Central and Peripheral Mechanisms in ApoE4-Driven Diabetic Pathology

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Alzheimer’s Disease and Apolipoprotein ε4
1.2. Alzheimer’s Disease and Type 2 Diabetes Miletus
1.3. ApoE4 and T2DM
2. Results
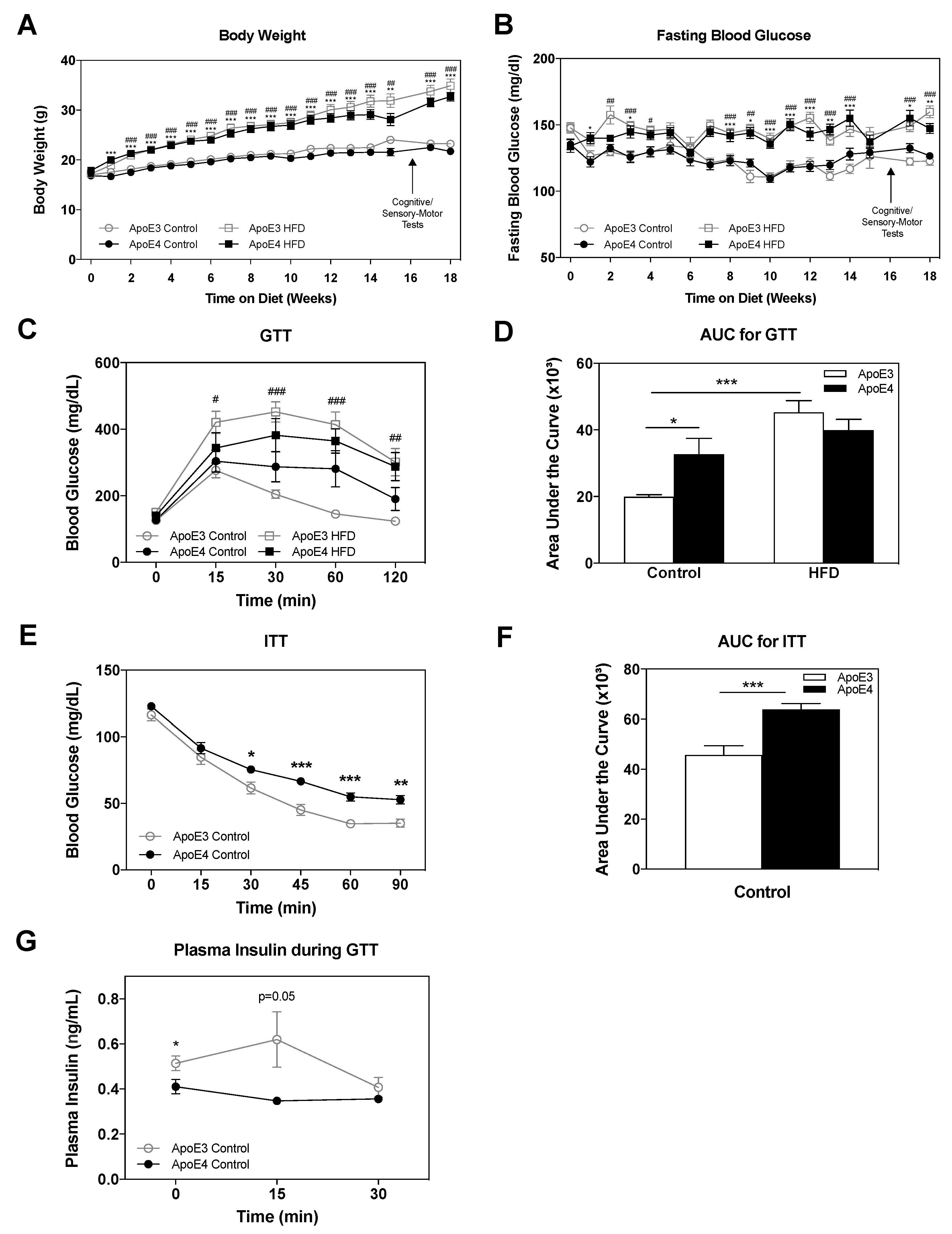
2.1. Body Weight and Glucose Metabolism
2.2. Cognitive and Sensorimotor Tests
2.3. PNS Skin Intra-Epidermal Nerve Fiber Density (IENFD) and Sciatic Nerve Electrophysiological Measurements
2.4. CNS Insulin Signaling
2.5. ApoE Lipidation and Levels in the Brain and Plasma
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Diets
4.3. Metabolic Tests
4.3.1. GTT
4.3.2. ITT
4.4. Behavioral Tests
4.4.1. Short-Term Memory Measurements
4.4.2. Motor Coordination and Learning Measurements
4.4.3. Thermal Pain Sensation Measurements
4.5. Electrophysiological Tests
4.6. Skin Biopsies for IENFD Measures
4.7. Immunoblots
4.7.1. Preparation of Plasma Protein Extracts
4.7.2. Preparation of Hippocampi Protein Extracts
4.7.3. SDS-Electrophoresis
4.7.4. Blue Native Gels
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| APOE | Apolipoprotein E gene |
| apoE4 | Apolipoprotein E4 isoform |
| PNS | Peripheral nervous system |
| NMJ | Neuromuscular Junction |
| T2DM | Type 2 Diabetes Miletus |
| DPN | Diabetic peripheral neuropathy |
| CNS | Central nervous system |
| IR | Insulin Receptor |
| GSK-3β | Glycogen synthase kinase-3β |
| GS | Glycogen Synthase |
| HFD | High Fat Diet |
| TR | Targeted replacement |
| PBS | Phosphate buffered saline |
| IP | Intraperitoneally |
| GTT | Glucose tolerance test |
| AUC | Area under the curve |
| ITT | Insulin tolerance test |
| NOR | Novel Object Recognition |
| MNCV | Motor nerve conduction velocity |
| IENFD | Intra-epidermal nerve fiber density |
| CMAP | Compound muscle action potential |
| WT | Wild Type |
| mTORC2 | Mechanistic target of rapamycin complex 2 |
References
- Haas, A.; Liraz, O.; Michaelson, D.M. The effects of apolipoproteins E3 and E4 on the transforming growth factor-β system in targeted replacement mice. Neurodegener. Dis. 2012, 10, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Hodson, R. Alzheimer’s disease. Nature 2018, 559, S1. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2013, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Chouraki, V.; Seshadri, S. Genetics of Alzheimer’s disease. Adv. Genet. 2014, 87, 245–294. [Google Scholar] [PubMed]
- Weisgraber, K.H.; Mahley, R.W. Human apolipoprotein E: The Alzheimer’s disease connection. FASEB J. 1996, 10, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.; Dean, R.; Beer, B.; Lippa, A. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Zhang, J.; Lin, Y.; Dai, X.; Fang, W.; Wu, X.; Chen, X. Metformin treatment improves the spatial memory of aged mice in an APOE genotype-dependent manner. FASEB J. 2019, 33, 7748–7759. [Google Scholar] [CrossRef]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef]
