Increase in Toxicity of Anticancer Drugs by PMTPV, a Claudin-1-Binding Peptide, Mediated via Down-Regulation of Claudin-1 in Human Lung Adenocarcinoma A549 Cells


Abstract
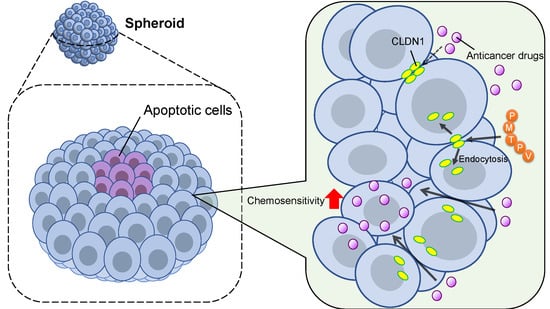
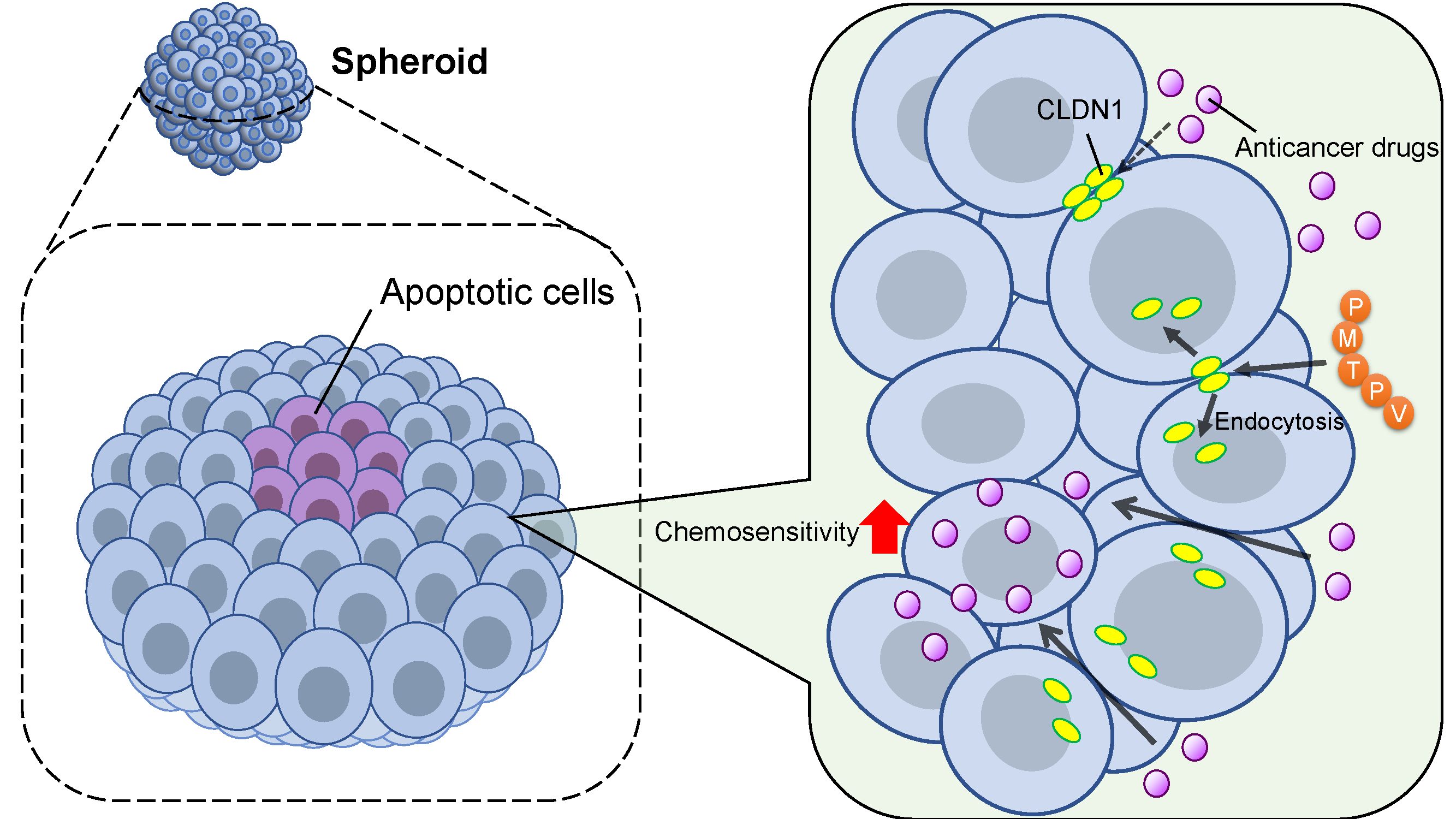{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
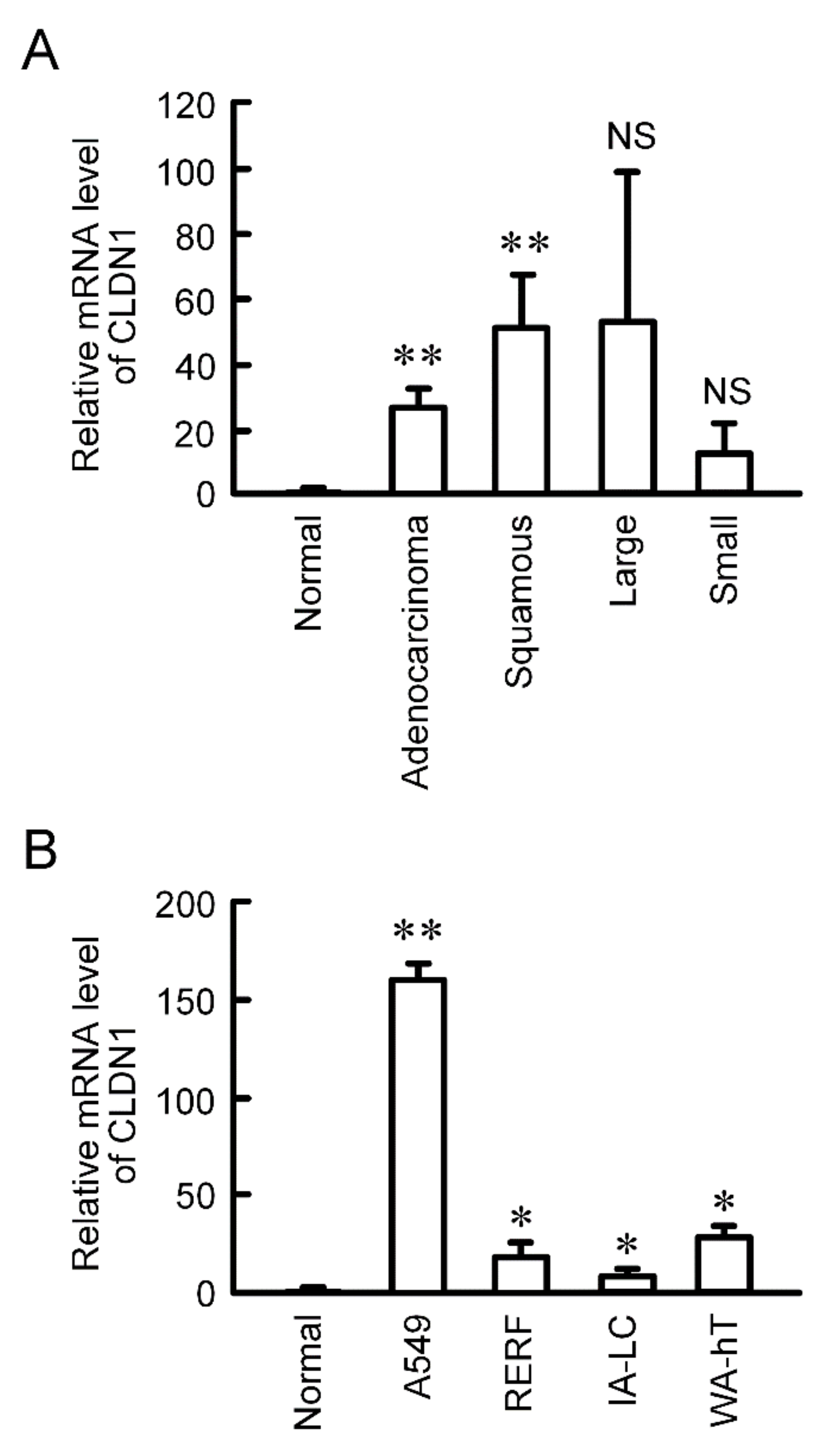
2.1. Expression of CLDN1 in Lung Tissues
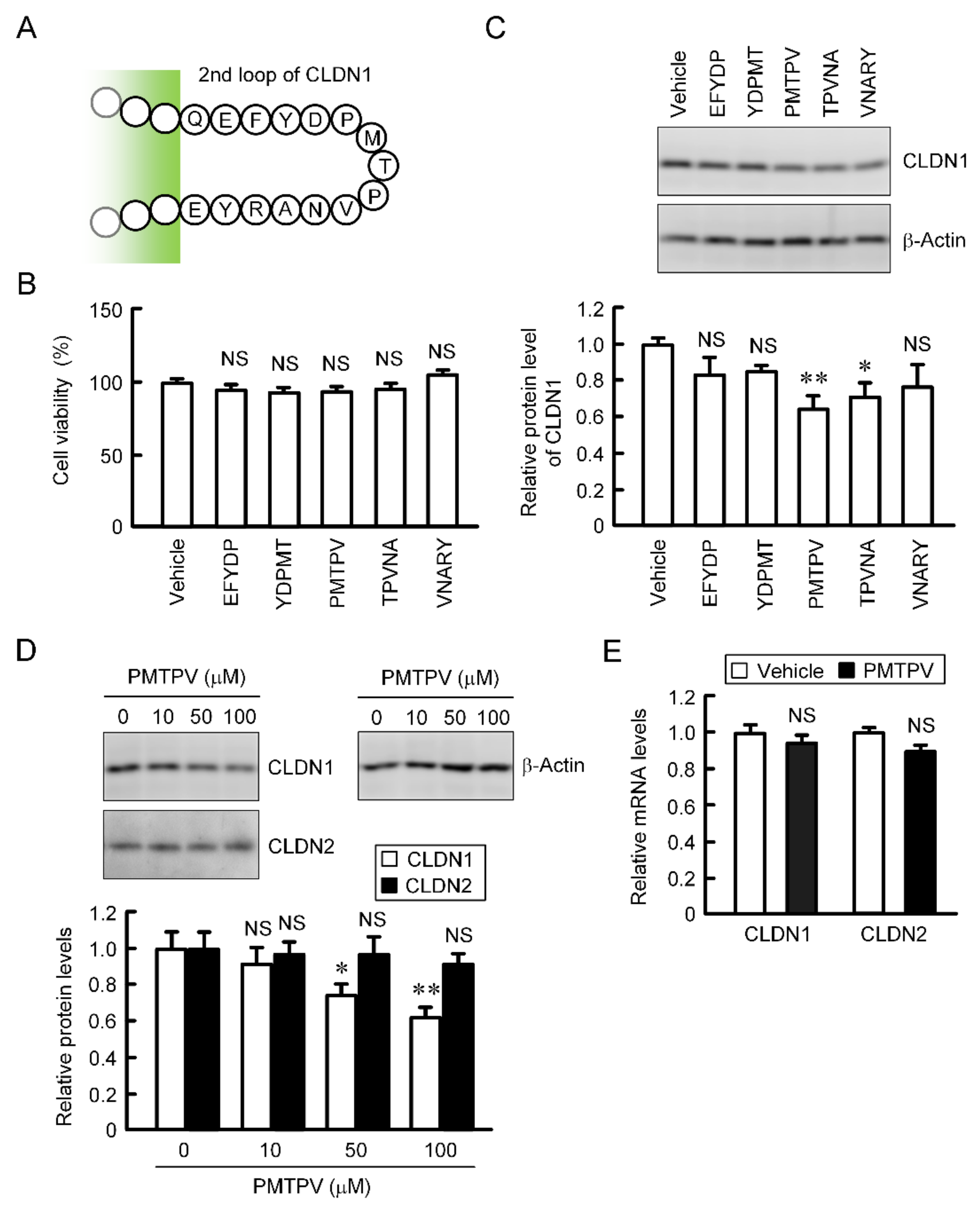
2.2. Decrease in Protein Level of CLDN1 by Short Peptides
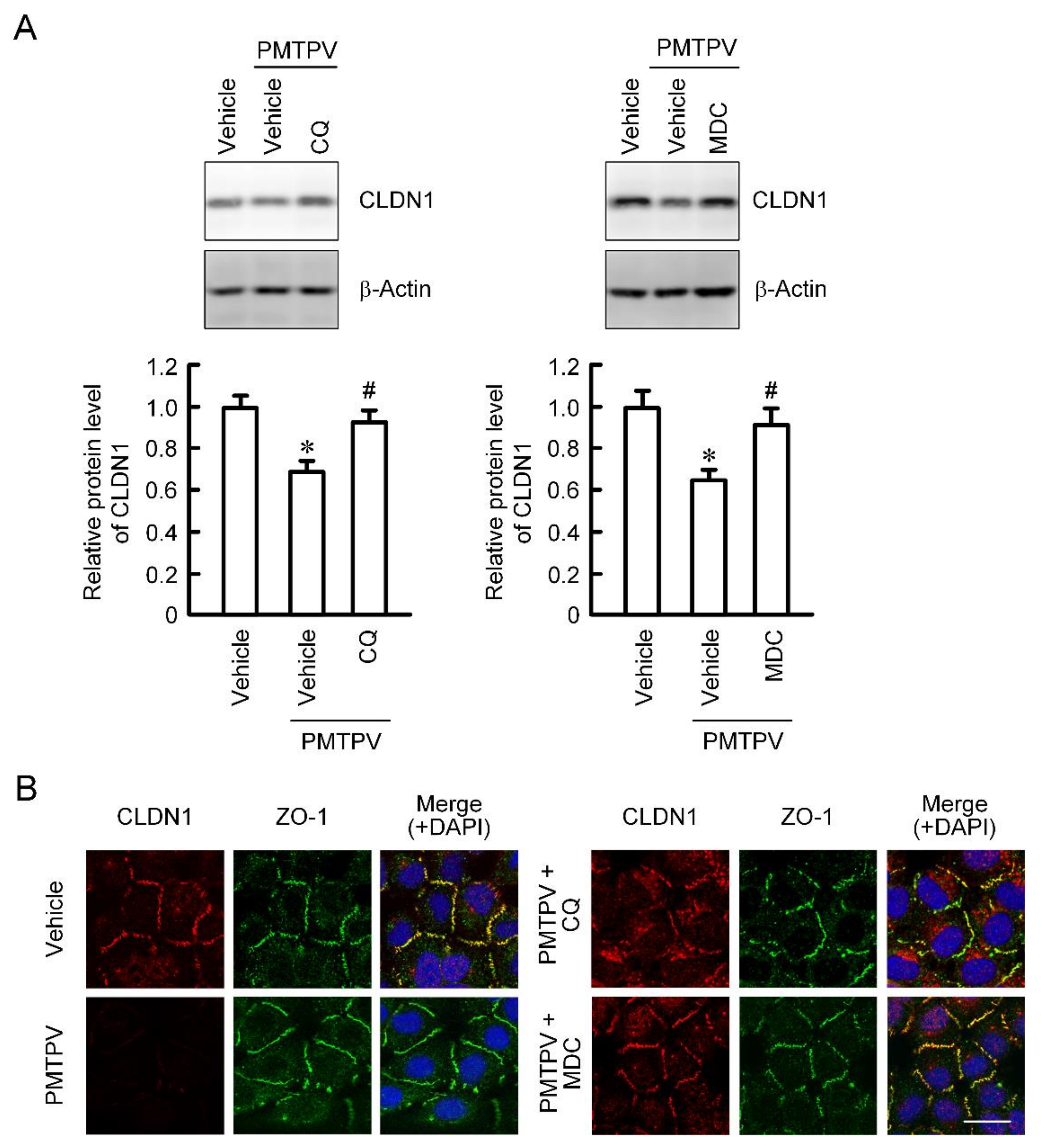
2.3. Rescue of PMTPV-Induced Decrease in CLDN1 Expression by Endocytosis and Lysosome Inhibitors
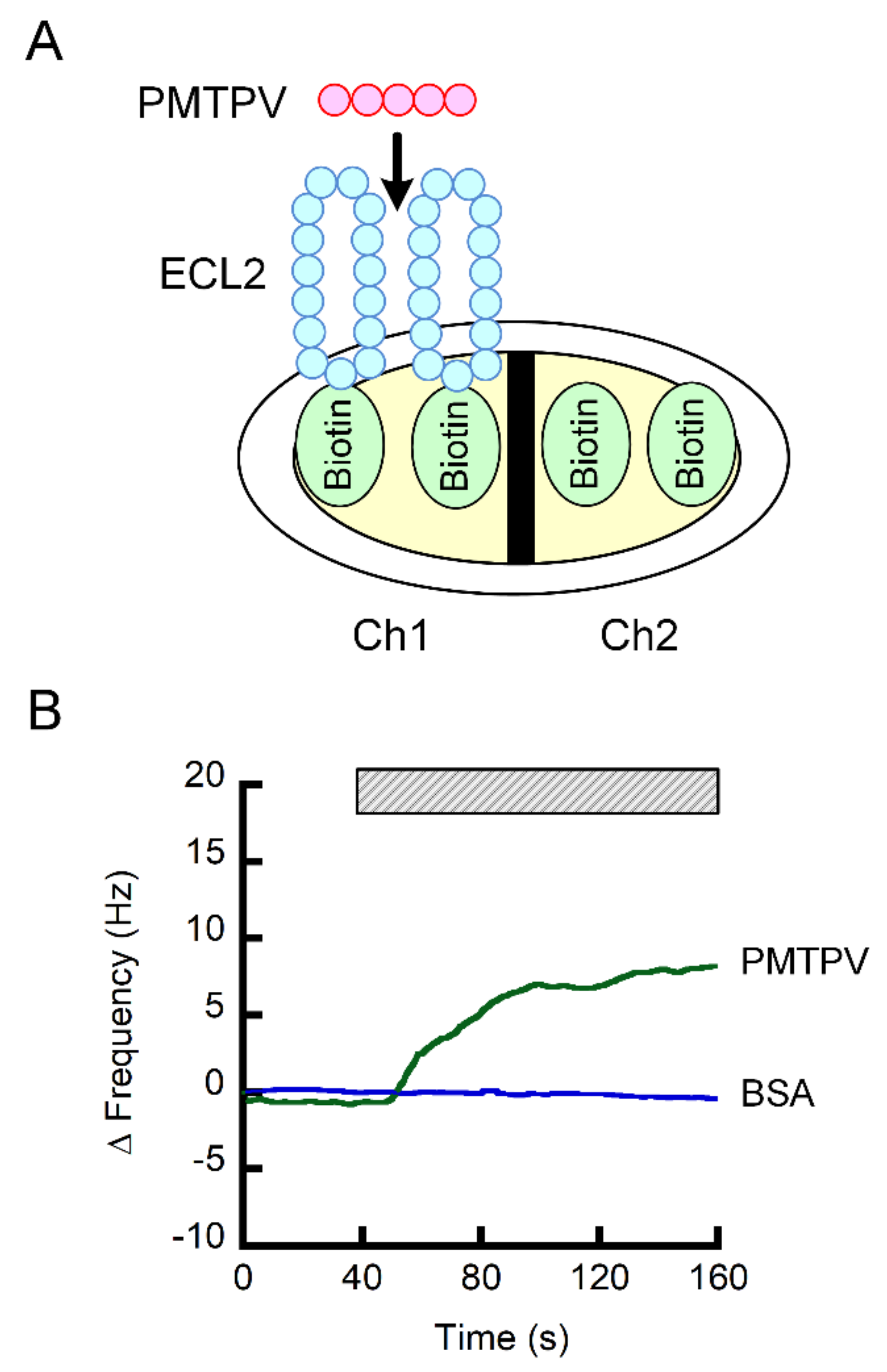
2.4. Interaction of PMTPV With ELC2 of CLDN1
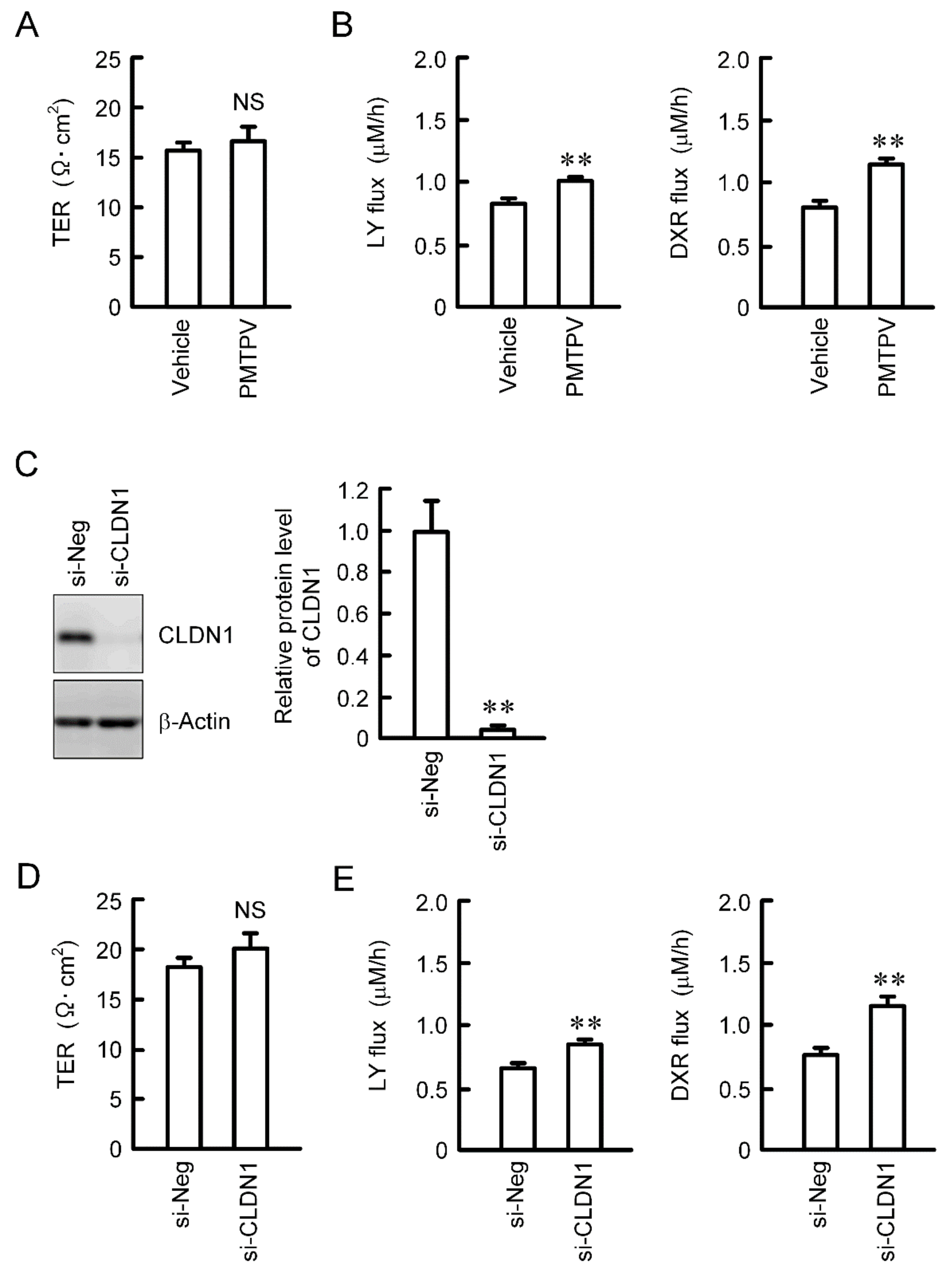
2.5. Effects of PMTPV and CLDN1 siRNA on Paracellular Permeability
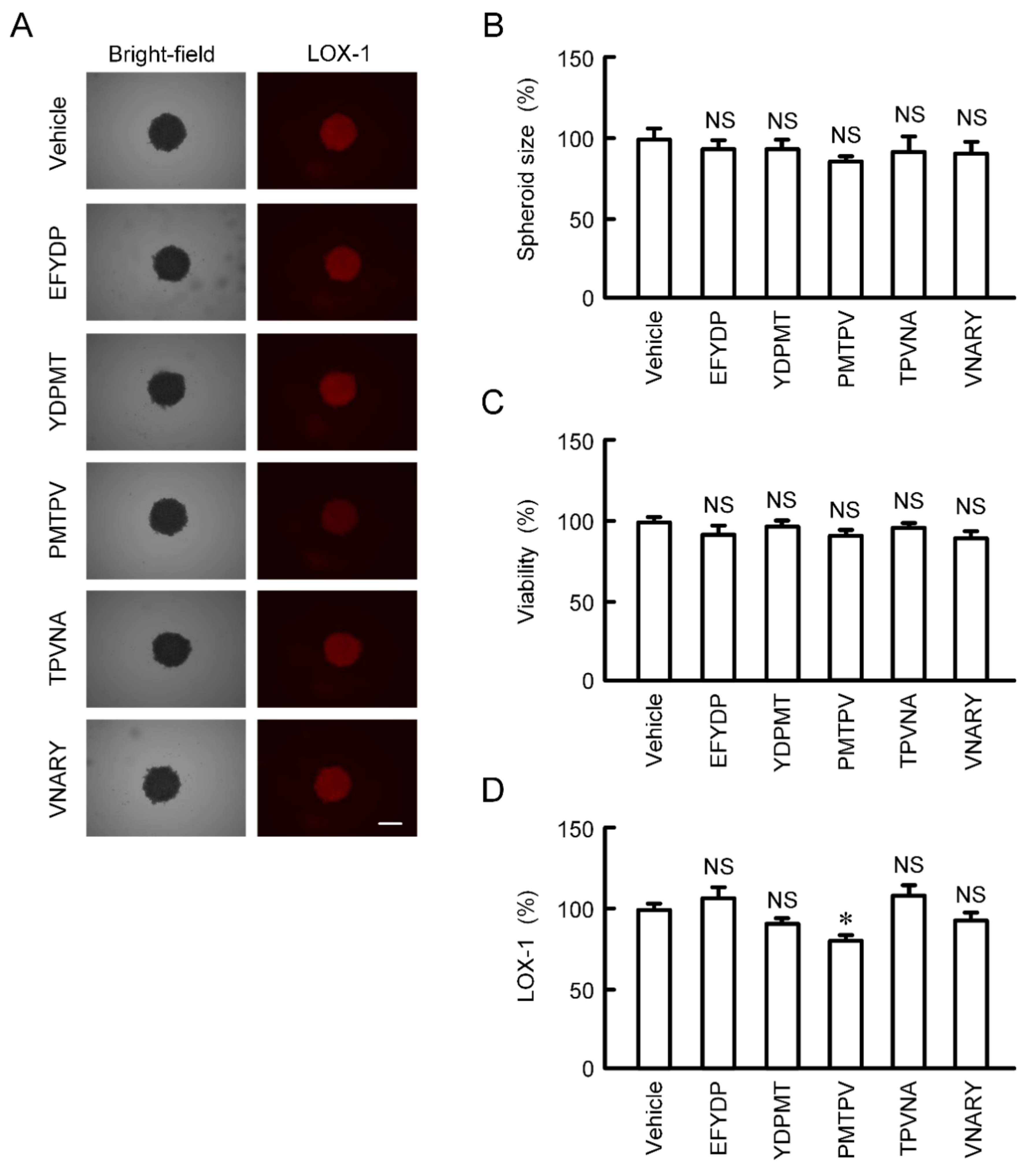
2.6. Effects of Peptides on the Characteristics of 3D Culture Model
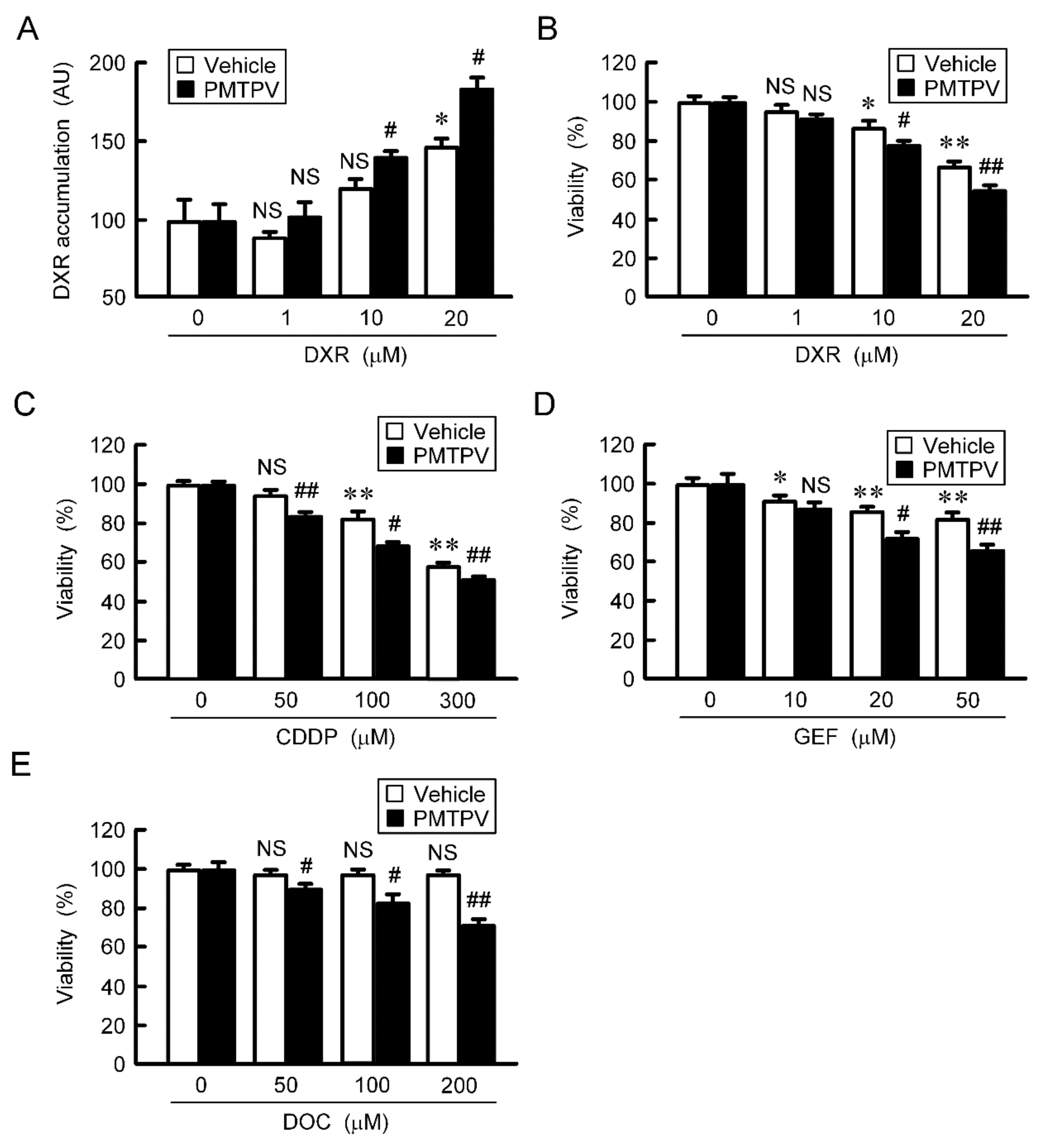
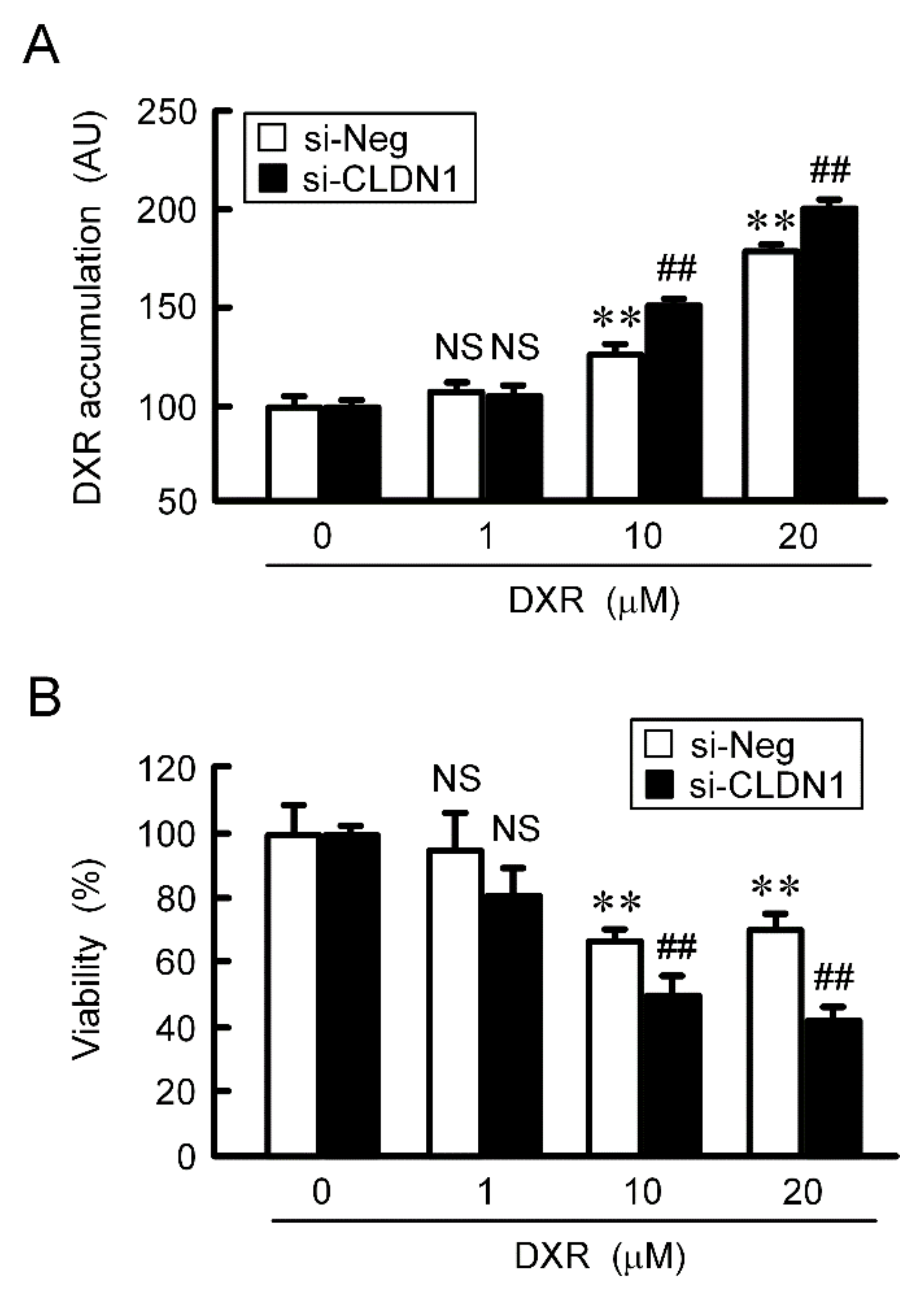
2.7. Rescue of Toxicity to Anticancer Drugs by PMTPV and CLDN1 siRNA in Spheroids
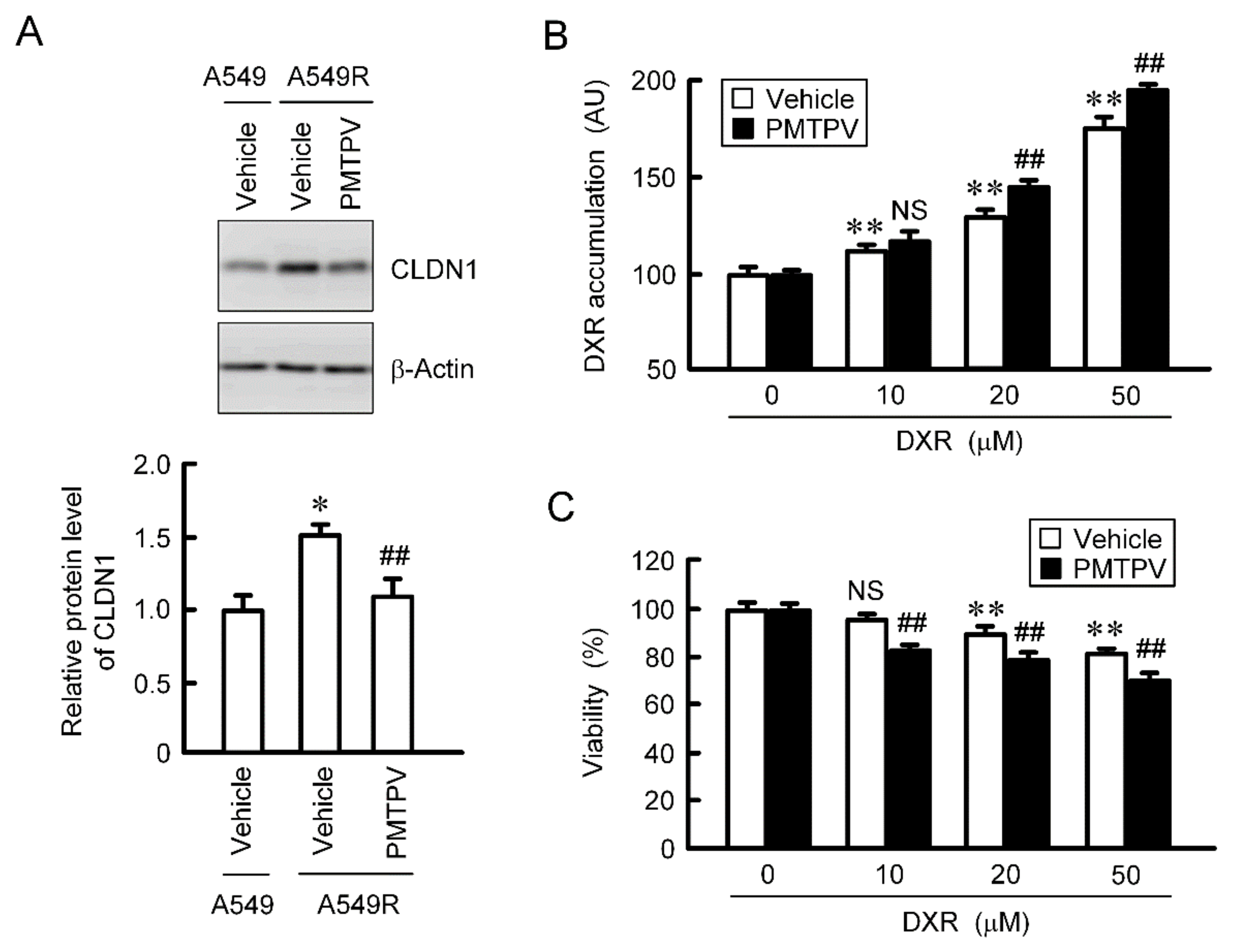
2.8. Rescue of Toxicity to Anticancer Drugs by PMTPV in the CDDP-Resistant Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Transfection
4.3. Western Blotting
4.4. Quantitative Reverse Transcription-PCR
4.5. Immunofluorescence Measurement
4.6. QCM Analysis
4.7. Paracellular Flux Assay
4.8. 3D Spheroid Model
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A549R | CDDP-resistant A549 |
| AU | arbitrary units |
