HMGA Genes and Proteins in Development and Evolution



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Developmental Expression of Hmga Genes
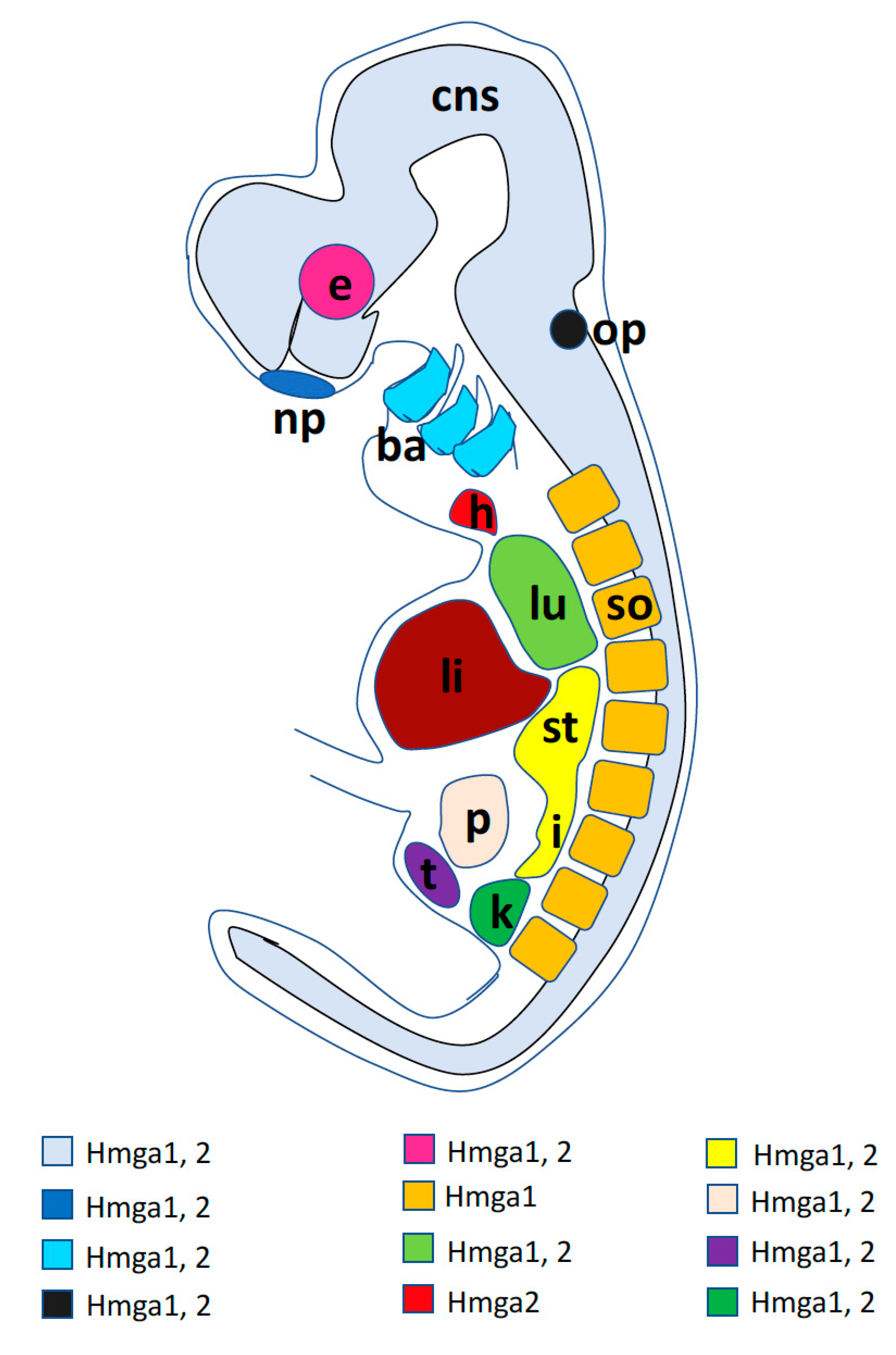
2.1. Hmga1 Developmental Expression
2.2. Hmga2 Developmental Expression
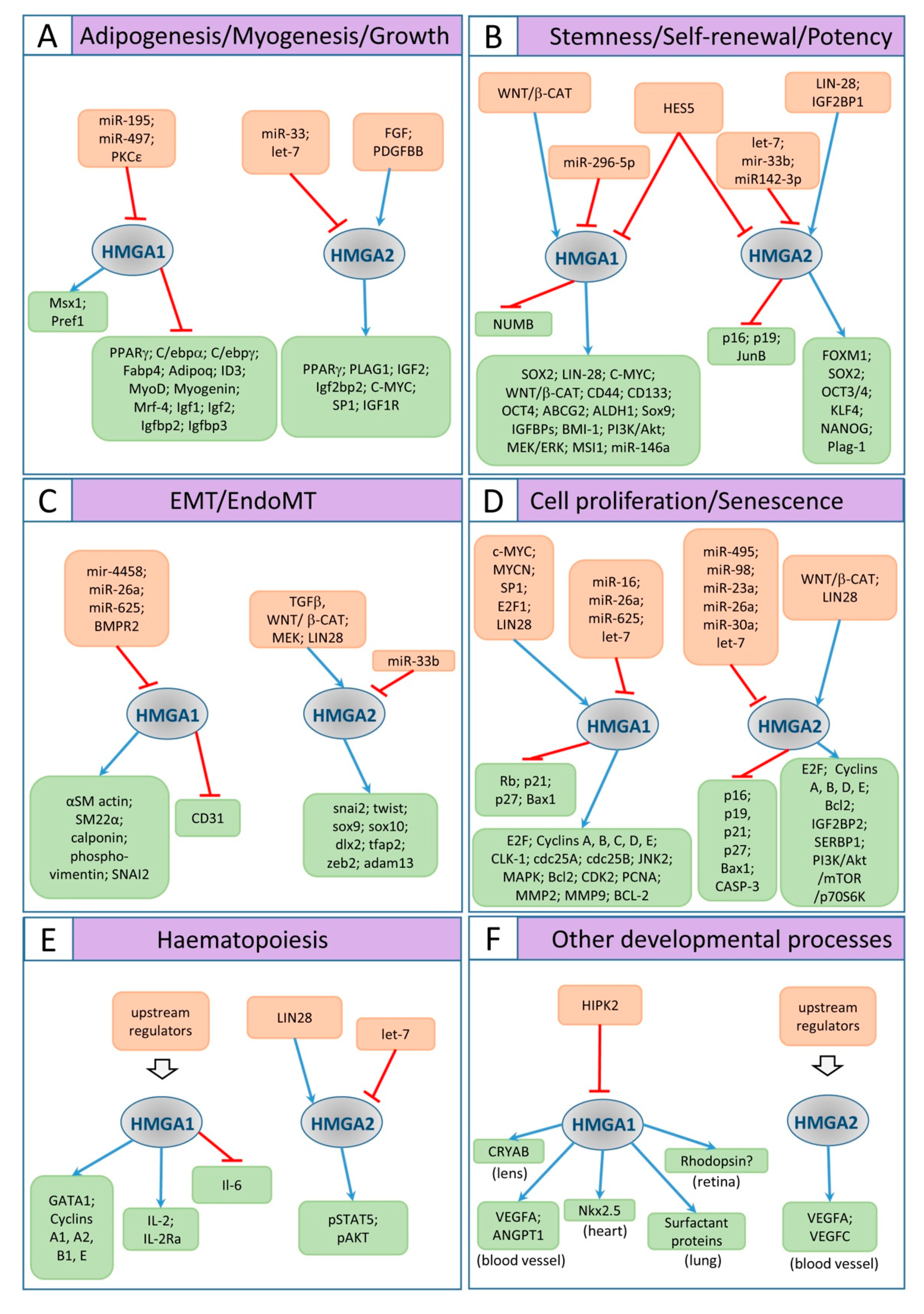
3. Developmental Roles of Hmga Genes and Proteins
3.1. HMGA Dysregulation and Its Impact on Body Size
3.2. Hmga1 and Hmga2 in the Development of the CNS
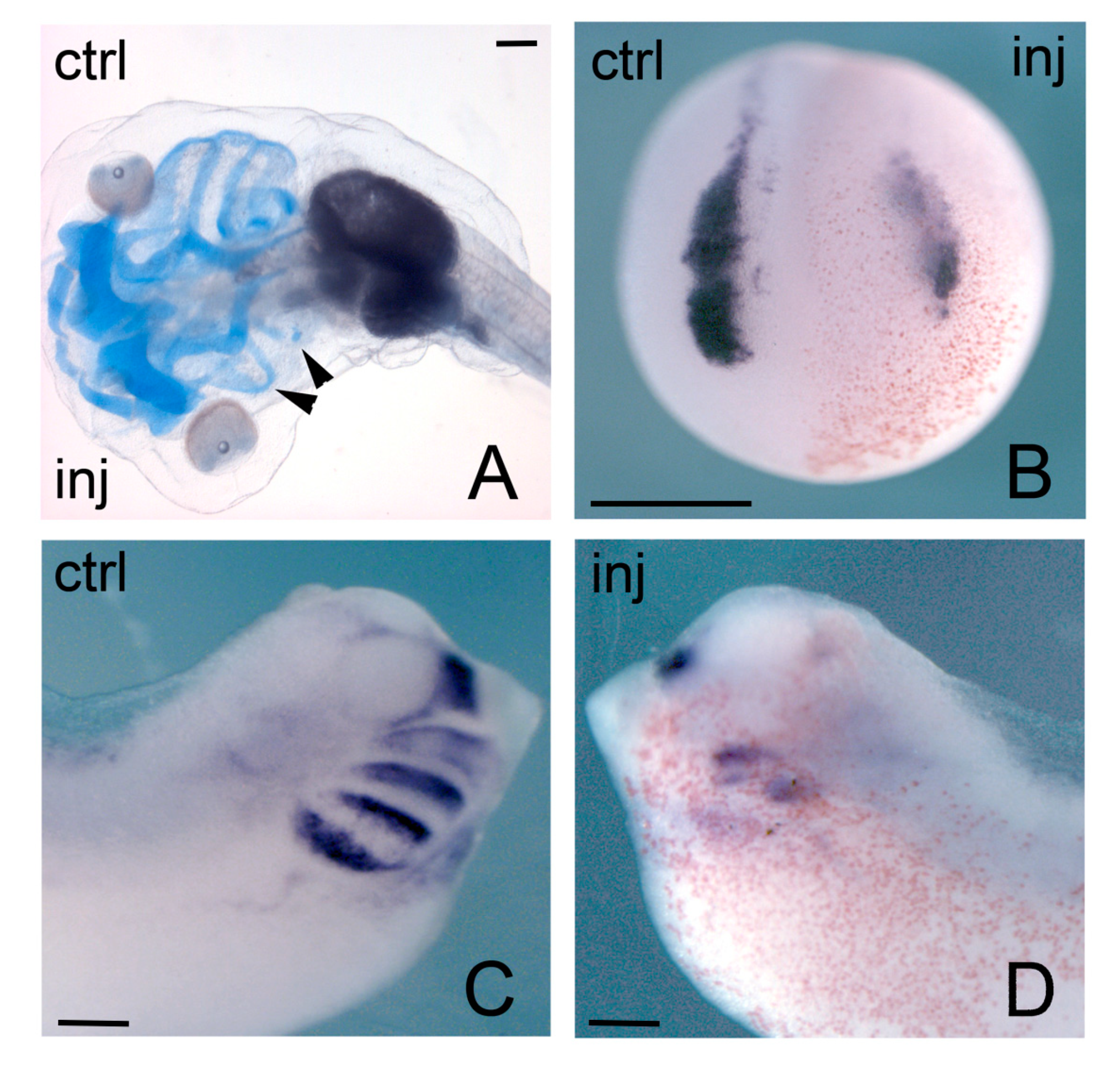
3.3. Hmga Genes in Xenopus Development: Focus on Neural Crest and Heart
3.4. HMGA, Proliferation, and the Cell Cycle
3.5. HMGA in EMT
3.6. HMGA in Stemness Maintenance
3.7. Other Developmental Effects of HMGA
4. HMGA and Evolution
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMP | bone morphogenetic protein |
| ChIP | chromatin immunoprecipitation |
| CNS | central nervous system |
| CTSCs | colon tumor stem cells |
| EMT | epithelial-mesenchymal transition |
| EndoMT | endothelial-to-mesenchymal transition |
| FGF | fibroblast growth factor |
| GBM | glioblastoma multiforme |
| GWAS | genome wide association studies |
| HDAC | histone deacetylase |
| hESCs | human embryonic stem cells |
| HMG | high mobility group |
| HMGA | high mobility group A proteins |
| IGF | insulin like-growth factor |
| IGFBP | insulin-like growth factor-binding protein |
| IL-2 | interleukin 2 |
| IL-6 | interleukin 6 |
| iPSCs | induced pluripotent stem cells |
| ISC | intestinal stem cell |
| MEFs | mouse embryonic fibroblasts |
| MNase | meganuclease |
| MO | morpholino |
| NBS | neural border specifiers |
| NCC | neural crest cells |
| NCS | neural crest specifiers |
| NPCs | neural precursor cells |
| NSCs | neural stem cells |
| PBAF | polybromo- and Brg1- associated factor-containing complex |
| PDGFBB | platelet-derived growth factor BB |
| PNS | peripheral nervous system |
| RPCs | retinal progenitor cells |
| SAHFs | senescence-associated heterochromatic foci |
| SRS | Silver-Russell syndrome |
| T-ALL | T-cell acute lymphoblastic leukemia/lymphoma |
| TGF | Transforming growth factor |
| UTR | untranslated region |
| VEGF | vascular endothelial growth factor |
| VZ | ventricular zone |
References
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur. J. Biochem. 1973, 38, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.; Holtlund, J.; Fredriksen, M.; Laland, S.G. On the presence of two new high mobility group-like proteins in HeLa S3 cells. FEBS Lett. 1983, 152, 163–167. [Google Scholar] [CrossRef]
- Strauss, F.; Varshavsky, A. A protein binds to a satellite DNA repeat at three specific sites that would be brought into mutual proximity by DNA folding in the nucleosome. Cell 1984, 37, 889–901. [Google Scholar] [CrossRef]
- Solomon, M.J.; Strauss, F.; Varshavsky, A. A mammalian high mobility group protein recognizes any stretch of six A.T base pairs in duplex DNA. Proc. Natl. Acad. Sci. USA 1986, 83, 1276–1280. [Google Scholar] [CrossRef]
- Reeves, R.; Nissen, M.S. The A.T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J. Biol. Chem. 1990, 265, 8573–8582. [Google Scholar]
- Mao, L.; Wertzler, K.J.; Maloney, S.C.; Wang, Z.; Magnuson, N.S.; Reeves, R. HMGA1 levels influence mitochondrial function and mitochondrial DNA repair efficiency. Mol. Cell. Biol. 2009, 29, 5426–5440. [Google Scholar] [CrossRef]
- Reeves, R. Nuclear functions of the HMG proteins. Biochim. Biophys. Acta 2010, 1799, 3–14. [Google Scholar] [CrossRef]
- Colombo, D.F.; Burger, L.; Baubec, T.; Schübeler, D. Binding of high mobility group A proteins to the mammalian genome occurs as a function of AT-content. PLoS Genet. 2017, 13, e1007102. [Google Scholar] [CrossRef]
- Huth, J.R.; Bewley, C.A.; Nissen, M.S.; Evans, J.N.; Reeves, R.; Gronenborn, A.M.; Clore, G.M. The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding motif. Nat. Struct. Biol. 1997, 4, 657–665. [Google Scholar] [CrossRef]
- Fedele, M.; Battista, S.; Manfioletti, G.; Croce, C.M.; Giancotti, V.; Fusco, A. Role of the high mobility group A proteins in human lipomas. Carcinogenesis 2001, 22, 1583–1591. [Google Scholar] [CrossRef]
- Reeves, R.; Beckerbauer, L. HMGI/Y proteins: Flexible regulators of transcription and chromatin structure. Biochim. Biophys. Acta 2001, 1519, 13–29. [Google Scholar] [CrossRef]
- Benecke, A.G.; Eilebrecht, S. RNA-mediated regulation of HMGA1 function. Biomolecules 2015, 5, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.; Nissen, M.S. Interaction of high mobility group-I (Y) nonhistone proteins with nucleosome core particles. J. Biol. Chem. 1993, 268, 21137–21146. [Google Scholar] [PubMed]
- Nissen, M.S.; Reeves, R. Changes in superhelicity are introduced into closed circular DNA by binding of high mobility group protein I/Y. J. Biol. Chem. 1995, 270, 4355–4360. [Google Scholar] [CrossRef] [PubMed]
- Noro, B.; Licheri, B.; Sgarra, R.; Rustighi, A.; Tessari, M.A.; Chau, K.Y.; Ono, S.J.; Giancotti, V.; Manfioletti, G. Molecular dissection of the architectural transcription factor HMGA2. Biochemistry 2003, 42, 4569–4577. [Google Scholar] [CrossRef]
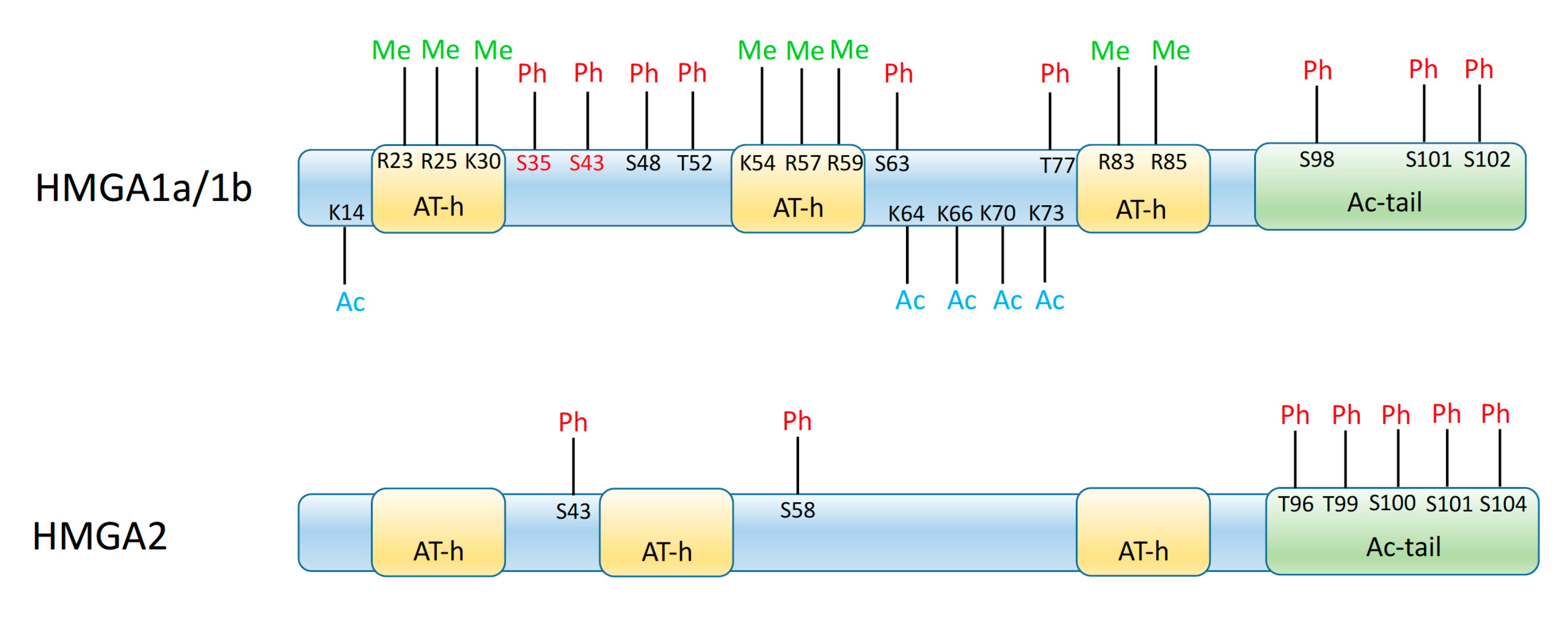
- Sgarra, R.; Maurizio, E.; Zammitti, S.; Lo Sardo, A.; Giancotti, V.; Manfioletti, G. Macroscopic differences in HMGA oncoproteins post-translational modifications: C-terminal phosphorylation of HMGA2 affects its DNA binding properties. J. Proteome Res. 2009, 8, 2978–2989. [Google Scholar] [CrossRef]
- Sgarra, R.; Zammitti, S.; Lo Sardo, A.; Maurizio, E.; Arnoldo, L.; Pegoraro, S.; Giancotti, V.; Manfioletti, G. HMGA molecular network: From transcriptional regulation to chromatin remodeling. Biochim. Biophys. Acta 2010, 1799, 37–47. [Google Scholar] [CrossRef]
- Reeves, R. Molecular biology of HMGA proteins: Hubs of nuclear function. Gene 2001, 277, 63–81. [Google Scholar] [CrossRef]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef]
- Cleynen, I.; Van de Ven, W.J. The HMGA proteins: A myriad of functions (Review). Int. J. Oncol. 2008, 32, 289–305. [Google Scholar] [CrossRef]
- Hammond, S.M.; Sharpless, N.E. HMGA2, microRNAs, and stem cell aging. Cell 2008, 135, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Fusco, A. HMGA and cancer. Biochim. Biophys. Acta 2010, 1799, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Pegoraro, S.; Ros, G.; Penzo, C.; Chiefari, E.; Foti, D.; Brunetti, A.; Manfioletti, G. High Mobility Group A (HMGA) proteins: Molecular instigators of breast cancer onset and progression. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 216–229. [Google Scholar] [CrossRef]
- Ozturk, N.; Singh, I.; Mehta, A.; Braun, T.; Barreto, G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front. Cell Dev. Biol. 2014, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Sumter, T.F.; Xian, L.; Huso, T.; Koo, M.; Chang, Y.T.; Almasri, T.N.; Chia, L.; Inglis, C.; Reid, D.; Resar, L.M. The High Mobility Group A1 (HMGA1) Transcriptome in cancer and development. Curr. Mol. Med. 2016, 16, 353–393. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Rustighi, A.; Tessari, M.A.; Di Bernardo, J.; Altamura, S.; Fusco, A.; Manfioletti, G.; Giancotti, V. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 2004, 574, 1–8. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y. HMG modifications and nuclear function. Biochim. Biophys. Acta 2010, 1799, 28–36. [Google Scholar] [CrossRef]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 pathway in cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef]
- Giancotti, V.; Bergamin, N.; Cataldi, P.; Rizzi, C. Epigenetic contribution of High-Mobility Group A proteins to stem cell properties. Int. J. Cell Biol. 2018, 2018, 3698078. [Google Scholar] [CrossRef]
- Resar, L.; Chia, L.; Xian, L. Lessons from the crypt: HMGA1-amping up Wnt for stem cells and tumor progression. Cancer Res. 2018, 78, 1890–1897. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, L.; Zheng, Y.; Guo, L. HMGA1 in cancer: Cancer classification by location. J. Cell. Mol. Med. 2019, 23, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, G.; Avantaggiato, V.; Visconti, R.; Fedele, M.; Battista, S.; Trapasso, F.; Merciai, B.M.; Fidanza, V.; Giancotti, V.; Santoro, M.; et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996, 13, 2439–2446. [Google Scholar] [PubMed]
- Moussavi Nik, S.H.; Newman, M.; Lardelli, M. The response of HMGA1 to changes in oxygen availability is evolutionarily conserved. Exp. Cell Res. 2011, 317, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Hirning-Folz, U.; Wilda, M.; Rippe, V.; Bullerdiek, J.; Hameister, H. The expression pattern of the Hmgic gene during development. Genes Chromosomes Cancer 1998, 23, 350–357. [Google Scholar] [CrossRef]
- Schiltz, J.F.; Rustighi, A.; Tessari, M.A.; Liu, J.; Braghetta, P.; Sgarra, R.; Stebel, M.; Bressan, G.M.; Altruda, F.; Giancotti, V.; et al. Hmga2 promoter analysis in transgenic mice. Biochem. Biophys. Res. Commun. 2003, 309, 718–723. [Google Scholar] [CrossRef]
