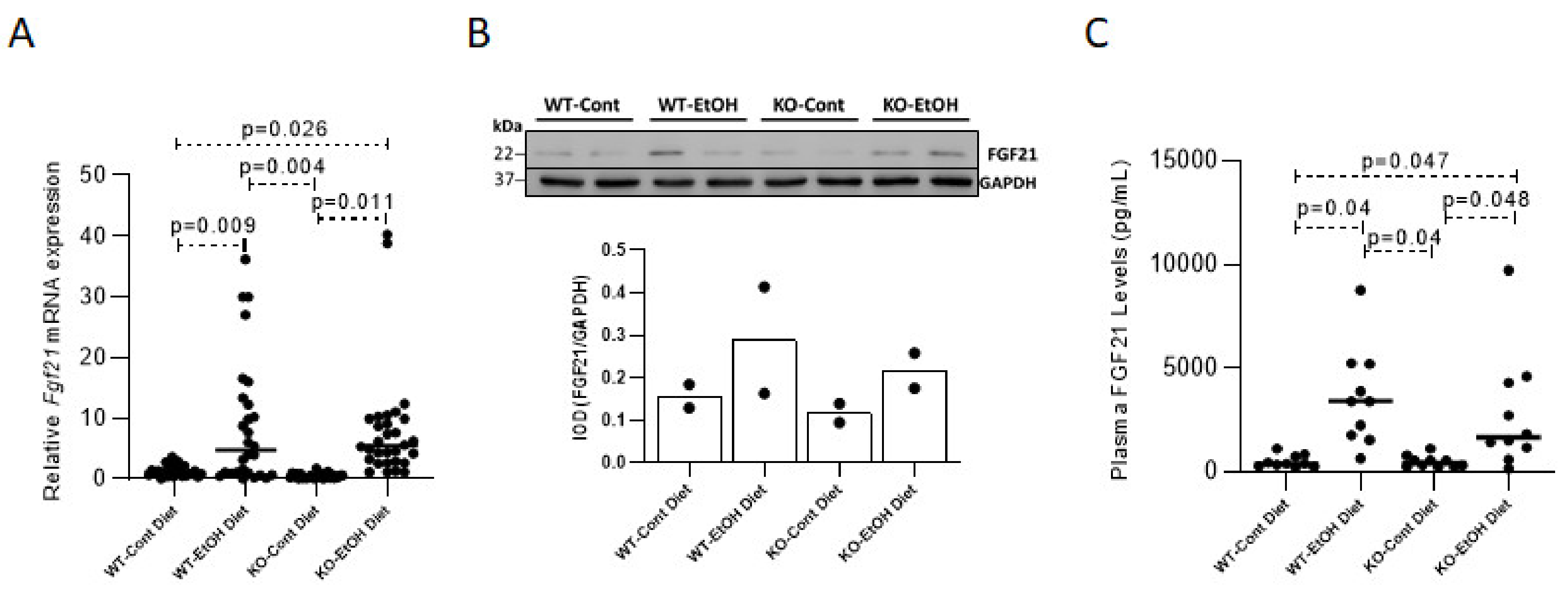
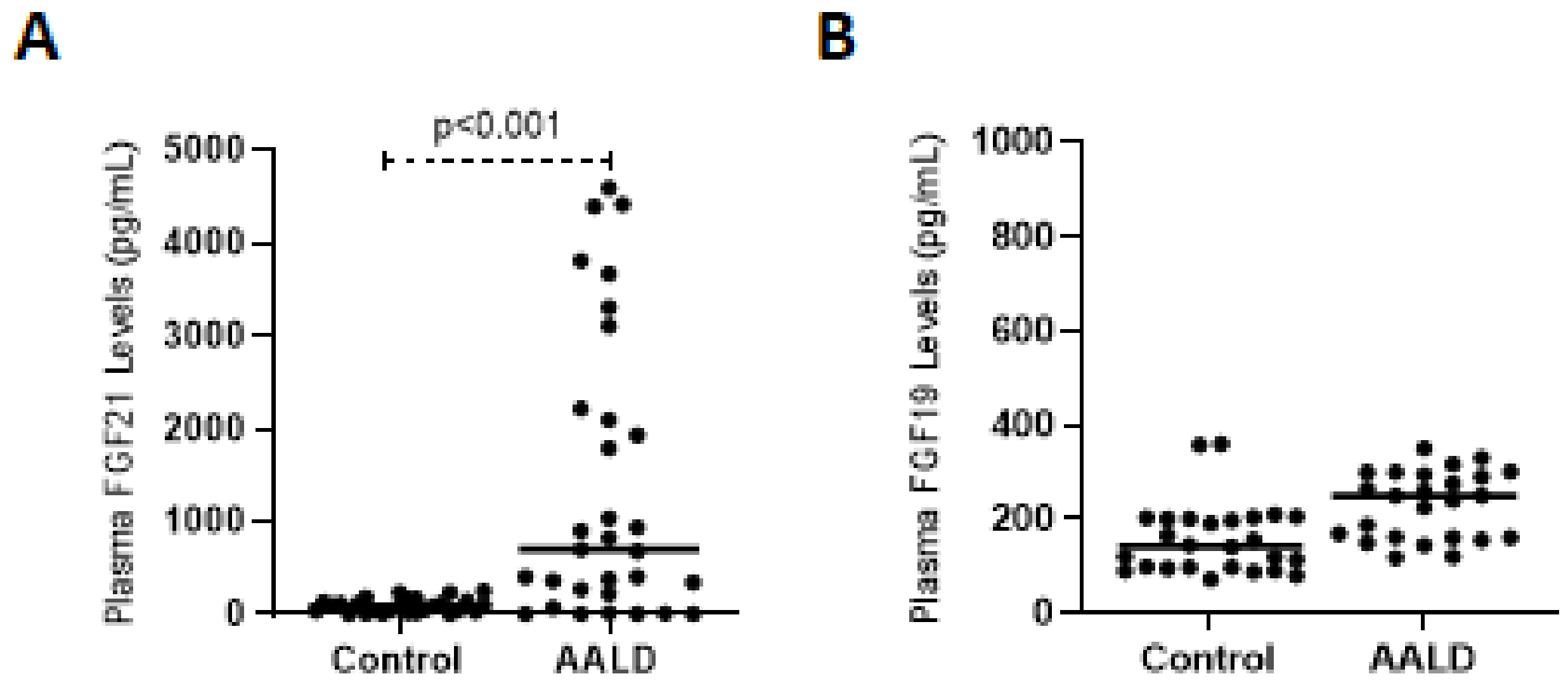
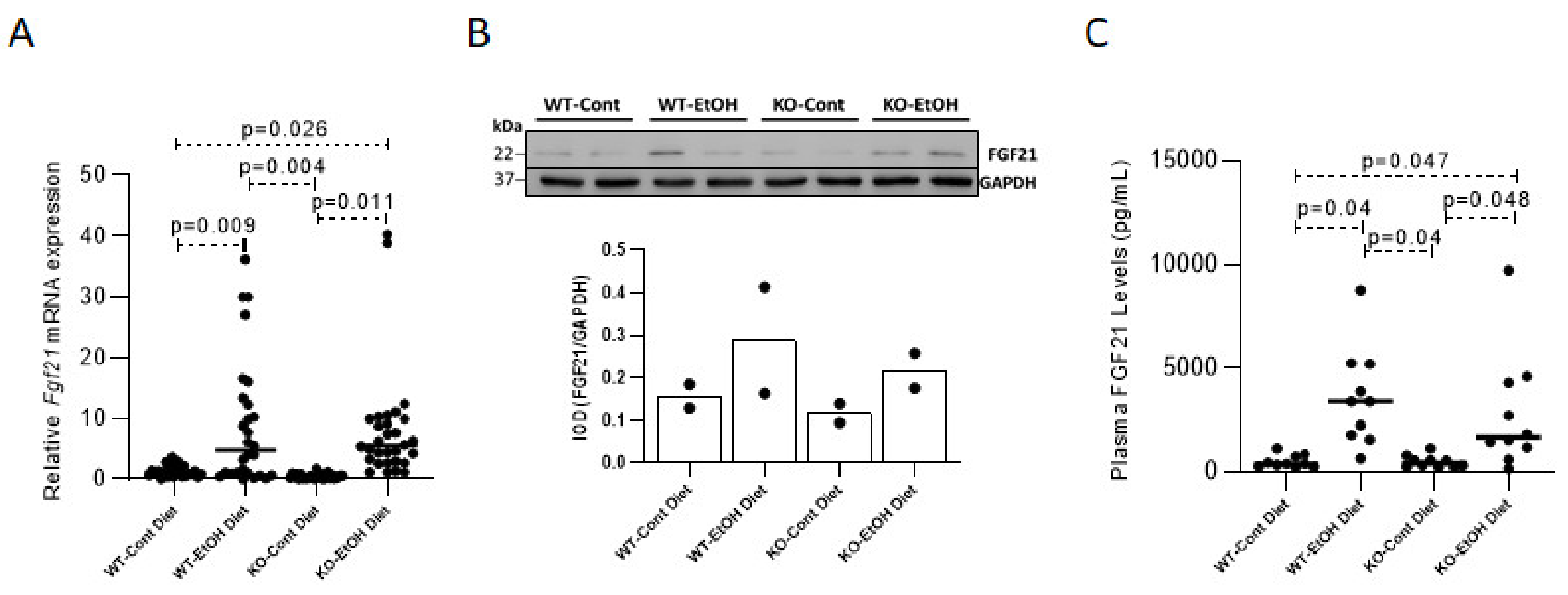
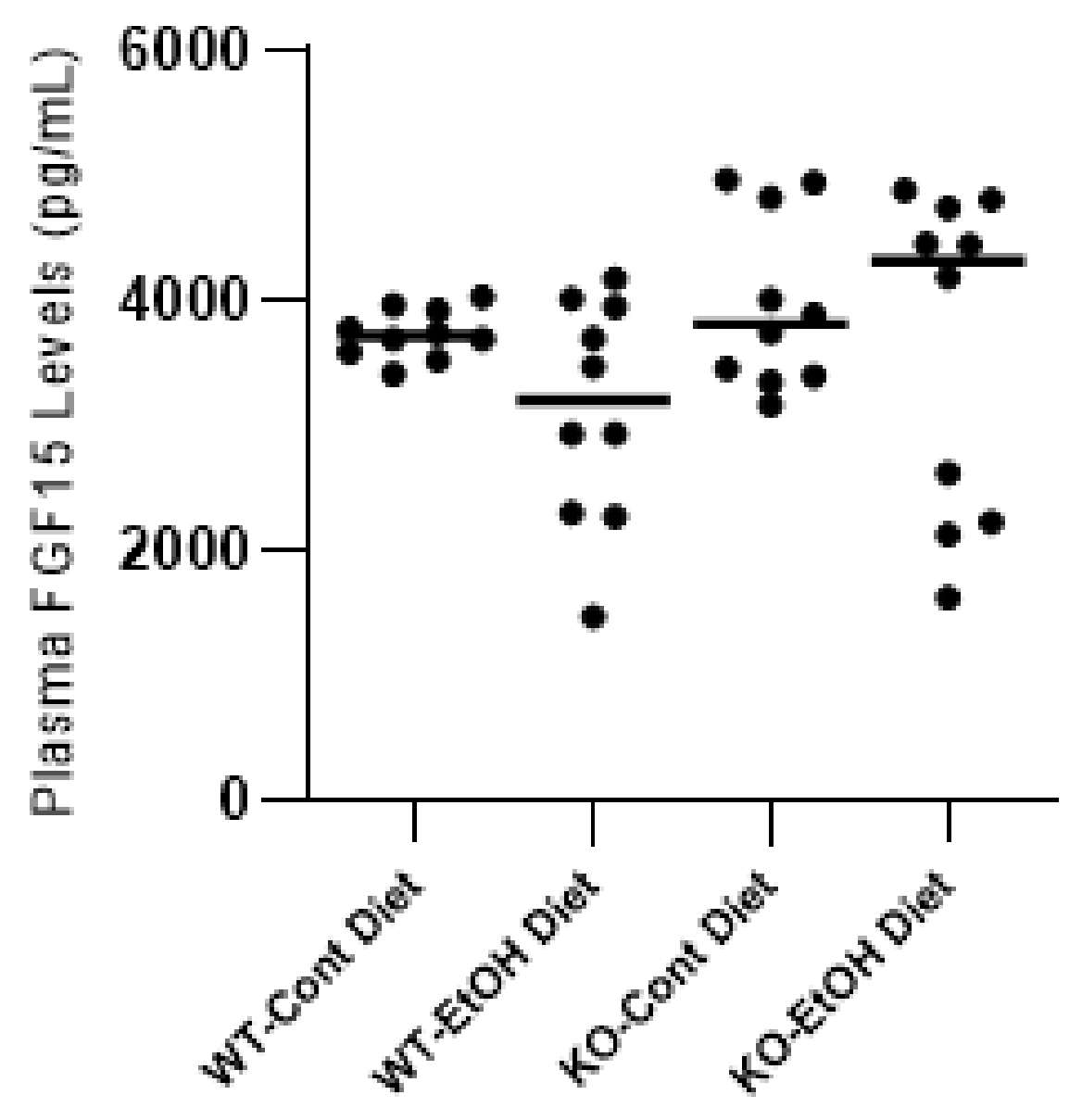
2.1.1. Effect of Ethanol Consumption on FGF21
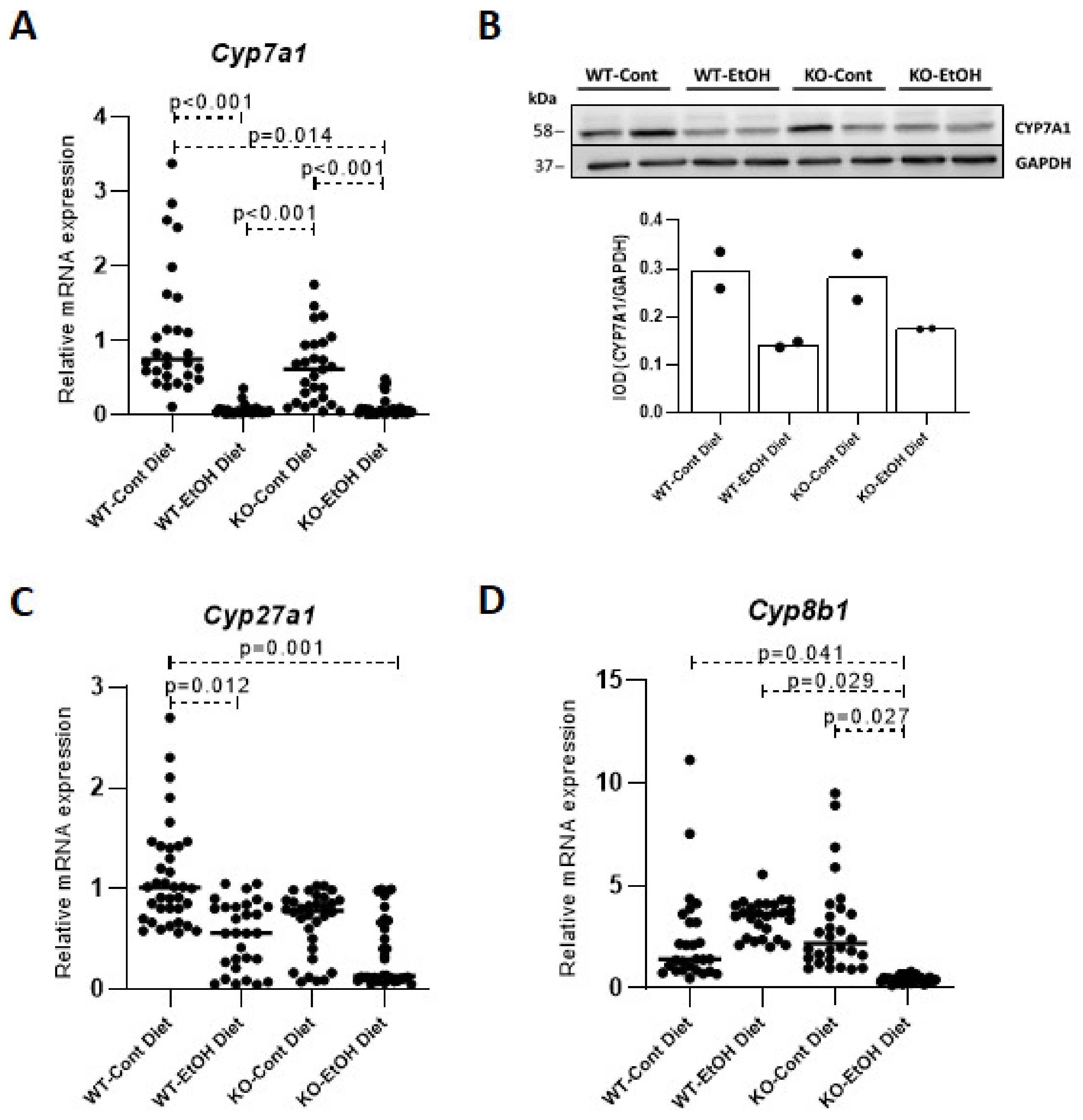
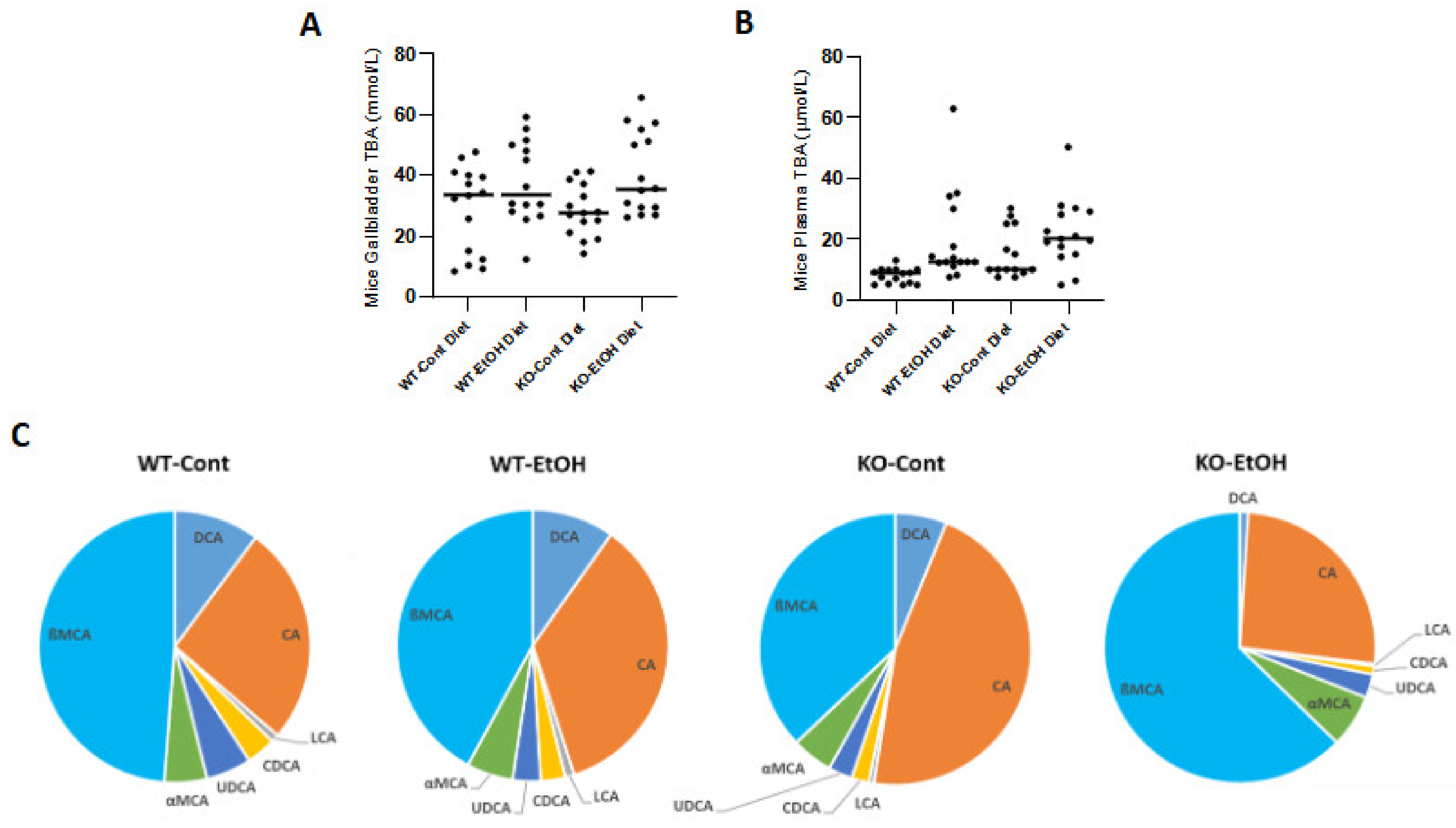
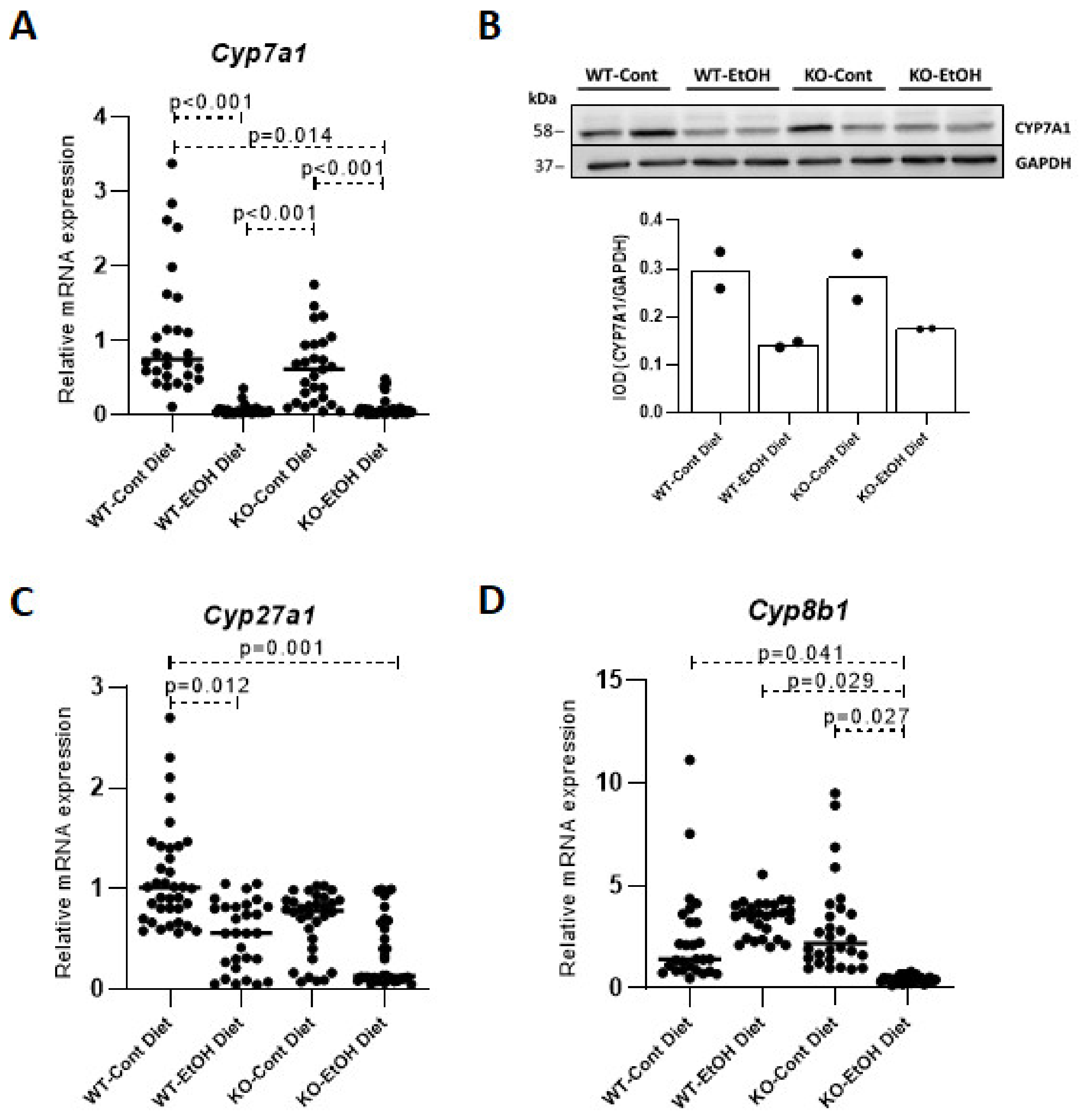
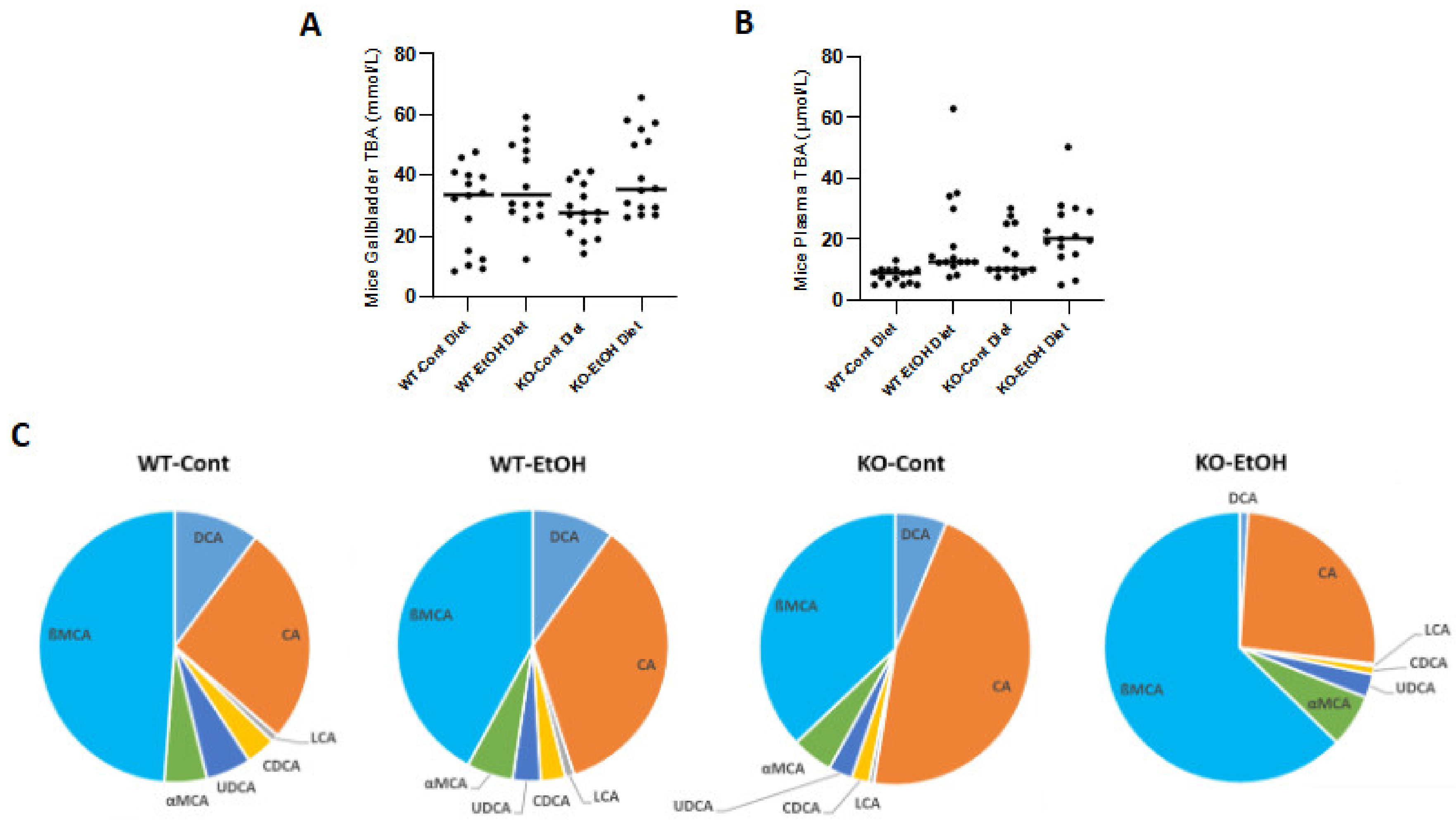
2.1.2. Relative Expression Levels of Bile Acid Synthesis Genes and Plasma and Gallbladder Total Bile Acid Levels
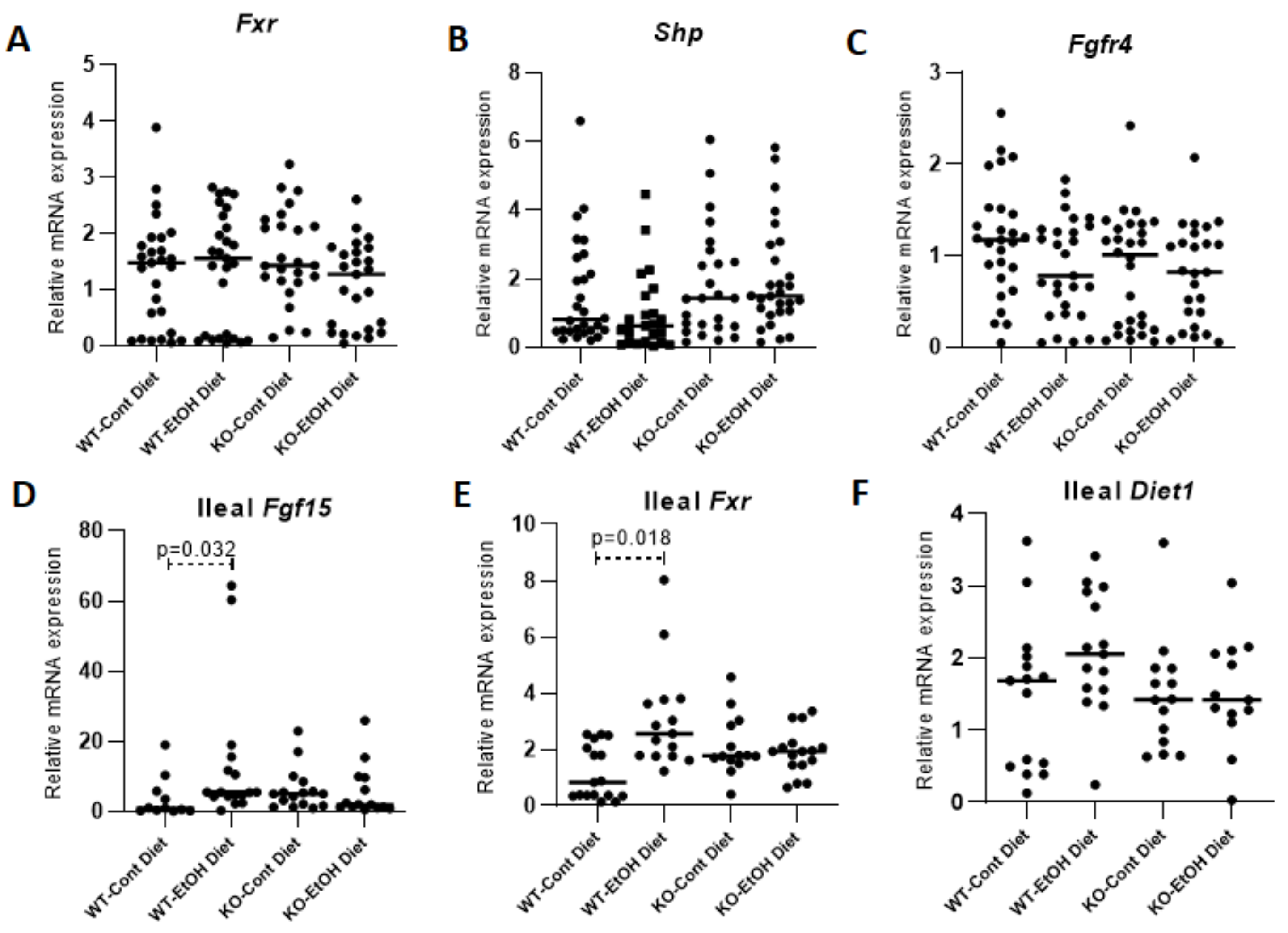
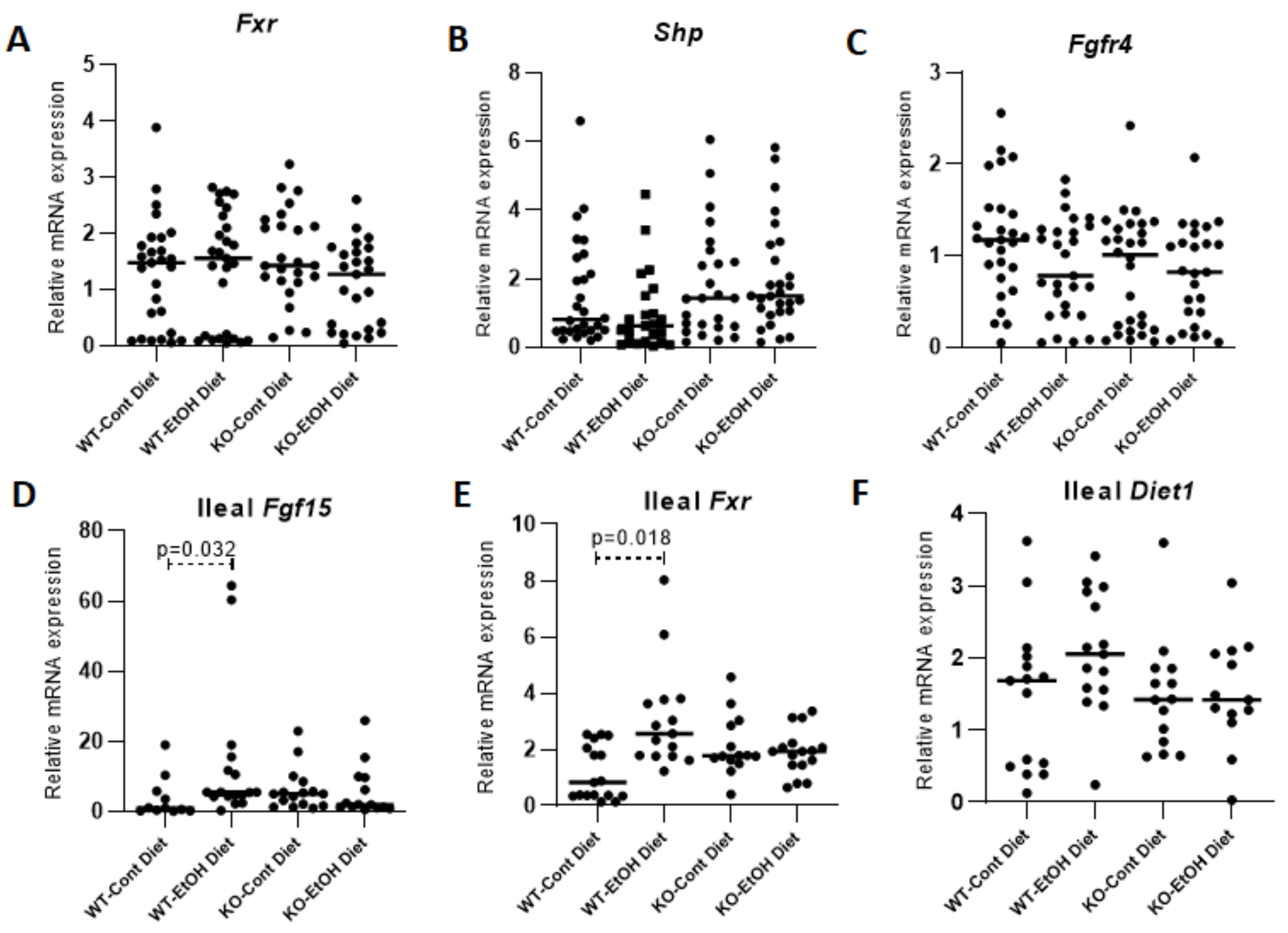
2.1.3. Hepatic FXR/SHP Pathway
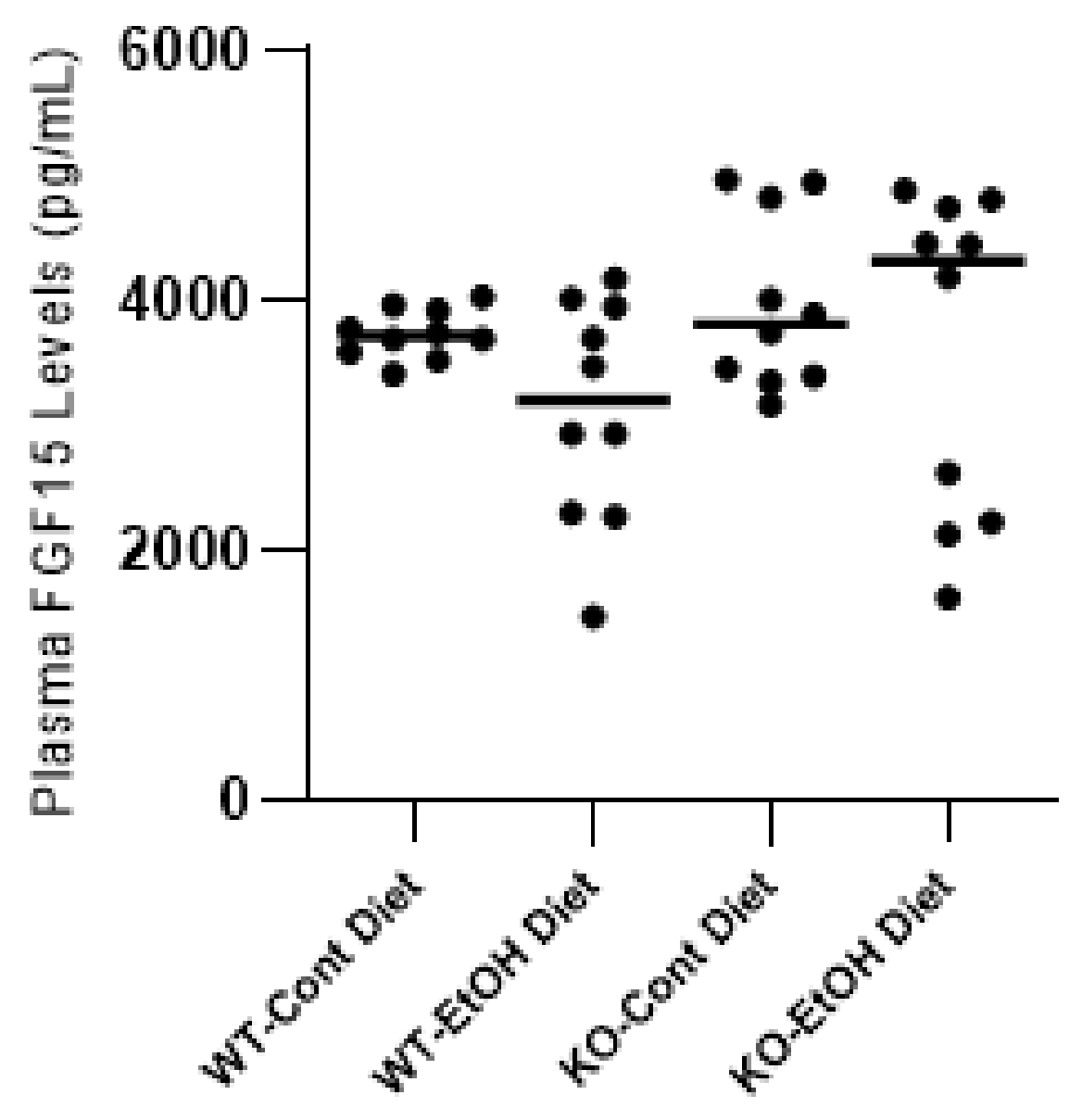
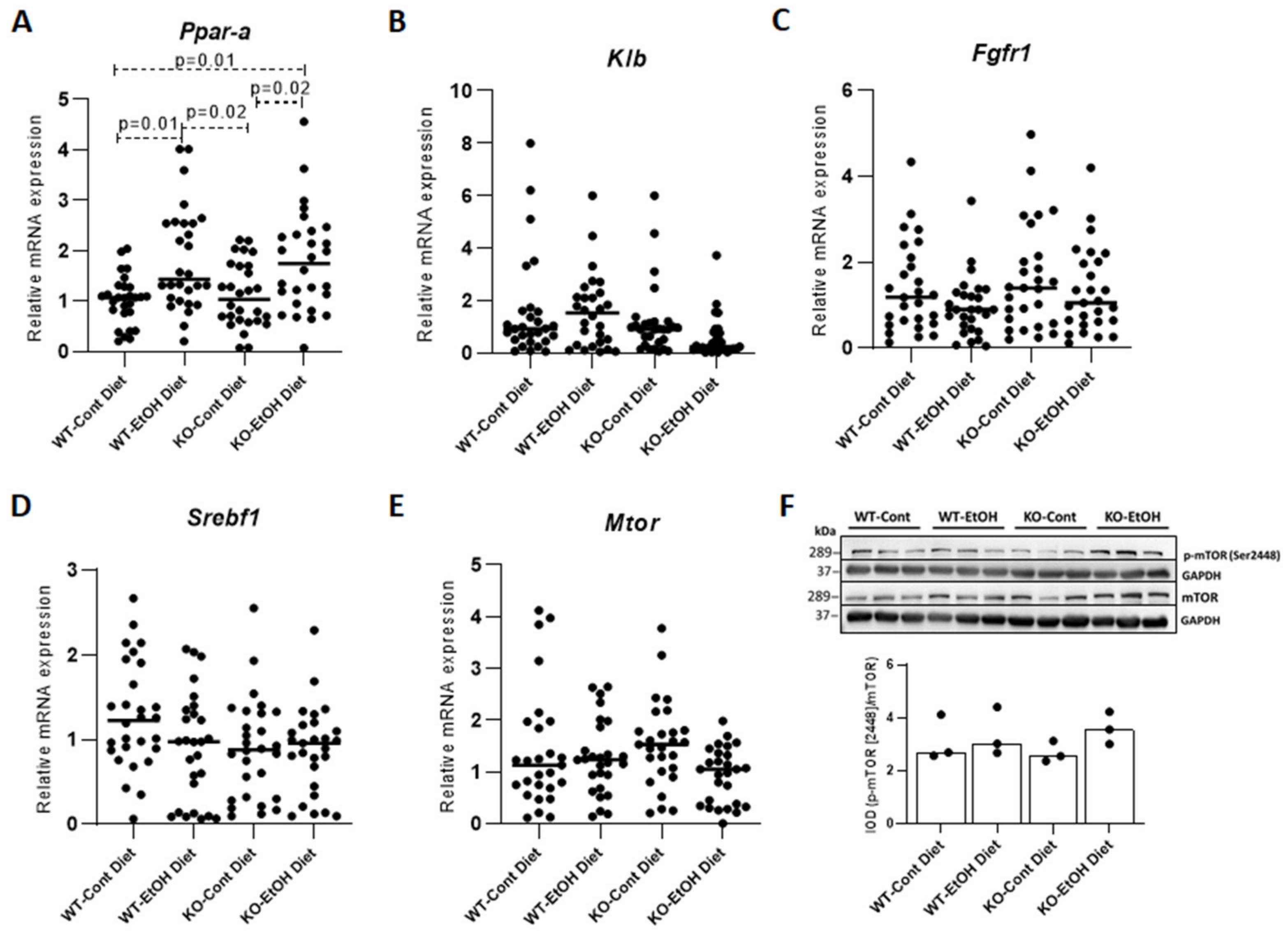
2.1.4. FXR/FGF15/FGFR4 Pathway
2.1.5. Total Bile Acid Concentrations and Composition in Mice
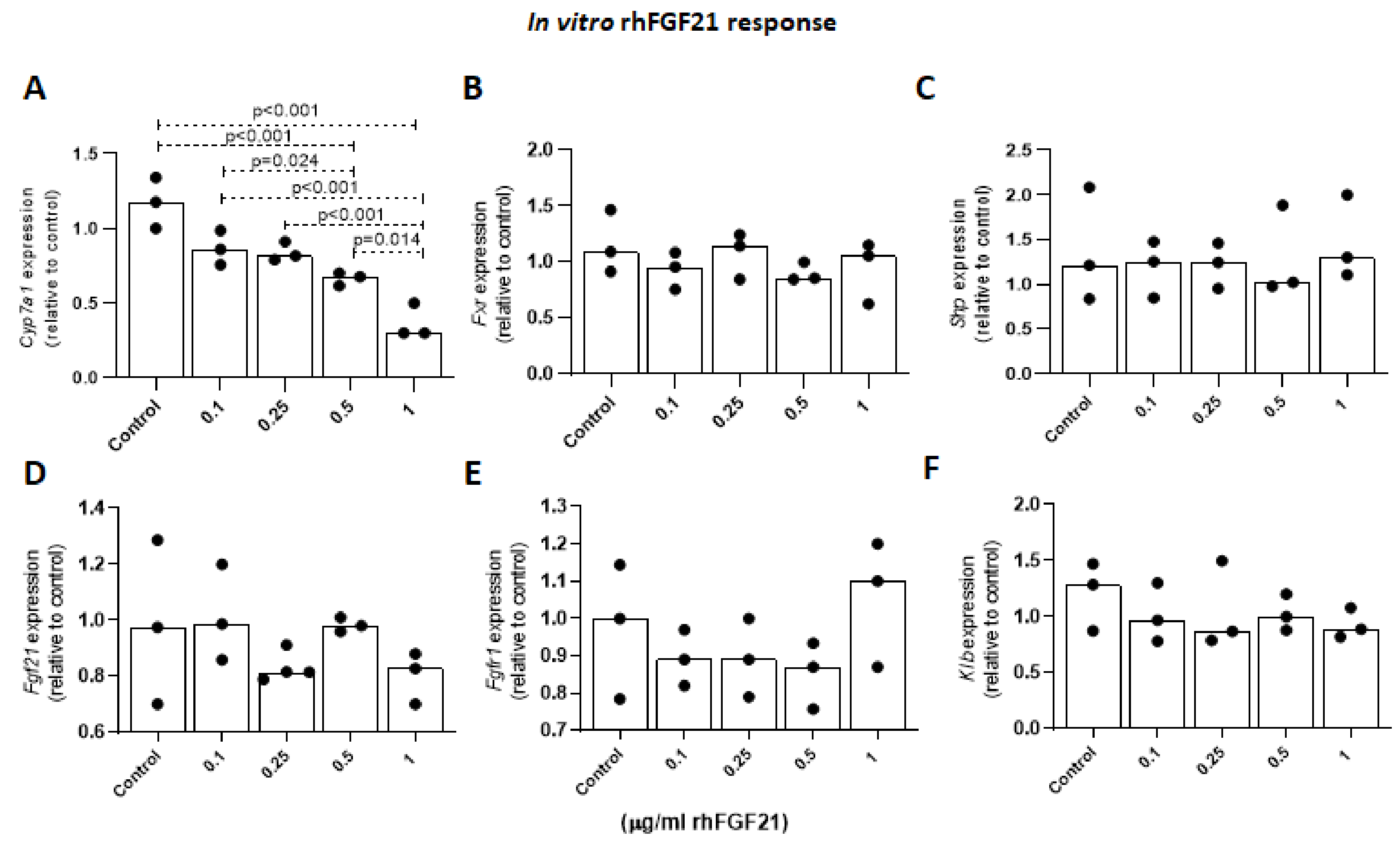
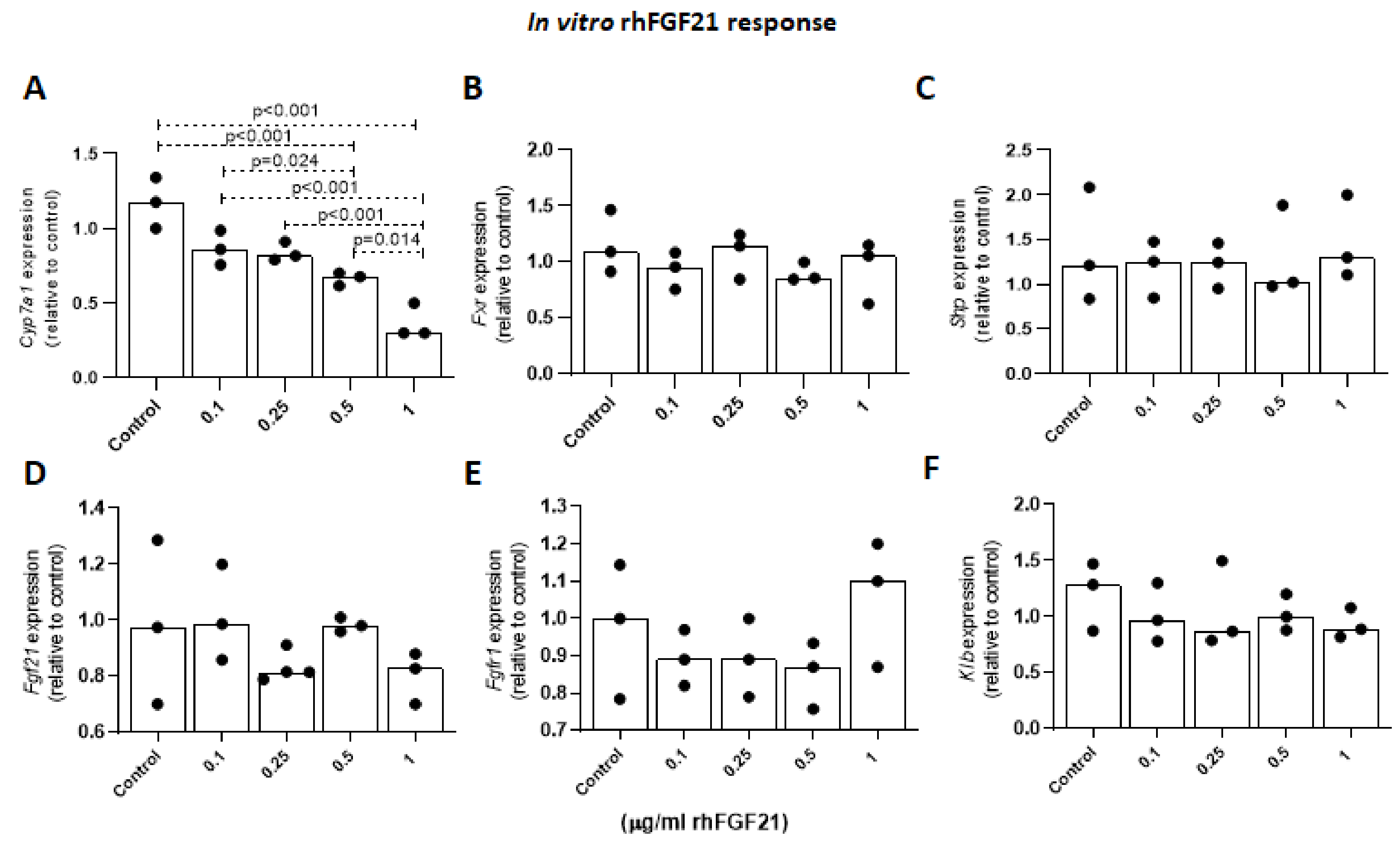
2.1.6. Effects of FGF21 on Cyp7a1 Expression Levels in Primary Mouse Hepatocytes
2.1.7. Plasma ALT, AST and Ethanol Levels in Mice
2.1.8. Histological Assessment of Murine Liver Injury
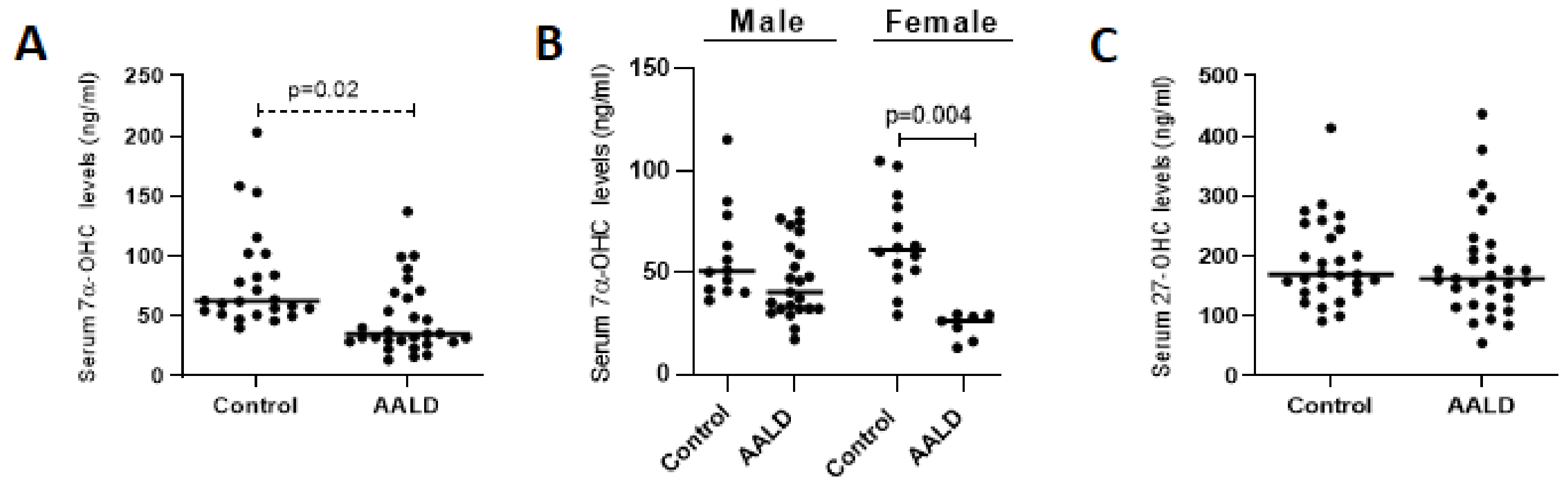
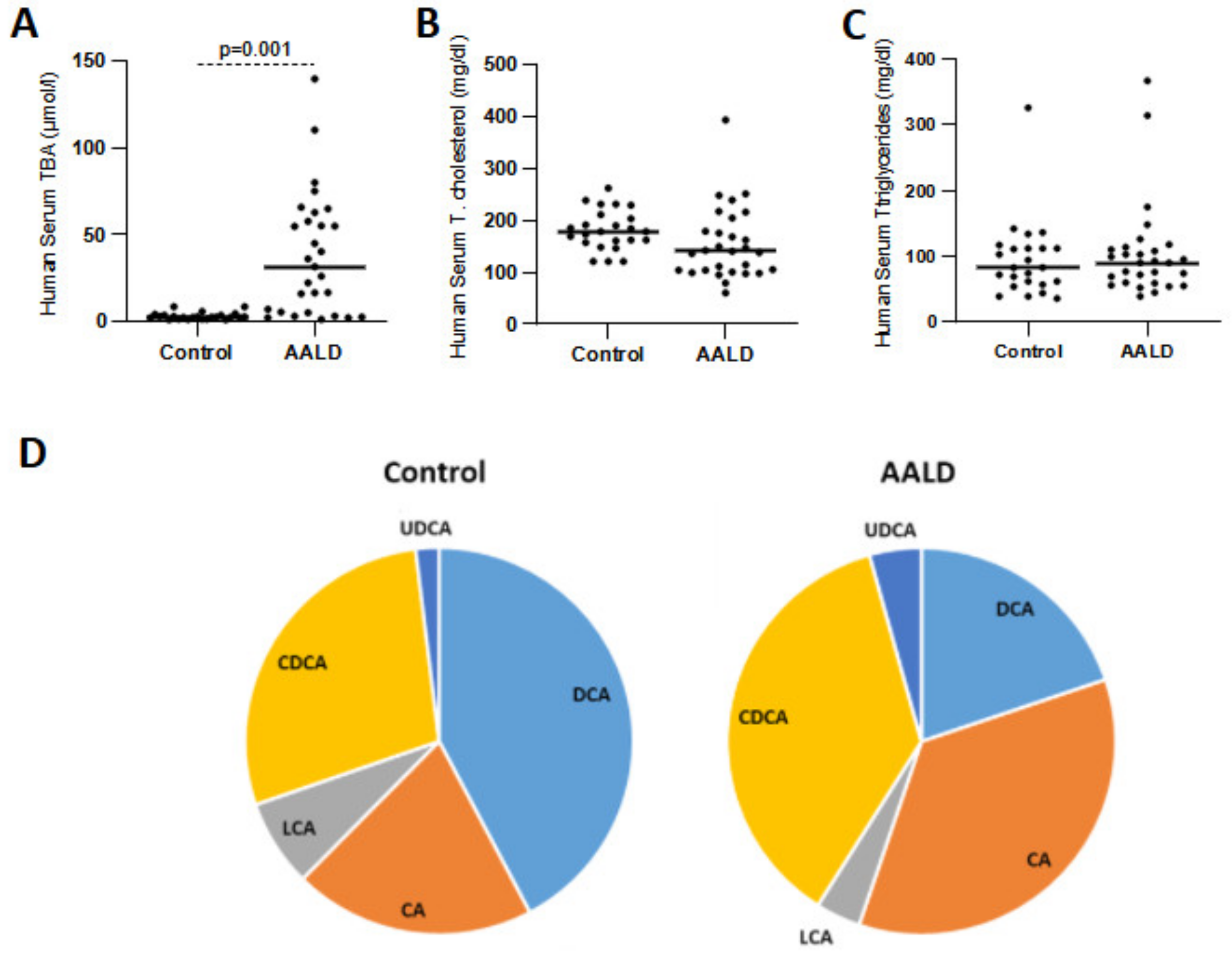
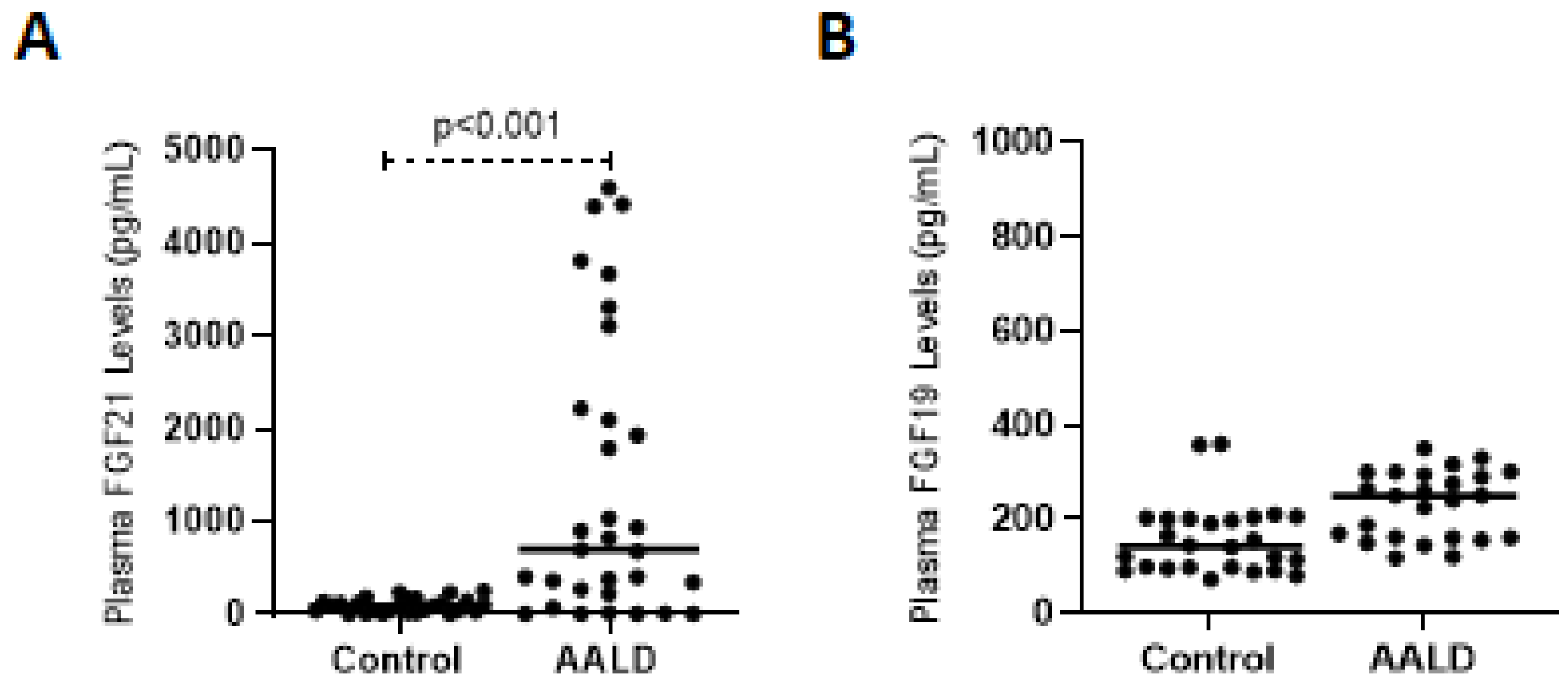
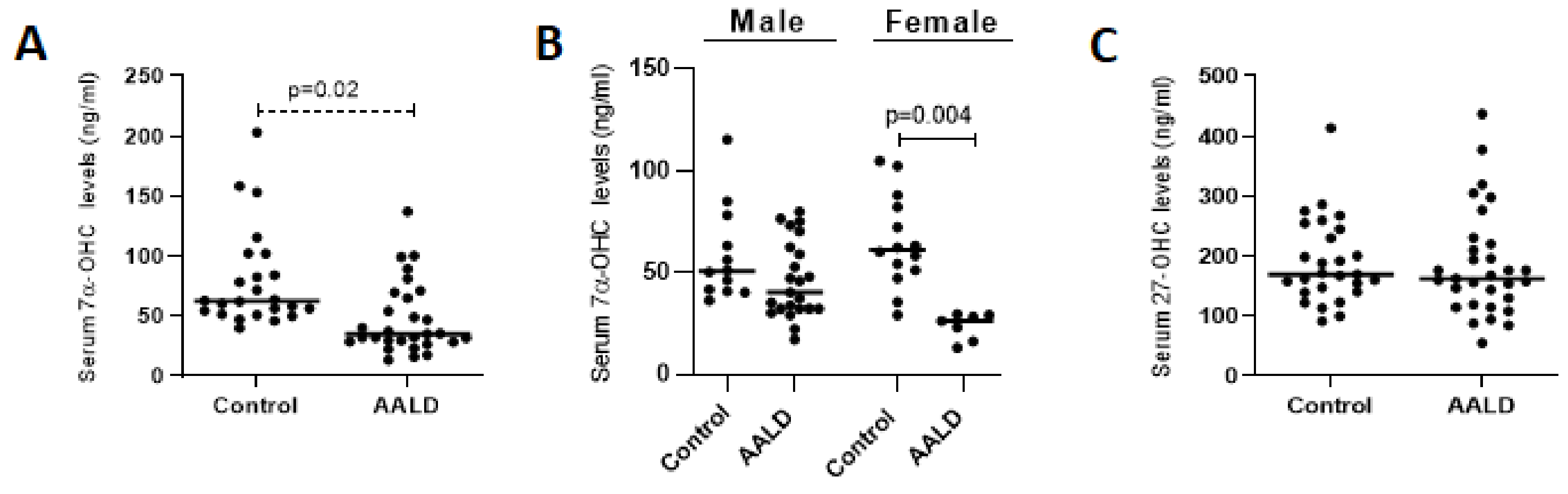
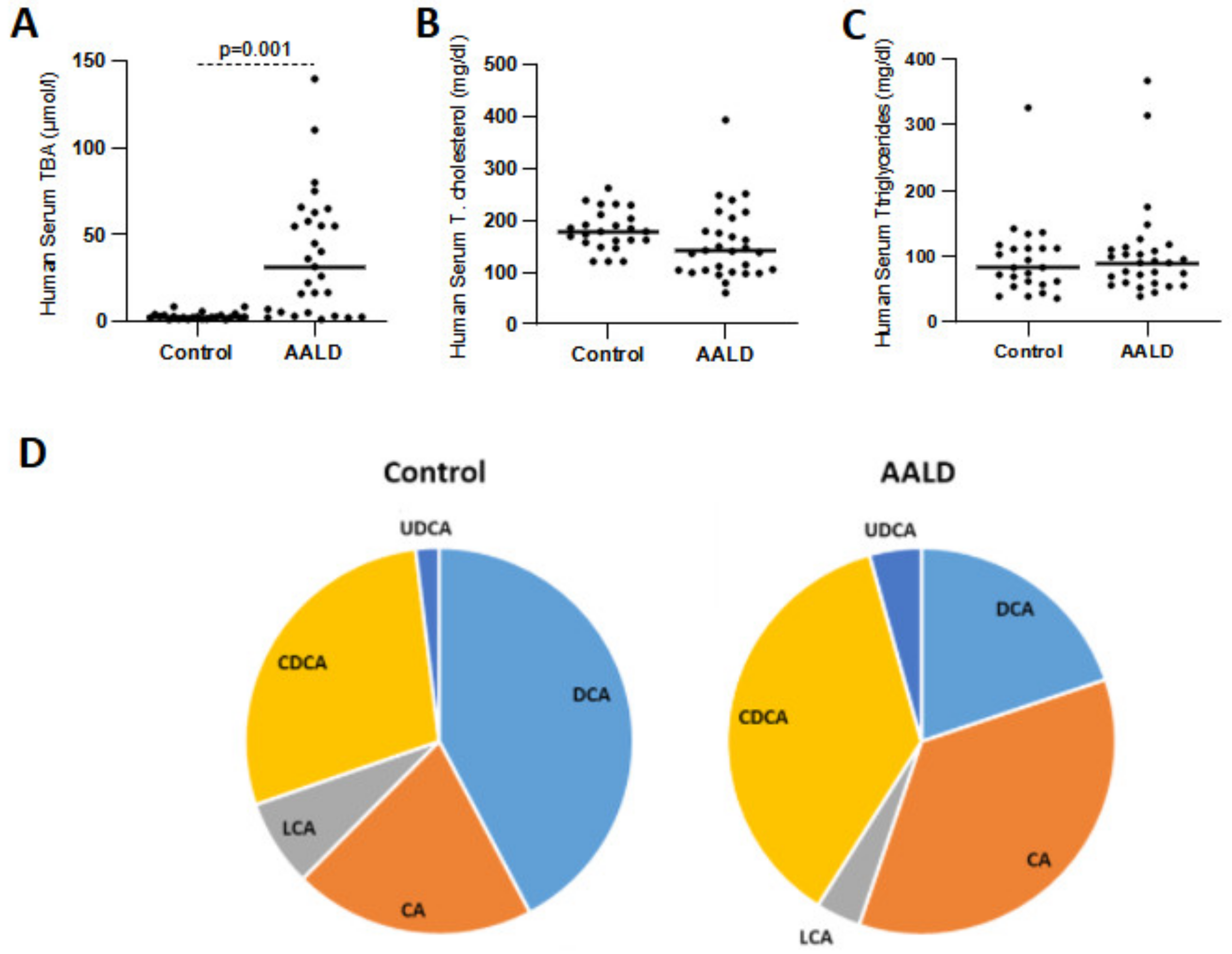
2.2. Human Studies
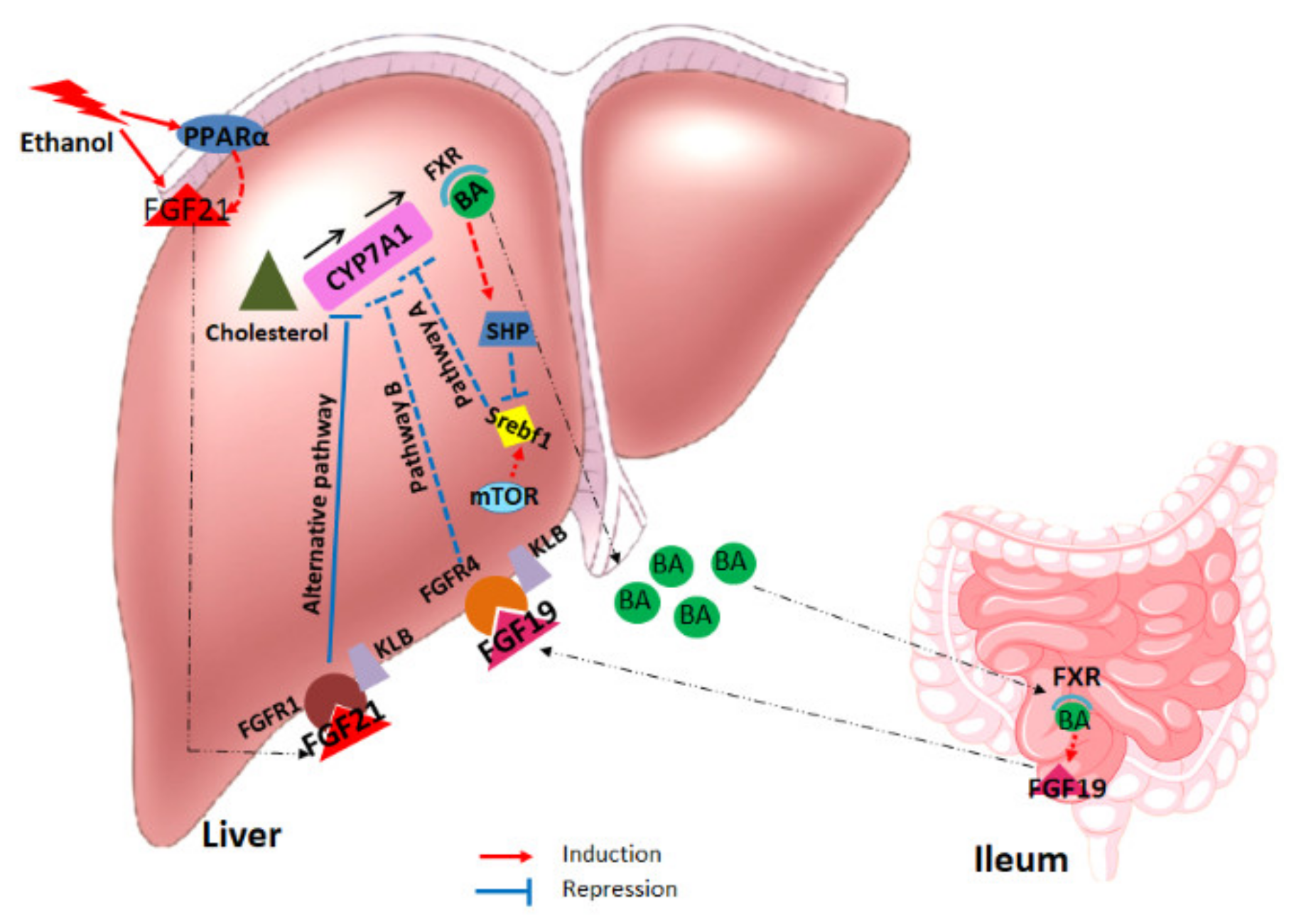
3. Discussion
4. Materials and Methods
4.1. Mouse Model
4.2. Human Subjects
4.3. Analysis of Plasma Biochemical Markers
4.4. Histology
Immunohistochemistry (IHC)
4.5. Determination of Plasma FGF15/FGF19 and FGF21 Levels
4.6. RNA Isolation and Quantitative Real-Time PCR Analysis
4.7. Western Blot Analysis
4.8. Total Bile Acid Measurements in Mouse Plasma and Bile
4.9. Bile Acid Profiling in Human and Murine Plasma Samples
4.10. Enzymatic Tests for the Analysis of Serum Total Bile Acid and Cholesterol Levels in Humans
4.11. Quantification of Human and Murine Serum Concentrations of Total Cholesterol and Oxysterols Using Gas-Chromatography Analyses
4.12. Primary Mouse Hepatocyte Isolation, Culture, and Treatment
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Miller, A.M.; Horiguchi, N.; Jeong, W.I.; Radaeva, S.; Gao, B. Molecular mechanisms of alcoholic liver disease: Innate immunity and cytokines. Alcohol. Clin. Exp. Res. 2011, 35, 787–793. [Google Scholar] [CrossRef]
