The Combination of the M2 Muscarinic Receptor Agonist and Chemotherapy Affects Drug Resistance in Neuroblastoma Cells



 and
and 
Abstract
1. Introduction
2. Results
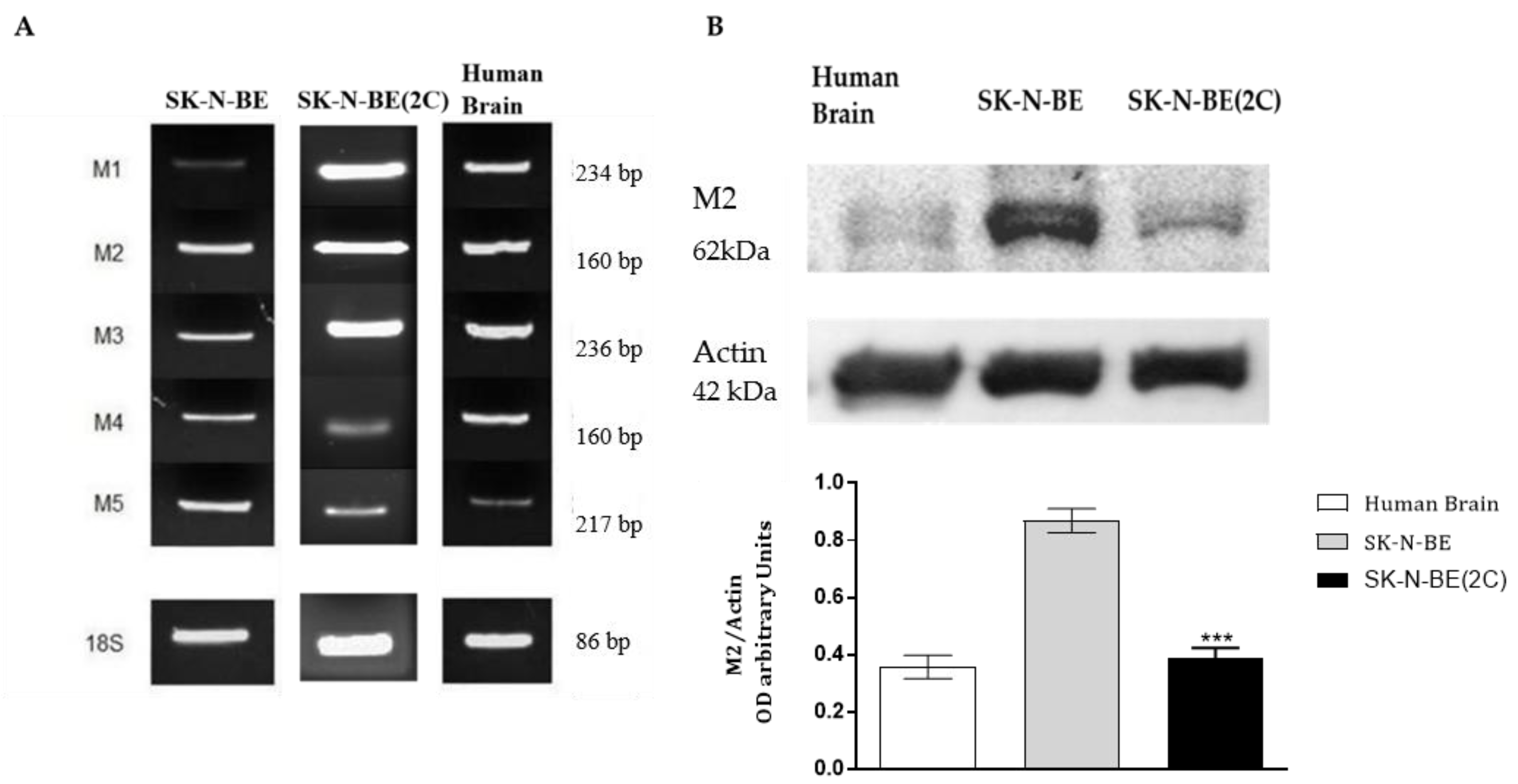
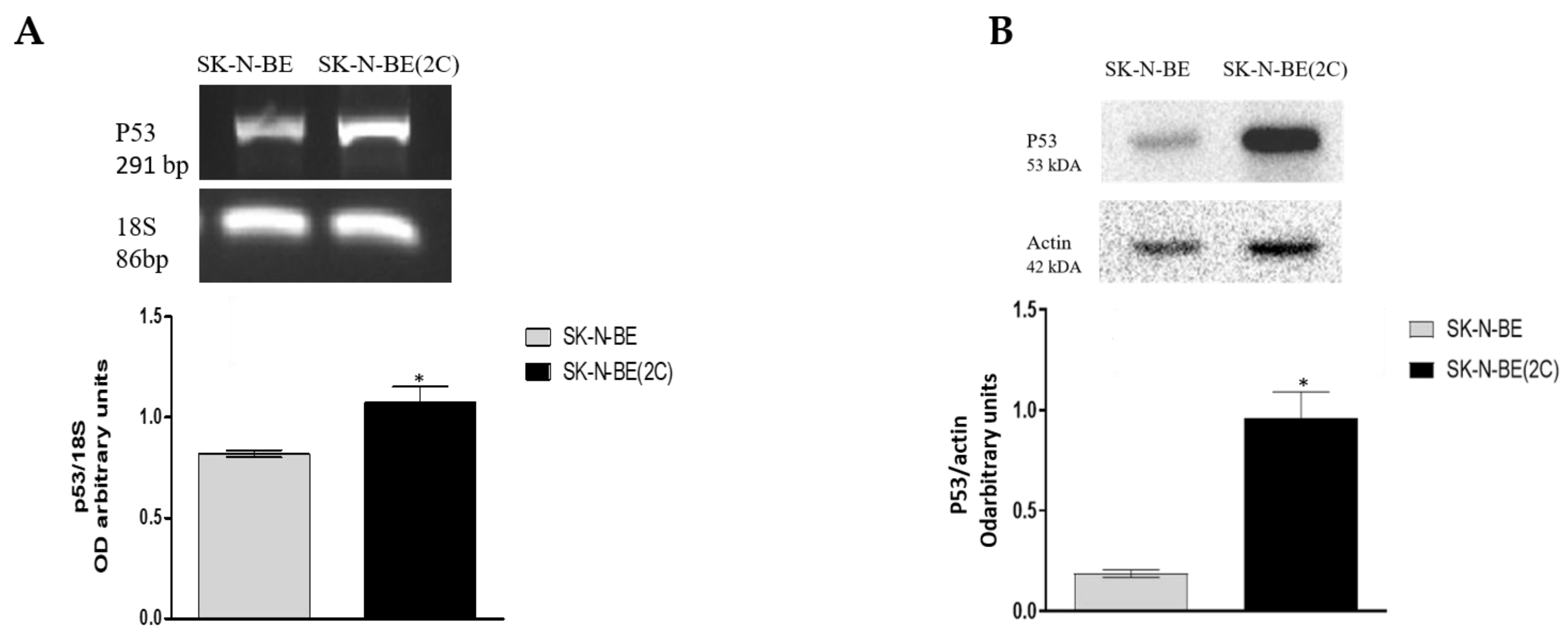
2.1. Muscarinic Receptor Expression in Neuroblastoma Cell Lines
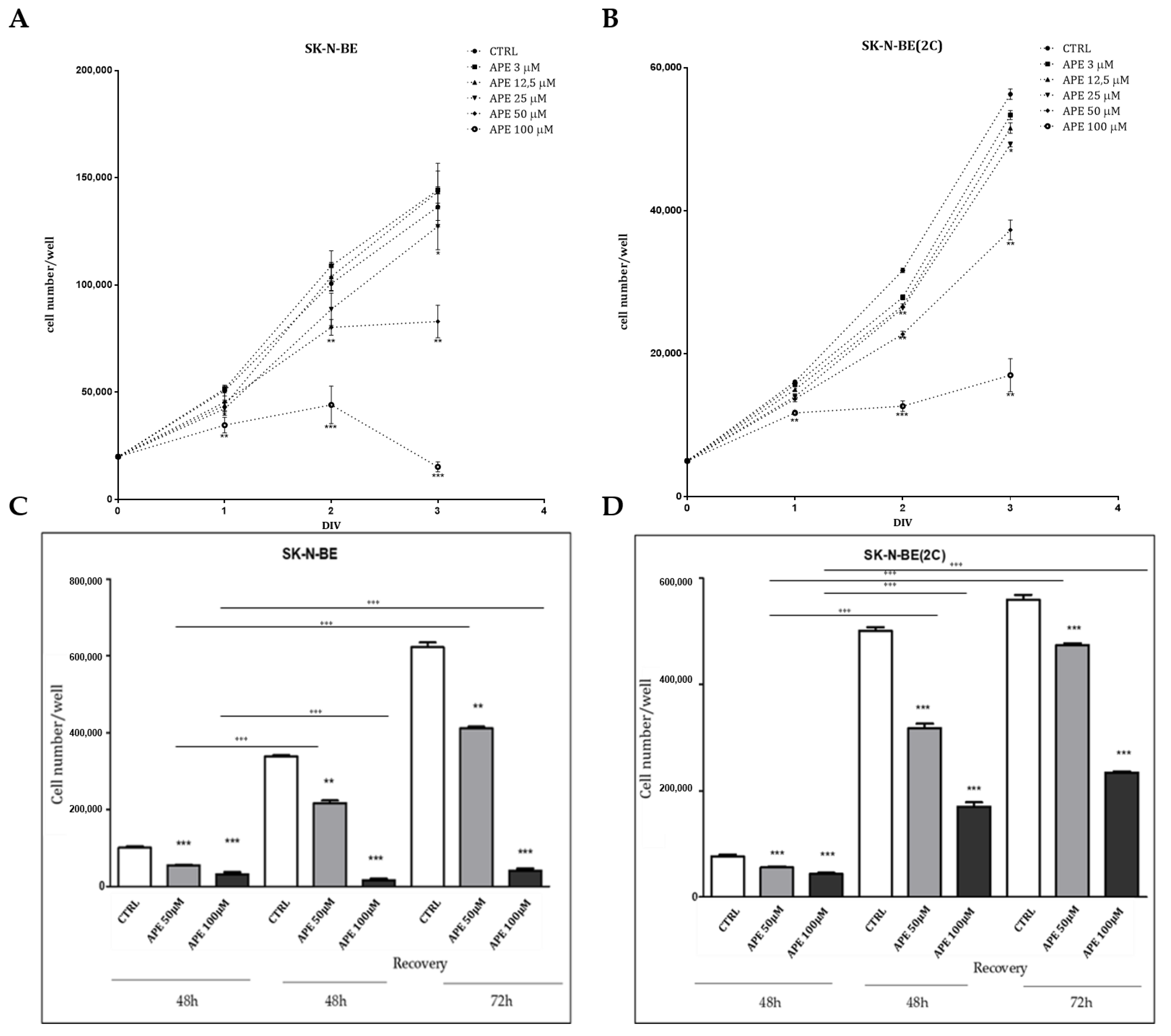
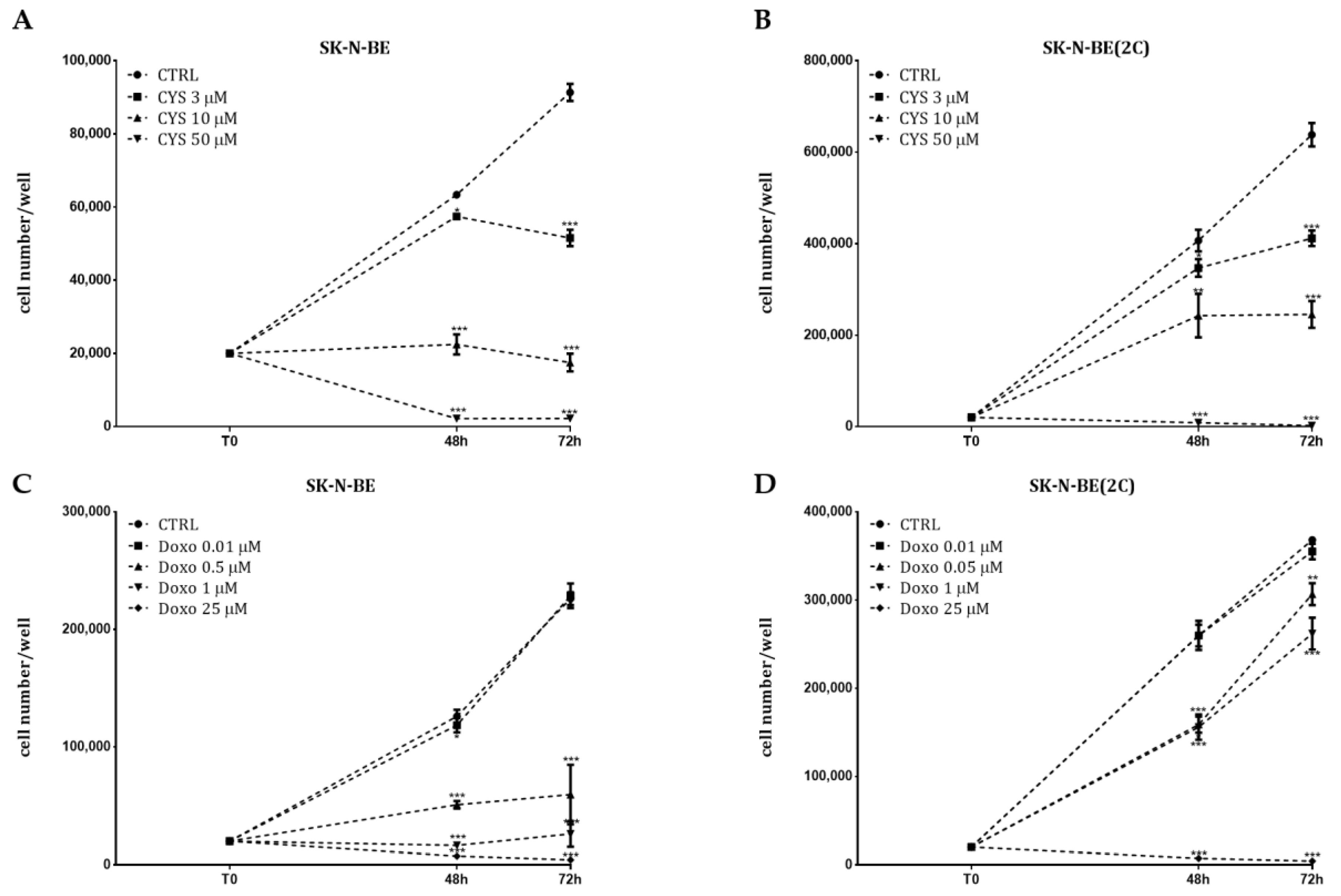
2.2. Analysis of Cell Proliferation
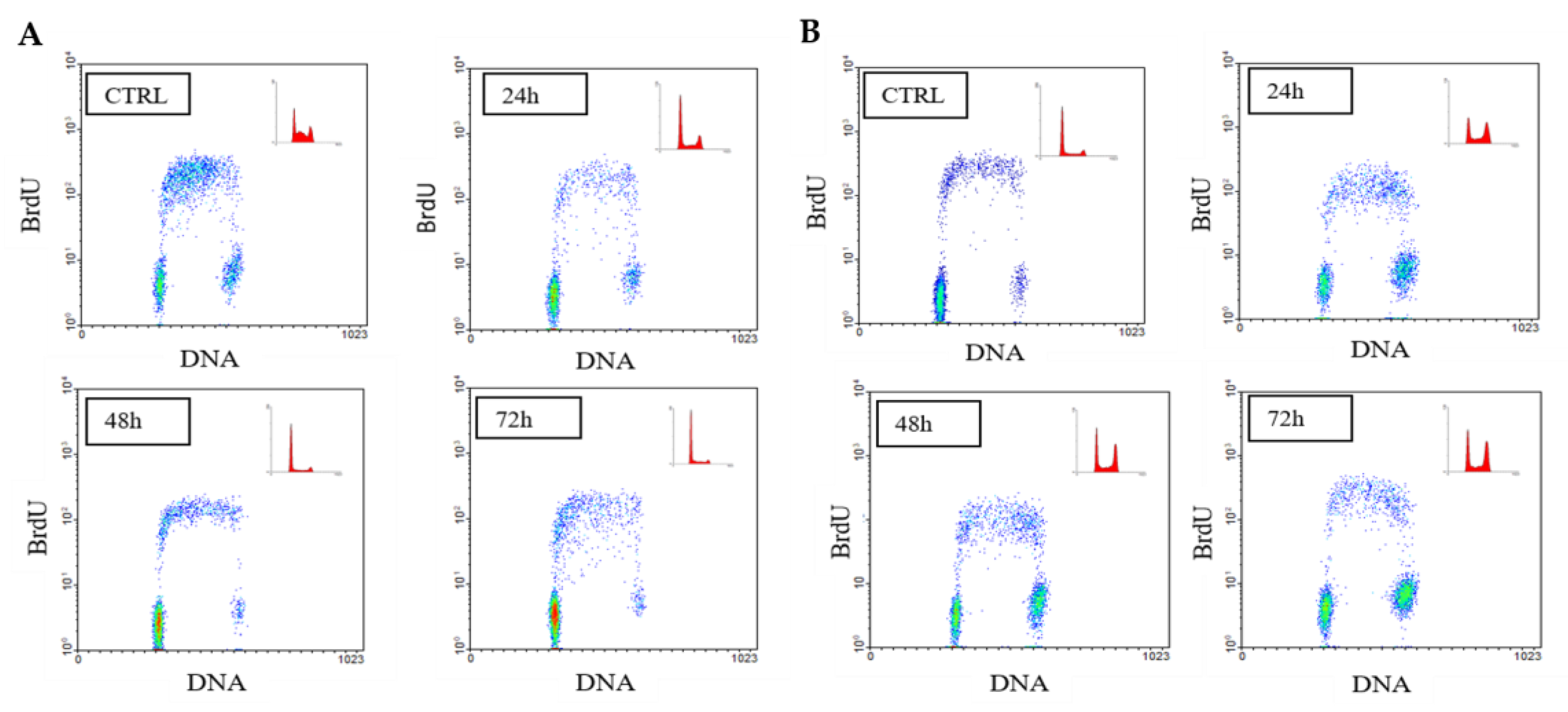
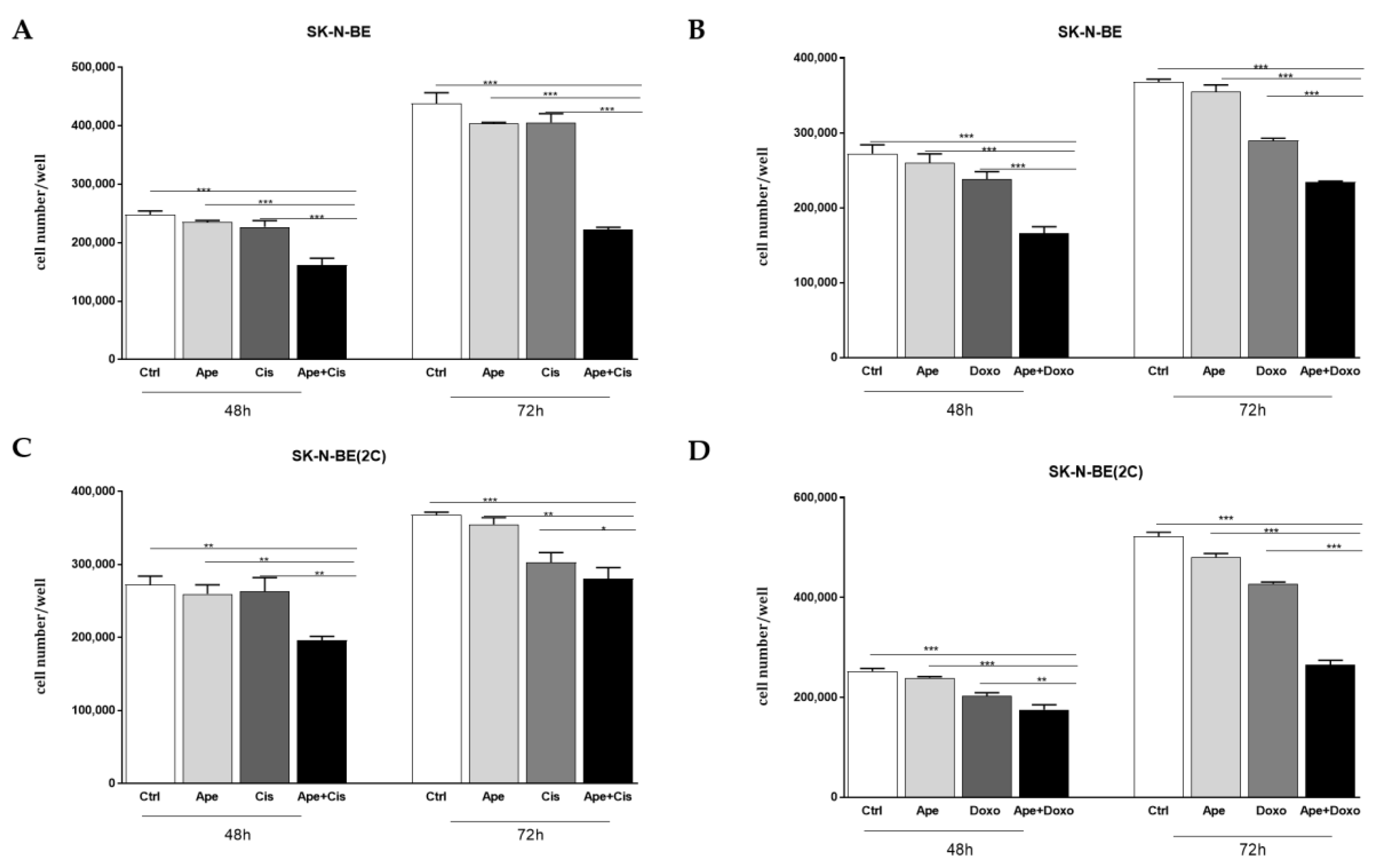
2.3. Chemosensitivity Test
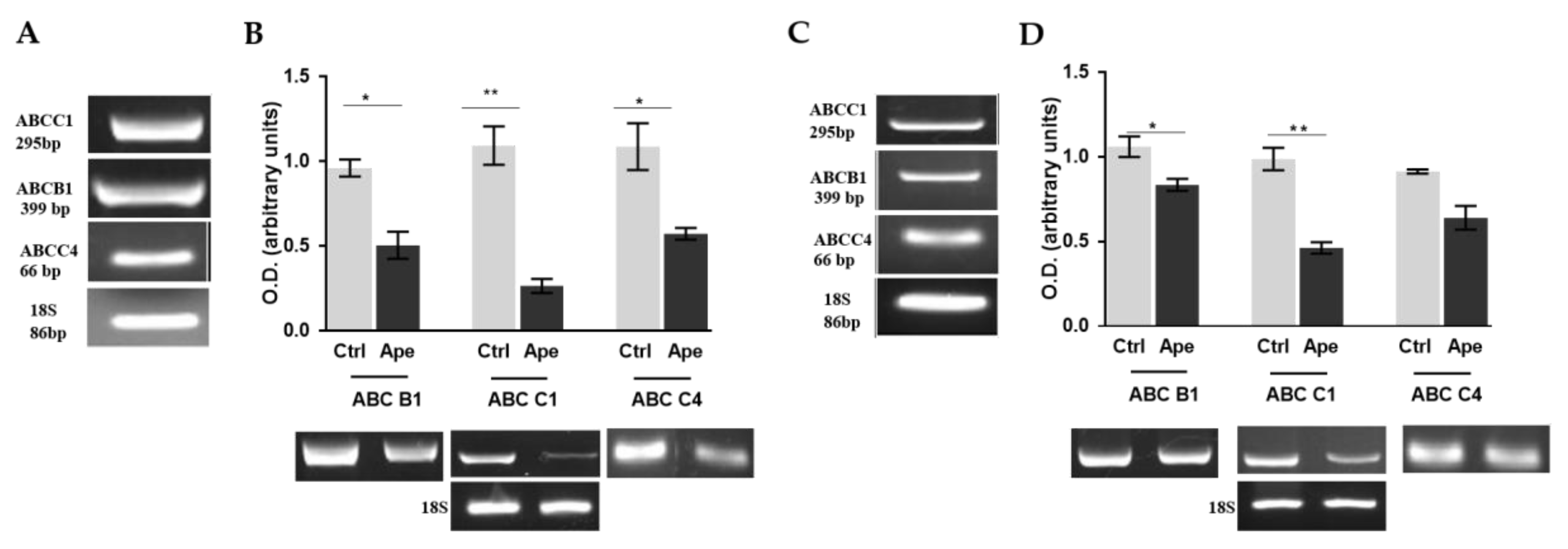
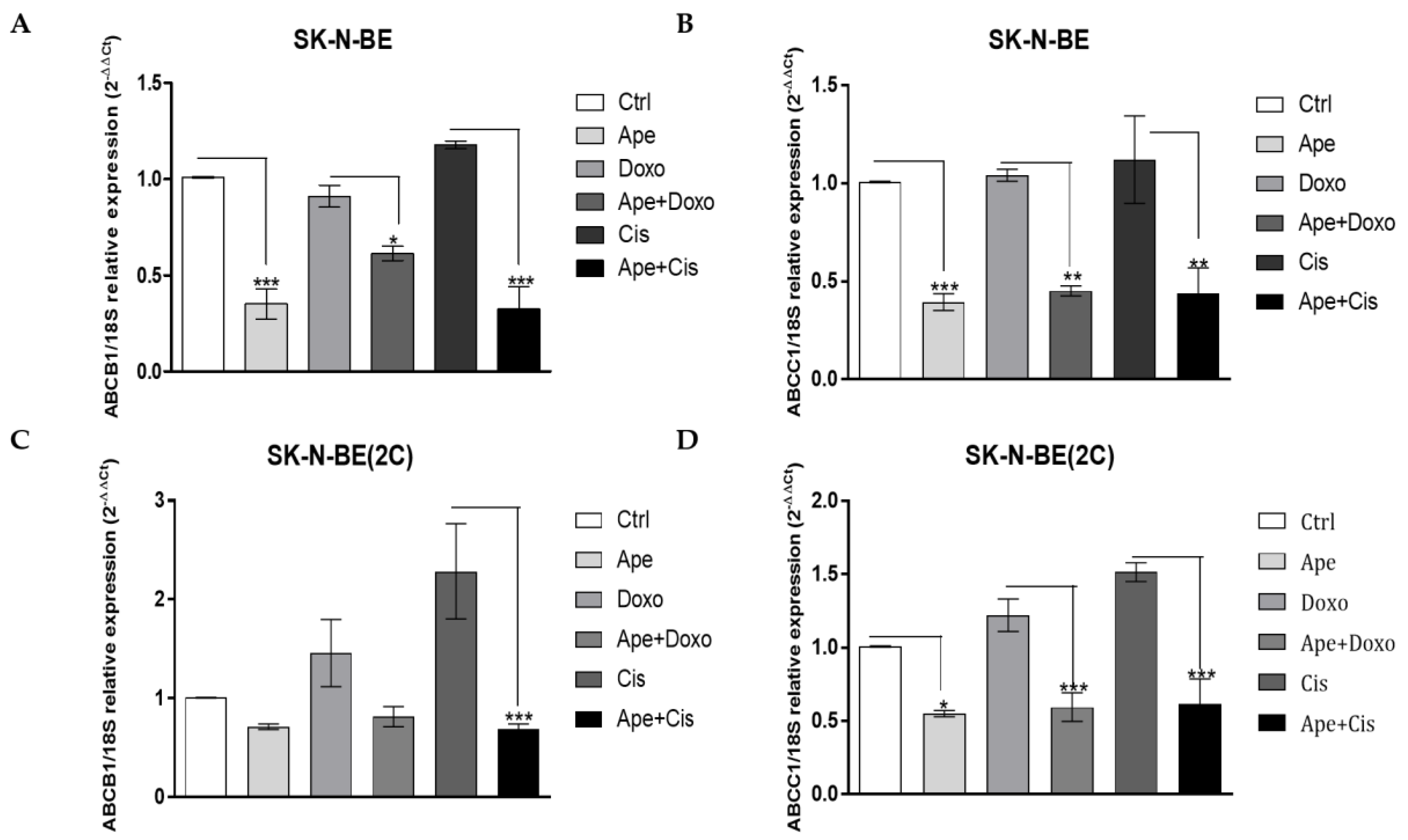
2.4. M2 Agonist Modulates the Efflux Pumps Expression
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Cell Viability Assays
4.3. Recovery Assay
4.4. Flow Cytometry Assay
4.5. Chemosensitivity Test
4.6. Western Blot
4.7. RNA Extraction RT-PCR and qRT-PCR Analysis
- CHRM1: (Acc. N. AW49504)
- CHRM2: (Acc. N. NM_000739.3)
- CHRM3: (Acc. N. NM_000740.4)
- CHRM4: (Acc. N. NM_000741.5)
- CHRM5 (Acc. N. NM_001320917.2)
- TP53 (Acc. N. NM_000546.6)
- 18S (Acc. N. NR_1461152)
- ABCB1: (Acc. N. NM_000927.5)
- ABCC1: (Acc. N. XM_017023237.1)
- ABCC4: (Acc. N. NM_001105515.3)
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kholodenko, I.V.; Kalinovsky, D.V.; Doronin, I.I.; Deyev, S.M.; Kholodenko, R.V. Neuroblastoma Origin and Therapeutic Targets for Immunotherapy. J. Immunol. Res. 2018, 2018, 1–25. [Google Scholar] [CrossRef]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; Mccarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Kachalaki, S.; Ebrahimi, M.; Khosroshahi, L.M.; Mohammadinejad, S.; Baradaran, B. Cancer chemoresistance; biochemical and molecular aspects: A brief overview. Eur. J. Pharm. Sci. 2016, 89, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Henderson, M.; Haber, M.; Norris, M. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life 2007, 59, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Nooter, K.; Stoter, G. Molecular Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Pathol. Res. Pr. 1996, 192, 768–780. [Google Scholar] [CrossRef]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Domenichini, A.; Adamska, A.; Falasca, M. ABC transporters as cancer drivers: Potential functions in cancer development. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 52–60. [Google Scholar] [CrossRef]
- Scotto, K.W. Transcriptional regulation of ABC drug transporters. Oncogene 2003, 22, 7496–7511. [Google Scholar] [CrossRef]
- Briz, O.; Perez-Silva, L.; Al-Abdulla, R.; Abete, L.; Reviejo, M.; Romero, M.R.; Marin, J.J. What “The Cancer Genome Atlas” database tells us about the role of ATP-binding cassette (ABC) proteins in chemoresistance to anticancer drugs. Expert Opin. Drug Metab. Toxicol. 2019, 15, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Peaston, A.; Gardaneh, M.; Franco, A.V.; E Hocker, J.; Murphy, K.M.; Farnsworth, M.L.; Catchpoole, D.R.; Haber, M.; Norris, M.D.; Lock, R.; et al. MRP1 gene expression level regulates the death and differentiation response of neuroblastoma cells. Br. J. Cancer 2001, 85, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Manohar, C.F.; A Bray, J.; Salwen, H.R.; Madafiglio, J.; Cheng, A.; Flemming, C.; Marshall, G.M.; Norris, M.D.; Haber, M.; Cohn, S.L. MYCN-mediated regulation of the MRP1 promoter in human neuroblastoma. Oncogene 2004, 23, 753–762. [Google Scholar] [CrossRef]