- Benini, F.; Onorati, M.; Altamura, S.; Manfioletti, G.; Vignali, R. Identification and developmental expression of Xenopus hmga2beta. Biochem. Biophys. Res. Commun. 2006, 351, 392–397. [Google Scholar] [CrossRef]
- Hock, R.; Witte, F.; Brocher, J.; Schütz, M.; Scheer, U. Expression of HMGA2 variants during oogenesis and early embryogenesis of Xenopus laevis. Eur. J. Cell Biol. 2006, 85, 519–528. [Google Scholar] [CrossRef]
- Monzen, K.; Ito, Y.; Naito, A.T.; Kasai, H.; Hiroi, Y.; Hayashi, D.; Shiojima, I.; Yamazaki, T.; Miyazono, K.; Asashima, M.; et al. A crucial role of a high mobility group protein HMGA2 in cardiogenesis. Nat. Cell Biol. 2008, 10, 567–574. [Google Scholar] [CrossRef]
- Macrì, S.; Simula, L.; Pellarin, I.; Pegoraro, S.; Onorati, M.; Sgarra, R.; Manfioletti, G.; Vignali, R. Hmga2 is required for neural crest cell specification in Xenopus laevis. Dev. Biol. 2016, 411, 25–37. [Google Scholar] [CrossRef]
- Shi, X.; Huang, T.; Wang, J.; Liang, Y.; Gu, C.; Xu, Y.; Sun, J.; Lu, Y.; Sun, K.; Chen, S.; et al. Next-generation sequencing identifies novel genes with rare variants in total anomalous pulmonary venous connection. EBioMedicine 2018, 38, 217–227. [Google Scholar] [CrossRef]
- Gattas, G.J.; Quade, B.J.; Nowak, R.A.; Morton, C.C. HMGIC expression in human adult and fetal tissues and in uterine leiomyomata. Genes Chromosomes Cancer 1999, 25, 316–322. [Google Scholar] [CrossRef]
- Chieffi, P.; Battista, S.; Barchi, M.; Di Agostino, S.; Pierantoni, G.M.; Fedele, M.; Chiariotti, L.; Tramontano, D.; Fusco, A. HMGA1 and HMGA2 protein expression in mouse spermatogenesis. Oncogene 2002, 21, 3644–3650. [Google Scholar] [CrossRef][Green Version]
- Di Agostino, S.; Fedele, M.; Chieffi, P.; Fusco, A.; Rossi, P.; Geremia, R.; Sette, C. Phosphorylation of high-mobility group protein A2 by Nek2 kinase during the first meiotic division in mouse spermatocytes. Mol. Biol. Cell 2004, 15, 1224–1232. [Google Scholar] [CrossRef]
- Nishino, J.; Kim, I.; Chada, K.; Morrison, S.J. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell 2008, 135, 227–239. [Google Scholar] [CrossRef]
- Nishino, J.; Kim, S.; Zhu, Y.; Zhu, H.; Morrison, S.J. A network of heterochronic genes including Imp1 regulates temporal changes in stem cell properties. eLife 2013, 2, e00924. [Google Scholar] [CrossRef]
- Parameswaran, S.; Xia, X.; Hegde, G.; Ahmad, I. Hmga2 regulates self-renewal of retinal progenitors. Development 2014, 141, 4087–4097. [Google Scholar] [CrossRef]
- Smeti, I.; Assou, S.; Savary, E.; Masmoudi, S.; Zine, A. Transcriptomic analysis of the developing and adult mouse cochlear sensory epithelia. PLoS ONE 2012, 7, e42987. [Google Scholar] [CrossRef] [PubMed]
- Smeti, I.; Watabe, I.; Savary, E.; Fontbonne, A.; Zine, A. HMGA2, the architectural transcription factor high mobility group, is expressed in the developing and mature mouse cochlea. PLoS ONE 2014, 9, e88757. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yang, S.L.; Herrlinger, S.; Liang, C.; Dzieciatkowska, M.; Hansen, K.C.; Desai, R.; Nagy, A.; Niswander, L.; Moss, E.G.; et al. Lin28 promotes the proliferative capacity of neural progenitor cells in brain development. Development 2015, 142, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Bansod, S.; Kageyama, R.; Ohtsuka, T. Hes5 regulates the transition timing of neurogenesis and gliogenesis in mammalian neocortical development. Development 2017, 144, 3156–3167. [Google Scholar] [CrossRef] [PubMed]
- Shu, P.; Wu, C.; Ruan, X.; Liu, W.; Hou, L.; Fu, H.; Wang, M.; Liu, C.; Zeng, Y.; Chen, P.; et al. Opposing gradients of microRNA expression temporally pattern layer formation in the developing neocortex. Dev. Cell 2019, 49, 764–785. [Google Scholar] [CrossRef] [PubMed]
- Thisse, C.; Thisse, B.; Schilling, T.F.; Postlethwait, J.H. Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 1993, 119, 1203–1215. [Google Scholar]
- Zhou, X.; Benson, K.F.; Ashar, H.R.; Chada, K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 1995, 376, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Benson, K.F.; Chada, K. Mini-mouse: Disruption of the pygmy locus in a transgenic insertional mutant. Science 1990, 247, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Benson, K.F.; Chada, K. Mini-mouse: Phenotypic characterization of a transgenic insertional mutant allelic to pygmy. Genet. Res. 1994, 64, 27–33. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Z.; Gilbert, J.A.; Zhang, Y.; Zhang, M.; Qiu, Q.; Ramanujan, K.; Shavlakadze, T.; Eash, J.K.; Scaramozza, A.; Goddeeris, M.M.; et al. An HMGA2-IGF2BP2 axis regulates myoblast proliferation and myogenesis. Dev. Cell. 2012, 23, 1176–1188, Erratum in 2013, 24, 112. [Google Scholar] [CrossRef]
- Chung, J.; Zhang, X.; Collins, B.; Sper, R.B.; Gleason, K.; Simpson, S.; Koh, S.; Sommer, J.; Flowers, W.L.; Petters, R.M.; et al. High mobility group A2 (HMGA2) deficiency in pigs leads to dwarfism, abnormal fetal resource allocation, and cryptorchidism. Proc. Natl. Acad. Sci. USA 2018, 115, 5420–5425. [Google Scholar] [CrossRef]
- Federico, A.; Forzati, F.; Esposito, F.; Arra, C.; Palma, G.; Barbieri, A.; Palmieri, D.; Fedele, M.; Pierantoni, G.M.; De Martino, I.; et al. Hmga1/Hmga2 double knock-out mice display a “superpygmy” phenotype. Biol. Open 2014, 3, 372–378. [Google Scholar] [CrossRef]
- Fedele, M.; Visone, R.; De Martino, I.; Troncone, G.; Palmieri, D.; Battista, S.; Ciarmiello, A.; Pallante, P.; Arra, C.; Melillo, R.M.; et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cells 2006, 9, 459–471. [Google Scholar] [CrossRef]
- Woo, R.A.; Poon, R.Y. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2003, 2, 316–324. [Google Scholar] [CrossRef]
- Anand, A.; Chada, K. In vivo modulation of Hmgic reduces obesity. Nat. Genet. 2000, 24, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Battista, S.; Fidanza, V.; Fedele, M.; Klein-Szanto, A.J.; Outwater, E.; Brunner, H.; Santoro, M.; Croce, C.M.; Fusco, A. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res. 1999, 59, 4793–4797. [Google Scholar] [PubMed]
- Arlotta, P.; Tai, A.K.; Manfioletti, G.; Clifford, C.; Jay, G.; Ono, S.J. Transgenic mice expressing a truncated form of the high mobility group I-C protein develop adiposity and an abnormally high prevalence of lipomas. J. Biol. Chem. 2000, 275, 14394–14400. [Google Scholar] [CrossRef] [PubMed]
- Ayoubi, T.A.; Jansen, E.; Meulemans, S.M.; Van de Ven, W.J. Regulation of HMGIC expression: An architectural transcription factor involved in growth control and development. Oncogene 1999, 18, 5076–5087. [Google Scholar] [CrossRef]
- Xi, Y.; Shen, W.; Ma, L.; Zhao, M.; Zheng, J.; Bu, S.; Hino, S.; Nakao, M. HMGA2 promotes adipogenesis by activating C/EBPβ-mediated expression of PPARγ. Biochem. Biophys. Res. Commun. 2016, 472, 617–623. [Google Scholar] [CrossRef]
- Yuan, Y.; Xi, Y.; Chen, J.; Zhu, P.; Kang, J.; Zou, Z.; Wang, F.; Bu, S. STAT3 stimulates adipogenic stem cell proliferation and cooperates with HMGA2 during the early stage of differentiation to promote adipogenesis. Biochem. Biophys. Res. Commun. 2017, 482, 1360–1366. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, Y.; Lei, W.; Zhang, K.; Shi, J.; Hu, Y.; Shu, G.; Song, J. Signal transducer and activator of transcription 3 (STAT3) regulates adipocyte differentiation via peroxisome-proliferator-activated receptor gamma (PPARgamma). Biol. Cell 2010, 102, 1–12. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, W.; Yang, Y.; Wu, J. JAK2/STAT3 pathway is involved in the early stage of adipogenesis through regulating C/EBPβ transcription. J. Cell. Biochem. 2011, 112, 488–497. [Google Scholar] [CrossRef]
- Mota de Sá, P.; Richard, A.J.; Hang, H.; Stephens, J.M. Transcriptional regulation of adipogenesis. Compr. Physiol. 2017, 7, 635–674. [Google Scholar]
- Price, N.; Holtrup, B.; Kwei, S.L.; Wabitsch, M.; Rodeheffer, M.; Bianchini, L.; Suárez, Y.; Fernández-Hernando, C. SREBP-1c/MicroRNA 33b Genomic loci control adipocyte differentiation. Mol. Cell. Biol. 2016, 36, 1180–1193. [Google Scholar] [CrossRef]
- Lee, J.; Schmidt, H.; Lai, B.; Ge, K. Transcriptional and epigenomic regulation of adipogenesis. Mol. Cell. Biol. 2019, 39, e00601–e00618. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Fu, M.; Bookout, A.L.; Kliewer, S.A.; Mangelsdorf, D.J. MicroRNA let-7 regulates 3T3-L1 adipogenesis. Mol. Endocrinol. 2009, 23, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Arce-Cerezo, A.; García, M.; Rodríguez-Nuevo, A.; Crosa-Bonell, M.; Enguix, N.; Peró, A.; Muñoz, S.; Roca, C.; Ramos, D.; Franckhauser, S.; et al. HMGA1 overexpression in adipose tissue impairs adipogenesis and prevents diet-induced obesity and insulin resistance. Sci. Rep. 2015, 5, 14487. [Google Scholar] [CrossRef] [PubMed]
