Complementary Transcriptome and Proteome Analyses Provide Insight into the Floral Transition in Bamboo (Dendrocalamus latiflorus Munro)


 , and
, and
Abstract
1. Introduction
2. Results
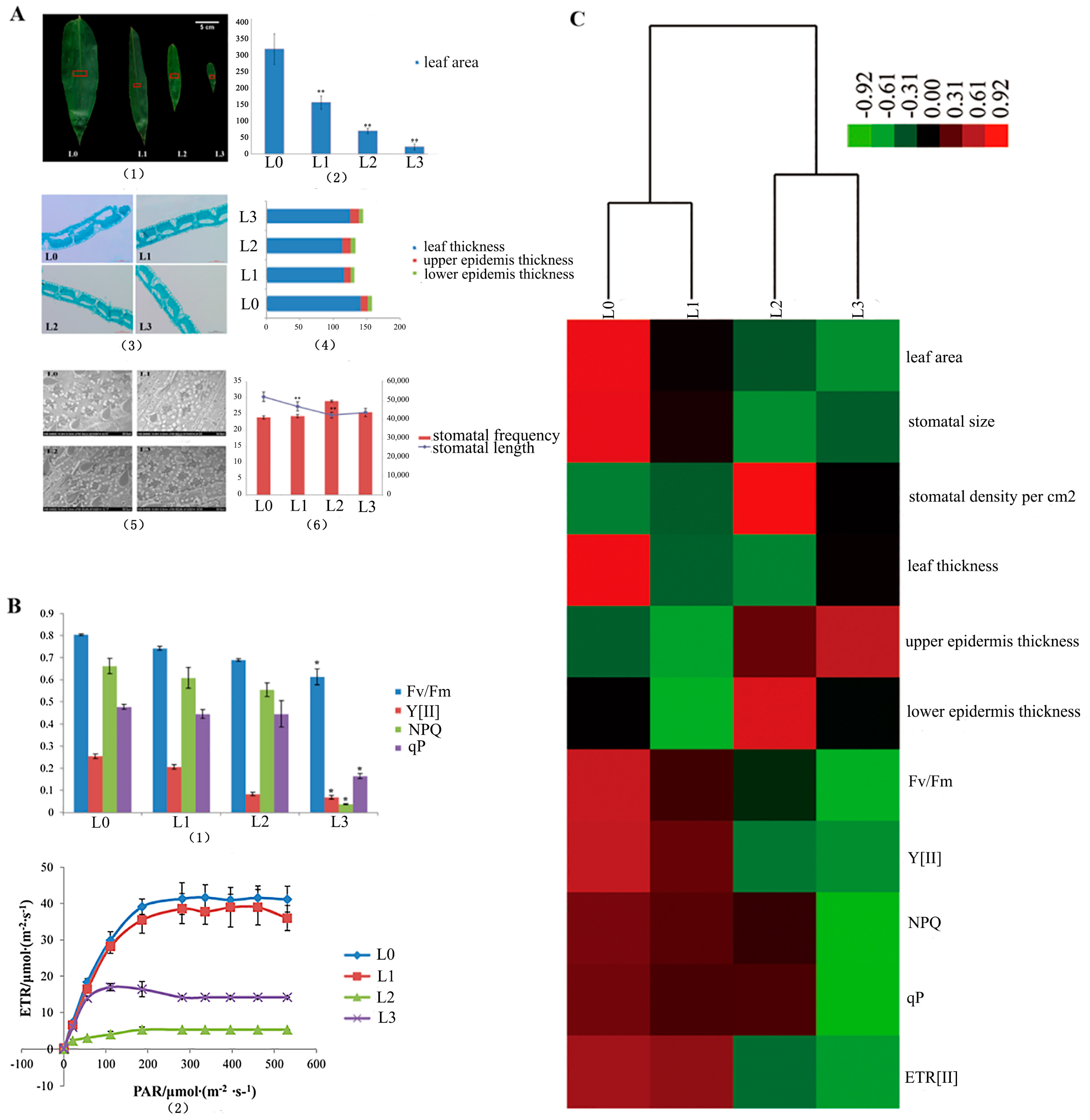
2.1. Leaf Microstructure and Chlorophyll Fluorescence of D. latiflorus
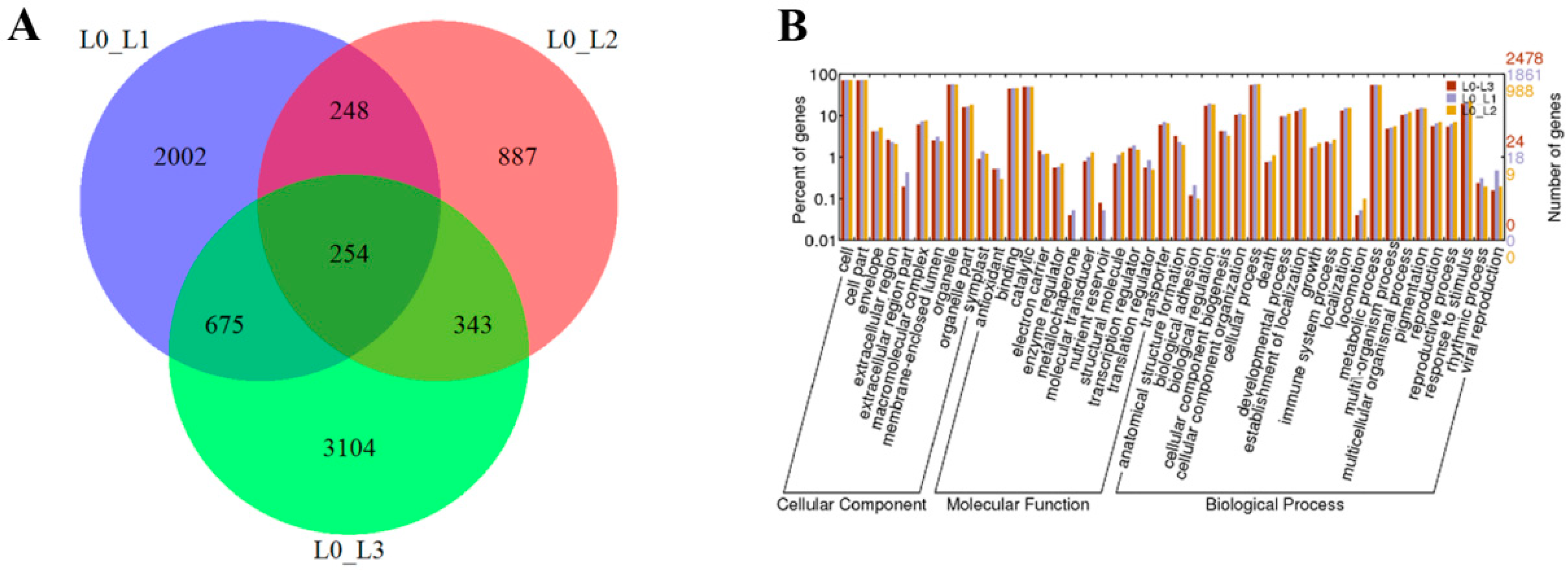
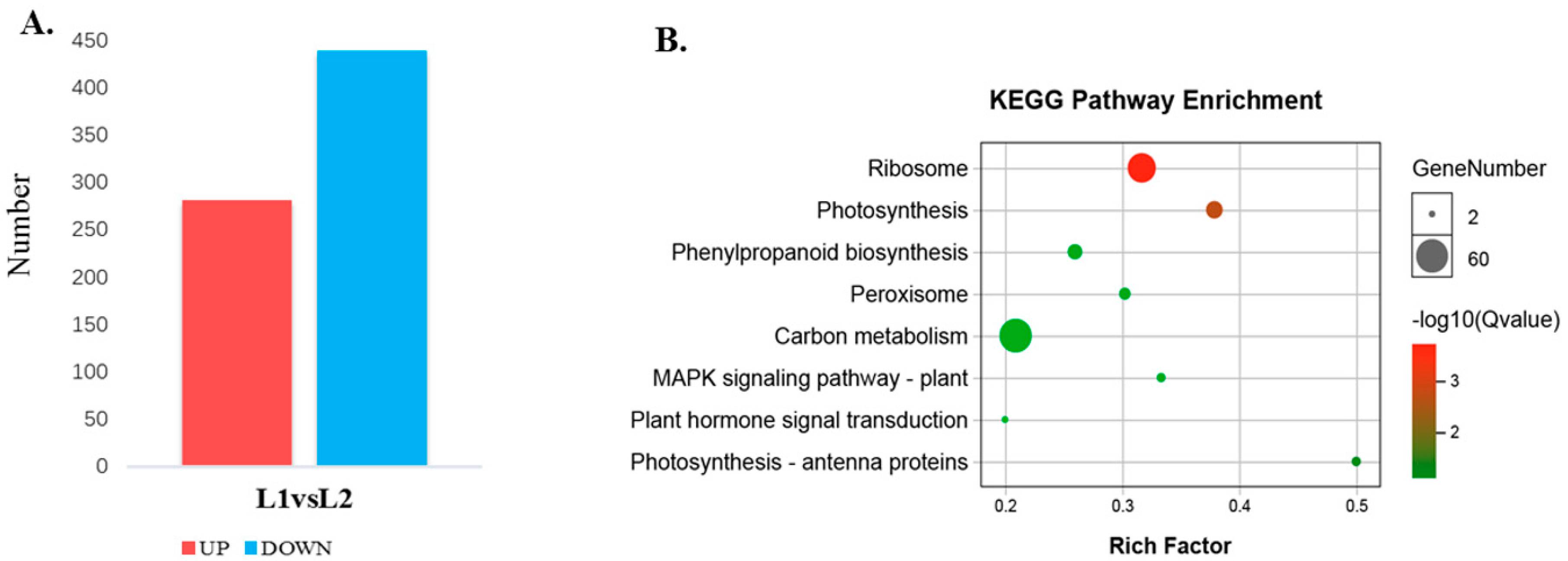
2.2. De Novo Assembly and Functional Annotation of the Differentially Expressed Genes (DEGs)
2.3. Screen Candidate Marker Genes
2.4. Identifying D. latiflorus Flowering Time-Associated Genes and Senescence-Associated Genes
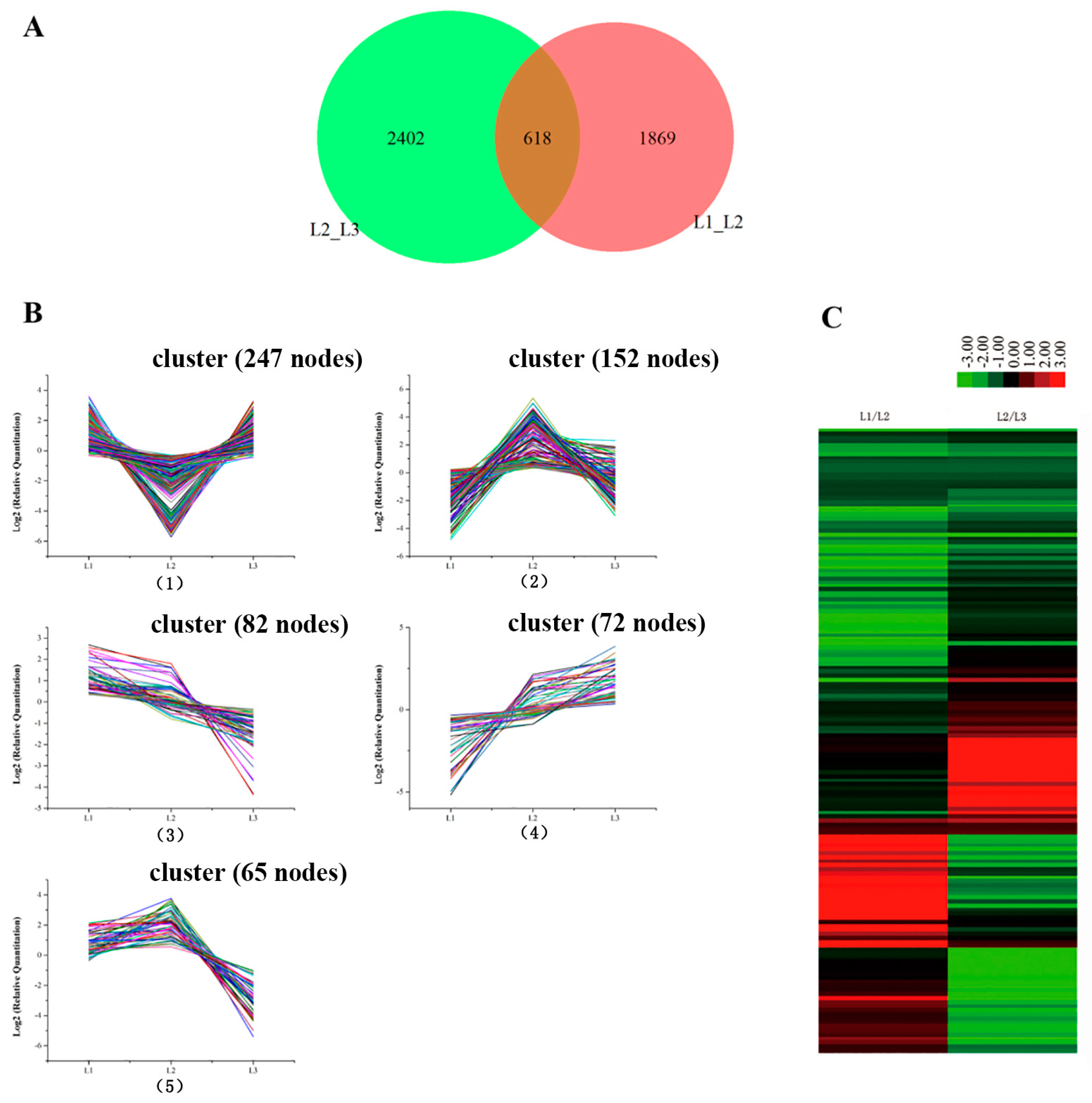
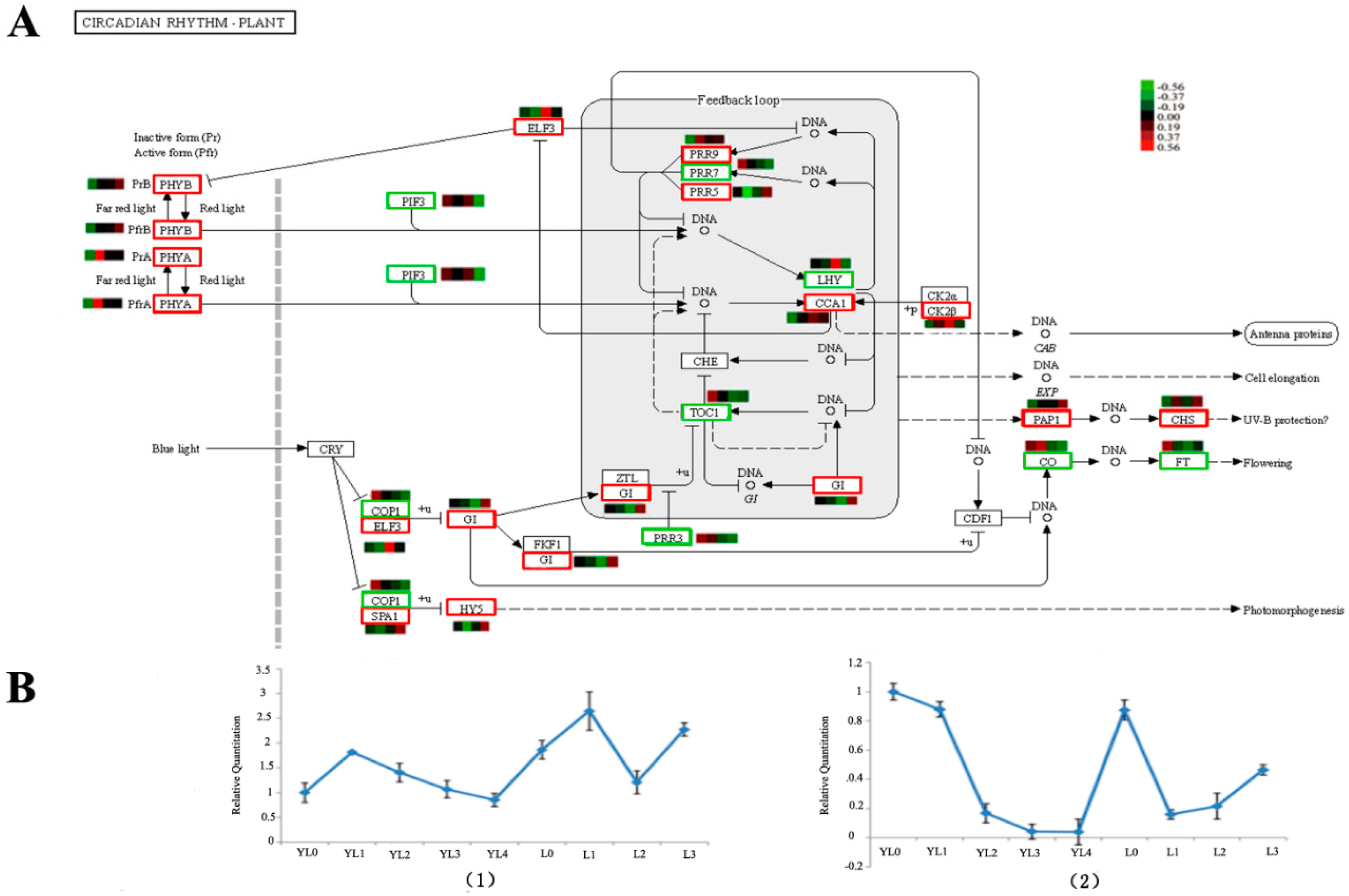
2.5. Pathways Involved during Floral Transition
2.6. Proteomics Analysis
2.7. Integrative Analysis of the Proteome and Transcriptome of L1 and L2
3. Discussion
3.1. Identifying the Genes Associated with D. latiflorus Flowering Time
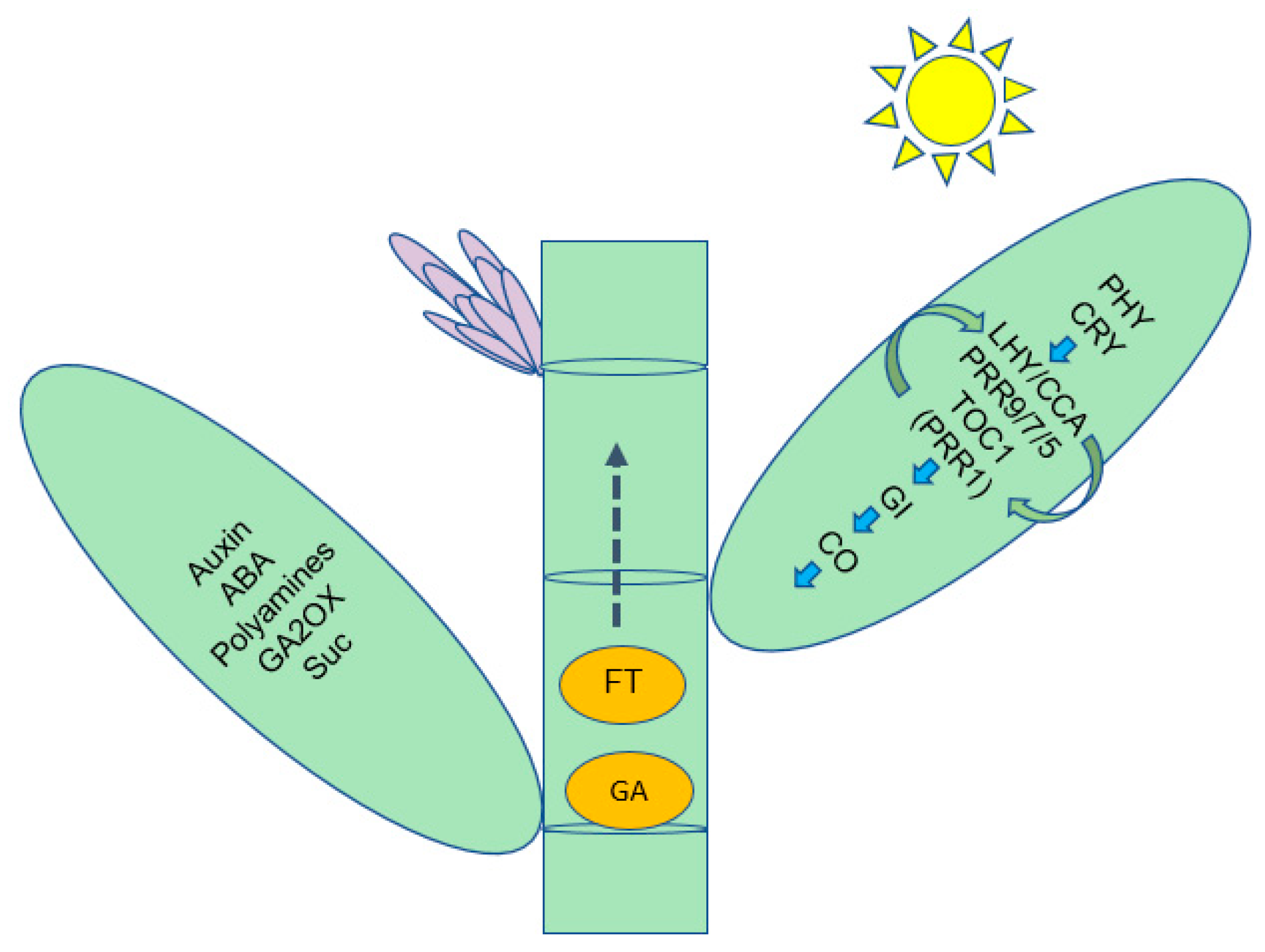
3.2. Identification of the Flowering Integration Genes and Metabolism Pathways Involved in Floral Transition
3.3. Screened Floral Biomarker Genes
3.4. Protein Changes and Metabolism Relate to Floral Transition
3.5. Proteome and Transcriptome Correlation Analysis
4. Materials and Methods
4.1. Sample Preparation
4.2. Measurement of Leaf Surface Area
4.3. Leaf Anatomy, Leaf Stomatal Size and Density Measurement
4.4. Chlorophyll Fluorescence Detection in the D. latiflorus
4.5. RNA-seq, Data Processing and Reference Preparation
4.6. Screening of DEGs
4.7. qRT-PCR Validation
4.8. iTRAQ Labeling and SCX Fractionation
4.9. LC-ESI-MS/MS Analysis Based on Q EXACTIVE
4.10. Data Analysis and Functional Annotation
4.11. Availability of Sequence Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RNA-seq | High-throughput RNA sequencing |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| SAM | Shoot apical meristem |
| ABA | Abscisic acid |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| ETR | Electron transfer reactions |