| BSA | bovine serum albumin |
| cCPE | carboxyl-terminal fragment of the bacterial Clostridium perfringens enterotoxin |
| CDDP | cisplatin |
| CLDN | claudin |
| CQ | chloroquine |
| 2D | two-dimensional |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DOC | docetaxel |
| DXR | doxorubicin |
| ECL | extracellular loop |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial mesenchymal transition |
| GEF | gefitinib |
| HIF-1 | hypoxia inducible factor-1 |
| LY | Lucifer yellow |
| MDC | monodansylcadaverine |
| NSCLC | non-small cell lung cancer |
| PCR | polymerase chain reaction |
| QCM | quartz crystal microbalance |
| siRNA | small interfering RNA |
| TKI | tyrosine kinase inhibitor |
| TJs | tight junctions |
References
- Johnson, A.M.; Hines, R.B.; Johnson, J.A., 3rd; Bayakly, A.R. Treatment and survival disparities in lung cancer: The effect of social environment and place of residence. Lung Cancer 2014, 83, 401–407. [Google Scholar] [CrossRef]
- Boulikas, T.; Vougiouka, M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs. Oncol. Rep. 2004, 11, 559–595. [Google Scholar] [CrossRef]
- Stewart, L.A. Non-small Cell Lung Cancer Collaborative Group. Chemotherapy in non-small cell lung cancer: A meta-analysis using updated data on individual patients from 52 randomised clinical trials. BMJ 1995, 311, 899–909. [Google Scholar]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; Siegel, P.M. The role of claudins in cancer metastasis. Oncogene 2017, 36, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Lu, Z.; Lu, Q.; Chen, Y.H. The claudin family of proteins in human malignancy: A clinical perspective. Cancer Manag. Res. 2013, 5, 367–375. [Google Scholar] [PubMed]
