Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease


 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Resv or Resv with ET Reduces Neuroinflammation in 3xTg-AD Mice
2.2. Resv or Resv with ET Attenuates the Accumulation of Toxic Conformational Species of Aβ
2.3. Resv or Resv with ET Increases Protein Expression of Neurotrophins and Synaptic Markers in 3xTg-AD Mice
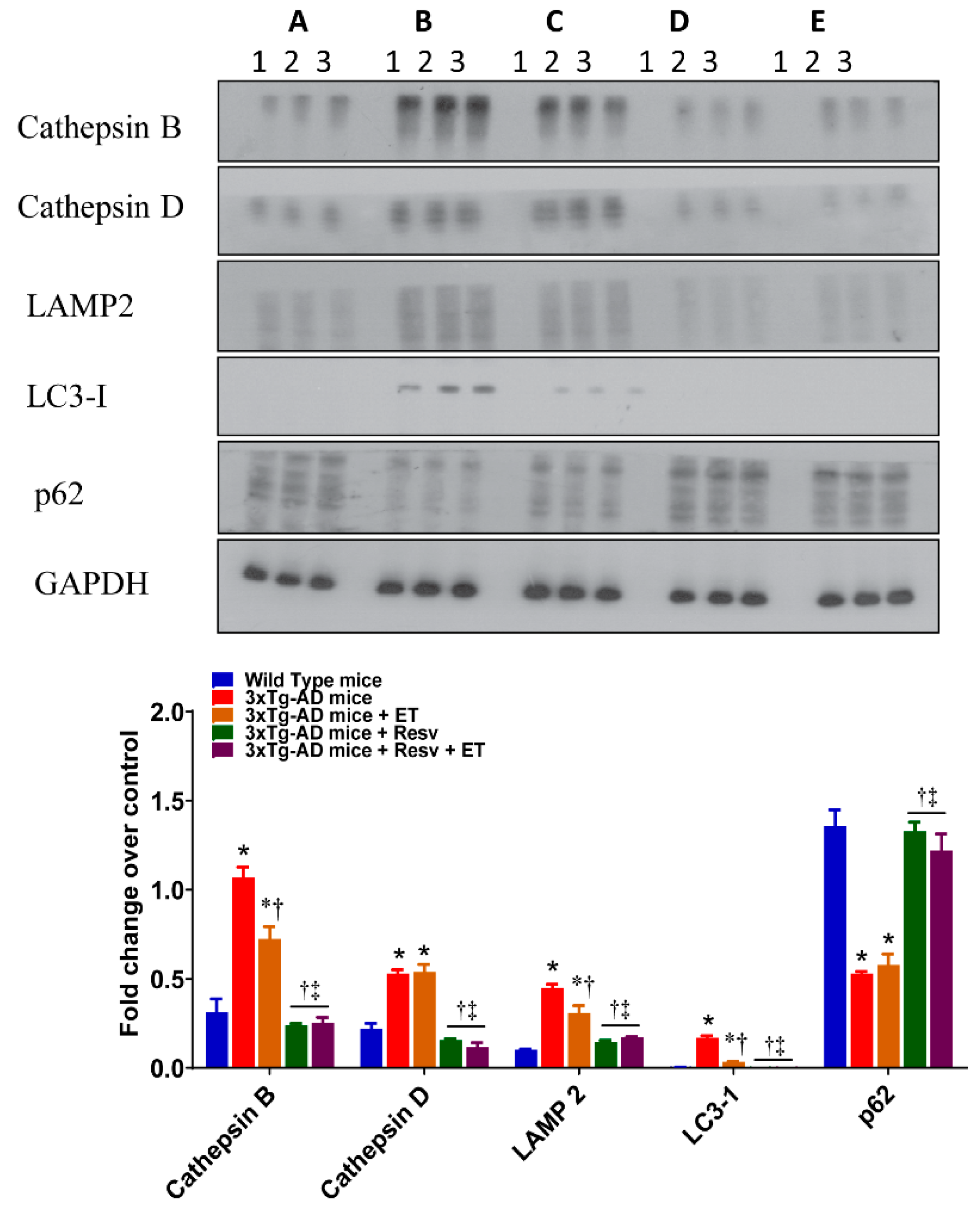
2.4. Resv or Resv with ET Reduces Apoptosis and Decreases Protein Expression of Autophagy and Accumulation of Ubiquitinated Proteins
2.5. Resv or Resv with ET Increases the Expression Level of SIRT1
3. Discussion
4. Materials and Methods
4.1. Mouse Model of AD and Treatment Protocols
4.2. Western Blot Analysis
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Adam 10 | disintegrin and metallopeptidase domain-containing protein 10 |
| AD | Alzheimer’s disease |
| Aβ | amyloid-beta |
| Ab | anti-body |
| AMPK | AMP-activated protein kinase |
| APP | amyloid precursor protein |
| APP-CTF | (carboxy terminal of amyloid precursor protein) |
| BACE1 | beta-secretase enzyme 1 |
| BBB | blood–brain barrier |
| BDNF | brain-derived neurotropic factor |
| ET | exercise training |
| GFAP | glial fibrillary acidic protein |
| GSK3β | glycogen synthase kinase beta |
| IRS-1 | insulin-receptor substrate |
| NF-κB | nuclear factor-kappa B |
| NGF | nerve growth factor |
| NFT | neurofibrillary tangles |
| PARP | poly (ADP-ribose) polymerase |
| PSD95 | postsynaptic density 95 |
| Resv | resveratrol |
| SIRT1 | silent information regulator 1 |
| UB | ubiquitinated proteins |
| UPS | ubiquitinated proteasome system |
References
- Sperling, R.A.; Dickerson, B.C.; Pihlajamaki, M.; Vannini, P.; LaViolette, P.S.; Vitolo, O.V.; Hedden, T.; Becker, J.A.; Rentz, D.M.; Selkoe, D.J.; et al. Functional Alterations in Memory Networks in Early Alzheimer’s Disease. Neuromol. Med. 2010, 12, 27–43. [Google Scholar] [CrossRef] [PubMed]
- World Alzheimer Report 2018. Available online: https://www.alz.co.uk/research/world-report-2018 (accessed on 18 July 2020).
- Harris, M.E.; Hensley, K.; Butterfield, D.A.; Leedle, R.A.; Carney, J.M. Direct evidence of oxidative injury produced by the Alzheimer’s beta-amyloid peptide (1-40) in cultured hippocampal neurons. Exp. Neurol. 1995, 131, 193–202. [Google Scholar] [CrossRef]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Rhein, V.; Müller-Spahn, F.; Götz, J.; Müller, W.E. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener. Dis. 2008, 5, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L. Synaptotoxic amyloid-β oligomers: A molecular basis for the cause, diagnosis, and treatment of Alzheimer’s disease? J. Alzheimers Dis. JAD 2013, 33 (Suppl. 1), S49–S65. [Google Scholar] [CrossRef]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. JAD 2013, 33 (Suppl. 1), S67–S78. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Ferrari, A.; Hoerndli, F.; Baechi, T.; Nitsch, R.M.; Götz, J. Beta-Amyloid induces paired helical filament-like tau filaments in tissue culture. J. Biol. Chem. 2003, 278, 40162–40168. [Google Scholar] [CrossRef]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Chabrier, M.A.; Blurton-Jones, M.; Agazaryan, A.A.; Nerhus, J.L.; Martinez-Coria, H.; LaFerla, F.M. Soluble aβ promotes wild-type tau pathology in vivo. J. Neurosci. 2012, 32, 17345–17350. [Google Scholar] [CrossRef] [PubMed]