| PAR | Photosynthetic active radiation |
| GO | Gene ontology |
| Nr | Non-redundant protein database |
| EC | Enzyme Commission |
| Fri | Frigida |
| PRR5 | Response regulator-like APRR5 |
| GAs | Gibberellins |
| GAI | Gibberellin insensitive |
| GI | Gigantea |
| CO | Constans |
| TOC1 | Timing of cab expression 1 |
| LHY | Late elongated hypocotyl |
| CCA1 | Circadian clock associated 1 |
| qRT-PCR | Quantitative real-time PCR |
| NADP-ME | NADP dependent malic enzyme |
| BSP | Bark Storage Proteins |
| DEGs | Differentially expressed genes |
| HCD | High-energy collision dissociation |
| AGC | Automatic gain control |
References
- Poethig, R.S. Phase Change and the Regulation of Shoot Morphogenesis in Plants. Science 1990, 250, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Cho, L.-H.; Yoon, J.; An, G. The control of flowering time by environmental factors. Plant J. 2017, 90, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Gómez-Ariza, J.; Brambilla, V.; Fornara, F. Molecular control of seasonal flowering in rice, arabidopsis and temperate cereals. Ann. Bot. 2014, 114, 1445–1458. [Google Scholar] [CrossRef] [PubMed]
- Fornara, F.; De Montaigu, A.; Coupland, G. SnapShot: Control of Flowering in Arabidopsis. Cell 2010, 141, 550–550.e2. [Google Scholar] [CrossRef]
- Komiya, R.; Ikegami, A.; Tamaki, S.; Yokoi, S.; Shimamoto, K. Hd3a and RFT1 are essential for flowering in rice. Development 2008, 135, 767–774. [Google Scholar] [CrossRef]
- Brandis, D. Biological Notes on Indian Bamboos; Nabu Press: Charleston, SC, USA, 1899. [Google Scholar]
- Böhlenius, H.; Huang, T.; Charbonnel-Campaa, L.; Brunner, A.M.; Jansson, S.; Strauss, S.H.; Nilsson, O. CO/FT Regulatory Module Controls Timing of Flowering and Seasonal Growth Cessation in Trees. Science 2006, 312, 1040–1043. [Google Scholar] [CrossRef]