- Brants, J.R.; Ayoubi, T.A.; Chada, K.; Marchal, K.; Van de Ven, W.J.; Petit, M.M. Differential regulation of the insulin-like growth factor II mRNA-binding protein genes by architectural transcription factor HMGA2. FEBS Lett. 2004, 569, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Huysmans, C.; Sasazuki, T.; Shirasawa, S.; Van de Ven, W.; Peeters, K. Transcriptional control of the human high mobility group A1 gene: Basal and oncogenic Ras-regulated expression. Cancer Res. 2007, 67, 4620–4629. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Kishi, Y.; Gotoh, Y. IMP2 regulates differentiation potentials of mouse neocortical neural precursor cells. Genes Cells 2013, 18, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.H.; Wold, B.J. c-myc inhibition of MyoD and myogenin-initiated myogenic differentiation. Mol. Cell. Biol. 1991, 11, 2842–2851. [Google Scholar] [CrossRef]
- La Rocca, S.A.; Crouch, D.H.; Gillespie, D.A. c-Myc inhibits myogenic differentiation and myoD expression by a mechanism which can be dissociated from celltransformation. Oncogene 1994, 9, 3499–3508. [Google Scholar]
- Alonso-Martin, S.; Rochat, A.; Mademtzoglou, D.; Morais, J.; de Reyniès, A.; Auradé, F.; Chang, T.H.; Zammit, P.S.; Relaix, F. Gene expression profiling of muscle stem cells identifies novel regulators of postnatal myogenesis. Front. Cell Dev. Biol. 2016, 4, 58. [Google Scholar] [CrossRef]
- Luo, W.; Chen, J.; Li, L.; Ren, X.; Cheng, T.; Lu, S.; Lawal, R.A.; Nie, Q.; Zhang, X. Olivier c-Myc inhibits myoblast differentiation and promotes myoblast proliferation and muscle fibre hypertrophy by regulating the expression of its target genes, miRNAs and lincRNAs. Cell Death Differ. 2019, 26, 426–442. [Google Scholar] [CrossRef]
- Brocher, J.; Vogel, B.; Hock, R. HMGA1 down-regulation is crucial for chromatin composition and a gene expression profile permitting myogenic differentiation. BMC Cell Biol. 2010, 11, 64. [Google Scholar] [CrossRef]
- Martinez Hoyos, J.; Fedele, M.; Battista, S.; Pentimalli, F.; Kruhoffer, M.; Arra, C.; Orntoft, T.F.; Croce, C.M.; Fusco, A. Identification of the genes up- and down-regulated by the high mobility group A1 (HMGA1) proteins: Tissue specificity of the HMGA1-dependent gene regulation. Cancer Res. 2004, 64, 5728–5735. [Google Scholar] [CrossRef]
- Qiu, H.; Zhong, J.; Luo, L.; Tang, Z.; Liu, N.; Kang, K.; Li, L.; Gou, D. Regulatory axis of miR-195/497 and HMGA1-Id3 governs muscle cell proliferation and differentiation. Int. J. Biol. Sci. 2017, 13, 157–166. [Google Scholar] [CrossRef][Green Version]
- Di Marcantonio, D.; Galli, D.; Carubbi, C.; Gobbi, G.; Queirolo, V.; Martini, S.; Merighi, S.; Vaccarezza, M.; Maffulli, N.; Sykes, S.M.; et al. PKCε as a novel promoter of skeletal muscle differentiation and regeneration. Exp. Cell Res. 2015, 339, 10–19. [Google Scholar] [CrossRef]
- Greene, H.S.; Hu, C.K.; Brown, W.H. A lethal dwarf mutation in the rabbit with stigmata of endocrine abnormality. Science 1934, 79, 487–488. [Google Scholar] [CrossRef]
- Greene, H.S. A dwarf mutation in the rabbit: The constitutional influence on homozygous and heterozygous individuals. J. Exp. Med. 1940, 71, 839–856. [Google Scholar] [CrossRef]
- Carneiro, M.; Hu, D.; Archer, J.; Feng, C.; Afonso, S.; Chen, C.; Blanco-Aguiar, J.A.; Garreau, H.; Boucher, S.; Ferreira, P.G.; et al. Dwarfism and altered craniofacial development in rabbits is caused by a 12.1 kb deletion at the HMGA2 locus. Genetics 2017, 205, 955–965. [Google Scholar] [CrossRef]
- Ruyter-Spira, C.P.; Herbergs, J.; Limpens, E.; Marsh, J.A.; van der Poel, J.J.; Ayoubi, T.A.; Groenen, M.A. Nucleotide sequence of the chicken HMGI-C cDNA and expression of the HMGI-C and IGF1 genes in autosomal dwarf chicken embryos. Biochim. Biophys. Acta 1998, 1399, 83–87. [Google Scholar] [CrossRef]
- Song, C.; Gu, X.; Feng, C.; Wang, Y.; Gao, Y.; Hu, X.; Li, N. Evaluation of SNPs in the chicken HMGA2 gene as markers for body weight gain. Anim. Genet. 2011, 42, 333–336. [Google Scholar] [CrossRef]
- Horikoshi, M.; Yaghootkar, H.; Mook-Kanamori, D.O.; Sovio, U.; Taal, H.R.; Hennig, B.J.; Bradfield, J.P.; St Pourcain, B.; Evans, D.M.; Charoen, P.; et al. New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat. Genet. 2013, 45, 76–82. [Google Scholar] [CrossRef]
- Weedon, M.N.; Lettre, G.; Freathy, R.M.; Lindgren, C.M.; Voight, B.F.; Perry, J.R.B.; Elliott, K.S.; Hackett, R.; Guiducci, C.; Shields, B.; et al. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat. Genet. 2007, 39, 1245–1250. [Google Scholar] [CrossRef]
- Weedon, M.N.; Lango, H.; Lindgren, C.M.; Wallace, C.; Evans, D.M.; Mangino, M.; Freathy, R.M.; Perry, J.R.B.; Stevens, S.; Hall, A.S.; et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008, 40, 575–583. [Google Scholar] [CrossRef]
- Mari, F.; Hermanns, P.; Giovannucci-Uzielli, M.L.; Galluzzi, F.; Scott, D.; Lee, B.; Renieri, A.; Unger, S.; Zabel, B.; Superti-Furga, A. Refinement of the 12q14 microdeletion syndrome: Primordial dwarfism and developmental delay with or without osteopoikilosis. Eur. J. Hum. Genet. 2009, 17, 1141–1147. [Google Scholar] [CrossRef]
- Liu, J.Z.; Medland, S.E.; Wright, M.J.; Henders, A.K.; Heath, A.C.; Madden, P.A.F.; Duncan, A.; Montgomery, G.W.; Martin, N.G.; McRae, A.F. Genome-wide association study of height and body mass index in Australian twin families. Twin Res. Hum. Genet. 2010, 13, 179–193. [Google Scholar] [CrossRef]
- Yang, T.L.; Guo, Y.; Zhang, L.S.; Tian, Q.; Yan, H.; Guo, Y.F.; Deng, H.W. HMGA2 is confirmed to be associated with human adult height. Ann. Hum. Genet. 2010, 74, 11–16. [Google Scholar] [CrossRef]
- Carty, C.L.; Johnson, N.A.; Hutter, C.M.; Reiner, A.P.; Peters, U.; Tang, H.; Kooperberg, C. Genome-wide association study of body height in African Americans: The Women’s Health Initiative SNP Health Association Resource (SHARe). Hum. Mol. Genet. 2012, 21, 711–720. [Google Scholar] [CrossRef]
- Fusco, I.; Babu, D.; Mellone, S.; Barizzone, N.; Prodam, F.; Fanelli, A.; Muniswamy, R.; Petri, A.; Bellone, S.; Bona, G.; et al. Variations in the high-mobility group-A2 gene (HMGA2) are associated with idiopathic short stature. Pediatr. Res. 2016, 79, 258–261. [Google Scholar] [CrossRef]
- Makvandi-Nejad, S.; Hoffman, G.E.; Allen, J.J.; Chu, E.; Gu, E.; Chandler, A.M.; Loredo, A.I.; Bellone, R.R.; Mezey, J.G.; Brooks, S.A.; et al. Four loci explain 83% of size variation in the horse. PLoS ONE 2012, 7, e39929. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Neuditschko, M.; Ricard, A.; Velie, B.; Lindgren, G.; Mesarič, M.; Cotman, M.; Horna, M.; Dobretsberger, M.; Brem, G.; et al. Genome-Wide Homozygosity Patterns and Evidence for Selection in a Set of European and Near Eastern Horse Breeds. Genes 2019, 10, 491. [Google Scholar] [CrossRef]
- Norton, E.M.; Avila, F.; Schultz, N.E.; Mickelson, J.R.; Geor, R.J.; McCue, M.E. Evaluation of an HMGA2 variant for pleiotropic effects on height and metabolic traits in ponies. J. Vet. Intern. Med. 2019, 33, 942–952. [Google Scholar] [CrossRef]
- Rimbault, M.; Beale, H.C.; Schoenebeck, J.J.; Hoopes, B.C.; Allen, J.J.; Kilroy-Glynn, P.; Wayne, R.K.; Sutter, N.B.; Ostrander, E.A. Derived variants at six genes explain nearly half of size reduction in dog breeds. Genome Res. 2013, 23, 1985–1995. [Google Scholar] [CrossRef]
- Abi Habib, W.; Brioude, F.; Edouard, T.; Bennett, J.T.; Lienhardt-Roussie, A.; Tixier, F.; Salem, J.; Yuen, T.; Azzi, S.; Le Bouc, Y.; et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 2018, 20, 250–258. [Google Scholar] [CrossRef]
- Leszinski, G.S.; Warncke, K.; Hoefele, J.; Wagner, M. A case report and review of the literature indicate that HMGA2 should be added as a disease gene for Silver-Russell syndrome. Gene 2018, 663, 110–114. [Google Scholar] [CrossRef]
- Takenouchi, T.; Enomoto, K.; Nishida, T.; Torii, C.; Okazaki, T.; Takahashi, T.; Kosaki, K. 12q14 microdeletion syndrome and short stature with or without relative macrocephaly. Am. J. Med. Genet. A 2012, 158A, 2542–2544. [Google Scholar] [CrossRef]
- Li, P.; Xiao, S.; Wei, N.; Zhang, Z.; Huang, R.; Gu, Y.; Guo, Y.; Ren, J.; Huang, L.; Chen, C. Fine mapping of a QTL for ear size on porcine chromosome 5 and identification of high mobility group AT-hook 2 (HMGA2) as a positional candidate gene. Genet. Sel. Evol. 2012, 44, 6. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Yan, H.; Liu, X.; Li, N.; Liang, J.; Pu, L.; Zhang, Y.; Shi, H.; Zhao, K.; et al. Genome-wide association studies identify the loci for 5 exterior traits in a Large White × Minzhu pig population. PLoS ONE 2014, 9, e103766. [Google Scholar] [CrossRef]
- Zhang, L.C.; Li, N.; Liu, X.; Liang, J.; Yan, H.; Zhao, K.B.; Pu, L.; Shi, H.B.; Zhang, Y.B.; Wang, L.G.; et al. A genome-wide association study of limb bone length using a Large White × Minzhu intercross population. Genet. Sel. Evol. 2014, 46, 56. [Google Scholar] [CrossRef]
- Guo, Y.; Hou, L.; Zhang, X.; Huang, M.; Mao, H.; Chen, H.; Ma, J.; Chen, C.; Ai, H.; Ren, J.; et al. A meta analysis of genome-wide association studies for limb bone lengths in four pig populations. BMC Genet. 2015, 16, 95. [Google Scholar] [CrossRef]
