Intramolecular Spodium Bonds in Zn(II) Complexes: Insights from Theory and Experiment


 ,
,  ,
,  and
and 
Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis
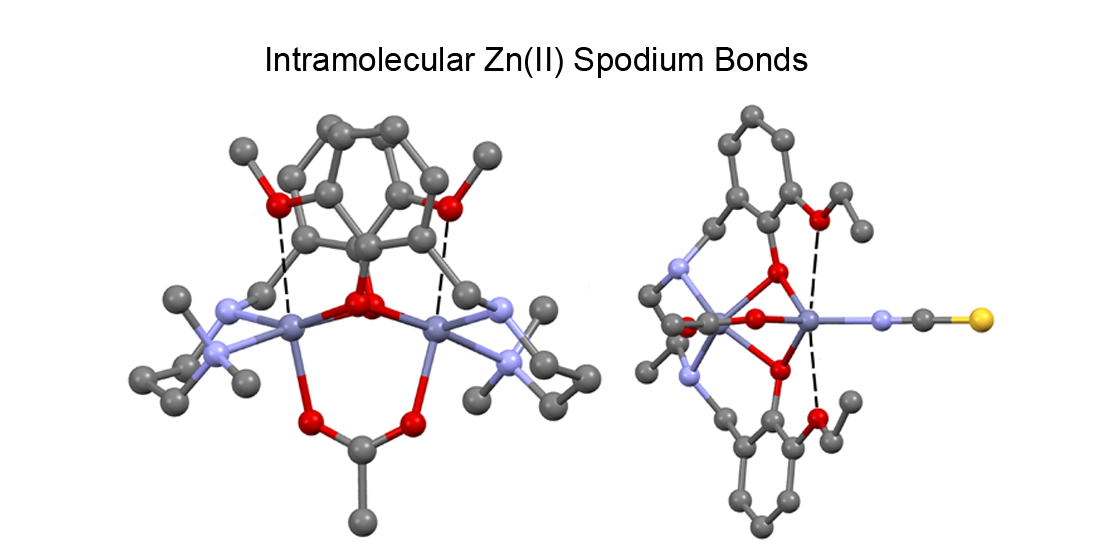
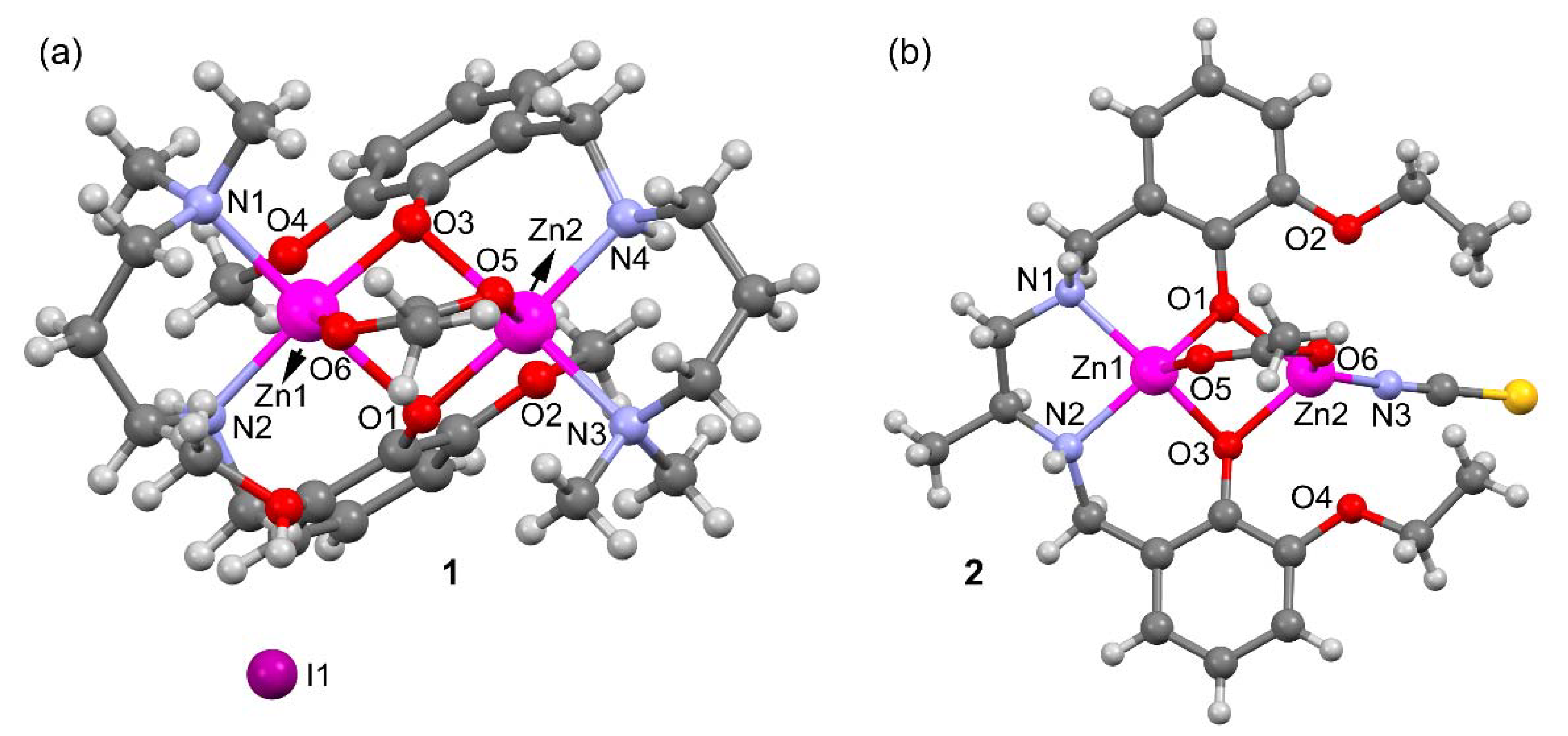
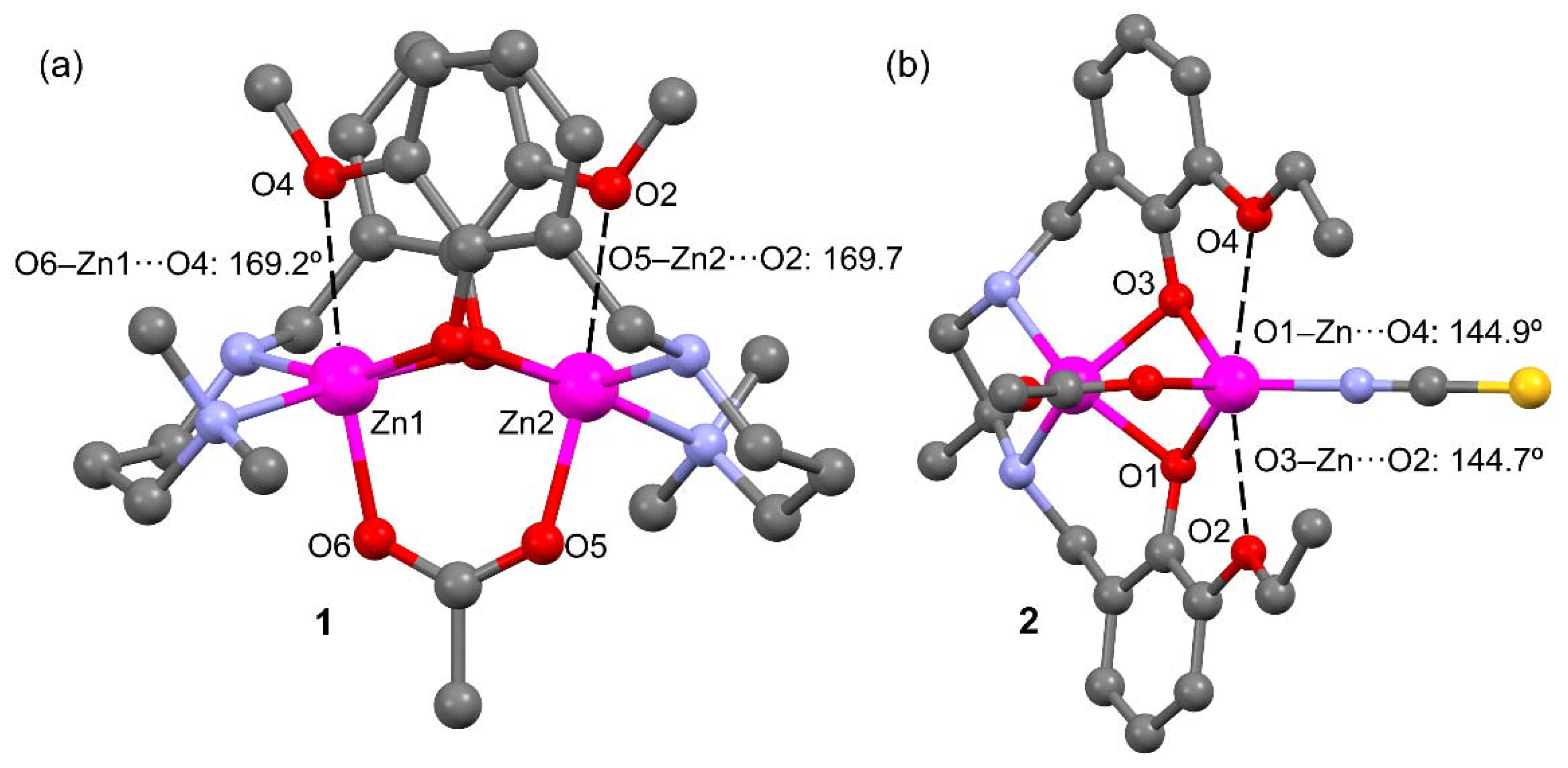
2.2. Description of Compounds 1 and 2
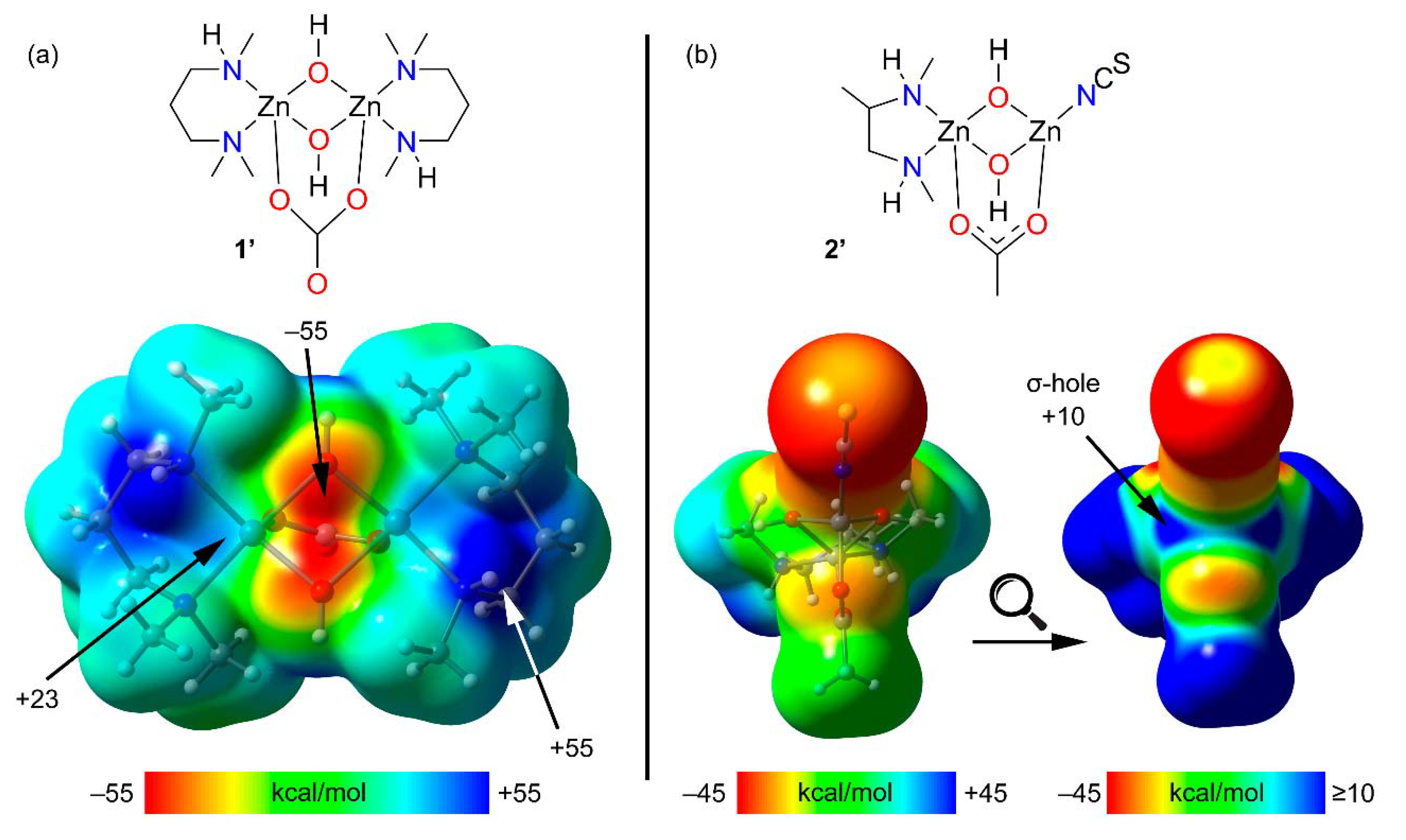
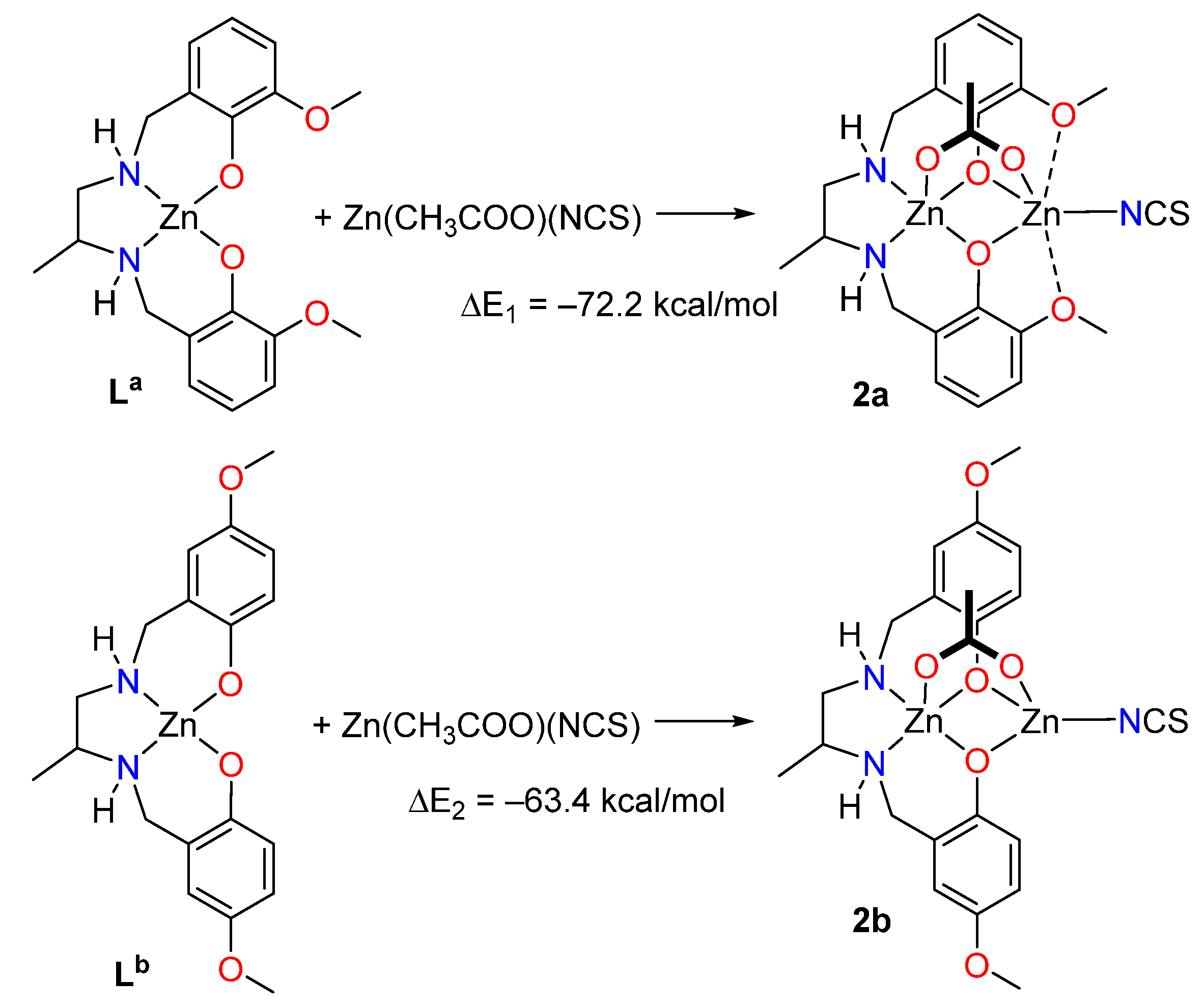
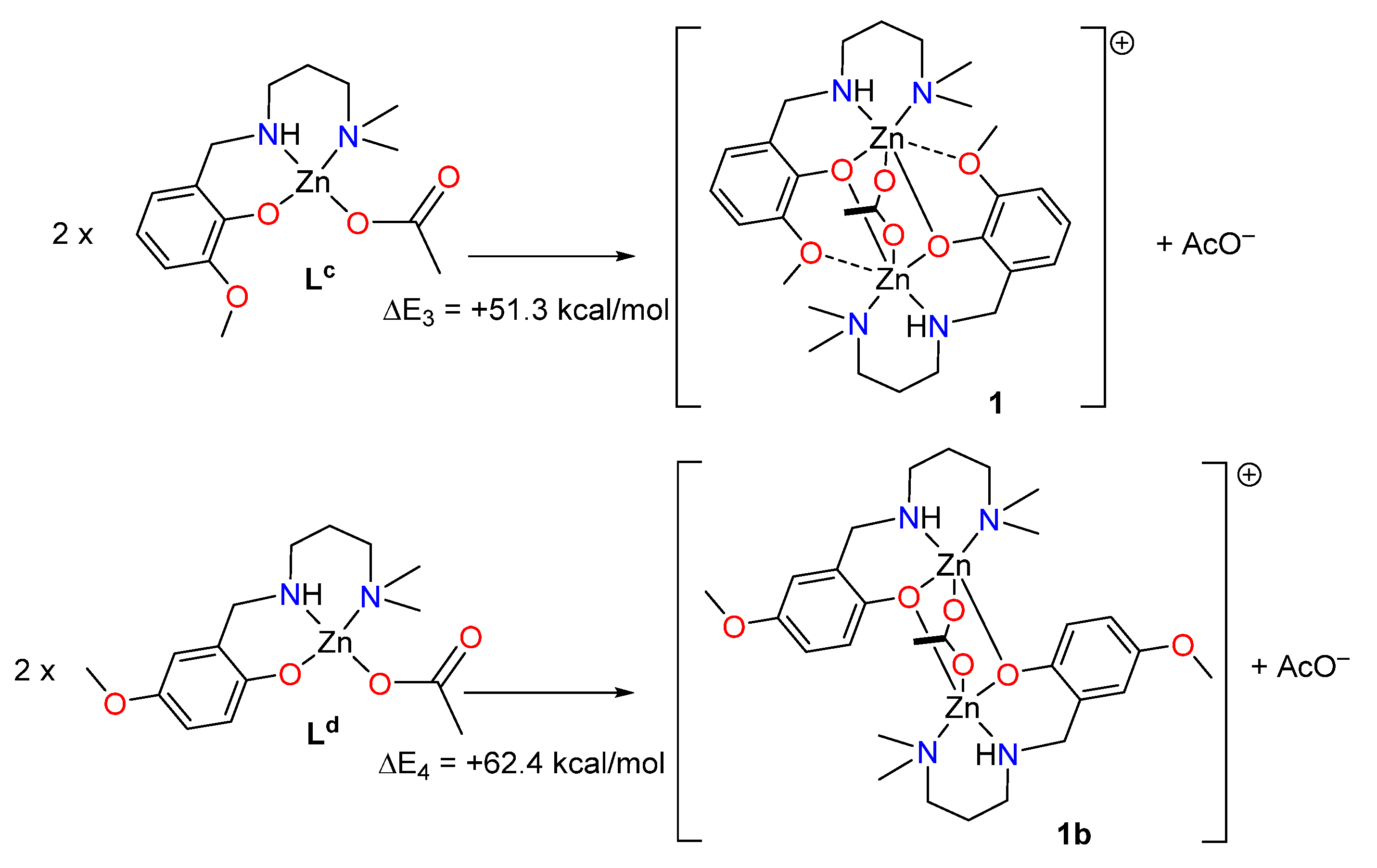
2.3. Theoretical DFT Study
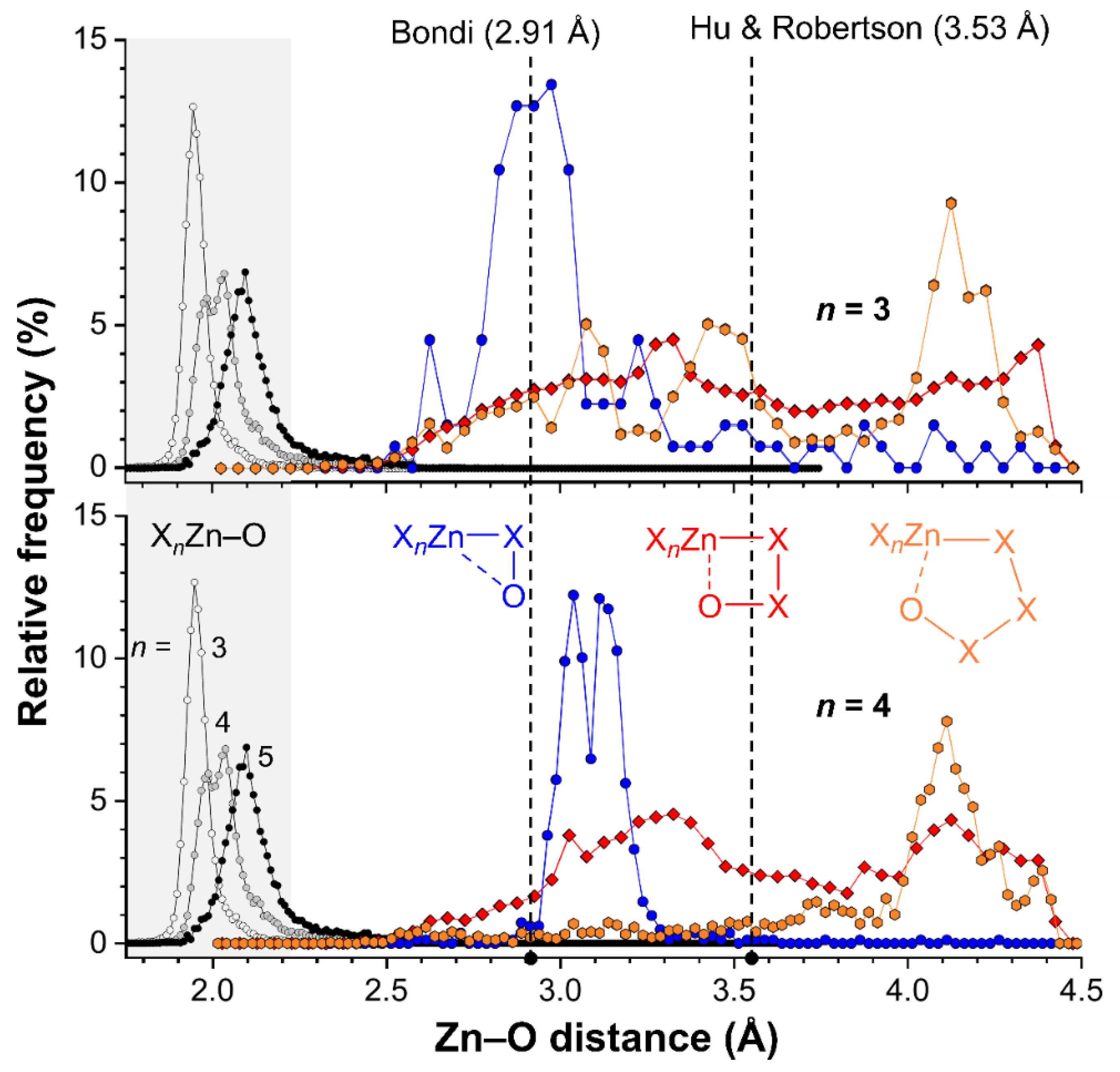
2.4. CSD Search
3. Materials and Methods
3.1. Materials
3.2. Preparation of Reduced Schiff Base Ligands
3.2.1. Preparation of 2-(((3-(dimethylamino)propyl)amino)methyl)-6-methoxyphenol (HL1)
3.2.2. Preparation of 2,2′-[(1-Methyl-1,2-ethanediyl)bis(iminomethylene)]bis[6-ethoxyphenol] (H2L2)
3.2.3. Preparation of the Complex [Zn2(µ1,3-OAc)(L1)2]I·MeOH (1)
3.2.4. Preparation of the Complex [Zn2(µ1,3-OAc)(L2)(NCS)] (2)
3.3. Details of Instrumentation
3.4. Crystal Data Collection and Refinement
3.5. Theoretical Methods
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muller-Dethlefs, K.; Hobza, P. Non-Covalent Interactions: Theory and Experiment. In Theoretical and Computational Chemistry Series, 1st ed.; Hirst, J., Jordan, K.D., Thiel, W., Lim, C., Eds.; RSC Publishing: Cambridge, UK, 2010; Volume 2. [Google Scholar]