- Karatayli, E.; Hall, R.A.; Weber, S.N.; Dooley, S.; Lammert, F. Effect of alcohol on the interleukin 6-mediated inflammatory response in a new mouse model of acute-on-chronic liver injury. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 298–307. [Google Scholar] [CrossRef]
- Moreau, R.; Jalan, R.; Gines, P.; Pavesi, M.; Angeli, P.; Cordoba, J.; Durand, F.; Gustot, T.; Saliba, F.; Domenicali, M.; et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 2013, 144, 1426–1437.e9. [Google Scholar] [CrossRef]
- Arteel, G.E. Animal models of alcoholic liver disease. Dig. Dis. 2010, 28, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Han, J.; Lee, C.; Yoon, M.; Jung, Y. Pathophysiological Aspects of Alcohol Metabolism in the Liver. Int. J. Mol. Sci. 2021, 22, 5717. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. The feeding of ethanol in liquid diets. Alcohol. Clin. Exp. Res. 1986, 10, 550–553. [Google Scholar] [CrossRef]
- Wilkin, R.J.; Lalor, P.F.; Parker, R.; Newsome, P.N. Murine Models of Acute Alcoholic Hepatitis and Their Relevance to Human Disease. Am. J. Pathol. 2016, 186, 748–760. [Google Scholar] [CrossRef] [Green Version]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Bertola, A.; Park, O.; Gao, B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: A critical role for E-selectin. Hepatology 2013, 58, 1814–1823. [Google Scholar] [CrossRef] [Green Version]
- Fickert, P.; Fuchsbichler, A.; Wagner, M.; Zollner, G.; Kaser, A.; Tilg, H.; Krause, R.; Lammert, F.; Langner, C.; Zatloukal, K.; et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004, 127, 261–274. [Google Scholar] [CrossRef]
- Smit, J.J.; Schinkel, A.H.; Oude Elferink, R.P.; Groen, A.K.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; Ottenhoff, R.; van der Lugt, N.M.; van Roon, M.A.; et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993, 75, 451–462. [Google Scholar] [CrossRef]
- Ruiz-Margáin, A.; Pohlmann, A.; Ryan, P.; Schierwagen, R.; Chi-Cervera, L.A.; Jansen, C.; Mendez-Guerrero, O.; Flores-García, N.C.; Lehmann, J.; Torre, A.; et al. Fibroblast growth factor 21 is an early predictor of acute-on-chronic liver failure in critically ill patients with cirrhosis. Liver Transplant. 2018, 24, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumann, G.; Liu, C.; O’Reilly, P.; Gao, H.; Song, P.; Xu, B.; Ruggeri, B.; Amin, N.; Jia, T.; Preis, S.; et al. KLB is associated with alcohol drinking, and its gene product β-Klotho is necessary for FGF21 regulation of alcohol preference. Proc. Natl. Acad. Sci. USA 2016, 113, 14372–14377. [Google Scholar] [CrossRef] [Green Version]