- Qiao, R.; Gao, J.; Zhang, Z.; Li, L.; Xie, X.; Fan, Y.; Cui, L.; Ma, J.; Ai, H.; Ren, J.; et al. Genome-wide association analyses reveal significant loci and strong candidate genes for growth and fatness traits in two pig populations. Genet. Sel. Evol. 2015, 47, 17. [Google Scholar] [CrossRef]
- Gong, H.; Xiao, S.; Li, W.; Huang, T.; Huang, X.; Yan, G.; Huang, Y.; Qiu, H.; Jiang, K.; Wang, X.; et al. Unravelling the genetic loci for growth and carcass traits in Chinese Bamaxiang pigs based on a 1.4 million SNP array. J. Anim. Breed Genet. 2019, 136, 3–14. [Google Scholar] [CrossRef]
- Iiritano, S.; Chiefari, E.; Ventura, V.; Arcidiacono, B.; Possidente, K.; Nocera, A.; Nevolo, M.T.; Fedele, M.; Greco, A.; Greco, M.; et al. The HMGA1-IGF-I/IGFBP system: A novel pathway for modulating glucose uptake. Mol. Endocrinol. 2012, 26, 1578–1589. [Google Scholar] [CrossRef][Green Version]
- Williams, M.J.; Almén, M.S.; Fredriksson, R.; Schiöth, H.B. What model organisms and interactomics can reveal about the genetics of human obesity. Cell. Mol. Life Sci. 2012, 69, 3819–3834. [Google Scholar] [CrossRef]
- Berndt, S.I.; Gustafsson, S.; Mägi, R.; Ganna, A.; Wheeler, E.; Feitosa, M.F.; Justice, A.E.; Monda, K.L.; Croteau-Chonka, D.C.; Day, F.R.; et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat. Genet. 2013, 45, 501–512. [Google Scholar] [CrossRef]
- Lango Allen, H.; Estrada, K.; Lettre, G.; Berndt, S.I.; Weedon, M.N.; Rivadeneira, F.; Willer, C.J.; Jackson, A.U.; Vedantam, S.; Raychaudhuri, S.; et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 2010, 467, 832–838. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Lango Allen, H.; Lindgren, C.M.; Luan, J.; Mägi, R.; et al. Association analyses of 249.;796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef]
- Passegué, E.; Wagner, E.F.; Weissman, I.L. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 2004, 119, 431–443. [Google Scholar] [CrossRef]
- Irelan, J.T.; Gutierrez Del Arroyo, A.; Gutierrez, A.; Peters, G.; Quon, K.C.; Miraglia, L.; Chanda, S.K. A functional screen for regulators of CKDN2A reveals MEOX2 as a transcriptional activator of INK4a. PLoS ONE 2009, 4, e5067. [Google Scholar] [CrossRef]
- Miller, F.D.; Gauthier, A.S. Timing is everything: Making neurons versus glia in the developing cortex. Neuron 2007, 54, 357–369. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Kageyama, R. Regulation of temporal properties of neural stem cells and transition timing of neurogenesis and gliogenesis during mammalian neocortical development. Semin. Cell Dev. Biol. 2019, 91, 4–11. [Google Scholar] [CrossRef]
- Kishi, Y.; Fujii, Y.; Hirabayashi, Y.; Gotoh, Y. HMGA regulates the global chromatin state and neurogenic potential in neocortical precursor cells. Nat. Neurosci. 2012, 15, 1127–1133. [Google Scholar] [CrossRef]
- Degrauwe, N.; Schlumpf, T.B.; Janiszewska, M.; Martin, P.; Cauderay, A.; Provero, P.; Riggi, N.; Suvà, M.L.; Paro, R.; Stamenkovic, I. The RNA Binding Protein IMP2 Preserves Glioblastoma Stem Cells by Preventing let-7 Target Gene Silencing. Cell Rep. 2016, 15, 1634–1647. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Chiou, G.Y.; Chien, C.S.; Wang, M.L.; Chen, M.T.; Yang, Y.P.; Yu, Y.L.; Chien, Y.; Chang, Y.C.; Shen, C.C.; Chio, C.C.; et al. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol. Cell. 2013, 52, 693–706. [Google Scholar] [CrossRef]
- Zhang, F.; Wu, A.; Wang, Y.; Liu, J. miR-490-3p functions as a tumor suppressor in glioma by inhibiting high-mobility group AT-hook 2 expression. Exp. Ther. Med. 2019, 18, 664–670. [Google Scholar] [CrossRef]
- Zhong, X.; Liu, X.; Li, Y.; Cheng, M.; Wang, W.; Tian, K.; Mu, L.; Zeng, T.; Liu, Y.; Jiang, X.; et al. HMGA2 sustains self-renewal and invasiveness of glioma-initiating cells. Oncotarget 2016, 7, 44365–44380. [Google Scholar] [CrossRef]
- Sakai, H.; Fujii, Y.; Kuwayama, N.; Kawaji, K.; Gotoh, Y.; Kishi, Y. Plag1 regulates neuronal gene expression and neuronal differentiation of neocortical neural progenitor cells. Genes Cells 2019, 24, 650–666. [Google Scholar] [CrossRef]
- Alam, S.; Zinyk, D.; Ma, L.; Schuurmans, C. Members of the Plag gene family are expressed in complementary and overlapping regions in the developing murine nervous system. Dev. Dyn. 2005, 234, 772–782. [Google Scholar] [CrossRef]
- Adnani, L.; Dixit, R.; Chen, X.; Balakrishnan, A.; Modi, H.; Touahri, Y.; Logan, C.; Schuurmans, C. Plag1 and Plagl2 have overlapping and distinct functions in telencephalic development. Biol. Open 2018, 7, bio038661. [Google Scholar] [CrossRef]
- Decembrini, S.; Andreazzoli, M.; Vignali, R.; Barsacchi, G.; Cremisi, F. Timing the generation of distinct retinal cells by homeobox proteins. PLoS Biol. 2006, 4, e272. [Google Scholar] [CrossRef]
- Decembrini, S.; Bressan, D.; Vignali, R.; Pitto, L.; Mariotti, S.; Rainaldi, G.; Wang, X.; Evangelista, M.; Barsacchi, G.; Cremisi, F. MicroRNAs couple cell fate and developmental timing in retina. Proc. Natl. Acad. Sci. USA 2009, 106, 21179–21184. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 regulates self-renewal and tumorigenicity of breast cancer cells. Cell 2007, 31, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Ahmad, I. let-7 microRNA regulates neurogliogenesis in the mammalian retina through Hmga2. Dev. Biol. 2016, 410, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Burger, C.A.; Casasent, A.K.; Albrecht, N.E.; Li, F.; Samuel, M.A. Spatiotemporal gene expression patterns reveal molecular relatedness between retinal laminae. J. Comp. Neurol. 2019. [Google Scholar] [CrossRef]
- Chau, K.Y.; Munshi, N.; Keane-Myers, A.; Cheung-Chau, K.W.; Tai, A.K.; Manfioletti, G.; Dorey, C.K.; Thanos, D.; Zack, D.J.; Ono, S.J. The architectural transcription factor high mobility group I(Y) participates in photoreceptor-specific gene expression. J. Neurosci. 2000, 20, 7317–7324. [Google Scholar] [CrossRef]
- Hellsten, U.; Harland, R.M.; Gilchrist, M.J.; Hendrix, D.; Jurka, J.; Kapitonov, V.; Ovcharenko, I.; Putnam, N.H.; Shu, S.; Taher, L.; et al. The genome of the Western clawed frog Xenopus tropicalis. Science 2010, 328, 633–636. [Google Scholar] [CrossRef]
- Session, A.M.; Uno, Y.; Kwon, T.; Chapman, J.A.; Toyoda, A.; Takahashi, S.; Fukui, A.; Hikosaka, A.; Suzuki, A.; Kondo, M.; et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 2016, 538, 336–343. [Google Scholar] [CrossRef]
- Macrì, S.; Sgarra, R.; Ros, G.; Maurizio, E.; Zammitti, S.; Milani, O.; Onorati, M.; Vignali, R.; Manfioletti, G. Expression and functional characterization of Xhmg-at-hook genes in Xenopus laevis. PLoS ONE 2013, 8, e69866. [Google Scholar] [CrossRef]
- Etchevers, H.C.; Dupin, E.; Le Douarin, N.M. The diverse neural crest: From embryology to human pathology. Development 2019, 146, dev169821. [Google Scholar] [CrossRef]
- Helms, J.A.; Schneider, R.A. Cranial skeletal biology. Nature 2003, 423, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal transition and cancer stem cells: At the crossroads of differentiation and dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell. 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.H.; Moustakas, A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef]
- Thuault, S.; Tan, E.J.; Peinado, H.; Cano, A.; Heldin, C.H.; Moustakas, A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef]
- Tan, E.J.; Thuault, S.; Caja, L.; Carletti, T.; Heldin, C.H.; Moustakas, A. Regulation of transcription factor Twist expression by the DNA architectural protein high mobility group A2 during epithelial-to-mesenchymal transition. J. Biol. Chem. 2012, 287, 7134–7145. [Google Scholar] [CrossRef]
- Tan, E.J.; Kahata, K.; Idås, O.; Thuault, S.; Heldin, C.H.; Moustakas, A. The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial-to-mesenchymal transition. Nucleic Acids Res. 2015, 43, 162–178. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef]
- Morishita, A.; Zaidi, M.R.; Mitoro, A.; Sankarasharma, D.; Szabolcs, M.; Okada, Y.; D’Armiento, J.; Chada, K. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013, 73, 4289–4299. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, X.D.; Ye, C.X.; Shen, Z.L.; Yang, Y.; Wang, B.; Guo, P.; Gao, Z.D.; Ye, Y.J.; Jiang, K.W.; et al. Long noncoding RNA HIT000218960 promotes papillary thyroid cancer oncogenesis and tumor progression by upregulating the expression of high mobility group AT-hook 2 (HMGA2) gene. Cell Cycle 2017, 16, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Hawsawi, O.; Henderson, V.; Burton, L.J.; Dougan, J.; Nagappan, P.; Odero-Marah, V. High mobility group A2 (HMGA2) promotes EMT via MAPK pathway in prostate cancer. Biochem. Biophys. Res. Commun. 2018, 504, 196–202. [Google Scholar] [CrossRef]