- Barnes, L.L.; Wilson, R.S.; Bienias, J.L.; Schneider, J.A.; Evans, D.A.; Bennett, D.A. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch. Gen. Psychiatry 2005, 62, 685–691. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef]
- Li, G.; Sorbi, S.; St George-Hyslop, P.H.; Foncin, J.-F.; Tanzi, R.E.; Chi, H.; Rommens, J.M.; Pinessi, L.; Ikeda, M.; Mar, L.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 2003, 375, 754–760. [Google Scholar]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 1996, 47, 387–400. [Google Scholar] [CrossRef]
- Lucotte, G.; Visvikis, S.; Leininger-Muler, B.; David, F.; Berriche, S.; Reveilleau, S.; Couderc, R.; Babron, M.C.; Aguillon, D.; Siest, G. Association of apolipoprotein E allele ε4 with late-onset sporadic Alzheimer’s disease. Am. J. Med. Genet. 1994, 43, 1467. [Google Scholar] [CrossRef]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef]
- Emi, M.; Wu, L.L.; Robertson, M.A.; Myers, R.L.; Hegele, R.A.; Williams, R.R.; White, R.; Lalouel, J.M. Genotyping and sequence analysis of apolipoprotein E isoforms. Genomics 1988, 3, 373–379. [Google Scholar] [CrossRef]
- Kamboh, M.I. Apolipoprotein E polymorphism and susceptibility to Alzheimer’s disease. Hum. Biol. 1995, 67, 195–215. [Google Scholar]
- Farrer, L.A. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease. JAMA 1997, 278, 1349. [Google Scholar] [CrossRef]
- Janssen, C.I.; Jansen, D.; Mutsaers, M.P.; Dederen, P.J.; Geenen, B.; Mulder, M.T.; Kiliaan, A.J. The Effect of a High-Fat Diet on Brain Plasticity, Inflammation and Cognition in Female ApoE4-Knockin and ApoE-Knockout Mice. PLoS ONE 2016, 11, e0155307. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y. Apolipoprotein E Sets the Stage: Response to Injury Triggers Neuropathology. Neuron 2012, 76, 871–885. [Google Scholar] [CrossRef]
- Arendt, T.; Schindler, C.; Brückner, M.K.; Eschrich, K.; Bigl, V.; Zedlick, D.; Marcova, L. Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein epsilon 4 allele. J. Neurosci. 1997, 17, 516–529. [Google Scholar] [CrossRef]
- Kutner, K.C.; Erlanger, D.M.; Tsai, J.; Jordan, B.; Relkin, N.R. Lower cognitive performance of older football players possessing apolipoprotein E e(element of)4. Neurosurgery 2000, 47, 651–658. [Google Scholar] [PubMed]
- Bedlack, R.S.; Strittmatter, W.J.; Morgenlander, J.C. Apolipoprotein E and neuromuscular disease: A critical review of the literature. Arch. Neurol. 2000, 57, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Geranmayeh, F.; Christian, L.; Turkheimer, F.E.; Gentleman, S.M.; O’Neill, K.S. A need to clarify the role of apolipoprotein E in peripheral nerve injury and repair. J. Peripher. Nerv. Syst. 2005, 10, 344–345. [Google Scholar] [CrossRef] [PubMed]