- Keeley, J.E.; Bond, W.J. Mast Flowering and Semelparity in Bamboos: The Bamboo Fire Cycle Hypothesis. Am. Nat. 1999, 154, 383–391. [Google Scholar] [CrossRef]
- Janzen, D.H. Why Bamboos Wait So Long to Flower. Annu. Rev. Ecol. Syst. 1976, 7, 347–391. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, X.; Fan, Z.; Liu, J.; Pimm, S.L.; Liu, L.; Garcia, C.; Songer, M.; Shao, X.; Skidmore, A.; et al. The next widespread bamboo flowering poses a massive risk to the giant panda. Biol. Conserv. 2019, 234, 180–187. [Google Scholar] [CrossRef]
- Guo, X.; Wang, Y.; Wang, Q.; Xu, Z.; Lin, X. Molecular characterization of FLOWERING LOCUS T(FT)genes from bamboo (Phyllostachys violascens). J. Plant Biochem. Biotechnol. 2015, 25, 168–178. [Google Scholar] [CrossRef]
- Shih, M.-C.; Chou, M.-L.; Yue, J.-J.; Hsu, C.-T.; Chang, W.-J.; Ko, S.-S.; Liao, D.-C.; Huang, Y.-T.; Chen, J.-W.; Yuan, J.-L.; et al. BeMADS1 is a key to delivery MADSs into nucleus in reproductive tissues-De novo characterization of Bambusa edulis transcriptome and study of MADS genes in bamboo floral development. BMC Plant Biol. 2014, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Hu, Q.; Zhu, Y.; Zhu, L.; Ma, T.; Zeng, H.; Zang, Q.; Li, X.; Lin, X. Comparative transcriptomic analysis of the flower induction and development of the Lei bamboo (Phyllostachys violascens). BMC Bioinform. 2019, 20, 687. [Google Scholar] [CrossRef] [PubMed]
- Gan, C.S.; Chong, P.K.; Pham, T.K.; Wright, P.C. Technical, Experimental, and Biological Variations in Isobaric Tags for Relative and Absolute Quantitation (iTRAQ). J. Proteome Res. 2007, 6, 821–827. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Zhao, L.; Guo, Z.-H. Morphology and Quantitative Monitoring of Gene Expression Patterns during Floral Induction and Early Flower Development in Dendrocalamus latiflorus. Int. J. Mol. Sci. 2014, 15, 12074–12093. [Google Scholar] [CrossRef] [PubMed]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A k-means clustering algorithm. JSTOR 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Johanson, U. Molecular Analysis of FRIGIDA, a Major Determinant of Natural Variation in Arabidopsis Flowering Time. Science 2000, 290, 344–347. [Google Scholar] [CrossRef]