- Hou, B.; Ishinaga, H.; Midorikawa, K.; Nakamura, S.; Hiraku, Y.; Oikawa, S.; Ma, N.; Takeuchi, K.; Murata, M. Let-7c inhibits migration and epithelial-mesenchymal transition in head and neck squamous cell carcinoma by targeting IGF1R and HMGA2. Oncotarget 2018, 9, 8927–8940. [Google Scholar] [CrossRef] [PubMed]
- Kou, B.; Liu, W.; Tang, X.; Kou, Q. HMGA2 facilitates epithelial-mesenchymal transition in renal cell carcinoma by regulating the TGF-β/Smad2 signaling pathway. Oncol. Rep. 2018, 39, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Zhang, Y.; Niu, C.; Guo, J.; Li, J.; Wei, X.; Jia, M.; Zhi, X.; Yao, L.; Meng, D. BTB and CNC homology 1 (Bach1) promotes human ovarian cancer cell metastasis by HMGA2-mediated epithelial-mesenchymal transition. Cancer Lett. 2019, 445, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Mechanisms of TGFβ-Induced Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [CrossRef]
- Leung, A.W.; Murdoch, B.; Salem, A.F.; Prasad, M.S.; Gomez, G.A.; García-Castro, M.I. WNT/β-catenin signaling mediates human neural crest induction via a pre-neural border intermediate. Development 2016, 143, 398–410. [Google Scholar] [CrossRef]
- Rothstein, M.; Bhattacharya, D.; Simoes-Costa, M. The molecular basis of neural crest axial identity. Dev. Biol. 2018, 444, S170–S180. [Google Scholar] [CrossRef]
- Pla, P.; Monsoro-Burq, A.H. The neural border: Induction, specification and maturation of the territory that generates neural crest cells. Dev. Biol. 2018, 444, S36–S46. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Xu, C.; Zhou, Z.; Zhu, Z.; You, T. HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett. 2014, 355, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Markowski, D.N.; Helmke, B.M.; Meyer, F.; von Ahsen, I.; Nimzyk, R.; Nolte, I.; Bullerdiek, J. BMP4 increases expression of HMGA2 in mesenchymal stem cells. Cytokine 2011, 56, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Wend, P.; Runke, S.; Wend, K.; Anchondo, B.; Yesayan, M.; Jardon, M.; Hardie, N.; Loddenkemper, C.; Ulasov, I.; Lesniak, M.S.; et al. WNT10B/β-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol. Med. 2013, 5, 264–279. [Google Scholar] [CrossRef]
- El Ayachi, I.; Fatima, I.; Wend, P.; Alva-Ornelas, J.A.; Runke, S.; Kuenzinger, W.L.; Silva, J.; Silva, W.; Gray, J.K.; Lehr, S.; et al. The wnt10b network is associated with survival and metastases in chemoresistant triple-negative breast cancer. Cancer Res. 2019, 79, 982–993. [Google Scholar] [CrossRef]
- Kalomoiris, S.; Cicchetto, A.C.; Lakatos, K.; Nolta, J.A.; Fierro, F.A. Fibroblast Growth Factor 2 regulates High Mobility Group A2 expression in human bone marrow-derived mesenchymal stem cells. J. Cell Biochem. 2016, 117, 2128–2137. [Google Scholar] [CrossRef]
- Wood, L.J.; Maher, J.F.; Bunton, T.E.; Resar, L.M. The oncogenic properties of the HMG-I gene family. Cancer Res. 2000, 60, 4256–4261. [Google Scholar]
- Giannini, G.; Cerignoli, F.; Mellone, M.; Massimi, I.; Ambrosi, C.; Rinaldi, C.; Gulino, A. Molecular mechanism of HMGA1 deregulation in human neuroblastoma. Cancer Lett. 2005, 228, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Watanabe, S.; Tei, S.; Saitoh, N.; Kuratsu, J.; Nakao, M. High mobility group protein HMGA1 inhibits retinoblastoma protein-mediated cellular G0 arrest. Cancer Sci. 2007, 98, 1893–1901. [Google Scholar] [CrossRef]
- Massimi, I.; Guerrieri, F.; Petroni, M.; Veschi, V.; Truffa, S.; Screpanti, I.; Frati, L.; Levrero, M.; Gulino, A.; Giannini, G. The HMGA1 protoncogene frequently deregulated in cancer is a transcriptional target of E2F1. Mol. Carcinog. 2013, 52, 526–534. [Google Scholar] [CrossRef]
- Reeves, R.; Edberg, D.D.; Li, Y. Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol. Cell. Biol. 2001, 21, 575–594. [Google Scholar] [CrossRef]
- Baldassarre, G.; Battista, S.; Belletti, B.; Thakur, S.; Pentimalli, F.; Trapasso, F.; Fedele, M.; Pierantoni, G.; Croce, C.M.; Fusco, A. Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol. Cell. Biol. 2003, 23, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Wang, T.; Wu, Z.; Feng, Y.; Wang, W.; Zhou, S.; Ma, X.; Wang, S. HMGA1 exacerbates tumor growth through regulating the cell cycle and accelerates migration/invasion via targeting miR-221/222 in cervical cancer. Cell Death Dis. 2018, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Battista, S.; Pentimalli, F.; Baldassarre, G.; Fedele, M.; Fidanza, V.; Croce, C.M.; Fusco, A. Loss of Hmga1 gene function affects embryonic stem cell lympho-hematopoietic differentiation. FASEB J. 2003, 17, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sumter, T.F.; Bhattacharya, R.; Tesfaye, A.; Fuchs, E.J.; Wood, L.J.; Huso, D.L.; Resar, L.M. The HMG-I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res. 2004, 64, 3371–3375. [Google Scholar] [CrossRef]
- Di Cello, F.; Shin, J.; Harbom, K.; Brayton, C. Knockdown of HMGA1 inhibits human breast cancer cell growth and metastasis in immunodeficient mice. Biochem. Biophys. Res. Commun. 2013, 434, 70–74. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schuldenfrei, A.; Belton, A.; Kowalski, J.; Talbot, C.C., Jr.; Di Cello, F.; Poh, W.; Tsai, H.L.; Shah, S.N.; Huso, T.H.; Huso, D.L.; et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genom. 2011, 12, 549. [Google Scholar] [CrossRef]
- Zhou, W.B.; Zhong, C.N.; Luo, X.P.; Zhang, Y.Y.; Zhang, G.Y.; Zhou, D.X.; Liu, L.P. miR-625 suppresses cell proliferation and migration by targeting HMGA1 in breast cancer. Biochem. Biophys. Res. Commun. 2016, 470, 838–844. [Google Scholar] [CrossRef]
- Kaddar, T.; Rouault, J.P.; Chien, W.W.; Chebel, A.; Gadoux, M.; Salles, G.; Ffrench, M.; Magaud, J.P. Two new miR-16 targets: Caprin-1 and HMGA1, proteins implicated in cell proliferation. Biol. Cell 2009, 101, 511–524. [Google Scholar] [CrossRef]
- Wang, B.; David, M.D.; Schrader, J.W. Absence of caprin-1 results in defects in cellular proliferation. J. Immunol. 2005, 175, 4274–4282. [Google Scholar] [CrossRef]
- Ikeda, K.; Mason, P.J.; Bessler, M. 3′UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood 2011, 117, 5860–5869. [Google Scholar] [CrossRef]
- Kumar, P.; Beck, D.; Galeev, R.; Thoms, J.A.I.; Talkhoncheh, M.S.; de Jong, I.; Unnikrishnan, A.; Baudet, A.; Subramaniam, A.; Pimanda, J.E.; et al. HMGA2 promotes long-term engraftment and myeloerythroid differentiation of human hematopoietic stem and progenitor cells. Blood Adv. 2019, 3, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Tessari, M.A.; Gostissa, M.; Altamura, S.; Sgarra, R.; Rustighi, A.; Salvagno, C.; Caretti, G.; Imbriano, C.; Mantovani, R.; Del Sal, G.; et al. Transcriptional activation of the cyclin A gene by the architectural transcription factor HMGA2. Mol. Cell. Biol. 2003, 23, 9104–9116. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Pierantoni, G.M.; Visone, R.; Fusco, A. Critical role of the HMGA2 gene in pituitary adenomas. Cell Cycle 2006, 5, 2045–2048. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fedele, M.; Paciello, O.; De Biase, D.; Monaco, M.; Chiappetta, G.; Vitiello, M.; Barbieri, A.; Rea, D.; Luciano, A.; Papparella, S.; et al. HMGA2 cooperates with either p27kip1 deficiency or Cdk4R24Cmutation in pituitary tumorigenesis. Cell Cycle 2018, 17, 580–588. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Zhao, S.G.; Wei, D.M.; Zhao, Y.R.; Huang, T.; Muhammad, T.; Yan, L.; Gao, F.; Li, L.; et al. The HMGA2-IMP2 pathway promotes granulosa cell proliferation in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 1049–1059. [Google Scholar] [CrossRef]
- Sun, L.; Yu, J.; Wang, P.; Shen, M.; Ruan, S. HIT000218960 promotes gastric cancer cell proliferation and migration through upregulation of HMGA2 expression. Oncol. Lett. 2019, 17, 4957–4963. [Google Scholar] [CrossRef]
- Shi, Z.; Li, X.; Wu, D.; Tang, R.; Chen, R.; Xue, S.; Sun, X. Silencing of HMGA2 suppresses cellular proliferation, migration, invasion, and epithelial-mesenchymal transition in bladder cancer. Tumour Biol. 2016, 37, 7515–7523. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Guo, W.; Ren, T.; Huang, Y.; Han, Y.; Zhang, H.; Zhang, J. Knockdown of HMGA2 regulates the level of autophagy via interactions between MSI2 and Beclin1 to inhibit NF1-associated malignant peripheral nerve sheath tumour growth. J. Exp. Clin. Cancer Res. 2019, 38, 185. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Zhang, H.; Li, J.; Zhao, H.D. microRNA-98 inhibits the proliferation, invasion, migration and promotes apoptosis of breast cancer cells by binding to HMGA2. Biosci. Rep. 2018, 38, BSR20180571. [Google Scholar] [CrossRef]