- Che, J.; Yang, Y.; Xiao, J.; Zhao, P.; Yan, B.; Dong, S.; Cao, B. Decreased expression of claudin-3 is associated with a poor prognosis and EMT in completely resected squamous cell lung carcinoma. Tumor Biol. 2015, 36, 6559–6568. [Google Scholar] [CrossRef] [PubMed]
- Shimobaba, S.; Taga, S.; Amizuki, R.; Hichino, A.; Endo, S.; Matsunaga, T.; Watanabe, R.; Yamaguchi, M.; Yamazaki, Y.; Sugatani, J.; et al. Claudin-18 inhibits cell proliferation and motility mediated by inhibition of phosphorylation of PDK1 and Akt in human lung adenocarcinoma A549 cells. Biochim. Biophys. Acta 2016, 1863, 1170–1178. [Google Scholar] [CrossRef]
- Kinugasa, T.; Akagi, Y.; Yoshida, T.; Ryu, Y.; Shiratuchi, I.; Ishibashi, N.; Shirouzu, K. Increased claudin-1 protein expression contributes to tumorigenesis in ulcerative colitis-associated colorectal cancer. Anticancer Res. 2010, 30, 3181–3186. [Google Scholar]
- Wu, Y.L.; Zhang, S.; Wang, G.R.; Chen, Y.P. Expression transformation of claudin-1 in the process of gastric adenocarcinoma invasion. World J. Gastroenterol. 2008, 14, 4943–4948. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, J.; Nemeth, Z.; Tatrai, P.; Peter, I.; Somoracz, A.; Szasz, A.M.; Kiss, A.; Schaff, Z. High expression of claudin-1 protein in papillary thyroid tumor and its regional lymph node metastasis. Pathol. Oncol. Res. 