- Mahmudov, K.T.; Kopylovich, M.N.; Pombeiro, A.J.L. (Eds.) Noncovalent Interactions in Catalysis, 1st ed.; RSC Publishing: Cambridge, UK, 2019. [Google Scholar]
- Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. (Eds.) Non-covalent Interactions in the Synthesis and Design of New Compounds, 1st ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the sigma-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ and π-hole interactions. Chem. Phys. Chem. 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Joy, J.; Jemmis, E.D. Contrasting Behavior of the Z Bonds in X–Z···Y Weak Interactions: Z = Main Group Elements Versus the Transition Metals. Inorg. Chem. 2017, 56, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Joy, J.; Jemmis, E.D. Designing M-bond (X-M···Y, M = transition metal): σ-hole and radial density distribution. J. Chem. Sci. 2019, 131, 117. [Google Scholar] [CrossRef]
- Bauzá, A.; Alkorta, I.; Elguero, J.; Mooibroek, T.J.; Frontera, A. Spodium Bonds: Noncovalent Interactions Involving Group 12 Elements. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Hazari, A.; Das, L.K.; Kadam, R.M.; Bauzá, A.; Frontera, A.; Ghosh, A. Unprecedented structural variations in trinuclear mixed valence Co(II/III) complexes: Theoretical studies, pnicogen bonding interactions and catecholase-like activities. Dalton Trans. 2015, 44, 3862–3876. [Google Scholar] [CrossRef]