- Wang, L.; Shen, H.; Zhu, D.; Feng, B.; Yu, L.; Tian, X.; Ren, C.; Gao, C.; Li, X.; Ma, D.; et al. Increased high mobility group A 2 expression promotes transition of cervical intraepithelial neoplasm into cervical cancer. Oncotarget 2018, 9, 7891–7901. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ogrodnik, M.; Salmonowicz, H.; Jurk, D.; Passos, J.F. Expansion and Cell-Cycle Arrest: Common Denominators of Cellular Senescence. Trends Biochem. Sci. 2019, 44, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Parry, A.J.; Narita, M. Old cells, new tricks: Chromatin structure in senescence. Mamm. Genome 2016, 27, 320–331. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nuñez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A novel role for High-Mobility Group A proteins in cellular senescence and heterochromatin formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Narita, M. Cellular senescence and chromatin organisation. Br. J. Cancer 2007, 96, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Parry, A.J.; Hoare, M.; Bihary, D.; Hänsel-Hertsch, R.; Smith, S.; Tomimatsu, K.; Mannion, E.; Smith, A.; D’Santos, P.; Russell, I.A.; et al. NOTCH-mediated non-cell autonomous regulation of chromatin structure during senescence. Nat. Commun. 2018, 9, 1840. [Google Scholar] [CrossRef]
- Lee, S.; Jung, J.W.; Park, S.B.; Roh, K.; Lee, S.Y.; Kim, J.H.; Kang, S.K.; Kang, K.S. Histone deacetylase regulates high mobility group A2-targeting microRNAs in human cord blood-derived multipotent stem cell aging. Cell. Mol. Life Sci. 2011, 68, 325–336. [Google Scholar] [CrossRef]
- Yu, K.R.; Park, S.B.; Jung, J.W.; Seo, M.S.; Hong, I.S.; Kim, H.S.; Seo, Y.; Kang, T.W.; Lee, J.Y.; Kurtz, A.; et al. HMGA2 regulates the in vitro aging and proliferation of human umbilical cord blood-derived stromal cells through the mTOR/p70S6K signaling pathway. Stem Cell Res. 2013, 10, 156–165. [Google Scholar] [CrossRef]
- Chen, Y.N.; Ren, C.C.; Yang, L.; Nai, M.M.; Xu, Y.M.; Zhang, F.; Liu, Y. MicroRNA let-7d-5p rescues ovarian cancer cell apoptosis and restores chemosensitivity by regulating the p53 signaling pathway via HMGA1. Int. J. Oncol. 2019, 54, 1771–1784. [Google Scholar] [CrossRef]
- Shi, X.; Tian, B.; Ma, W.; Zhang, N.; Qiao, Y.; Li, X.; Zhang, Y.; Huang, B.; Lu, J. A novel anti-proliferative role of HMGA2 in induction of apoptosis through caspase 2 in primary human fibroblast cells. Biosci. Rep. 2015, 35, e00169. [Google Scholar] [CrossRef]
- Petroni, M.; Veschi, V.; Gulino, A.; Giannini, G. Molecular mechanisms of MYCN-dependent apoptosis and the MDM2-p53 pathway: An Achille’s heel to be exploited for the therapy of MYCN-amplified neuroblastoma. Front. Oncol. 2012, 2, 141. [Google Scholar] [CrossRef] [PubMed]
- Zaatiti, H.; Abdallah, J.; Nasr, Z.; Khazen, G.; Sandler, A.; Abou-Antoun, T.J. Tumorigenic proteins upregulated in the MYCN-amplified IMR-32 human neuroblastoma cells promote proliferation and migration. Int. J. Oncol. 2018, 52, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, S.; Ros, G.; Piazza, S.; Sommaggio, R.; Ciani, Y.; Rosato, A.; Sgarra, R.; Del Sal, G.; Manfioletti, G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013, 4, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.N.; Cope, L.; Poh, W.; Belton, A.; Roy, S.; Talbot, C.C., Jr.; Sukumar, S.; Huso, D.L.; Resar, L.M. HMGA1: A master regulator of tumor progression in triple-negative breast cancer cells. PLoS ONE 2013, 8, e63419. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, X.; Chen, S.; Du, B.; Li, Y. MicroRNA-4458 suppresses migration and epithelial-mesenchymal transition via targeting HMGA1 in non-small-cell lung cancer cells. Cancer Manag. Res. 2019, 11, 637–649. [Google Scholar] [CrossRef]
- Sekimoto, N.; Suzuki, A.; Suzuki, Y.; Sugano, S. Expression of miR-26A exhibits a negative correlation with HMGA1 and regulates cancer progression by targeting HMGA1 in lung adenocarcinoma cells. Mol. Med. Rep. 2017, 15, 534–542. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, H.; Jiang, X. MiR-1297 promotes apoptosis and inhibits the proliferation and invasion of hepatocellular carcinoma cells by targeting HMGA2. Int. J. Mol. Med. 2015, 36, 1345–1352. [Google Scholar] [CrossRef]
- Qin, M.M.; Chai, X.; Huang, H.B.; Feng, G.; Li, X.N.; Zhang, J.; Zheng, R.; Liu, X.C.; Pu, C. let-7i inhibits proliferation and migration of bladder cancer cells by targeting HMGA1. BMC Urol. 2019, 19, 53. [Google Scholar] [CrossRef]
- Teng, K.; Wei, S.; Zhang, C.; Chen, J.; Chen, J.; Xiao, K.; Liu, J.; Dai, M.; Guan, X.; Yun, J.; et al. KIFC1 is activated by TCF-4 and promotes hepatocellular carcinoma pathogenesis by regulating HMGA1 transcriptional activity. J. Exp. Clin. Cancer Res. 2019, 38, 329, Erratum in: J. Exp. Clin. Cancer Res. 2019, 38, 442. [Google Scholar] [CrossRef]
- Hopper, R.K.; Moonen, J.R.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef]
- Watanabe, S.; Ueda, Y.; Akaboshi, S.; Hino, Y.; Sekita, Y.; Nakao, M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am. J. Pathol. 2009, 174, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ru, N.Y.; Zhang, Y.; Li, Y.; Wei, D.; Ren, Z.; Huang, X.F.; Chen, Z.N.; Bian, H. HAb18G/CD147 promotes epithelial-mesenchymal transition through TGF-β signaling and is transcriptionally regulated by Slug. Oncogene 2011, 30, 4410–4427. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Xu, B.; Zi, L.; Chen, X. miR-625 reverses multidrug resistance in gastric cancer cells by directly targeting ALDH1A1. Cancer Manag. Res. 2019, 11, 6615–6624. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sells, E.; Pandey, R.; Abril, E.R.; Hsu, C.H.; Krouse, R.S.; Nagle, R.B.; Pampalakis, G.; Sotiropoulou, G.; Ignatenko, N.A. Kallikrein 6 protease advances colon tumorigenesis via induction of the high mobility group A2 protein. Oncotarget 2019, 10, 6062–6078. [Google Scholar] [CrossRef] [PubMed]
- Kolliopoulos, C.; Lin, C.Y.; Heldin, C.H.; Moustakas, A.; Heldin, P. Has2 natural antisense RNA and Hmga2 promote Has2 expression during TGFβ-induced EMT in breast cancer. Matrix Biol. 2019, 80, 29–45. [Google Scholar] [CrossRef]
- Li, W.; Wang, Z.; Zha, L.; Kong, D.; Liao, G.; Li, H. HMGA2 regulates epithelial-mesenchymal transition and the acquisition of tumor stem cell properties through TWIST1 in gastric cancer. Oncol. Rep. 2017, 37, 185–192. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Naghizadeh, S.; Gjerstorff, M.; Shanehbandi, D.; Shirjang, S.; Najafi, S.; Holmskov, U.; Khaze, V.; Duijf, P.H.G.; et al. miR-330 suppresses EMT and induces apoptosis by downregulating HMGA2 in human colorectal cancer. J. Cell. Physiol. 2019, 235, 1–12. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, J.; Wu, H.; Yi, C. High glucose promotes epithelial-mesenchymal transition, migration and invasion in A20 murine diffuse large b-cell lymphoma cells through increased expression of High Mobility Group AT-Hook 2 (HMGA2). Med. Sci. Monit. 2019, 25, 3860–3868. [Google Scholar] [CrossRef]
- Gong, J.; Wang, Y.; Jiang, B.; Xu, B.; Hu, C. Impact of high-mobility-group A2 overexpression on epithelial-mesenchymal transition in pancreatic cancer. Cancer Manag. Res. 2019, 11, 4075–4084. [Google Scholar] [CrossRef]
- Saika, S.; Kono-Saika, S.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Flanders, K.C.; Yoo, J.; Anzano, M.; Liu, C.Y.; et al. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am. J. Pathol. 2004, 164, 651–663. [Google Scholar] [CrossRef]
- Cho, H.J.; Baek, K.E.; Saika, S.; Jeong, M.J.; Yoo, J. Snail is required for transforming growth factor-beta-induced epithelial-mesenchymal transition by activating PI3 kinase/Akt signal pathway. Biochem. Biophys. Res. Commun. 2007, 353, 337–343. [Google Scholar] [CrossRef]
- Hou, M.; Bao, X.; Luo, F.; Chen, X.; Liu, L.; Wu, M. HMGA2 modulates the TGFβ/Smad, TGFβ/ERK and Notch signaling pathways in human lens Epithelial-Mesenchymal Transition. Curr. Mol. Med. 2018, 18, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.N.; Kerr, C.; Cope, L.; Zambidis, E.; Liu, C.; Hillion, J.; Belton, A.; Huso, D.L.; Resar, L.M. HMGA1 reprograms somatic cells into pluripotent stem cells by inducing stem cell transcriptional networks. PLoS ONE 2012, 7, e48533. [Google Scholar] [CrossRef] [PubMed]
- Pells, S.; Koutsouraki, E.; Morfopoulou, S.; Valencia-Cadavid, S.; Tomlinson, S.R.; Kalathur, R.; Futschik, M.E.; De Sousa, P.A. Novel human embryonic stem cell regulators identified by conserved and distinct CpG island methylation state. PLoS ONE 2015, 10, e0131102. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.R.; Shin, J.H.; Kim, J.J.; Koog, M.G.; Lee, J.Y.; Choi, S.W.; Kim, H.S.; Seo, Y.; Lee, S.; Shin, T.H.; et al. rapid and efficient direct conversion of human adult somatic cells into neural stem cells by HMGA2/let-7b. Cell Rep. 2015, 10, 441–452. [Google Scholar] [CrossRef]
- Colamaio, M.; Tosti, N.; Puca, F.; Mari, A.; Gattordo, R.; Kuzay, Y.; Federico, A.; Pepe, A.; Sarnataro, D.; Ragozzino, E.; et al. HMGA1 silencing reduces stemness and temozolomide resistance in glioblastoma stem cells. Expert Opin. Ther. Targets 2016, 20, 1169–1179. [Google Scholar] [CrossRef]
- Lopez-Bertoni, H.; Lal, B.; Michelson, N.; Guerrero-Cázares, H.; Quiñones-Hinojosa, A.; Li, Y.; Laterra, J. Epigenetic modulation of a miR-296-5p:HMGA1 axis regulates Sox2 expression and glioblastoma stem cells. Oncogene 2016, 35, 4903–4913. [Google Scholar] [CrossRef]