- Liu, S.N.; Zhu, L.F.; Lin, X.; Ma, L. Overexpression of the repressor gene PvFRI-L from Phyllostachys violascens delays flowering time in transgenic Arabidopsis thaliana. Biol. Plant. 2016, 60, 401–409. [Google Scholar] [CrossRef]
- Sakamoto, T.; Miura, K.; Itoh, H.; Tatsumi, T.; Ueguchi-Tanaka, M.; Ishiyama, K.; Kobayashi, M.; Agrawal, G.K.; Takeda, S.; Abe, K.; et al. An Overview of Gibberellin Metabolism Enzyme Genes and Their Related Mutants in Rice. Plant Physiol. 2004, 134, 1642–1653. [Google Scholar] [CrossRef]
- Murase, K.; Hirano, Y.; Sun, T.-P.; Hakoshima, T. Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nat. Cell Biol. 2008, 456, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, S.; Kimizu, M.; Sugita, M.; Miyao, A.; Hirochika, H.; Uchida, E.; Nagato, Y.; Yoshida, H. MOSAIC FLORAL ORGANS1, an AGL6-Like MADS Box Gene, Regulates Floral Organ Identity and Meristem Fate in Rice. Plant Cell 2009, 21, 3008–3025. [Google Scholar] [CrossRef] [PubMed]
- Sung, Z.R.; Belachew, A.; Shunong, B.; Bertrand-Garcia, R. EMF, an Arabidopsis Gene Required for Vegetative Shoot Development. Science 1992, 258, 1645–1647. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.-H.; Ma, P.-F.; Yang, G.-Q.; Hu, J.-Y.; Liu, Y.-L.; Xia, E.-H.; Zhong, M.-C.; Zhao, L.; Sun, G.-L.; Xu, Y.-X.; et al. Genome Sequences Provide Insights into the Reticulate Origin and Unique Traits of Woody Bamboos. Mol. Plant 2019, 12, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Theissen, G.; Michaels, S.D.; Ditta, G.; Gustafson-Brown, C.; Pelaz, S.; Yanofsky, M.; Amasino, R.M. Faculty Opinions recommendation of AGL24 acts as a promoter of flowering in Arabidopsis and is positively regulated by vernalization. Fac. Opin. Publ. Peer Rev. Biomed. Lit. 2003, 33, 867–874. [Google Scholar] [CrossRef]