- Haber, M.; Smith, J.; Bordow, S.B.; Flemming, C.; Cohn, S.L.; London, W.B.; Marshall, G.M.; Norris, M.D. Association of High-Level MRP1 Expression With Poor Clinical Outcome in a Large Prospective Study of Primary Neuroblastoma. J. Clin. Oncol. 2006, 24, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Perini, G.; Diolaiti, D.; Porro, A.; Della Valle, G. In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation. Proc. Natl. Acad. Sci. USA 2005, 102, 12117–12122. [Google Scholar] [CrossRef]
- Piovesana, R.; Faroni, A.; Taggi, M.; Matera, A.; Soligo, M.; Canipari, R.; Manni, L.; Reid, A.J.; Tata, A.M. Muscarinic receptors modulate Nerve Growth Factor production in rat Schwann-like adipose-derived stem cells and in Schwann cells. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Piovesana, R.; Melfi, S.; Fiore, M.; Magnaghi, V.; Tata, A.M. M2 muscarinic receptor activation inhibits cell proliferation and migration of rat adipose-mesenchymal stem cells. J. Cell. Physiol. 2018, 233, 5348–5360. [Google Scholar] [CrossRef] [PubMed]
- Uggenti, C.; De Stefano, M.E.; Costantino, M.; Loreti, S.; Pisano, A.; Avallone, B.; Talora, C.; Magnaghi, V.; Tata, A.M. M2 muscarinic receptor activation regulates schwann cell differentiation and myelin organization. Dev. Neurobiol. 2014, 74, 676–691. [Google Scholar] [CrossRef]
- Confaloni, A.; Tosto, G.; Tata, A.M. Promising Therapies for Alzheimer’s Disease. Curr. Pharm. Des. 2016, 22, 1. [Google Scholar] [CrossRef]