2010, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Akizuki, R.; Maruhashi, R.; Eguchi, H.; Kitabatake, K.; Tsukimoto, M.; Furuta, T.; Matsunaga, T.; Endo, S.; Ikari, A. Decrease in paracellular permeability and chemosensitivity to doxorubicin by claudin-1 in spheroid culture models of human lung adenocarcinoma A549 cells. Biochim. Biophys. Acta 2018, 1865, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Lian, H.; Su, M.; Zhu, Y.; Zhou, Y.; Soomro, S.H.; Fu, H. Protein kinase CK2, a potential therapeutic target in carcinoma management. Asian Pac. J. Cancer Prev. 2019, 20, 23–32. [Google Scholar] [CrossRef]
- Singla, N.; Ghandour, R.A.; Raj, G.V. Investigational luteinizing hormone releasing hormone (LHRH) agonists and other hormonal agents in early stage clinical trials for prostate cancer. Expert Opin. Investig. Drugs 2019, 28, 249–259. [Google Scholar] [CrossRef]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Rae Bressler, L.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef]
- Baumgartner, H.K.; Beeman, N.; Hodges, R.S.; Neville, M.C. A D-peptide analog of the second extracellular loop of claudin-3 and -4 leads to mislocalized claudin and cellular apoptosis in mammary epithelial cells. Chem. Biol. Drug Des. 2011, 77, 124–136. [Google Scholar] [CrossRef]
- Nasako, H.; Akizuki, R.; Takashina, Y.; Ishikawa, Y.; Shinoda, T.; Shirouzu, M.; Asai, T.; Matsunaga, T.; Endo, S.; Ikari, A. Claudin-2 binding peptides, VPDSM and DSMKF, down-regulate claudin-2 expression and anticancer resistance in human lung adenocarcinoma A549 cells. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118642. [Google Scholar] [CrossRef]