- Akaaboune, M.; Villanova, M.; Festoff, B.W.; Verdière-Sahuqué, M.; Hantaï, D. Apolipoprotein E expression at neuromuscular junctions in mouse, rat and human skeletal muscle. FEBS Lett. 1994, 351, 246–248. [Google Scholar] [CrossRef]
- Boyles, J.K.; Pitas, R.E.; Wilson, E.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Investig. 1985, 76, 1501–1513. [Google Scholar] [CrossRef]
- Comley, L.H.; Fuller, H.R.; Wishart, T.M.; Mutsaers, C.A.; Thomson, D.; Wright, A.K.; Ribchester, R.R.; Morris, G.E.; Parson, S.H.; Horsburgh, K.; et al. ApoE isoform-specific regulation of regeneration in the peripheral nervous system. Hum. Mol. Genet. 2011, 20, 2406–2421. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Murano, T.; Watanabe, H.; Itoh, Y.; Miyashita, Y.; Shirai, K. [The examination of apoE phenotypes in diabetic patients with peripheral neuropathy]. Rinsho Byori. 1998, 46, 829–833. [Google Scholar]
- Bedlack, R.S.; Edelman, D.; Gibbs, J.W.; Kelling, D.; Strittmatter, W.; Saunders, A.M.; Morgenlander, J. APOE genotype is a risk factor for neuropathy severity in diabetic patients. Neurology 2003, 60, 1022–1024. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, N.-Q.; Yan, F.; Jin, H.; Zhou, S.-Y.; Shi, J.-S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Haan, M.N. Therapy Insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S. V Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Woung, L.C.; Tsai, M.T.; Liu, C.C.; Su, Y.H.; Li, C.Y. Risk of Alzheimer’s disease in relation to diabetes: A population-based cohort study. Neuroepidemiology 2012, 38, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, E. An Update on Type 2 Diabetes Mellitus as a Risk Factor for Dementia. J. Alzheimer’s Dis. 2016, 53, 393–402. [Google Scholar] [CrossRef]
- Khorami, S.A.H.; Movahedi, A.; Huzwah, K.; Sokhini, A.M.M. Review Article; PI3K/AKT pathway in modulating glucose homeostasis and its alteration in Diabetes. Ann. Med. Biomed. Sci. 2015, 1, 2. [Google Scholar]
- Tesfaye, S.; Boulton, A.J.M.; Dyck, P.J.; Freeman, R.; Horowitz, M.; Kempler, P.; Lauria, G.; Malik, R.A.; Spallone, V.; Vinik, A.; et al. Diabetic neuropathies: Update on definitions, diagnostic criteria, estimation of severity, and treatments. Proc. Diabetes Care 2010, 33, 2285–2293. [Google Scholar] [CrossRef]
- Mojto, V.; Chládeková, A.; Komlósi, M.; Timárová, G. Diabetic neuropathy. Lek. Obz. 2019, 68, 73–77. [Google Scholar]
- Mittal, K.; Katare, D.P. Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab. Syndr. 2016, 10, 144–149. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Seiça, R.; Oliveira, C.R. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J. Neurol. Sci. 2007, 257, 206–214. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Sena, C.; Nunes, E.; Seiça, R.; Oliveira, C.R. CoQ10 therapy attenuates amyloid beta-peptide toxicity in brain mitochondria isolated from aged diabetic rats. Exp. Neurol. 2005, 196, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Steculorum, S.M.; Solas, M.; Brüning, J.C. The paradox of neuronal insulin action and resistance in the development of aging-associated diseases. Alzheimer’s Dement. 2014, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef]