- Søberg, S.; Andersen, E.S.; Dalsgaard, N.B.; Jarlhelt, I.; Hansen, N.L.; Hoffmann, N.; Vilsbøll, T.; Chenchar, A.; Jensen, M.; Grevengoed, T.J.; et al. FGF21, a liver hormone that inhibits alcohol intake in mice, increases in human circulation after acute alcohol ingestion and sustained binge drinking at Oktoberfest. Mol. Metab. 2018, 11, 96–103. [Google Scholar] [CrossRef]
- Talukdar, S.; Owen, B.M.; Song, P.; Hernandez, G.; Zhang, Y.; Zhou, Y.; Scott, W.T.; Paratala, B.; Turner, T.; Smith, A.; et al. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab. 2016, 23, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Holstein-Rathlou, S.; Gillum, M.P. Fibroblast growth factor 21: An endocrine inhibitor of sugar and alcohol appetite. J. Physiol. 2019, 597, 3539–3548. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Zechner, C.; Hernandez, G.; Cánovas, J.; Xie, Y.; Sondhi, V.; Wagner, M.; Stadlbauer, V.; Horvath, A.; Leber, B.; et al. The Hormone FGF21 Stimulates Water Drinking in Response to Ketogenic Diet and Alcohol. Cell Metab. 2018, 27, 1338–1347.e4. [Google Scholar] [CrossRef] [Green Version]
- Fisher, F.M.; Chui, P.C.; Nasser, I.A.; Popov, Y.; Cunniff, J.C.; Lundasen, T.; Kharitonenkov, A.; Schuppan, D.; Flier, J.S.; Maratos-Flier, E. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology 2014, 147, 1073–1083.e6. [Google Scholar] [CrossRef] [Green Version]
- Desai, B.N.; Singhal, G.; Watanabe, M.; Stevanovic, D.; Lundasen, T.; Fisher, F.M.; Mather, M.L.; Vardeh, H.G.; Douris, N.; Adams, A.C.; et al. Fibroblast growth factor 21 (FGF21) is robustly induced by ethanol and has a protective role in ethanol associated liver injury. Mol. Metab. 2017, 6, 1395–1406. [Google Scholar] [CrossRef]
- Ye, D.; Wang, Y.; Li, H.; Jia, W.; Man, K.; Lo, C.M.; Wang, Y.; Lam, K.S.; Xu, A. Fibroblast growth factor 21 protects against acetaminophen-induced hepatotoxicity by potentiating peroxisome proliferator-activated receptor coactivator protein-1α-mediated antioxidant capacity in mice. Hepatology 2014, 60, 977–989. [Google Scholar] [CrossRef]