- De De Angelis, F.; Tata, A. Analgesic Effects Mediated by Muscarinic Receptors: Mechanisms and Pharmacological Approaches. Central Nerv. Syst. Agents Med. Chem. 2016, 16, 1. [Google Scholar] [CrossRef]
- Gatta, V.; Mengod, G.; Reale, M.; Tata, A.M. Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis. Biomedicines 2020, 8, 153. [Google Scholar] [CrossRef]
- Tata, A.M. Muscarinic Acetylcholine Receptors: New Potential Therapeutic Targets in Antinociception and in Cancer Therapy. Recent Patents CNS Drug Discov. 2008, 3, 94–103. [Google Scholar] [CrossRef]
- Shah, N.; Khurana, S.; Cheng, K.; Raufman, J.-P. Muscarinic receptors and ligands in cancer. Am. J. Physiol. cell Physiol. 2009, 296, C221–C232. [Google Scholar] [CrossRef]
- Frucht, H.; Jensen, R.T.; Dexter, D.; Yang, W.L.; Xiao, Y. Human colon cancer cell proliferation mediated by the M3 muscarinic cholinergic receptor. Clin. Cancer Res. 1999, 5, 2532–2539. [Google Scholar]
- Tolaymat, M.; Larabee, S.M.; Hu, S.; Xie, G.; Raufman, J.-P. The Role of M3 Muscarinic Receptor Ligand-Induced Kinase Signaling in Colon Cancer Progression. Cancers 2019, 11, 308. [Google Scholar] [CrossRef]
- Oppitz, M.; Möbus, V.; Brock, S.; Drews, U. Muscarinic Receptors in Cell Lines from Ovarian Carcinoma: Negative Correlation with Survival of Patients. Gynecol. Oncol. 2002, 85, 159–164. [Google Scholar] [CrossRef]
- Rayford, W.; Noble, M.; Austenfeld, M.; Weigel, J.; Mebust, W.; Shah, G. Muscarinic cholinergic receptors promote growth of human prostate cancer cells. Prostate 1997, 30, 160–166. [Google Scholar] [CrossRef]
- Guo, L.; Liu, Y.; Ding, Z.; Sun, W.; Yuan, M. Signal transduction by M3 muscarinic acetylcholine receptor in prostate cancer. Oncol. Lett. 2016, 11, 385–392. [Google Scholar] [CrossRef][Green Version]