- Banerjee, A.; Frontera, A.; Chattopadhyay, S. Methylene spacer regulated variation in molecular and crystalline architectures of cobalt(iii) complexes with reduced Schiff base ligands: A combined experimental and theoretical study. Dalton Trans. 2019, 48, 11433–11447. [Google Scholar] [CrossRef]
- Mondal, I.; Basak, T.; Banerjee, S.; Chattopadhyay, S. A theoretical insight on the rigid hydrogen-bonded network in the solid state structure of two zinc(ii) complexes and their strong fluorescence behaviors. Cryst. Eng. Comm. 2020, 22, 3005–3019. [Google Scholar] [CrossRef]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Supplement. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d- and f-block metals. J. Chem. Soc. Dalton. Trans. 1989, S1–S83. [Google Scholar] [CrossRef]
- Roeleveld, J.J.; Lekanne Deprez, S.J.; Verhoofstad, A.; Frontera, A.; van der Vlugt, J.I.; Mooibroek, T.J. Engineering Crystals Using sp3-C Centred Tetrel Bonding Interactions. Chem. Eur. J. 2020, 26, 10126–10132. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals volumes and radii of metals in covalent compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Hu, S.-Z.; Zhou, Z.-H.; Robertson, B.E. Consistent approaches to van der Waals radii for the metallic elements. Z. Kristallogr. 2009, 224, 375–383. [Google Scholar] [CrossRef]