- Chen, M.M.; Hale, C.; Stanislaus, S.; Xu, J.; Véniant, M.M. FGF21 acts as a negative regulator of bile acid synthesis. J. Endocrinol. 2018, 237, 139–152. [Google Scholar] [CrossRef]
- Zhang, J.; Gupte, J.; Gong, Y.; Weiszmann, J.; Zhang, Y.; Lee, K.J.; Richards, W.G.; Li, Y. Chronic Over-expression of Fibroblast Growth Factor 21 Increases Bile Acid Biosynthesis by Opposing FGF15/19 Action. EBioMedicine 2017, 15, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.W. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009, 50, S120–S125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, D.; Kim, Y.H.; Li, T.; Misra, J.; Kim, D.K.; Kim, J.R.; Kwon, J.; Jeong, W.I.; Ahn, S.H.; Park, T.S.; et al. Hepatic cannabinoid receptor type 1 mediates alcohol-induced regulation of bile acid enzyme genes expression via CREBH. PLoS ONE 2013, 8, e68845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Zhu, B.; Peng, X.; Zhou, M.; Jia, D.; Gu, J. Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease. Biochem. Biophys. Res. Commun. 2014, 443, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhong, W.; Li, H.; Li, Q.; Qiu, Y.; Zheng, X.; Chen, H.; Zhao, X.; Zhang, S.; Zhou, Z.; et al. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J. 2013, 27, 3583–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massimi, M.; Lear, S.R.; Huling, S.L.; Jones, A.L.; Erickson, S.K. Cholesterol 7alpha-hydroxylase (CYP7A): Patterns of messenger RNA expression during rat liver development. Hepatology 1998, 28, 1064–1072. [Google Scholar] [CrossRef]
- Liang, H.; Huang, H.; Tan, P.Z.; Liu, Y.; Nie, J.H.; Zhang, Y.T.; Zhang, K.L.; Diao, Y.; He, Q.; Hou, B.Y.; et al. Effect of iron on cholesterol 7α-hydroxylase expression in alcohol-induced hepatic steatosis in mice. J. Lipid Res. 2017, 58, 1548–1560. [Google Scholar] [CrossRef] [Green Version]
- Donepudi, A.C.; Ferrell, J.M.; Boehme, S.; Choi, H.S.; Chiang, J.Y.L. Deficiency of cholesterol 7α-hydroxylase in bile acid synthesis exacerbates alcohol-induced liver injury in mice. Hepatol. Commun. 2018, 2, 99–112. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003, 17, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Hochrath, K.; Ehnert, S.; Ackert-Bicknell, C.L.; Lau, Y.; Schmid, A.; Krawczyk, M.; Hengstler, J.G.; Dunn, J.; Hiththetiya, K.; Rathkolb, B.; et al. Modeling hepatic osteodystrophy in Abcb4 deficient mice. Bone 2013, 55, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamall, I.S.; Finelli, V.N.; Que Hee, S.S. A simple method to determine nanogram levels of 4-hydroxyproline in biological tissues. Anal. Biochem. 