- Xu, C.; Yin, Q.-Q.; Xu, L.-H.; Zhang, M. Muscarinic acetylcholine receptor M1 mediates prostate cancer cell migration and invasion through hedgehog signaling. Asian J. Androl. 2018, 20, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Gu, X.; Zhang, C.; Lu, Q.; Chen, H.; Xu, L. Blocking M2 muscarinic receptor signaling inhibits tumor growth and reverses epithelial-mesenchymal transition (EMT) in non-small cell lung cancer (NSCLC). Cancer Biol. Ther. 2015, 16, 634–643. [Google Scholar] [CrossRef]
- Xu, R.; Shang, C.; Zhao, J.; Han, Y.; Liu, J.; Chen, K.; Shi, W. Activation of M3 muscarinic receptor by acetylcholine promotes non-small cell lung cancer cell proliferation and invasion via EGFR/PI3K/AKT pathway. Tumor Biol. 2015, 36, 4091–4100. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, E.; Montiel, M. Activation of MAP kinase by muscarinic cholinergic receptors induces cell proliferation and protein synthesis in human breast cancer cells. J. Cell. Physiol. 2005, 204, 678–686. [Google Scholar] [CrossRef]
- Sales, M.E.; Español, A.J.; Salem, A.R.; Pulido, P.M.; Sanchez, Y.; Sanchez, F.; Martínez, P.P. Role of Muscarinic Acetylcholine Receptors in Breast Cancer: Design of Metronomic Chemotherapy. Curr. Clin. Pharmacol. 2019, 14, 91–100. [Google Scholar] [CrossRef]
- Fiszman, G.L.; Middonno, M.C.; De La Torre, E.; Farina, M.; Español, A.J.; Sales, M.E. Activation of muscarinic cholinergic receptors induces MCF-7 cells proliferation and angiogenesis by stimulating nitric oxide synthase activity. Cancer Biol. Ther. 2007, 6, 1106–1113. [Google Scholar] [CrossRef]