- Searle, I.; Coupland, G. Induction of flowering by seasonal changes in photoperiod. EMBO J. 2004, 23, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Hayama, R.; Coupland, G. Shedding light on the circadian clock and the photoperiodic control of flowering. Curr. Opin. Plant Biol. 2003, 6, 13–19. [Google Scholar] [CrossRef]
- Alabadí, D.; Oyama, T.; Yanovsky, M.J.; Harmon, F.G.; Más, P.; Kay, S.A. Reciprocal Regulation Between TOC1 and LHY/CCA1 Within the Arabidopsis Circadian Clock. Science 2001, 293, 880–883. [Google Scholar] [CrossRef]
- Dutta, S.; Biswas, P.; Chakraborty, S.; Mitra, D.; Pal, A.; Das, M. Identification, characterization and gene expression analyses of important flowering genes related to photoperiodic pathway in bamboo. BMC Genom. 2018, 19, 190. [Google Scholar] [CrossRef]
- Hisamoto, Y.; Kobayashi, M. Flowering habit of two bamboo species, Phyllostachys meyeri and Shibataea chinensis, analyzed with flowering gene expression. Plant Species Biol. 2012, 28, 109–117. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Weigel, D. Move on up, it’s time for change—Mobile signals controlling photoperiod-dependent flowering. Genes Dev. 2007, 21, 2371–2384. [Google Scholar] [CrossRef] [PubMed]
- Samach, A.; Onouchi, H.; Gold, S.E.; Ditta, G.S.; Schwarz-Sommer, Z.; Yanofsky, M.F.; Coupland, G. Distinct Roles of CONSTANS Target Genes in Reproductive Development of Arabidopsis. Science 2000, 288, 1613–1616. [Google Scholar] [CrossRef] [PubMed]
- Bernier, G.; Périlleux, C. A physiological overview of the genetics of flowering time control. Plant Biotechnol. J. 2005, 3, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Flexas, J.; Badger, M.; Chow, W.S.; Medrano, H.; Osmond, C.B. Analysis of the Relative Increase in Photosynthetic O2 Uptake When Photosynthesis in Grapevine Leaves Is Inhibited following Low Night Temperatures and/or Water Stress. Plant Physiol. 1999, 121, 675–684. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, J.; Kramer, P.; Musser, R. Temperature-Induced Water Stress in Chilling-Sensitive Plants. Funct. Plant Biol. 1982, 9, 343–352. [Google Scholar] [CrossRef]