- Kaarteenaho, R.; Merikallio, H.; Lehtonen, S.; Harju, T.; Soini, Y. Divergent expression of claudin -1, -3, -4, -5 and -7 in developing human lung. Respir. Res. 2010, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Taga, S.; Watanabe, R.; Sato, T.; Shimobaba, S.; Sonoki, H.; Endo, S.; Matsunaga, T.; Sakai, H.; Yamaguchi, M.; et al. Clathrin-dependent endocytosis of claudin-2 by DFYSP peptide causes lysosomal damage in lung adenocarcinoma A549 cells. Biochim. Biophys. Acta 2015, 1848, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Syed, N.; Therachiyil, L.; Nisar, S.; Hashem, S.; Macha, M.A.; Yadav, S.K.; Krishnankutty, R.; Muralitharan, S.; Al-Naemi, H.; et al. Claudin-1, A Double-Edged Sword in Cancer. Int. J. Mol. Sci. 2020, 21, 569. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Sharma, A.; Smith, J.J.; Krishnan, M.; Chen, X.; Eschrich, S.; Washington, M.K.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. Claudin-1 up-regulates the repressor ZEB-1 to inhibit E-cadherin expression in colon cancer cells. Gastroenterology 2011, 141, 2140–2153. [Google Scholar] [CrossRef]
- Higashi, Y.; Suzuki, S.; Sakaguchi, T.; Nakamura, T.; Baba, S.; Reinecker, H.C.; Nakamura, S.; Konno, H. Loss of claudin-1 expression correlates with malignancy of hepatocellular carcinoma. J. Surg. Res. 2007, 139, 68–76. [Google Scholar] [CrossRef]
- Sun, B.S.; Yao, Y.Q.; Pei, B.X.; Zhang, Z.F.; Wang, C.L. Claudin-1 correlates with poor prognosis in lung adenocarcinoma. Thorac. Cancer 2016, 7, 556–563. [Google Scholar] [CrossRef]