1981, 112, 70–75. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Lütjohann, D.; Hahn, C.; Prange, W.; Sudhop, T.; Axelson, M.; Sauerbruch, T.; von Bergmann, K.; Reichel, C. Influence of rifampin on serum markers of cholesterol and bile acid synthesis in men. Int. J. Clin. Pharmacol. Ther. 2004, 42, 307–313. [Google Scholar] [CrossRef]
- Mackay, D.S.; Jones, P.J.; Myrie, S.B.; Plat, J.; Lütjohann, D. Methodological considerations for the harmonization of non-cholesterol sterol bio-analysis. J. Chromatogr. B 2014, 957, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Šošić-Jurjević, B.; Lütjohann, D.; Renko, K.; Filipović, B.; Radulović, N.; Ajdžanović, V.; Trifunović, S.; Nestorović, N.; Živanović, J.; Manojlović Stojanoski, M.; et al. The isoflavones genistein and daidzein increase hepatic concentration of thyroid hormones and affect cholesterol metabolism in middle-aged male rats. J. Steroid Biochem. Mol. Biol. 2019, 190, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingmüller, U.; Bauer, A.; Bohl, S.; Nickel, P.J.; Breitkopf, K.; Dooley, S.; Zellmer, S.; Kern, C.; Merfort, I.; Sparna, T.; et al. Primary mouse hepatocytes for systems biology approaches: A standardized in vitro system for modelling of signal transduction pathways. Syst. Biol. 2006, 153, 433–447. [Google Scholar] [CrossRef] [Green Version]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Minard, A.Y.; Tan, S.X.; Yang, P.; Fazakerley, D.J.; Domanova, W.; Parker, B.L.; Humphrey, S.J.; Jothi, R.; Stöckli, J.; James, D.E. mTORC1 Is a Major Regulatory Node in the FGF21 Signaling Network in Adipocytes. Cell Rep. 2016, 17, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Katafuchi, T.; Esterházy, D.; Lemoff, A.; Ding, X.; Sondhi, V.; Kliewer, S.A.; Mirzaei, H.; Mangelsdorf, D.J. Detection of FGF15 in plasma by stable isotope standards and capture by anti-peptide antibodies and targeted mass spectrometry. Cell Metab. 