- Song, S.; Qi, T.; Fan, M.; Zhang, X.; Gao, H.; Huang, H.; Wu, D.; Guo, H.; Xie, D. The bHLH Subgroup IIId Factors Negatively Regulate Jasmonate-Mediated Plant Defense and Development. PLoS Genet. 2013, 9, e1003653. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yasuno, N.; Sato, Y.; Yoda, M.; Yamazaki, R.; Kimizu, M.; Yoshida, H.; Nagamura, Y.; Kyozuka, J. Inflorescence meristem identity in rice is specified by overlapping functions of three AP1/FUL-like MADS box genes and PAP2, a SEPALLATA MADS box gene. Plant Cell 2012, 24, 1848–1859. [Google Scholar] [CrossRef]
- Gallardo, K.; Firnhaber, C.; Zuber, H.; Héricher, D.; Belghazi, M.; Henry, C.; Küster, H.; Thompson, R. A Combined Proteome and Transcriptome Analysis of Developing Medicago truncatula Seeds. Mol. Cell. Proteom. 2007, 6, 2165–2179. [Google Scholar] [CrossRef]
- Griffin, T.J.; Gygi, S.P.; Ideker, T.; Rist, B.; Eng, J.; Hood, L.; Aebersold, R. Complementary Profiling of Gene Expression at the Transcriptome and Proteome Levels in Saccharomyces cerevisiae. Mol. Cell. Proteom. 2002, 1, 323–333. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nat. Cell Biol. 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Wildhagen, H.; Dürr, J.; Ehlting, B.; Rennenberg, H. Seasonal nitrogen cycling in the bark of field-grown Grey poplar is correlated with meteorological factors and gene expression of bark storage proteins. Tree Physiol. 2010, 30, 1096–1110. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.E.K.; Weih, M. Nitrogen storage and seasonal nitrogen cycling in Populus: Bridging molecular physiology and ecophysiology. New Phytol. 2005, 167, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Hattersley, P.W. Characterization of C4 Type Leaf Anatomy in Grasses (Poaceae). Mesophyll: Bundle Sheath Area Ratios. Ann. Bot. 1984, 53, 163–180. [Google Scholar] [CrossRef]