- Pensalfini, A.; Albay, R.; Rasool, S.; Wu, J.W.; Hatami, A.; Arai, H.; Margol, L.; Milton, S.; Poon, W.W.; Corrada, M.M.; et al. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol. Dis. 2014, 71, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Scarmeas, N.; Luchsinger, J.A.; Schupf, N.; Brickman, A.M.; Cosentino, S.; Tang, M.X.; Stern, Y. Physical activity, diet, and risk of Alzheimer disease. JAMA 2009, 302, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Martin, B.; Maudsley, S. Anti-inflammatory effects of physical activity in relationship to improved cognitive status in humans and mouse models of Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.C.; Filho, C.B.; Goes, A.T.R.; Fabbro, L.D.; de Gomes, M.G.; Savegnago, L.; Oliveira, M.S.; Jesse, C.R. Neuroprotective effect of physical exercise in a mouse model of Alzheimer’s disease induced by β-amyloid1-40 peptide. Neurotox. Res. 2013, 24, 148–163. [Google Scholar] [CrossRef]
- García-Mesa, Y.; López-Ramos, J.C.; Giménez-Llort, L.; Revilla, S.; Guerra, R.; Gruart, A.; Laferla, F.M.; Cristòfol, R.; Delgado-García, J.M.; Sanfeliu, C. Physical exercise protects against Alzheimer’s disease in 3xTg-AD mice. J. Alzheimers Dis. JAD 2011, 24, 421–454. [Google Scholar] [CrossRef]
- Pena, G.S.; Paez, H.G.; Johnson, T.K.; Halle, J.L.; Carzoli, J.P.; Visavadiya, N.P.; Zourdos, M.C.; Whitehurst, M.A.; Khamoui, A.V. Hippocampal Growth Factor and Myokine Cathepsin B Expression following Aerobic and Resistance Training in 3xTg-AD Mice. Int. J. Chronic Dis. 2020, 2020, 5919501. [Google Scholar] [CrossRef]
- Liu, Y.; Chu, J.M.T.; Yan, T.; Zhang, Y.; Chen, Y.; Chang, R.C.C.; Wong, G.T.C. Short-term resistance exercise inhibits neuroinflammation and attenuates neuropathological changes in 3xTg Alzheimer’s disease mice. J. Neuroinflammation 2020, 17, 4. [Google Scholar] [CrossRef]
- Kim, D.; Cho, J.; Kang, H. Protective effect of exercise training against the progression of Alzheimer’s disease in 3xTg-AD mice. Behav. Brain Res. 2019, 374, 112105. [Google Scholar] [CrossRef]
- Do, K.; Laing, B.T.; Landry, T.; Bunner, W.; Mersaud, N.; Matsubara, T.; Li, P.; Yuan, Y.; Lu, Q.; Huang, H. The effects of exercise on hypothalamic neurodegeneration of Alzheimer’s disease mouse model. PLoS ONE 2018, 13, e0190205. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Cho, J.; Lee, I.; Jin, Y.; Kang, H. Exercise Attenuates High-Fat Diet-induced Disease Progression in 3xTg-AD Mice. Med. Sci. Sports Exerc. 