- Hill, J.M.; Lesniak, M.A.; Pert, C.B.; Roth, J. Autoradiographic localization of insulin receptors in rat brain: Prominence in olfactory and limbic areas. Neuroscience 1986, 17, 1127–1138. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.-C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129. [Google Scholar] [CrossRef]
- Patel, S.; Doble, B.; Woodgett, J.R. Glycogen Synthase Kinase-3 in Insulin and Wnt Signalling: A Double-edged Sword? Biochem. Soc. Trans. 2004, 32, 803–808. [Google Scholar] [CrossRef]
- Krishnankutty, A.; Kimura, T.; Saito, T.; Aoyagi, K.; Asada, A.; Takahashi, S.I.; Ando, K.; Ohara-Imaizumi, M.; Ishiguro, K.; Hisanaga, S.I. In vivo regulation of glycogen synthase kinase 3β activity in neurons and brains. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain–Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Chiu, S.-L.; Chen, C.-M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef]
- Hanson, A.J.; Craft, S.; Banks, W.A. The APOE genotype: Modification of therapeutic responses in Alzheimer’s disease. Curr. Pharm. Des. 2015, 21, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Torres, E.R.; Impey, S.; Stevens, J.F.; Raber, J. Apolipoprotein E4 and Insulin Resistance Interact to Impair Cognition and Alter the Epigenome and Metabolome. Sci Rep. 2017, 7, 43701. [Google Scholar] [CrossRef] [PubMed]
- El-Lebedy, D.; Raslan, H.M.; Mohammed, A.M. Erratum to: Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc. Diabetol. 2016. [Google Scholar] [CrossRef]
- Chaudhary, R.; Likidlilid, A.; Peerapatdit, T.; Tresukosol, D.; Srisuma, S.; Ratanamaneechat, S.; Sriratanasathavorn, C. Apolipoprotein E gene polymorphism: Effects on plasma lipids and risk of type 2 diabetes and coronary artery disease. Cardiovasc. Diabetol. 2012, 11, 36. [Google Scholar] [CrossRef]
- Malek-Ahmadi, M.; Beach, T.; Obradov, A.; Sue, L.; Belden, C.; Davis, K.; Walker, D.G.; Lue, L.; Adem, A.; Sabbagh, M.N. Type 2 Diabetes Is Associated With Increased Alzheimer’s Disease Neuropathology in ApoE epsilon4 Carriers. Curr. Alzheimer Res. 2013, 10, 654. [Google Scholar] [CrossRef]
- Ravona-Springer, R.; Heymann, A.; Schmeidler, J.; Sano, M.; Preiss, R.; Koifman, K.; Hoffman, H.; Silverman, J.M.; Beeri, M.S. The ApoE4 genotype modifies the relationship of long-term glycemic control with cognitive functioning in elderly with type 2 diabetes. Eur. Neuropsychopharmacol. 2014, 24, 1303–1308. [Google Scholar] [CrossRef][Green Version]
- Weykamp, C. HbA1c: A review of analytical and clinical aspects. Ann. Lab. Med. 2013, 33, 393–400. [Google Scholar] [CrossRef]