- Karmakar, M.; Frontera, A.; Chattopadhyay, S. Methylene spacer regulated variation in supramolecular assembly of zinc(II) dicyanamide complexes with reduced Schiff base ligands: Synthesis, structure and DFT study. Cryst. Eng. Comm. 2020. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, V2014/5, Software for Empirical Absorption Correction; University of Göttingen, Institute fur Anorganische Chemie der Universitat: Gottingen, Germany, 1999. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.; Keith, T.K. (Eds.) AIMAll (Version 13.05.06); Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zelenkov, L.E.; Ivanov, D.M.; Sadykov, E.K.; Bokach, N.A.; Galmés, B.; Frontera, A.; Kukushkin, V.Y. Semicoordination Bond Breaking and Halogen Bond Making Changes the Supramolecular Architecture of Metal-containing Aggregates. Cryst. Growth Des. 2020. [Google Scholar] [CrossRef]
- Thakur, S.; Gil, D.M.; Frontera, A.; Chattopadhyay, S. Exploration of Br⋯O halogen bonding interactions in dinuclear vanadium (V) complexes with Schiff base ligands. Polyhedron 2020, 187, 114676. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ananyev, I.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. π-Hole···dz2 [PtII] Interactions with Electron-Deficient Arenes Enhance the Phosphorescence of PtII-Based Luminophores. Inorg. Chem. 2020, 59, 9308–9314. [Google Scholar]
- Navarro-García, E.; Galmés, B.; Velasco, M.D.; Frontera, A.; Caballero, A. Anion Recognition by Neutral Chalcogen Bonding Receptors: Experimental and Theoretical Investigations. Chem. Eur. J. 2020, 26, 4706–4713. [Google Scholar] [CrossRef]
- Galmés, B.; Juan-Bals, A.; Frontera, A.; Resnati, G. Charge assisted chalcogen bonds: CSD and DFT analyses and biological implication in glucosidase inhibitors. Chem. Eur. J. 2020, 26, 4599–4606. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Haukka, M.; Levin, O.V.; Frontera, A.; Kukushkin, V.Y. Supramolecular Assembly of Metal Complexes by (Aryl) I⋯dz2[PtII] Halogen Bond. Chem. Eur. J. 2020, 26, 7692–7701. [Google Scholar] [CrossRef]
- Bofill, L.; Prohens, R.; Barbas, R.; Frontera, A. DFT analysis of uncommon π···H-bond array interaction in a new pterostilbene/theophylline cocrystal. Cryst. Growth Des. 2020. [Google Scholar] [CrossRef]
- Soldatova, N.S.; Postnikov, P.S.; Suslonov, V.V.; Kissler, T.Y.; Ivanov, D.M.; Yusubov, M.S.; Galmés., B.; Frontera, A.; Kukushkin, V.Y. Diaryliodonium as a double σ-hole donor: The dichotomy of thiocyanate halogen bonding provides divergent solid state arylation by diaryliodonium cations. Org. Chem. Front. 2020, 7, 2230–2242. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | |||