- Minaxi; Saxena, J. Disease suppression and crop improvement in moong beans (Vigna radiata) through Pseudomonas and Burkholderia strains isolated from semi arid region of Rajasthan, India. BioControl 2010, 55, 799–810. [Google Scholar] [CrossRef]
- Marrie, T.J.; Nelligan, J.; Costerton, J.W. A scanning and transmission electron microscopic study of an infected endocardial pacemaker lead. Circulation 1982, 66, 1339–1341. [Google Scholar] [CrossRef]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Nedbal, L.; Soukupová, J.; Kaftan, D.; Whitmarsh, J.; Trtílek, M. Kinetic imaging of chlorophyll fluorescence using modulated light. Photosynth. Res. 2000, 66, 3–12. [Google Scholar] [CrossRef]
- Genty, B.; Briantais, J.-M.; Baker, N.R. The relationship between the quantum yield of photosynthetic electron transport and quenching of chlorophyll fluorescence. Biochim. Biophys. Acta (BBA) Gen. Subj. 1989, 990, 87–92. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Zhao, L.; Larson-Rabin, Z.; Li, D.-Z.; Guo, Z.-H. De Novo Sequencing and Characterization of the Floral Transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS ONE 2012, 7, e42082. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Liu, M.; Qiao, G.; Jiang, J.; Yang, H.; Xie, L.; Xie, J.; Zhuo, R. Transcriptome Sequencing and De Novo Analysis for Ma Bamboo (Dendrocalamus latiflorus Munro) Using the Illumina Platform. PLoS ONE 2012, 7, e46766. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- De Hoon, M.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef]
- Wickham, H.; Chang, W. ggplot2: An Implementation of the Grammar of Graphics. R Package Version 07. Available online: http://cran R-project org/package= ggplot2 (accessed on 8 November 2020).