- Español, A.J.; Salem, A.; Di Bari, M.; Cristofaro, I.; Sanchez, Y.; Tata, A.M.; Sales, M.E. The metronomic combination of paclitaxel with cholinergic agonists inhibits triple negative breast tumor progression. Participation of M2 receptor subtype. PLoS ONE 2020, 15, e0226450. [Google Scholar] [CrossRef]
- Oppitz, M.; Busch, C.; Garbe, C.; Drews, U. Distribution of muscarinic receptor subtype M3 in melanomas and their metastases. J. Cutan. Pathol. 2008, 35, 809–815. [Google Scholar] [CrossRef]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Ponti, D.; Calogero, A.; Tata, A. M2 muscarinic receptors inhibit cell proliferation in human glioblastoma cell lines. Life Sci. 2012, 91, 1134–1137. [Google Scholar] [CrossRef]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Ponti, D.; Ruggieri, P.; Raco, A.; Ricordy, R.; et al. M2 receptor activation inhibits cell cycle progression and survival in human glioblastoma cells. J. Cell. Mol. Med. 2013, 17, 552–566. [Google Scholar] [CrossRef]
- Alessandrini, F.; Cristofaro, I.; Di Bari, M.; Zasso, J.; Conti, L.; Tata, A.M. The activation of M2 muscarinic receptor inhibits cell growth and survival in human glioblastoma cancer stem cells. Int. Immunopharmacol. 2015, 29, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Tombolillo, V.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Iorio, E.; Carpinelli, G.; Ricordy, R.; Fiore, M.; Degrassi, F.; et al. Cytotoxic and genotoxic effects mediated by M2 muscarinic receptor activation in human glioblastoma cells. Neurochem. Int. 2015, 90, 261–270. [Google Scholar] [CrossRef]