2017, 49, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gong, Q.; Dong, H.; Shi, J. Resveratrol, a neuroprotective supplement for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, K.W.; Lee, H.J. Protective effects of piceatannol against beta-amyloid-induced neuronal cell death. Ann. N. Y. Acad. Sci. 2007, 1095, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.-L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ho, L.; Zhao, W.; Ono, K.; Rosensweig, C.; Chen, L.; Humala, N.; Teplow, D.B.; Pasinetti, G.M. Grape-derived polyphenolics prevent Abeta oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 6388–6392. [Google Scholar] [CrossRef]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol mitigates lipopolysaccharide- and Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef]
- Ladiwala, A.R.A.; Lin, J.C.; Bale, S.S.; Marcelino-Cruz, A.M.; Bhattacharya, M.; Dordick, J.S.; Tessier, P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Abeta into off-pathway conformers. J. Biol. Chem. 2010, 285, 24228–24237. [Google Scholar] [CrossRef]
- Rahvar, M.; Nikseresht, M.; Shafiee, S.M.; Naghibalhossaini, F.; Rasti, M.; Panjehshahin, M.R.; Owji, A.A. Effect of oral resveratrol on the BDNF gene expression in the hippocampus of the rat brain. Neurochem. Res. 2011, 36, 761–765. [Google Scholar] [CrossRef]
- Shojaei, S.; Panjehshahin, M.R.; Shafiee, S.M.; Khoshdel, Z.; Borji, M.; Ghasempour, G.; Owji, A.A. Differential Effects of Resveratrol on the Expression of Brain-Derived Neurotrophic Factor Transcripts and Protein in the Hippocampus of Rat Brain. Iran. J. Med. Sci. 2017, 42, 32–39. [Google Scholar]
- Park, H.R.; Kong, K.H.; Yu, B.P.; Mattson, M.P.; Lee, J. Resveratrol inhibits the proliferation of neural progenitor cells and hippocampal neurogenesis. J. Biol. Chem. 2012, 287, 42588–42600. [Google Scholar] [CrossRef] [PubMed]
- Cowansage, K.K.; LeDoux, J.E.; Monfils, M.-H. Brain-derived neurotrophic factor: A dynamic gatekeeper of neural plasticity. Curr. Mol. Pharmacol. 2010, 3, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Esfandiarei, M.; Hoxha, B.; Talley, N.A.; Anderson, M.R.; Alkhouli, M.F.; Squire, M.A.; Eckman, D.M.; Babu, J.R.; Lopaschuk, G.D.; Broderick, T.L. Beneficial effects of resveratrol and exercise training on cardiac and aortic function and structure in the 3xTg mouse model of Alzheimer’s disease. Drug Des. Devel. Ther. 2019, 13, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Alkhouli, M.F.; Hung, J.; Squire, M.; Anderson, M.; Castro, M.; Babu, J.R.; Al-Nakkash, L.; Broderick, T.L.; Plochocki, J.H. Exercise and resveratrol increase fracture resistance in the 3xTg-AD mouse model of Alzheimer’s disease. BMC Complement. Altern. Med. 2019, 19, 39. [Google Scholar] [CrossRef] [PubMed]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Zheng, C.; Geetha, T.; Gearing, M.; Babu, J.R. Amyloid β-abrogated TrkA ubiquitination in PC12 cells analogous to Alzheimer’s disease. J. Neurochem. 2015, 133, 919–925. [Google Scholar] [CrossRef]
- Crews, L.; Tsigelny, I.; Hashimoto, M.; Masliah, E. Role of synucleins in Alzheimer’s disease. Neurotox. Res. 2009, 16, 306–317. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H.; Alafuzoff, I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2012, 96, 87–95. [Google Scholar] [CrossRef]