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Liu, M.; Jiang, J.; Han, X.; Qiao, G.; Zhuo, R. Validation of Reference Genes Aiming Accurate Normalization of qRT-PCR Data in Dendrocalamus latiflorus Munro. PLoS ONE 2014, 9, e87417. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary | Number |
|---|---|
| Total number of unigenes | 155,494 |
| Total length | 229,730,264 |
| Mean unigene length | 1477 |
| N50 (bp) | 2069 |
| N90 (bp) | 779 |
| Gene ID | Gene Name | Predicted Protein | Pathway | Log2FC |
|---|---|---|---|---|
| Unigene2563 | FT | Putative kinase inhibitor | Pathway integrator | −2.81 |
| Unigene123271 | SOC1 | MADS-box | Pathway integrator | −8.71 |
| Unigene39652 | AGL24 | MADS-box | Pathway integrator | 7.37 |
| Unigene57034 | Fri | Coiled-coil domain | Vernalization | −6.04 |
| Unigene110221 | VIN3 | PHD, VID-domain | Vernalization | 6.69 |
| Unigene120387 | FCA | RNA-binding | Autonomous | 10.74 |
| Unigene132444 | FY | Polyadenylation factor | Autonomous | 2.64 |
| Unigene132606 | GID1 | Gibberellin receptor GID1 | Gibberellin | 8.79 |
| Unigene33012 | GA20ox | Gibberellin 20 oxidase 1-B | Gibberellin | −6.88 |
| Unigene29825 | SPL9 | Squamosa promoter-binding-like protein 9 | Age pathway | −6.11 |
| Unigene30392 | MADS14 | MADS-box | Meristem identity, Floral organ identity | −8.11 |
| Unigene108692 | AGL6 | MADS-box | Other flowering gene | 10.02 |
| Unigene72731 | EMF1 | Polycomb-group (Pc-G) proteins | Other flowering gene | 7.43 |
| Gene ID | Gene Annotation | FPKM | Log2FC | |
|---|---|---|---|---|
| L0 | L3 | |||
| Unigene42305 | Phytochrome A (Phy A) | 0.01 | 1.09 | 6.77 |
| Unigene28224 | Phytochrome B (Phy B) | 0.21 | 4.23 | 4.33 |
| Unigene81404 | Cryptochrome (Cry) | 9.01 | 54.54 | 2.60 |
| Unigene10798 | Gigantea (GI) | 0.42 | 5.57 | 3.73 |
| Unigene118524 | Two-component response regulator-like APRR5 (PRR5) | 0.01 | 14.92 | 10.54 |
| Unigene4579 | Two-component response regulator-like APRR7 (PRR7) | 5.65 | 0.01 | −9.14 |
| Unigene105685 | Two-component response regulator-like APRR9 (PRR9) | 0.01 | 9.9 | 9.95 |
| Unigene91042 | Two-component response regulator-like APRR9 (PRR3) | 3.98 | 0.01 | −8.64 |
| Unigene105693 | Two-component response regulator-like APRR1 (PRR1/TOC1) | 83.05 | 21.21 | −1.97 |
| Unigene99793 | Homeobox-leucine zipper protein HDG6 (FWA) | 0.82 | 4.4 | 2.42 |
| Unigene2906 | protein LHY | 0.01 | 4.02 | 8.65 |
| Unigene7391 | EARLY FLOWERING 3 (ELF3) | 0.01 | 2.13 | 7.73 |
| Unigene24623 | EARLY FLOWERING 4 (ELF4) | 0.98 | 0.01 | −6.61 |
| Unigene5825 | Protein CCA1 | 0.69 | 4.78 | 2.79 |
| Unigene56852 | COP1 | 13.94 | 2.17 | −2.68 |
| Unigene102233 | CRY | 4.46 | 0.82 | −2.44 |
| Unigene26943 | PIF3 | 2.41 | 0.01 | −7.91 |
| Unigene26128 | SPA1 | 0.06 | 2.59 | 5.43 |
| Unigene75776 | PAP1 | 0.58 | 15.43 | 4.73 |
| Unigene148518 | CHS | 13.97 | 121.68 | 3.12 |
| Unigene56158 | CK2β | 0.24 | 3.56 | 3.89 |
| Unigene129590 | HY5 | 1.45 | 15.35 | 3.40 |
| Unigene ID | Protein ID | Proteins log2FC | Transcripts log2FC | Description |
|---|---|---|---|---|
| Unigene36630 | P1053 | 0.41 | 1.51 | psbP; photosystem II oxygen-evolving enhancer protein 2 |
| Unigene124766 | P3937 | −1.33 | −0.23 | PsbS protein |
| Unigene62245 | P536 | −0.77 | −2.94 | psaH; photosystem I subunit VI |
| Unigene9416 | P3559 | 1.00 | 8.83 | NADP dependent malic enzyme |
| Unigene117107 | P1873 | −0.30 | −0.45 | glgC; glucose-1-phosphate adenylyltransferase |
| Unigene148716 | P639 | −3.27 | −2.47 | Chitinase |
| Unigene111502 | P1156 | −1.81 | −0.16 | Armadillo-like |
| Unigene129955 | P977 | −1.10 | −1.15 | vegetative storage protein PNI288 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, Y.; Yang, G.; Zhao, L.; Zhang, X.; Li, D.; Guo, Z. Complementary Transcriptome and Proteome Analyses Provide Insight into the Floral Transition in Bamboo (Dendrocalamus latiflorus Munro). Int. J. Mol. Sci. 2020, 21, 8430. https://doi.org/10.3390/ijms21228430
Wang X, Wang Y, Yang G, Zhao L, Zhang X, Li D, Guo Z. Complementary Transcriptome and Proteome Analyses Provide Insight into the Floral Transition in Bamboo (Dendrocalamus latiflorus Munro). International Journal of Molecular Sciences. 2020; 21(22):8430. https://doi.org/10.3390/ijms21228430
Chicago/Turabian StyleWang, Xiaoyan, Yujiao Wang, Guoqian Yang, Lei Zhao, Xuemei Zhang, Dezhu Li, and Zhenhua Guo. 2020. "Complementary Transcriptome and Proteome Analyses Provide Insight into the Floral Transition in Bamboo (Dendrocalamus latiflorus Munro)" International Journal of Molecular Sciences 21, no. 22: 8430. https://doi.org/10.3390/ijms21228430
APA StyleWang, X., Wang, Y., Yang, G., Zhao, L., Zhang, X., Li, D., & Guo, Z. (2020). Complementary Transcriptome and Proteome Analyses Provide Insight into the Floral Transition in Bamboo (Dendrocalamus latiflorus Munro). International Journal of Molecular Sciences, 21(22), 8430. https://doi.org/10.3390/ijms21228430