- Raghav, K.P.; Gonzalez-Angulo, A.M.; Blumenschein, G.R., Jr. Role of HGF/MET axis in resistance of lung cancer to contemporary management. Transl. Lung Cancer Res. 2012, 1, 179–193. [Google Scholar]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell. Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef]
- Ng, I.K.; Kumarakulasinghe, N.B.; Syn, N.L.; Soo, R.A. Development, internal validation and calibration of a risk score to predict survival in patients with EGFR-mutant non-small cell lung cancer. J. Clin. Pathol. 2020. [Google Scholar] [CrossRef]
- Sekine, I.; Yamamoto, N.; Nishio, K.; Saijo, N. Emerging ethnic differences in lung cancer therapy. Br. J. Cancer 2008, 99, 1757–1762. [Google Scholar] [CrossRef]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Klaassen, C.D. Structure, function, expression, genomic organization, and single nucleotide polymorphisms of human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) efflux transporters. Int. J. Toxicol. 2006, 25, 231–259. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.D.; Stratford, I.; Harris, A.L. The role of hypoxia-inducible factor-1 in three-dimensional tumor growth, apoptosis, and regulation by the insulin-signaling pathway. Cancer Res. 2005, 65, 4147–4152. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, J.; Jiang, Y.; Xu, W.; Li, X.; Jing, W. CLDN1 Increases drug resistance of non-small cell lung cancer by activating autophagy via up-regulation of ULK1 phosphorylation. Med. Sci. Monit. 2017, 23, 2906–2916. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Shin, J.H.; Shim, H.S.; Lee, C.Y.; Kim, D.J.; Kim, Y.S.; Chung, K.Y. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir. Res. 2015, 16, 138. [Google Scholar] [CrossRef]