| Zn1–N1 (coord.) 1 | 2.169(5) | Zn2–N3 (coord.) | 2.160(5) |
| Zn1–N2 (coord.) | 2.074(4) | Zn2–N4 (coord.) | 2.085(4) |
| Zn1–O1 (coord.) 2 | 2.212(4) | Zn2–O1 (coord.) | 1.983(4) |
| Zn1–O3 (coord.) | 1.977(3) | Zn2–O3 (coord.) | 2.165(3) |
| Zn1–O6 (coord.) | 2.051(4) | Zn2–O5 (coord.) | 2.075(4) |
| Zn1–O4 (SpB) | 2.688(5) | Zn2–O2 (SpB) | 2.667(4) |
| Zn1···Zn2 | 3.047(1) | ||
| 2 | |||
| Zn1–N1 (coord.) | 2.091(2) | Zn2–N3 (coord.)a | 1.922(2) |
| Zn1–N2 (coord.) | 2.099(2) | Zn2–O1 (coord.) | 2.017(1) |
| Zn1–O1 (coord.) | 2.058(2) | Zn2–O3 (coord.) | 2.007(2) |
| Zn1–O3 (coord.) | 2.042(2) | Zn2–O6 (coord.) | 1.976(2) |
| Zn1–O5 (coord.) | 1.978(2) | Zn2–O2 (SpB) | 2.692(2) |
| Zn1···Zn2 | 2.9025(5) | Zn2–O4 (SpB) | 2.664(2) |
| CP | ρ(r) | ∇2ρ(r) | V(r) | G(r) | H(r) | DI |
|---|---|---|---|---|---|---|
| 1 | ||||||
| a | 0.017 | 0.055 | −0.014 | 0.014 | 0.000 | 0.072 |
| b | 0.018 | 0.057 | −0.015 | 0.015 | 0.000 | 0.075 |
| c | 0.081 | 0.428 | −0.136 | 0.121 | −0.015 | 0.370 |
| d | 0.076 | 0.313 | −0.107 | 0.093 | −0.014 | 0.394 |
| 2 | ||||||
| a | 0.016 | 0.055 | −0.014 | 0.014 | 0.000 | 0.059 |