2015, 21, 898–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Ong, J.R.; Vergnes, L.; de Aguiar Vallim, T.Q.; Nolan, J.; Cantor, R.M.; Walters, J.R.F.; Reue, K. Diet1, bile acid diarrhea, and FGF15/19: Mouse model and human genetic variants. J. Lipid Res. 2018, 59, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Johansson, H.; Mörk, L.M.; Li, M.; Sandblom, A.L.; Björkhem, I.; Höijer, J.; Ericzon, B.G.; Jorns, C.; Gilg, S.; Sparrelid, E.; et al. Circulating Fibroblast Growth Factor 19 in Portal and Systemic Blood. J. Clin. Exp. Hepatol. 2018, 8, 162–168. [Google Scholar] [CrossRef]
- Lundåsen, T.; Gälman, C.; Angelin, B.; Rudling, M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 2006, 260, 530–536. [Google Scholar] [CrossRef]
- Hahn, C.; Reichel, C.; von Bergmann, K. Serum concentration of 7 alpha-hydroxycholesterol as an indicator of bile acid synthesis in humans. J. Lipid Res. 1995, 36, 2059–2066. [Google Scholar] [CrossRef]
- Wu, L.; Pan, Q.; Wu, G.; Qian, L.; Zhang, J.; Zhang, L.; Fang, Q.; Zang, G.; Wang, Y.; Lau, G.; et al. Diverse Changes of Circulating Fibroblast Growth Factor 21 Levels in Hepatitis B Virus-Related Diseases. Sci. Rep. 2017, 7, 16482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelps, T.; Snyder, E.; Rodriguez, E.; Child, H.; Harvey, P. The influence of biological sex and sex hormones on bile acid synthesis and cholesterol homeostasis. Biol. Sex Differ. 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Al-Aqil, F.A.; Monte, M.J.; Peleteiro-Vigil, A.; Briz, O.; Rosales, R.; González, R.; Aranda, C.J.; Ocón, B.; Uriarte, I.; de Medina, F.S.; et al. Interaction of glucocorticoids with FXR/FGF19/FGF21-mediated ileum-liver crosstalk. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 2927–2937. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3281–3282. [Google Scholar] [CrossRef] [Green Version]
- Bortolami, M.; Comparato, A.; Benna, C.; Errico, A.; Maretto, I.; Pucciarelli, S.; Cillo, U.; Farinati, F. Gene and protein expression of mTOR and LC3 in hepatocellular carcinoma, colorectal liver metastasis and "normal" liver tissues. PLoS ONE 2020, 15, e0244356. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Ding, W. Role of farnesoid X receptor and bile acids in alcoholic liver disease. Acta Pharm. Sin. B 2015, 5, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Trinchet, J.C.; Gerhardt, M.F.; Balkau, B.; Munz, C.; Poupon, R.E. Serum bile acids and cholestasis in alcoholic hepatitis. Relationship with usual liver tests and histological features. J. Hepatol. 1994, 21, 235–240. [Google Scholar] [CrossRef]