- Mao, X.G.; Hütt-Cabezas, M.; Orr, B.A.; Weingart, M.; Taylor, I.; Rajan, A.K.; Odia, Y.; Kahlert, U.; Maciaczyk, J.; Nikkhah, G.; et al. LIN28A facilitates the transformation of human neural stem cells and promotes glioblastoma tumorigenesis through a pro-invasive genetic program. Oncotarget 2013, 4, 1050–1064. [Google Scholar] [CrossRef]
- Wang, Z.; Park, H.J.; Carr, J.R.; Chen, Y.J.; Zheng, Y.; Li, J.; Tyner, A.L.; Costa, R.H.; Bagchi, S.; Raychaudhuri, P. FoxM1 in tumorigenicity of the neuroblastoma cells and renewal of the neural progenitors. Cancer Res. 2011, 71, 4292–4302. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, K.H.; Kim, D.G.; Cho, H.J.; Kim, Y.; Rheey, J.; Shin, K.; Seo, Y.J.; Choi, Y.S.; Lee, J.I.; et al. FoxM1 Promotes Stemness and Radio-Resistance of Glioblastoma by Regulating the Master Stem Cell Regulator Sox2. PLoS ONE 2015, 10, e0137703. [Google Scholar] [CrossRef]
- Luo, W.; Gao, F.; Li, S.; Liu, L. FoxM1 Promotes Cell Proliferation, Invasion, and Stem Cell Properties in Nasopharyngeal Carcinoma. Front. Oncol. 2018, 8, 483. [Google Scholar] [CrossRef] [PubMed]
- Besharat, Z.M.; Abballe, L.; Cicconardi, F.; Bhutkar, A.; Grassi, L.; Le Pera, L.; Moretti, M.; Chinappi, M.; D’Andrea, D.; Mastronuzzi, A.; et al. Foxm1 controls a pro-stemness microRNA network in neural stem cells. Sci. Rep. 2018, 8, 3523. [Google Scholar] [CrossRef]
- Chiu, W.T.; Huang, Y.F.; Tsai, H.Y.; Chen, C.C.; Chang, C.H.; Huang, S.C.; Hsu, K.F.; Chou, C.Y. FOXM1 confers to epithelial-mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget 2015, 6, 2349–2365. [Google Scholar] [CrossRef]
- Xian, L.; Georgess, D.; Huso, T.; Cope, L.; Belton, A.; Chang, Y.T.; Kuang, W.; Gu, Q.; Zhang, X.; Senger, S.; et al. HMGA1 amplifies Wnt signalling and expands the intestinal stem cell compartment and Paneth cell niche. Nat. Commun. 2017, 8, 15008. [Google Scholar] [CrossRef] [PubMed]
- Belton, A.; Gabrovsky, A.; Bae, Y.K.; Reeves, R.; Iacobuzio-Donahue, C.; Huso, D.L.; Resar, L.M. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS ONE 2012, 7, e30034. [Google Scholar] [CrossRef] [PubMed]
- Puca, F.; Colamaio, M.; Federico, A.; Gemei, M.; Tosti, N.; Bastos, A.U.; Del Vecchio, L.; Pece, S.; Battista, S.; Fusco, A. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget 2014, 5, 3234–3245. [Google Scholar] [CrossRef]
- Akaboshi, S.; Watanabe, S.; Hino, Y.; Sekita, Y.; Xi, Y.; Araki, K.; Yamamura, K.; Oshima, M.; Ito, T. HMGA1 is induced by Wnt/beta-catenin pathway and maintains cell proliferation in gastric cancer. Am. J. Pathol. 2009, 175, 1675–1685. [Google Scholar] [CrossRef]
- Bush, B.M.; Brock, A.T.; Deng, J.A.; Nelson, R.A.; Sumter, T.F. The Wnt/β-catenin/T-cell factor 4 pathway up-regulates high-mobility group A1 expression in colon cancer. Cell Biochem. Funct. 2012, 31, 228–236. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Austin, C.R.; Vincan, E.; Phesse, T.J. Wnt Signalling in Gastrointestinal Epithelial Stem Cells. Genes 2018, 9, 178. [Google Scholar] [CrossRef]
- Kretzschmar, K.; Clevers, H. Wnt/β-catenin signaling in adult mammalian epithelial stem cells. Dev. Biol. 2017, 428, 273–282. [Google Scholar] [CrossRef]
- Wei, F.; Zhang, T.; Deng, S.C.; Wei, J.C.; Yang, P.; Wang, Q.; Chen, Z.P.; Li, W.L.; Chen, H.C.; Hu, H.; et al. PD-L1 promotes colorectal cancer stem cell expansion by activating HMGA1-dependent signaling pathways. Cancer Lett. 2019, 450, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, A.Y.; Fan, C.; Zheng, H.; Li, Y.; Zhang, C.; Wu, S.; Yu, D.; Huang, Z.; Liu, F.; et al. MicroRNA-33b Inhibits Breast Cancer Metastasis by Targeting HMGA2, SALL4 and Twist1. Sci. Rep. 2015, 5, 9995. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Seo, E.J.; Choi, E.J.; Lee, S.I.; Kwon, Y.W.; Jang, I.H.; Kim, S.C.; Kim, K.H.; Suh, D.S.; Seong-Jang, K.; et al. Crucial role of HMGA1 in the self-renewal and drug resistance of ovarian cancer stem cells. Exp. Mol. Med. 2016, 48, e255. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.S.; Wang, M.L.; Chu, P.Y.; Chang, Y.L.; Liu, W.H.; Yu, C.C.; Lan, Y.T.; Huang, P.I.; Lee, Y.Y.; Chen, Y.W.; et al. Lin28B/Let-7 Regulates Expression of Oct4 and Sox2 and Reprograms Oral Squamous Cell Carcinoma Cells to a Stem-like State. Cancer Res. 2015, 75, 2553–2565. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, R.; Drago-Ferrante, R.; Pentimalli, F.; Di Marzo, D.; Forte, I.M.; Carlisi, D.; De Blasio, A.; Tesoriere, G.; Giordano, A.; Vento, R. Let-7d miRNA Shows Both Antioncogenic and Oncogenic Functions in Osteosarcoma-Derived 3AB-OS Cancer Stem Cells. J. Cell. Physiol. 2016, 231, 1832–1841. [Google Scholar] [CrossRef]
- Puca, F.; Tosti, N.; Federico, A.; Kuzay, Y.; Pepe, A.; Morlando, S.; Savarese, T.; D’Alessio, F.; Colamaio, M.; Sarnataro, D.; et al. HMGA1 negatively regulates NUMB expression at transcriptional and post transcriptional level in glioblastoma stem cells. Cell Cycle 2019, 18, 1446–1457. [Google Scholar] [CrossRef]
- Okano, H.; Imai, T.; Okabe, M. Musashi: A translational regulator of cell fate. J. Cell Sci. 2002, 115, 1355–1359. [Google Scholar]
- Shen, Q.; Zhong, W.; Jan, Y.N.; Temple, S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development 2002, 129, 4843–4853. [Google Scholar]
- Wang, J.; Sullenger, B.A.; Rich, J.N. Notch signaling in cancer stem cells. Adv. Exp. Med. Biol. 2012, 727, 174–185. [Google Scholar] [PubMed]
- Hu, Y.Y.; Zheng, M.H.; Cheng, G.; Li, L.; Liang, L.; Gao, F.; Wei, Y.N.; Fu, L.A.; Han, H. Notch signaling contributes to the maintenance of both normal neural stem cells and patient-derived glioma stem cells. BMC Cancer 2011, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Himes, S.R.; Reeves, R.; Attema, J.; Nissen, M.; Li, Y.; Shannon, M.F. The role of high-mobility group I(Y) proteins in expression of IL-2 and T cell proliferation. J. Immunol. 2000, 164, 3157–3168. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Robbins, C.M.; Leonard, W.J. An IL-2 response element in the human IL-2 receptor-a chain promoter is a composite element that binds Stat5, Elf-1, HMG-I(Y) and a GATA family protein. EMBO J. 1996, 15, 5627. [Google Scholar] [CrossRef] [PubMed]
- Copley, M.R.; Babovic, S.; Benz, C.; Knapp, D.J.; Beer, P.A.; Kent, D.G.; Wohrer, S.; Treloar, D.Q.; Day, C.; Rowe, K.; et al. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol. 2013, 15, 916–925. [Google Scholar] [CrossRef]
- Lam, K.; Muselman, A.; Du, R.; Harada, Y.; Scholl, A.G.; Yan, M.; Matsuura, S.; Weng, S.; Harada, H.; Zhang, D.E. Hmga2 is a direct target gene of RUNX1 and regulates expansion of myeloid progenitors in mice. Blood 2014, 124, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Wang, L.D.; Coma, S.; Han, A.; Mathieu, R.; Pearson, D.S.; Ross, S.; Sousa, P.; Nguyen, P.T.; Rodriguez, A.; et al. Developmental regulation of myeloerythroid progenitor function by the Lin28b-let-7-Hmga2 axis. J. Exp. Med. 2016, 213, 1497–1512. [Google Scholar] [CrossRef]
- Gasparini, G.; De Gori, M.; Paonessa, F.; Chiefari, E.; Brunetti, A.; Galasso, O. Functional relationship between high mobility group A1 (HMGA1) protein and insulin-like growth factor-binding protein 3 (IGFBP-3) in human chondrocytes. Arthritis Res. Ther. 2012, 14, R207. [Google Scholar] [CrossRef]
- Pierantoni, G.M.; Fedele, M.; Pentimalli, F.; Benvenuto, G.; Pero, R.; Viglietto, G.; Santoro, M.; Chiariotti, L.; Fusco, A. High mobility group I (Y) proteins bind HIPK2, a serine-threonine kinase protein which inhibits cell growth. Oncogene 2001, 20, 6132–6141. [Google Scholar] [CrossRef]
- Gerlini, R.; Amendola, E.; Conte, A.; Valente, V.; Tornincasa, M.; Credendino, S.C.; Cammarota, F.; Gentile, C.; Di Guida, L.; Paladino, S.; et al. Double knock-out of Hmga1 and Hipk2 genes causes perinatal death associated to respiratory distress and thyroid abnormalities in mice. Cell Death Dis. 2019, 10, 747. [Google Scholar] [CrossRef]
- Camós, S.; Gubern, C.; Sobrado, M.; Rodríguez, R.; Romera, V.G.; Moro, M.A.; Lizasoain, I.; Serena, J.; Mallolas, J.; Castellanos, M. The high-mobility group I-Y transcription factor is involved in cerebral ischemia and modulates the expression of angiogenic proteins. Neuroscience 2014, 269, 112–130. [Google Scholar] [CrossRef]
- Zanin, R.; Pegoraro, S.; Ros, G.; Ciani, Y.; Piazza, S.; Bossi, F.; Bulla, R.; Zennaro, C.; Tonon, F.; Lazarevic, D.; et al. HMGA1 promotes breast cancer angiogenesis supporting the stability, nuclear localization and transcriptional activity of FOXM1. J. Exp. Clin. Cancer Res. 2019, 38, 313. [Google Scholar] [CrossRef] [PubMed]
- Sakata, J.; Hirosue, A.; Yoshida, R.; Kawahara, K.; Matsuoka, Y.; Yamamoto, T.; Nakamoto, M.; Hirayama, M.; Takahashi, N.; Nakamura, T.; et al. HMGA2 contributes to distant metastasis and poor prognosis by promoting angiogenesis in oral squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 2473. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Frost, R.J.A.; Anderson, C.; Zhao, F.; Ma, J.; Yu, B.; Wang, S. let-7 contributes to diabetic retinopathy but represses pathological ocular angiogenesis. Mol. Cell. Biol. 2017, 37, e00001–e00017. [Google Scholar] [CrossRef] [PubMed]