- Foley, J.M.; Salat, D.H.; Stricker, N.H.; McGlinchey, R.E.; Milberg, W.P.; Grande, L.J.; Leritz, E.C. Glucose Dysregulation Interacts With APOE-∊4 to Potentiate Temporoparietal Cortical Thinning. Am. J. Alzheimers. Dis. Other Demen. 2016, 31, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Altmann, A.; Ng, B.; Landau, S.M.; Jagust, W.J.; Greicius, M.D. Alzheimer’s Disease Neuroimaging Initiative Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 2015, 138, 3734–3746. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.; Wolf, A.B.; Chavira, B.; Shonebarger, D.; Meckel, J.P.; Leung, L.; Ballina, L.; Ly, S.; Saini, A.; Jones, T.B.; et al. Altered Energy Metabolism Pathways in the Posterior Cingulate in Young Adult Apolipoprotein e ε 4 Carriers. J. Alzheimer’s Dis. 2016, 53, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Ong, Q.R.; Chan, E.S.; Lim, M.L.; Cole, G.M.; Wong, B.S. Reduced phosphorylation of brain insulin receptor substrate and Akt proteins in apolipoprotein-E4 targeted replacement mice. Sci. Rep. 2014, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Ahrén, B. The high-fat diet-fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Proc. Diabetes 2004, 53, 215–219. [Google Scholar] [CrossRef]
- Yuan, T.; Chu, C.; Shi, R.; Cui, T.; Zhang, X.; Zhao, Y.; Shi, X.; Hui, Y.; Pan, J.; Qian, R.; et al. ApoE-Dependent Protective Effects of Sesamol on High-Fat Diet-Induced Behavioral Disorders: Regulation of the Microbiome-Gut-Brain Axis. J. Agric. Food Chem. 2019, 67, 6190–6201. [Google Scholar] [CrossRef]
- Le Floch, J.P.; Escuyer, P.; Baudin, E.; Baudon, D.; Perlemuter, L. Blood glucose area under the curve. Methodological aspects. Diabetes Care 1990, 13, 172–175. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Salomon-Zimri, S.; Koren, A.; Angel, A.; Ben-Zur, T.; Offen, D.; Michaelson, D.M. The Role of MAPK’s Signaling in Mediating ApoE4-Driven Pathology In Vivo. Curr. Alzheimer Res. 2019, 16, 281–292. [Google Scholar] [CrossRef]
- Bar, R.; Boehm-Cagan, A.; Luz, I.; Kleper-Wall, Y.; Michaelson, D.M. The effects of apolipoprotein E genotype, alpha-synuclein deficiency, and sex on brain synaptic and Alzheimer’s disease-related pathology. Alzheimers Dement. 2018, 10, 1–11. [Google Scholar]
- Boehm-Cagan, A.; Bar, R.; Harats, D.; Shaish, A.; Levkovitz, H.; Bielicki, J.K.; Johansson, J.O.; Michaelson, D.M. Differential Effects of apoE4 and Activation of ABCA1 on Brain and Plasma Lipoproteins. PLoS ONE 2016, 11, e0166195. [Google Scholar] [CrossRef] [PubMed]
- Boehm-Cagan, A.; Bar, R.; Liraz, O.; Bielicki, J.K.; Johansson, J.O.; Michaelson, D.M. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J. Alzheimers Dis. 2016, 54, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar] [PubMed]
- Lee, Y.C.; Lu, S.C.; Hsieh, Y.L. Establishing a Mouse Model of a Pure Small Fiber Neuropathy with the Ultrapotent Agonist of Transient Receptor Potential Vanilloid Type 1. J. Vis. Exp. 2018, 56651. [Google Scholar] [CrossRef]