- Horvatits, T.; Drolz, A.; Roedl, K.; Rutter, K.; Ferlitsch, A.; Fauler, G.; Trauner, M.; Fuhrmann, V. Serum bile acids as marker for acute decompensation and acute-on-chronic liver failure in patients with non-cholestatic cirrhosis. Liver Transplant. 2017, 37, 224–231. [Google Scholar] [CrossRef]
- You, M.; Arteel, G.E. Effect of ethanol on lipid metabolism. J. Hepatol. 2019, 70, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liao, X.; Yang, C.; Huang, K.; Yu, T.; Yu, L.; Han, C.; Zhu, G.; Zeng, X.; Liu, Z.; et al. Identification of prognostic biomarkers for patients with hepatocellular carcinoma after hepatectomy. Oncol. Rep. 2019, 41, 1586–1602. [Google Scholar] [CrossRef]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Poh, W.; Wong, W.; Ong, H.; Aung, M.O.; Lim, S.G.; Chua, B.T.; Ho, H.K. Klotho-beta overexpression as a novel target for suppressing proliferation and fibroblast growth factor receptor-4 signaling in hepatocellular carcinoma. Mol. Cancer 2012, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Guo, Y.; Zhang, Q.; Chen, W.; Hua, X.; Liu, W.; Yang, Y.; Chen, G. βKlotho suppresses tumor growth in hepatocellular carcinoma by regulating Akt/GSK-3β/cyclin D1 signaling pathway. PLoS ONE 2013, 8, e55615. [Google Scholar] [CrossRef] [Green Version]
- Sugita, T.; Amano, K.; Nakano, M.; Masubuchi, N.; Sugihara, M.; Matsuura, T. Analysis of the serum bile Acid composition for differential diagnosis in patients with liver disease. Gastroenterol. Res. Pract. 2015, 2015, 717431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Chiang, J.Y. Transcriptional regulation of the human sterol 12alpha-hydroxylase gene (CYP8B1): Roles of heaptocyte nuclear factor 4alpha in mediating bile acid repression. J. Biol. Chem. 2001, 276, 41690–41699. [Google Scholar] [CrossRef] [Green Version]
- Fujino, T.; Une, M.; Imanaka, T.; Inoue, K.; Nishimaki-Mogami, T. Structure-activity relationship of bile acids and bile acid analogs in regard to FXR activation. J. Lipid Res. 2004, 45, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y. Recent advances in understanding bile acid homeostasis. F1000Research 2017, 6, 2029. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % (mean ± SD) | CA | CDCA | DCA | LCA | UDCA | ßMCA | αMCA | Primary BA | Secondary BA |