- Piontek, J.; Krug, S.M.; Protze, J.; Krause, G.; Fromm, M. Molecular architecture and assembly of the tight junction backbone. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183279. [Google Scholar] [CrossRef]
- Nakamura, S.; Irie, K.; Tanaka, H.; Nishikawa, K.; Suzuki, H.; Saitoh, Y.; Tamura, A.; Tsukita, S.; Fujiyoshi, Y. Morphologic determinant of tight junctions revealed by claudin-3 structures. Nat. Commun. 2019, 10, 816. [Google Scholar] [CrossRef]
- Vecchio, A.J.; Stroud, R.M. Claudin-9 structures reveal mechanism for toxin-induced gut barrier breakdown. Proc. Natl. Acad. Sci. USA 2019, 116, 17817–17824. [Google Scholar] [CrossRef]
- Suzuki, H.; Nishizawa, T.; Tani, K.; Yamazaki, Y.; Tamura, A.; Ishitani, R.; Dohmae, N.; Tsukita, S.; Nureki, O.; Fujiyoshi, Y. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 2014, 344, 304–307. [Google Scholar] [CrossRef]
- Shinoda, T.; Shinya, N.; Ito, K.; Ohsawa, N.; Terada, T.; Hirata, K.; Kawano, Y.; Yamamoto, M.; Kimura-Someya, T.; Yokoyama, S.; et al. Structural basis for disruption of claudin assembly in tight junctions by an enterotoxin. Sci. Rep. 2016, 6, 33632. [Google Scholar] [CrossRef]