- Di Bari, M.; Bevilacqua, V.; De Jaco, A.; Laneve, P.; Piovesana, R.; Trobiani, L.; Talora, C.; Caffarelli, E.; Tata, A.M. Mir-34a-5p Mediates Cross-Talk between M2 Muscarinic Receptors and Notch-1/EGFR Pathways in U87MG Glioblastoma Cells: Implication in Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 1631. [Google Scholar] [CrossRef]
- Cristofaro, I.; Alessandrini, F.; Spinello, Z.; Guerriero, C.; Fiore, M.; Caffarelli, E.; Laneve, P.; Dini, L.; Conti, L.; Tata, A.M. Cross Interaction between M2 Muscarinic Receptor and Notch1/EGFR Pathway in Human Glioblastoma Cancer Stem Cells: Effects on Cell Cycle Progression and Survival. Cells 2020, 9, 657. [Google Scholar] [CrossRef]
- Lucianò, A.M.; Mattei, F.; Damo, E.; Panzarini, E.; Dini, L.; Tata, A.M. Effects mediated by M2 muscarinic orthosteric agonist on cell growth in human neuroblastoma cell lines. Pure Appl. Chem. 2019, 91, 1641–1650. [Google Scholar] [CrossRef]
- Tweddle, D.A.; Malcolm, A.J.; Bown, N.; Pearson, A.D.; Lunec, J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. 2001, 61, 8–13. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
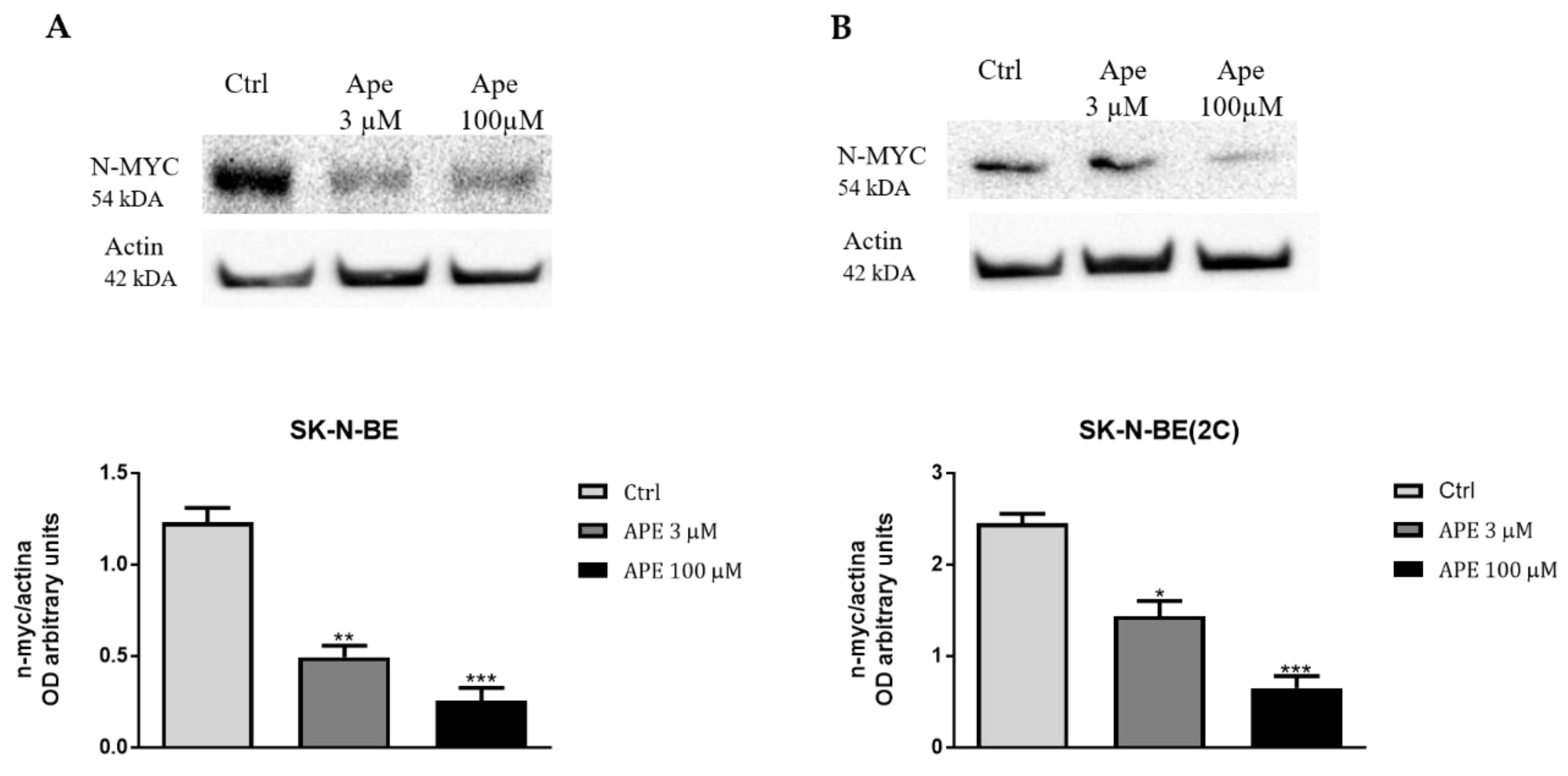{kind=link}
| (A) | |||||||||
| SK-N-BE | %G1 | ±SEM | p | %S | ±SEM | p | %G2/M | ±SEM | p |
| CTRL | 33.2 | 0.9815 | 52.25 | 2.569 | 12.55 | 3.551 | |||
| APE 24 h | 63.2 | 1.039 | 0.0001 | 28.85 | 0.7217 | 0.0007 | 7.95 | 0.3175 | 0.2665 |
| CTRL 48 h | 45.55 | 2.223 | 50.3 | 2.309 | 4.15 | 0.8666 | |||
| APE 48 h | 78.85 | 1.992 | 0.0005 | 17 | 17.312 | 0.0003 | 7.15 | 0.2598 | 0.0004 |
| CTRL 72 h | 58.6 | 0.4619 | 32.55 | 1.415 | 8.85 | 0.9526 | |||
| APE 72 h | 82.6 | 0.4819 | 0.001 | 14.45 | 0.5485 | 0.0003 | 2.95 | 0.866 | 0.0035 |
| (B) | |||||||||
| SK-N-BE(2C) | %G1 | ±SEM | p | %S | ±SEM | p | %G2/M | ±SEM | p |