- Su, H.; Wang, X. Autophagy and p62 in cardiac protein quality control. Autophagy 2011, 7, 1382–1383. [Google Scholar] [CrossRef]
- Caccamo, A.; Ferreira, E.; Branca, C.; Oddo, S. p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 2017, 22, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Vidoni, E.D.; Johnson, D.K.; Van Sciver, A.; Mahnken, J.D.; Honea, R.A.; Wilkins, H.M.; Brooks, W.M.; Billinger, S.A.; Swerdlow, R.H.; et al. Aerobic exercise for Alzheimer’s disease: A randomized controlled pilot trial. PLoS ONE 2017, 12, e0170547. [Google Scholar] [CrossRef] [PubMed]
- Cass, S.P. Alzheimer’s Disease and Exercise: A Literature Review. Curr. Sports Med. Rep. 2017, 16, 19–22. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’Banion, M.K. Inflammatory processes in Alzheimer’s disease. J. Neuroimmunol. 2007, 184, 69–91. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Ospina, G.G.; Jimenez del Rio, M. Abeta[25-35] peptide and iron promote apoptosis in lymphocytes by an oxidative stress mechanism: Involvement of H2O2, caspase-3, NF-kappaB, p53 and c-Jun. Neurotoxicology 2002, 23, 351–365. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Kayed, R. Astrocytes contain amyloid-β annular protofibrils in Alzheimer’s disease brains. FEBS Lett. 2011, 585, 3052–3057. [Google Scholar] [CrossRef]
- Saiko, P.; Szakmary, A.; Jaeger, W.; Szekeres, T. Resveratrol and its analogs: Defense against cancer, coronary disease and neurodegenerative maladies or just a fad? Mutat. Res. 2008, 658, 68–94. [Google Scholar] [CrossRef]
- Alexe, D.-M.; Syridou, G.; Petridou, E.T. Determinants of early life leptin levels and later life degenerative outcomes. Clin. Med. Res. 2006, 4, 326–335. [Google Scholar] [CrossRef]
- Knight, E.M.; Martins, I.V.A.; Gümüsgöz, S.; Allan, S.M.; Lawrence, C.B. High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol. Aging 2014, 35, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Moraska, A.; Deak, T.; Spencer, R.L.; Roth, D.; Fleshner, M. Treadmill running produces both positive and negative physiological adaptations in Sprague-Dawley rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1321–R1329. [Google Scholar] [CrossRef] [PubMed]
- Lyons, C.E.; Bartolomucci, A. Stress and Alzheimer’s disease: A senescence link? Neurosci. Biobehav. Rev. 2020, 115, 285–298. [Google Scholar] [CrossRef] [PubMed]
- de la Rubia Ortí, J.E.; Sancho Castillo, S.; Benlloch, M.; Julián Rochina, M.; Corchón Arreche, S.; García-Pardo, M.P. Impact of the Relationship of Stress and the Immune System in the Appearance of Alzheimer’s Disease. J. Alzheimers Dis. JAD 2017, 55, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 1946–1959. [Google Scholar] [CrossRef]
- Ovsepian, S.V.; O’Leary, V.B.; Zaborsky, L.; Ntziachristos, V.; Dolly, J.O. Synaptic vesicle cycle and amyloid β: Biting the hand that feeds. Alzheimers Dement. 2018, 14, 502–513. [Google Scholar] [CrossRef]