| b | 0.017 | 0.059 | −0.015 | 0.015 | 0.000 | 0.064 |
| c | 0.073 | 0.388 | −0.117 | 0.107 | −0.010 | 0.327 |
| d | 0.099 | 0.475 | −0.171 | 0.145 | −0.026 | 0.510 |
| Entry | Search | CIFs | Hits |
|---|---|---|---|
| 1a | SpT4X4 | 22,055 | |
| 1b | ⮡ X3ZnT4–O | 9557 | 37,255 |
| 1ca | ⮡ X3ZnT4–X–O (2 bonds) | 62 | 134 |
| 1d a,b | ⮡ X3ZnT4–X–X–O (3 bonds) | 7880 | 31,120 |
| 1ea | ⮡ X3ZnT4–X–X–X–O (4 bonds) | 724 | 2123 |
| 2a | SpT5X5 | 22,449 | |
| 2b | ⮡ X4ZnT5–O | 8649 | 36,519 |
| 2ca | ⮡ X4ZnT5–X–O (2 bonds) | 240 | 818 |
| 2da,c | ⮡ X4ZnT5–X–X–O (3 bonds) | 6377 | 21,227 |
| 2ea | ⮡ X4ZnT5–X–X–X–O (4 bonds) | 1136 | 2463 |
| 3a | SpT6X6 | 13,505 | |
| 3b | ⮡ X5ZnT6–O | 10,347 | 44,770 |
| Complex | 1 | 2 |
|---|---|---|
| Formula | C29H49N4O7Zn2I | C24H31N3O6SZn2 |
| Formula Weight | 823.40 | 620.37 |
| Temperature (K) | 273(2) | 273(2) |
| Crystal System | Orthorhombic | Triclinic |
| Space group | P212121 | P |
| a (Å) | 12.744(6) | 10.5900(8) |
| b (Å) | 16.260(7) | 11.7781(9) |
| c (Å) | 17.008(8) | 12.0470(9) |
| β (°) | 90 | 90.588(2) |
| β (°) | 90 | 101.556(2) |
| γ (°) | 90 | 109.376(2) |
| Z | 4 | 2 |
| dcal (g cm−3) | 1.552 | 1.489 |
| μ(mm−1) | 2.284 | 1.850 |
| F(000) | 1680.0 | 640 |
| Total reflection | 29800 | 49447 |
| Unique Reflections | 7136 | 6157 |