|---|---|---|---|---|---|---|---|---|---|
| Wt-Cont | 26.3 ± 2.1 | 3.5 ± 1.7 | 10.3 ± 3.3 | 0.9 ± 0.1 | 5.4 ± 1.8 | 48.5 ± 8.7 | 5.1 ± 1.2 | 83.4% | 16.6% |
| Wt-EtOH | 35.3 ± 2.3 | 2.9 ± 0.6 | 9.8 ± 3.1 | 1.1 ± 0.2 | 3.3 ± 0.9 | 42.1 ± 0.7 | 5.5 ± 1.5 | 85.8% | 14.2% |
| KO-Cont | 46.2 ± 4.7 | 2.1 ± 1.2 | 6.5 ± 1.6 | 0.6 ± 0.2 | 2.8 ± 1.0 | 36.9 ± 3.1 | 4.9 ± 1.0 | 90.1% | 9.9% |
| KO-EtOH | 25.9 ± 1.3 | 1.0 ± 0.2 | 1.0 ± 0.2 | 0.3 ± 0.1 | 2.8 ± 0.2 | 62.7 ± 2.2 | 6.3 ± 1.1 | 95.9% | 4.1% |
| % (mean ± SD) | CA | CDCA | DCA | LCA | UDCA | Primary BA | Secondary BA |
|---|---|---|---|---|---|---|---|
| Control | 20.6 ± 5.3 | 29.1 ± 2.1 | 40.8 ± 5.8 | 7.5 ± 1.8 | 2.0 ± 0.4 | 49.7% | 50.3% |
| AALD | 35.5 ± 7.6 | 36.7 ± 3.6 | 19.8 ± 4.3 | 3.7 ± 1.3 | 4.3 ± 1.3 | 72.2% | 27.8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christidis, G.; Karatayli, E.; Hall, R.A.; Weber, S.N.; Reichert, M.C.; Hohl, M.; Qiao, S.; Boehm, U.; Lütjohann, D.; Lammert, F.; et al. Fibroblast Growth Factor 21 Response in a Preclinical Alcohol Model of Acute-on-Chronic Liver Injury. Int. J. Mol. Sci. 2021, 22, 7898. https://doi.org/10.3390/ijms22157898
Christidis G, Karatayli E, Hall RA, Weber SN, Reichert MC, Hohl M, Qiao S, Boehm U, Lütjohann D, Lammert F, et al. Fibroblast Growth Factor 21 Response in a Preclinical Alcohol Model of Acute-on-Chronic Liver Injury. International Journal of Molecular Sciences. 2021; 22(15):7898. https://doi.org/10.3390/ijms22157898
Chicago/Turabian StyleChristidis, Grigorios, Ersin Karatayli, Rabea A. Hall, Susanne N. Weber, Matthias C. Reichert, Mathias Hohl, Sen Qiao, Ulrich Boehm, Dieter Lütjohann, Frank Lammert, and et al. 2021. "Fibroblast Growth Factor 21 Response in a Preclinical Alcohol Model of Acute-on-Chronic Liver Injury" International Journal of Molecular Sciences 22, no. 15: 7898. https://doi.org/10.3390/ijms22157898
APA StyleChristidis, G., Karatayli, E., Hall, R. A., Weber, S. N., Reichert, M. C., Hohl, M., Qiao, S., Boehm, U., Lütjohann, D., Lammert, F., & Karatayli, S. C. (2021). Fibroblast Growth Factor 21 Response in a Preclinical Alcohol Model of Acute-on-Chronic Liver Injury. International Journal of Molecular Sciences, 22(15), 7898. https://doi.org/10.3390/ijms22157898