- Ovsepian, S.V.; O’Leary, V.B.; Zaborsky, L.; Ntziachristos, V.; Dolly, J.O. Amyloid plaques of Alzheimer’s disease as hotspots of glutamatergic activity. Neuroscientist 2019, 25, 288–297. [Google Scholar] [CrossRef]
- Li, J.; Feng, L.; Xing, Y.; Wang, Y.; Du, L.; Xu, C.; Cao, J.; Wang, Q.; Fan, S.; Liu, Q.; et al. Radioprotective and antioxidant effect of resveratrol in hippocampus by activating Sirt1. Int. J. Mol. Sci. 2014, 15, 5928–5939. [Google Scholar] [CrossRef]
- Rege, S.D.; Geetha, T.; Broderick, T.L.; Babu, J.R. Resveratrol protects β amyloid-induced oxidative damage and memory associated proteins in H19-7 hippocampal neuronal cells. Curr. Alzheimer Res. 2015, 12, 147–156. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef]
- Pallàs, M.; Ortuño-Sahagún, D.; Andrés-Benito, P.; Ponce-Regalado, M.D.; Rojas-Mayorquín, A.E. Resveratrol in epilepsy: Preventive or treatment opportunities? Front. Biosci. Landmark Ed. 2014, 19, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zeng, Y.; Kermode, A.R. A plant cell-based system that predicts aβ42 misfolding: Potential as a drug discovery tool for Alzheimer’s disease. Mol. Genet. Metab. 2012, 107, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Soylemez, S.; Gurdal, H.; Sepici, A.; Akar, F. The effect of long-term resveratrol treatment on relaxation to estrogen in aortae from male and female rats: Role of nitric oxide and superoxide. Vascul. Pharmacol. 2008, 49, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.C.; Connell, B.J.; Saleh, T.M. Resveratrol induced neuroprotection is mediated via both estrogen receptor subtypes, ER(α) and ER(β). Neurosci. Lett. 2013, 548, 217–221. [Google Scholar] [CrossRef]
- Feng, Y.; Cui, Y.; Gao, J.-L.; Li, R.; Jiang, X.-H.; Tian, Y.-X.; Wang, K.-J.; Li, M.-H.; Zhang, H.-A.; Cui, J.-Z. Neuroprotective effects of resveratrol against traumatic brain injury in rats: Involvement of synaptic proteins and neuronal autophagy. Mol. Med. Rep. 2016, 13, 5248–5254. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Schweiger, S.; Matthes, F.; Posey, K.; Kickstein, E.; Weber, S.; Hettich, M.M.; Pfurtscheller, S.; Ehninger, D.; Schneider, R.; Krauß, S. Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci. Rep. 2017, 7, 13753. [Google Scholar] [CrossRef]
- Giménez-Llort, L.; García, Y.; Buccieri, K.; Revilla, S.; Suñol, C.; Cristofol, R.; Sanfeliu, C. Gender-Specific Neuroimmunoendocrine Response to Treadmill Exercise in 3xTg-AD Mice. Int. J. Alzheimers Dis. 2010, 2010, 128354. [Google Scholar] [CrossRef][Green Version]
- Ruschak, A.M.; Miranker, A.D. The role of prefibrillar structures in the assembly of a peptide amyloid. J. Mol. Biol. 2009, 393, 214–226. [Google Scholar] [CrossRef]
- Lee, J.; Culyba, E.K.; Powers, E.T.; Kelly, J.W. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [Google Scholar] [CrossRef]