- Duncan, B.; Zhao, K. HMGA1 mediates the activation of the CRYAB promoter by BRG1. DNA Cell Biol. 2007, 26, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Foti, D.; Iuliano, R.; Chiefari, E.; Brunetti, A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol. Cell. Biol. 2003, 23, 2720–2732. [Google Scholar] [CrossRef] [PubMed]
- Foti, D.; Chiefari, E.; Fedele, M.; Iuliano, R.; Brunetti, L.; Paonessa, F.; Manfioletti, G.; Barbetti, F.; Brunetti, A.; Croce, C.M.; et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat. Med. 2005, 11, 765–773. [Google Scholar] [CrossRef]
- Chiefari, E.; Iiritano, S.; Paonessa, F.; Le Pera, I.; Arcidiacono, B.; Filocamo, M.; Foti, D.; Liebhaber, S.A.; Brunetti, A. Pseudogene-mediated posttranscriptional silencing of HMGA1 can result in insulin resistance and type 2 diabetes. Nat. Commun. 2010, 1, 40. [Google Scholar] [CrossRef]
- Chiefari, E.; Nevolo, M.T.; Arcidiacono, B.; Maurizio, E.; Nocera, A.; Iiritano, S.; Sgarra, R.; Possidente, K.; Palmieri, C.; Paonessa, F.; et al. HMGA1 is a novel downstream nuclear target of the insulin receptor signaling pathway. Sci. Rep. 2012, 2, 251. [Google Scholar] [CrossRef]
- Chiefari, E.; Foti, D.P.; Sgarra, R.; Pegoraro, S.; Arcidiacono, B.; Brunetti, F.S.; Greco, M.; Manfioletti, G.; Brunetti, A. Transcriptional Regulation of Glucose Metabolism: The Emerging Role of the HMGA1 Chromatin Factor. Front. Endocrinol. (Lausanne) 2018, 9, 357. [Google Scholar] [CrossRef]
- Xue, W.; Huang, J.; Chen, H.; Zhang, Y.; Zhu, X.; Li, J.; Zhang, W.; Yuan, Y.; Wang, Y.; Zheng, L.; et al. Histone methyltransferase G9a modulates hepatic insulin signaling via regulating HMGA1. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 338–346. [Google Scholar] [CrossRef]
- Grasser, K.D. Chromatin-associated HMGA and HMGB proteins: Versatile co-regulators of DNA-dependent processes. Plant Mol. Biol. 2003, 53, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, M.; Macrì, S.; Calzia, D.; Sgarra, R.; Manfioletti, G.; Ramoino, P.; Lacalli, T.; Vignali, R.; Pestarino, M.; Candiani, S. The HMGA gene family in chordates: Evolutionary perspectives from amphioxus. Dev. Genes Evol. 2017, 227, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.; Stricker, S.; Wiecha, U.; Stiege, A.; Panopoulou, G.; Podsiadlowski, L.; Poustka, A.J.; Dieterich, C.; Ehrich, S.; Suvorova, J.; et al. Evolution of a core gene network for skeletogenesis in chordates. PLoS Genet. 2008, 4, e1000025. [Google Scholar] [CrossRef] [PubMed]
- Jandzik, D.; Garnett, A.T.; Square, T.A.; Cattell, M.V.; Yu, J.K.; Medeiros, D.M. Evolution of the new vertebrate head by co-option of an ancient chordate skeletal tissue. Nature 2015, 518, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Yong, L.W.; Yu, J.K. Tracing the evolutionary origin of vertebrate skeletal tissues: Insights from cephalochordate amphioxus. Curr. Opin. Genet. Dev. 2016, 39, 55–62. [Google Scholar] [CrossRef]
- Rychel, A.L.; Swalla, B.J. Development and evolution of chordate cartilage. J. Exp. Zool. B Mol. Dev. Evol. 2007, 308, 325–335. [Google Scholar] [CrossRef]
- Richter, A.; Hauschild, G.; Murua Escobar, H.; Nolte, I.; Bullerdiek, J. Application of high-mobility-group-A proteins increases the proliferative activity of chondrocytes in vitro. Tissue Eng. Part A 2009, 15, 473–477. [Google Scholar] [CrossRef]
- Richter, A.; Lübbing, M.; Frank, H.G.; Nolte, I.; Bullerdiek, J.C.; von Ahsen, I. High-mobility group protein HMGA2-derived fragments stimulate the proliferation of chondrocytes and adipose tissue-derived stem cells. Eur. Cells Mater. 2011, 21, 355–363. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Han, F.; Webster, M.T.; Andersson, L.; Grant, B.R.; Grant, P.R. Rapid hybrid speciation in Darwin’s finches. Science 2018, 359, 224–228. [Google Scholar] [CrossRef]
- Grant, B.R.; Grant, P.R. 40 Years of Evolution: Darwin’s Finches on Daphne Major Island; Princeton University Press: Princeton, NJ, USA, 2014; 432p. [Google Scholar]
- Chaves, J.A.; Cooper, E.A.; Hendry, A.P.; Podos, J.; De León, L.F.; Raeymaekers, J.A.; MacMillan, W.O.; Uy, J.A. Genomic variation at the tips of the adaptive radiation of Darwin’s finches. Mol. Ecol. 2016, 25, 5282–5295. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Han, F.; Berglund, J.; Wang, C.; Almén, M.S.; Webster, M.T.; Grant, B.R.; Grant, P.R.; Andersson, L. A beak size locus in Darwin’s finches facilitated character displacement during a drought. Science 2016, 352, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Mundy, N.I. Population genomics fits the bill: Genetics of adaptive beak variation in Darwin’s finches. Mol. Ecol. 2016, 25, 5265–5266. [Google Scholar] [CrossRef] [PubMed]
- Ligon, A.H.; Moore, S.D.; Parisi, M.A.; Mealiffe, M.E.; Harris, D.J.; Ferguson, H.L.; Quade, B.J.; Morton, C.C. Constitutional rearrangement of the architectural factor HMGA2: A novel human phenotype including overgrowth and lipomas. Am. J. Hum. Genet. 2005, 76, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.L.; Medland, S.E.; Vasquez, A.A.; Hibar, D.P.; Senstad, R.E.; Winkler, A.M.; Toro, R.; Appel, K.; Bartecek, R.; Bergmann, Ø.; et al. Identification of common variants associated with human hippocampal and intracranial volumes. Nat. Genet. 2012, 44, 552–561. [Google Scholar] [CrossRef]
- Taal, H.R.; Pourcain, B.S.; Thiering, E.; Das, S.; Mook-Kanamori, D.O.; Warrington, N.M.; Kaakinen, M.; Kreiner-Møller, E.; Bradfield, J.P.; Freathy, R.M.; et al. Common variants at 12q15 and 12q24 are associated with infant head circumference. Nat. Genet. 2012, 44, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Fatemifar, G.; Hoggart, C.J.; Paternoster, L.; Kemp, J.P.; Prokopenko, I.; Horikoshi, M.; Wright, V.J.; Tobias, J.H.; Richmond, S.; Zhurov, A.I.; et al. Genome-wide association study of primary tooth eruption identifies pleiotropic loci associated with height and craniofacial distances. Hum. Mol. Genet. 2013, 22, 3807–3817. [Google Scholar] [CrossRef] [PubMed]
- Moreno Uribe, L.M.; Ray, A.; Blanchette, D.R.; Dawson, D.V.; Southard, T.E. Phenotype-genotype correlations of facial width and height proportions in patients with Class II malocclusion. Orthod. Craniofac. Res. 2015, 18 (Suppl. 1), 100–108. [Google Scholar] [CrossRef]
- Diana, F.; Sgarra, R.; Manfioletti, G.; Rustighi, A.; Sciortino, M.T.; Mastino, A.; Giancotti, V. A link between apoptosis and degree of phosphorylation of high mobility groul 1a protein in leukemic cells. J. Biol. Chem. 2001, 276, 11354–11361. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Rothstein, M.; Azambuja, A.P.; Simoes-Costa, M. Control of neural crest multipotency by Wnt signaling and the Lin28/let-7 axis. Elife 2018, 7, e40556. [Google Scholar] [CrossRef]
- Robinton, D.A.; Chal, J.; Lummertz da Rocha, E.; Han, A.; Yermalovich, A.V.; Oginuma, M.; Schlaeger, T.M.; Sousa, P.; Rodriguez, A.; Urbach, A.; et al. The Lin28/let-7 Pathway Regulates the Mammalian Caudal Body Axis Elongation Program. Dev. Cell. 2019, 48, 396–405. [Google Scholar] [CrossRef]
- Bajpai, R.; Chen, D.A.; Rada-Iglesias, A.; Zhang, J.; Xiong, Y.; Helms, J.; Chang, C.P.; Zhao, Y.; Swigut, T.; Wysocka, J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010, 463, 958–962. [Google Scholar] [CrossRef] [PubMed]
- De Crozé, N.; Maczkowiak, F.; Monsoro-Burq, A.H. Reiterative AP2a activity controls sequential steps in the neural crest gene regulatory network. Proc. Natl. Acad. Sci. USA 2011, 108, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Prescott, S.; Brugmann, S.A.; Swigut, T.; Wysocka, J. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell 2012, 11, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, R.S.; Long, H.K.; Swigut, T.; Wysocka, J. Single Amino Acid Change Underlies Distinct Roles of H2A.Z Subtypes in Human Syndrome. Cell 2019, 178, 1421–1436. [Google Scholar] [CrossRef]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vignali, R.; Marracci, S. HMGA Genes and Proteins in Development and Evolution. Int. J. Mol. Sci. 2020, 21, 654. https://doi.org/10.3390/ijms21020654
Vignali R, Marracci S. HMGA Genes and Proteins in Development and Evolution. International Journal of Molecular Sciences. 2020; 21(2):654. https://doi.org/10.3390/ijms21020654
Chicago/Turabian StyleVignali, Robert, and Silvia Marracci. 2020. "HMGA Genes and Proteins in Development and Evolution" International Journal of Molecular Sciences 21, no. 2: 654. https://doi.org/10.3390/ijms21020654
APA StyleVignali, R., & Marracci, S. (2020). HMGA Genes and Proteins in Development and Evolution. International Journal of Molecular Sciences, 21(2), 654. https://doi.org/10.3390/ijms21020654