- Schulz, A.; Walther, C.; Morrison, H.; Bauer, R. In vivo electrophysiological measurements on mouse sciatic nerves. J. Vis. Exp. 2014, 86, e51181. [Google Scholar] [CrossRef]
- Chan, E.S.; Chen, C.; Soong, T.W.; Wong, B.S. Differential Binding of Human ApoE Isoforms to Insulin Receptor is Associated with Aberrant Insulin Signaling in AD Brain Samples. Neuromol. Med. 2018, 20, 124–132. [Google Scholar] [CrossRef]
- Segev, Y.; Livne, A.; Mints, M.; Rosenblum, K. Concurrence of High Fat Diet and APOE Gene Induces Allele Specific Metabolic and Mental Stress Changes in a Mouse Model of Alzheimer’s Disease. Front. Behav. Neurosci. 2016, 10, 170. [Google Scholar] [CrossRef]
- Johnson, L.A.; Torres, E.R.; Weber Boutros, S.; Patel, E.; Akinyeke, T.; Alkayed, N.J.; Raber, J. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J. Cereb. Blood Flow Metab. 2019, 39, 770–781. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Herz, J. High-Fat Diet Changes Hippocampal Apolipoprotein E (ApoE) in a Genotype- and Carbohydrate-Dependent Manner in Mice. PLoS ONE 2016, 11, e0148099. [Google Scholar] [CrossRef]
- Brandon, J.A.; Farmer, B.C.; Williams, H.C.; Johnson, L.A. APOE and alzheimer’s disease: Neuroimaging of metabolic and cerebrovascular dysfunction. Front. Aging Neurosci. 2018, 10, 180. [Google Scholar] [CrossRef]
- Salomon-Zimri, S.; Boehm-Cagan, A.; Liraz, O.; Michaelson, D.M. Hippocampus-related cognitive impairments in young apoE4 targeted replacement mice. Neurodegener Dis. 2014, 13, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Kornecook, T.J.; McKinney, A.P.; Ferguson, M.T.; Dodart, J.C. Isoform-specific effects of apolipoprotein E on cognitive performance in targeted-replacement mice overexpressing human APP. ; Genes Brain Behav. 2010, 9, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.S.; Shetty, M.S.; Sajikumar, S.; Chen, C.; Soong, T.W.; Wong, B.S. ApoE4 expression accelerates hippocampus-dependent cognitive deficits by enhancing Aβ impairment of insulin signaling in an Alzheimer’s disease mouse model. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Berteau-Pavy, F.; Park, B.; Raber, J. Effects of sex and APOE epsilon4 on object recognition and spatial navigation in the elderly. Neuroscience 2007, 147, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Haley, G.E.; Berteau-Pavy, F.; Parkv, B.; Raber, J.; Park, B. Effects of ε4 on Object Recognition in the Non-Demented Elderly. Curr. Aging Sci. 2010, 3, 127–137. [Google Scholar] [CrossRef]
- Gainey, S.J.; Kwakwa, K.A.; Bray, J.K.; Pillote, M.M.; Tir, V.L.; Towers, A.E.; Freund, G.G. Short-term high-fat diet (HFD) induced anxiety-like behaviors and cognitive impairment are improved with treatment by glyburide. Front. Behav. Neurosci. 2016, 10, 156. [Google Scholar] [CrossRef]