- Saitoh, Y.; Suzuki, H.; Tani, K.; Nishikawa, K.; Irie, K.; Ogura, Y.; Tamura, A.; Tsukita, S.; Fujiyoshi, Y. Tight junctions. Structural insight into tight junction disassembly by Clostridium perfringens enterotoxin. Science 2015, 347, 775–778. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasako, H.; Takashina, Y.; Eguchi, H.; Ito, A.; Ishikawa, Y.; Matsunaga, T.; Endo, S.; Ikari, A. Increase in Toxicity of Anticancer Drugs by PMTPV, a Claudin-1-Binding Peptide, Mediated via Down-Regulation of Claudin-1 in Human Lung Adenocarcinoma A549 Cells. Int. J. Mol. Sci. 2020, 21, 5909. https://doi.org/10.3390/ijms21165909
Nasako H, Takashina Y, Eguchi H, Ito A, Ishikawa Y, Matsunaga T, Endo S, Ikari A. Increase in Toxicity of Anticancer Drugs by PMTPV, a Claudin-1-Binding Peptide, Mediated via Down-Regulation of Claudin-1 in Human Lung Adenocarcinoma A549 Cells. International Journal of Molecular Sciences. 2020; 21(16):5909. https://doi.org/10.3390/ijms21165909
Chicago/Turabian StyleNasako, Haruka, Yui Takashina, Hiroaki Eguchi, Ayaka Ito, Yoshinobu Ishikawa, Toshiyuki Matsunaga, Satoshi Endo, and Akira Ikari. 2020. "Increase in Toxicity of Anticancer Drugs by PMTPV, a Claudin-1-Binding Peptide, Mediated via Down-Regulation of Claudin-1 in Human Lung Adenocarcinoma A549 Cells" International Journal of Molecular Sciences 21, no. 16: 5909. https://doi.org/10.3390/ijms21165909
APA StyleNasako, H., Takashina, Y., Eguchi, H., Ito, A., Ishikawa, Y., Matsunaga, T., Endo, S., & Ikari, A. (2020). Increase in Toxicity of Anticancer Drugs by PMTPV, a Claudin-1-Binding Peptide, Mediated via Down-Regulation of Claudin-1 in Human Lung Adenocarcinoma A549 Cells. International Journal of Molecular Sciences, 21(16), 5909. https://doi.org/10.3390/ijms21165909