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 2011, 286, 23063–23076. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res. 2006, 54, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Maudsley, S.; Martin, B. BDNF and 5-HT: A dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004, 27, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Holsinger, R.M.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in Alzheimer’s disease. Brain Res. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Müller-Spahn, F.; Huber, P.; Riesen, W.; Nitsch, R.M.; Otten, U. Increased cerebrospinal fluid levels of neurotrophin 3 (NT-3) in elderly patients with major depression. Mol. Psychiatry 2000, 5, 510–513. [Google Scholar] [CrossRef]
- Michalski, B.; Fahnestock, M. Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer’s disease. Brain Res. Mol. Brain Res. 2003, 111, 148–154. [Google Scholar] [CrossRef]
- Yasutake, C.; Kuroda, K.; Yanagawa, T.; Okamura, T.; Yoneda, H. Serum BDNF, TNF-alpha and IL-1beta levels in dementia patients: Comparison between Alzheimer’s disease and vascular dementia. Eur. Arch. Psychiatry Clin. Neurosci. 2006, 256, 402–406. [Google Scholar] [CrossRef]
- Parikh, V.; Howe, W.M.; Welchko, R.M.; Naughton, S.X.; D’Amore, D.E.; Han, D.H.; Deo, M.; Turner, D.L.; Sarter, M. Diminished trkA receptor signaling reveals cholinergic-attentional vulnerability of aging. Eur. J. Neurosci. 2013, 37, 278–293. [Google Scholar] [CrossRef]
- Hellweg, R.; Gericke, C.A.; Jendroska, K.; Hartung, H.D.; Cervós-Navarro, J. NGF content in the cerebral cortex of non-demented patients with amyloid-plaques and in symptomatic Alzheimer’s disease. Int. J. Dev. Neurosci. 1998, 16, 787–794. [Google Scholar] [CrossRef]
- Gylys, K.H.; Fein, J.A.; Yang, F.; Wiley, D.J.; Miller, C.A.; Cole, G.M. Synaptic changes in Alzheimer’s disease: Increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am. J. Pathol. 2004, 165, 1809–1817. [Google Scholar] [CrossRef]
- Love, S.; Siew, L.K.; Dawbarn, D.; Wilcock, G.K.; Ben-Shlomo, Y.; Allen, S.J. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol. Aging 2006, 27, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.T.; Coulson, E.J.; Dodd, P.R. Reduction in post-synaptic scaffolding PSD-95 and SAP-102 protein levels in the Alzheimer inferior temporal cortex is correlated with disease pathology. J. Alzheimers Dis. JAD 2010, 21, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Okabe, S. Molecular anatomy of the postsynaptic density. Mol. Cell. Neurosci. 2007, 34, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Kim, M.J. Postsynaptic signaling and plasticity mechanisms. Science 2002, 298, 776–780. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Arendt, T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. (Berl.) 2009, 118, 167–179. [Google Scholar] [CrossRef]
- Neves, G.; Cooke, S.F.; Bliss, T.V.P. Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nat. Rev. Neurosci. 2008, 9, 65–75. [Google Scholar] [CrossRef]