| CTRL | 40.4 | 0.6 | 49.9 | 1.155 | 9.7 | 0.8083 | |||
| APE 24 h | 47 | 0.6 | 0.0166 | 41.3 | 0.0333 | 0.0628 | 11.7 | 0.3464 | 0.0853 |
| CTRL 48 h | 32.8 | 2.2 | 58.75 | 3.118 | 8.45 | 0.8083 | |||
| APE 48 h | 40 | 2.4 | 0.0644 | 30.55 | 1.819 | 0.0025 | 29 | 0.7217 | 0.0058 |
| CTRL 72 h | 35.45 | 0.95 | 58.85 | 0.8372 | 5.7 | 0.1155 | |||
| APE 72 h | 48 | 2.13 | 0.0025 | 22.75 | 2.281 | 0.0006 | 29.25 | 0.1443 | 0.0004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucianò, A.M.; Perciballi, E.; Fiore, M.; Del Bufalo, D.; Tata, A.M. The Combination of the M2 Muscarinic Receptor Agonist and Chemotherapy Affects Drug Resistance in Neuroblastoma Cells. Int. J. Mol. Sci. 2020, 21, 8433. https://doi.org/10.3390/ijms21228433
Lucianò AM, Perciballi E, Fiore M, Del Bufalo D, Tata AM. The Combination of the M2 Muscarinic Receptor Agonist and Chemotherapy Affects Drug Resistance in Neuroblastoma Cells. International Journal of Molecular Sciences. 2020; 21(22):8433. https://doi.org/10.3390/ijms21228433
Chicago/Turabian StyleLucianò, Anna Maria, Elisa Perciballi, Mario Fiore, Donatella Del Bufalo, and Ada Maria Tata. 2020. "The Combination of the M2 Muscarinic Receptor Agonist and Chemotherapy Affects Drug Resistance in Neuroblastoma Cells" International Journal of Molecular Sciences 21, no. 22: 8433. https://doi.org/10.3390/ijms21228433
APA StyleLucianò, A. M., Perciballi, E., Fiore, M., Del Bufalo, D., & Tata, A. M. (2020). The Combination of the M2 Muscarinic Receptor Agonist and Chemotherapy Affects Drug Resistance in Neuroblastoma Cells. International Journal of Molecular Sciences, 21(22), 8433. https://doi.org/10.3390/ijms21228433