- Sah, S.K.; Lee, C.; Jang, J.-H.; Park, G.H. Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 493, 731–736. [Google Scholar] [CrossRef]
- Hwang, L.L.; Wang, C.H.; Li, T.L.; Chang, S.D.; Lin, L.C.; Chen, C.P.; Chen, C.T.; Liang, K.C.; Ho, I.K.; Yang, W.S.; et al. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity 2010, 18, 463–469. [Google Scholar] [CrossRef]
- Takase, K.; Tsuneoka, Y.; Oda, S.; Kuroda, M.; Funato, H. High-fat diet feeding alters olfactory-, social-, and reward-related behaviors of mice independent of obesity. Obesity 2016, 24, 886–894. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Keeney, J.T.R.; Ibrahimi, S.; Zhao, L. Human ApoE Isoforms Differentially Modulate Glucose and Amyloid Metabolic Pathways in Female Brain: Evidence of the Mechanism of Neuroprotection by ApoE2 and Implications for Alzheimer’s Disease Prevention and Early Intervention. J. Alzheimer’s Dis. 2015, 48, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.S.; Chen, C.; Cole, G.M.; Wong, B.-S. Differential interaction of Apolipoprotein-E isoforms with insulin receptors modulates brain insulin signaling in mutant human amyloid precursor protein transgenic mice. Sci. Rep. 2015, 5, 13842. [Google Scholar] [CrossRef]
- Traversy, M.-T.; Vandal, M.; Tremblay, C.; Tournissac, M.; Giguère-Rancourt, A.; Bennett, A.D.; Calon, F. Altered cerebral insulin response in transgenic mice expressing the epsilon-4 allele of the human apolipoprotein E gene. Psychoneuroendocrinology 2017, 77, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Cho, K.A.; Song, J.; Kim, Y.K. Transcriptomic analysis of high fat diet fed mouse brain cortex. Front. Genet. 2019, 10, 83. [Google Scholar] [CrossRef]
- Fordahl, S.C.; Jones, S.R. High-Fat-Diet-Induced Deficits in Dopamine Terminal Function Are Reversed by Restoring Insulin Signaling. ACS Chem. Neurosci. 2017, 8, 290–299. [Google Scholar] [CrossRef]
- DiLoreto, R.; Murphy, C.T. The cell biology of aging. Mol. Biol. Cell 2015, 26, 4524–4531. [Google Scholar] [CrossRef]
- Rocha, E.M.; Hirata, A.E.; Carneiro, E.M.; Saad, M.J.A.; Velloso, L.A. Impact of gender on insulin signaling pathway in lacrimal and salivary glands of rats. Endocrine 2002, 18, 191–199. [Google Scholar] [CrossRef]
- Marks, J.L.; Porte, D.; Stahl, W.L.; Baskin, D.G. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 1990, 127, 3234–3236. [Google Scholar] [CrossRef]
- Xu, W.H.; Huber, R.; Riepe, M.W. Gender- and region-specific expression of insulin receptor protein in mouse brain: Effect of mild inhibition of oxidative phosphorylation. J. Neural Transm. 2007, 114, 373–377. [Google Scholar] [CrossRef]
- Luz, I.; Liraz, O.; Michaelson, D.M. An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr. Alzheimer Res. 2016, 13, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.M.; Mezdour, H.; Aratani, Y.; Knouff, C.; Najib, J.; Reddick, R.L.; Quarfordt, S.H.; Maeda, N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 1997, 272, 17972–17980. [Google Scholar] [CrossRef] [PubMed]