- Griffin, É.W.; Mullally, S.; Foley, C.; Warmington, S.A.; O’Mara, S.M.; Kelly, A.M. Aerobic exercise improves hippocampal function and increases BDNF in the serum of young adult males. Physiol. Behav. 2011, 104, 934–941. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Marcellino, B.K.; Yue, Z. Cell “self-eating” (autophagy) mechanism in Alzheimer’s disease. Mt. Sinai J. Med. N. Y. 2010, 77, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, P.A.; Wyss-Coray, T. Beclin 1 complex in autophagy and Alzheimer disease. Arch. Neurol. 2010, 67, 1181–1184. [Google Scholar] [CrossRef]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, Y.; Zhang, X.; Bu, G.; Xu, H.; Zhang, Y. Trafficking regulation of proteins in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 1993, 10, 1151–1160. [Google Scholar] [CrossRef]
- Ramesh Babu, J.; Lamar Seibenhener, M.; Peng, J.; Strom, A.-L.; Kemppainen, R.; Cox, N.; Zhu, H.; Wooten, M.C.; Diaz-Meco, M.T.; Moscat, J.; et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 2008, 106, 107–120. [Google Scholar] [CrossRef]
- Kuusisto, E.; Salminen, A.; Alafuzoff, I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 2001, 12, 2085–2090. [Google Scholar] [CrossRef]
- Du, Y.; Wooten, M.C.; Gearing, M.; Wooten, M.W. Age-associated oxidative damage to the p62 promoter: Implications for Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 492–501. [Google Scholar] [CrossRef]
- Høydal, M.A.; Wisløff, U.; Kemi, O.J.; Ellingsen, O. Running speed and maximal oxygen uptake in rats and mice: Practical implications for exercise training. Eur. J. Cardiovasc. Prev. Rehabil. 2007, 14, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Jones, K.E.; Sidhu, R.S.; Haykowsky, M.; Czubryt, M.P.; Gordon, T.; Dyck, J.R.B. Improvements in skeletal muscle strength and cardiac function induced by resveratrol during exercise training contribute to enhanced exercise performance in rats. J. Physiol. 2012, 590, 2783–2799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, H.; Wu, Q.; Lu, Y.; Nie, J.; Xie, X.; Shi, J. Resveratrol protects cortical neurons against microglia-mediated neuroinflammation. Phytother. Res. PTR 2013, 27, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broderick, T.L.; Rasool, S.; Li, R.; Zhang, Y.; Anderson, M.; Al-Nakkash, L.; Plochocki, J.H.; Geetha, T.; Babu, J.R. Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7337. https://doi.org/10.3390/ijms21197337
Broderick TL, Rasool S, Li R, Zhang Y, Anderson M, Al-Nakkash L, Plochocki JH, Geetha T, Babu JR. Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(19):7337. https://doi.org/10.3390/ijms21197337
Chicago/Turabian StyleBroderick, Tom L., Suhail Rasool, Rongzi Li, Yuxian Zhang, Miranda Anderson, Layla Al-Nakkash, Jeffrey H. Plochocki, Thangiah Geetha, and Jeganathan Ramesh Babu. 2020. "Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 19: 7337. https://doi.org/10.3390/ijms21197337
APA StyleBroderick, T. L., Rasool, S., Li, R., Zhang, Y., Anderson, M., Al-Nakkash, L., Plochocki, J. H., Geetha, T., & Babu, J. R. (2020). Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 21(19), 7337. https://doi.org/10.3390/ijms21197337