| Observe data[I>2σ(I)] | 6500 | 5165 |
| R(int) | 0.052 | 0.032 |
| R1, wR2 (all data) | 0.0392, 0.0675 | 0.0408, 0.1052 |
| R1, wR2 [I>2σ(I)] | 0.0338, 0.0655 | 0.0308, 0.0922 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karmakar, M.; Frontera, A.; Chattopadhyay, S.; Mooibroek, T.J.; Bauzá, A. Intramolecular Spodium Bonds in Zn(II) Complexes: Insights from Theory and Experiment. Int. J. Mol. Sci. 2020, 21, 7091. https://doi.org/10.3390/ijms21197091
Karmakar M, Frontera A, Chattopadhyay S, Mooibroek TJ, Bauzá A. Intramolecular Spodium Bonds in Zn(II) Complexes: Insights from Theory and Experiment. International Journal of Molecular Sciences. 2020; 21(19):7091. https://doi.org/10.3390/ijms21197091
Chicago/Turabian StyleKarmakar, Mainak, Antonio Frontera, Shouvik Chattopadhyay, Tiddo J. Mooibroek, and Antonio Bauzá. 2020. "Intramolecular Spodium Bonds in Zn(II) Complexes: Insights from Theory and Experiment" International Journal of Molecular Sciences 21, no. 19: 7091. https://doi.org/10.3390/ijms21197091
APA StyleKarmakar, M., Frontera, A., Chattopadhyay, S., Mooibroek, T. J., & Bauzá, A. (2020). Intramolecular Spodium Bonds in Zn(II) Complexes: Insights from Theory and Experiment. International Journal of Molecular Sciences, 21(19), 7091. https://doi.org/10.3390/ijms21197091