- Levi, O.; Jongen-Relo, A.L.; Feldon, J.; Roses, A.D.; Michaelson, D.M. ApoE4 impairs hippocampal plasticity isoform-specifically and blocks the environmental stimulation of synaptogenesis and memory. Neurobiol. Dis. 2003, 13, 273–282. [Google Scholar] [CrossRef]
- Heydemann, A. An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2016, 2902351. [Google Scholar] [CrossRef] [PubMed]
- Vinué, Á.; González-Navarro, H. Glucose and insulin tolerance tests in the mouse. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2015; Volume 1339, pp. 247–254. [Google Scholar]
- Ayala, J.E.; Samuel, V.T.; Morton, G.J.; Obici, S.; Croniger, C.M.; Shulman, G.I.; Wasserman, D.H.; McGuinness, O.P. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. DMM Dis. Model. Mech. 2010, 3, 525–534. [Google Scholar] [CrossRef]
- Prado, V.F.; Martins-Silva, C.; de Castro, B.M.; Lima, R.F.; Barros, D.M.; Amaral, E.; Ramsey, A.J.; Sotnikova, T.D.; Ramirez, M.R.; Kim, H.G.; et al. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron 2006, 51, 601–612. [Google Scholar] [CrossRef]
- Lauria, G.; Hsieh, S.T.; Johansson, O.; Kennedy, W.R.; Leger, J.M.; Mellgren, S.I.; Nolano, M.; Merkies, I.S.J.; Polydefkis, M.; Smith, A.G.; et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Eur. J. Neurol. 2010, 17, 903–912. [Google Scholar] [CrossRef]
- Salomon-Zimri, S.; Glat, M.J.; Barhum, Y.; Luz, I.; Boehm-Cagan, A.; Liraz, O.; Ben-Zur, T.; Offen, D.; Michaelson, D.M. Reversal of ApoE4-Driven Brain Pathology by Vascular Endothelial Growth Factor Treatment. J. Alzheimers Dis. 2016, 53, 1443–1458. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koren-Iton, A.; Salomon-Zimri, S.; Smolar, A.; Shavit-Stein, E.; Dori, A.; Chapman, J.; Michaelson, D.M. Central and Peripheral Mechanisms in ApoE4-Driven Diabetic Pathology. Int. J. Mol. Sci. 2020, 21, 1289. https://doi.org/10.3390/ijms21041289
Koren-Iton A, Salomon-Zimri S, Smolar A, Shavit-Stein E, Dori A, Chapman J, Michaelson DM. Central and Peripheral Mechanisms in ApoE4-Driven Diabetic Pathology. International Journal of Molecular Sciences. 2020; 21(4):1289. https://doi.org/10.3390/ijms21041289
Chicago/Turabian StyleKoren-Iton, Amit, Shiran Salomon-Zimri, Alex Smolar, Efrat Shavit-Stein, Amir Dori, Joab Chapman, and Daniel M. Michaelson. 2020. "Central and Peripheral Mechanisms in ApoE4-Driven Diabetic Pathology" International Journal of Molecular Sciences 21, no. 4: 1289. https://doi.org/10.3390/ijms21041289
APA StyleKoren-Iton, A., Salomon-Zimri, S., Smolar, A., Shavit-Stein, E., Dori, A., Chapman, J., & Michaelson, D. M. (2020). Central and Peripheral Mechanisms in ApoE4-Driven Diabetic Pathology. International Journal of Molecular Sciences, 21(4), 1289. https://doi.org/10.3390/ijms21041289