Tilivalline- and Tilimycin-Independent Effects of Klebsiella oxytoca on Tight Junction-Mediated Intestinal Barrier Impairment

 ,
,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
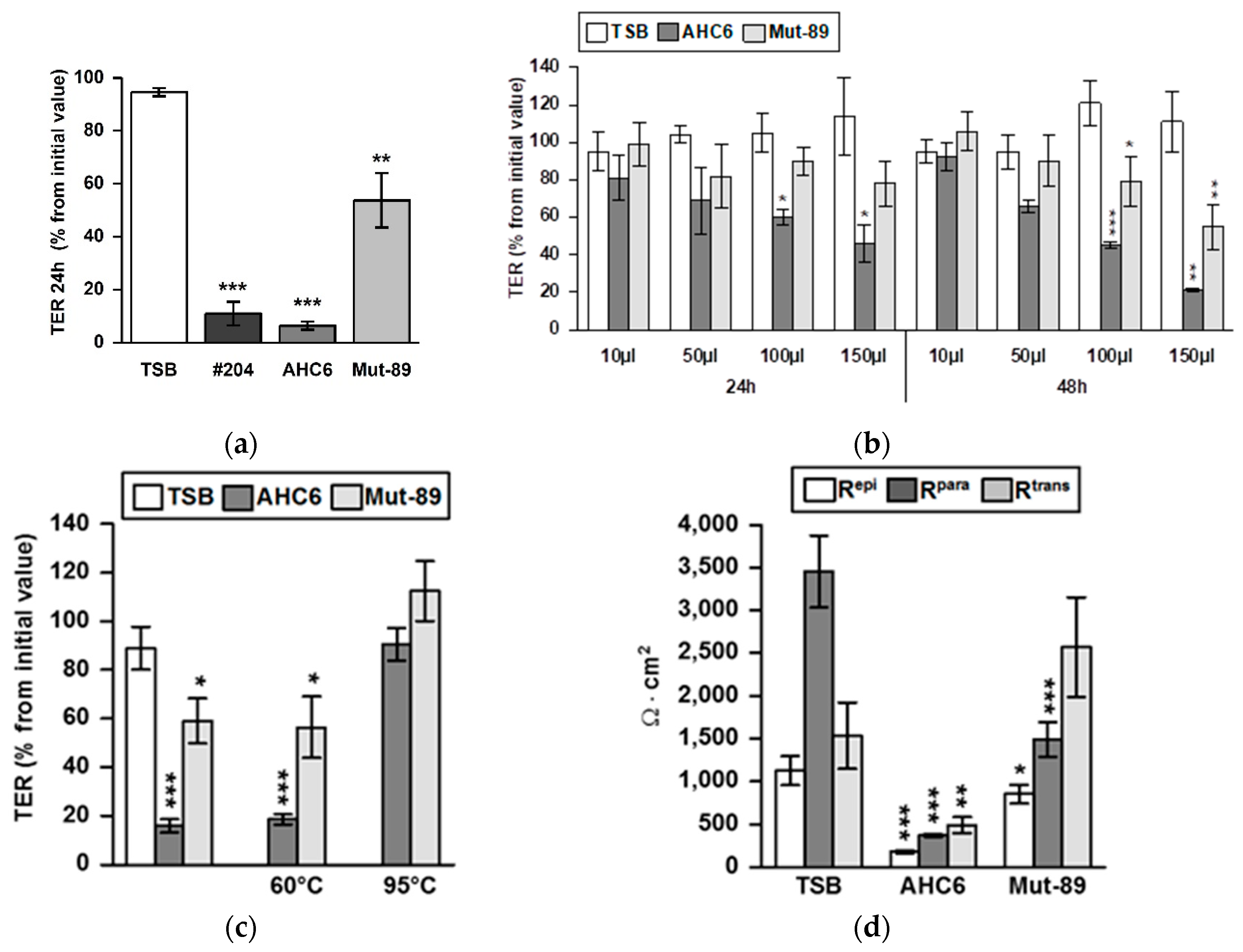
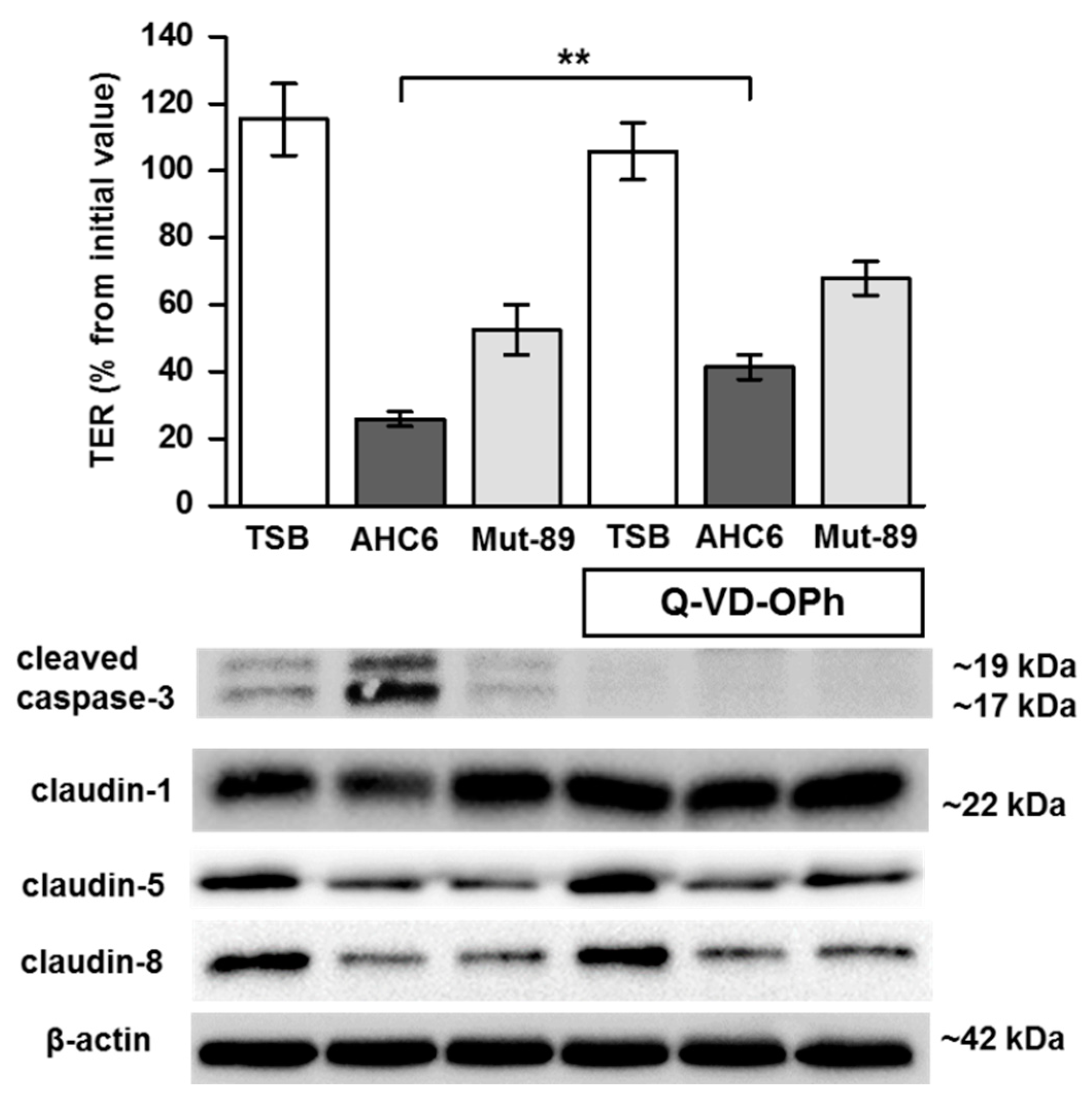
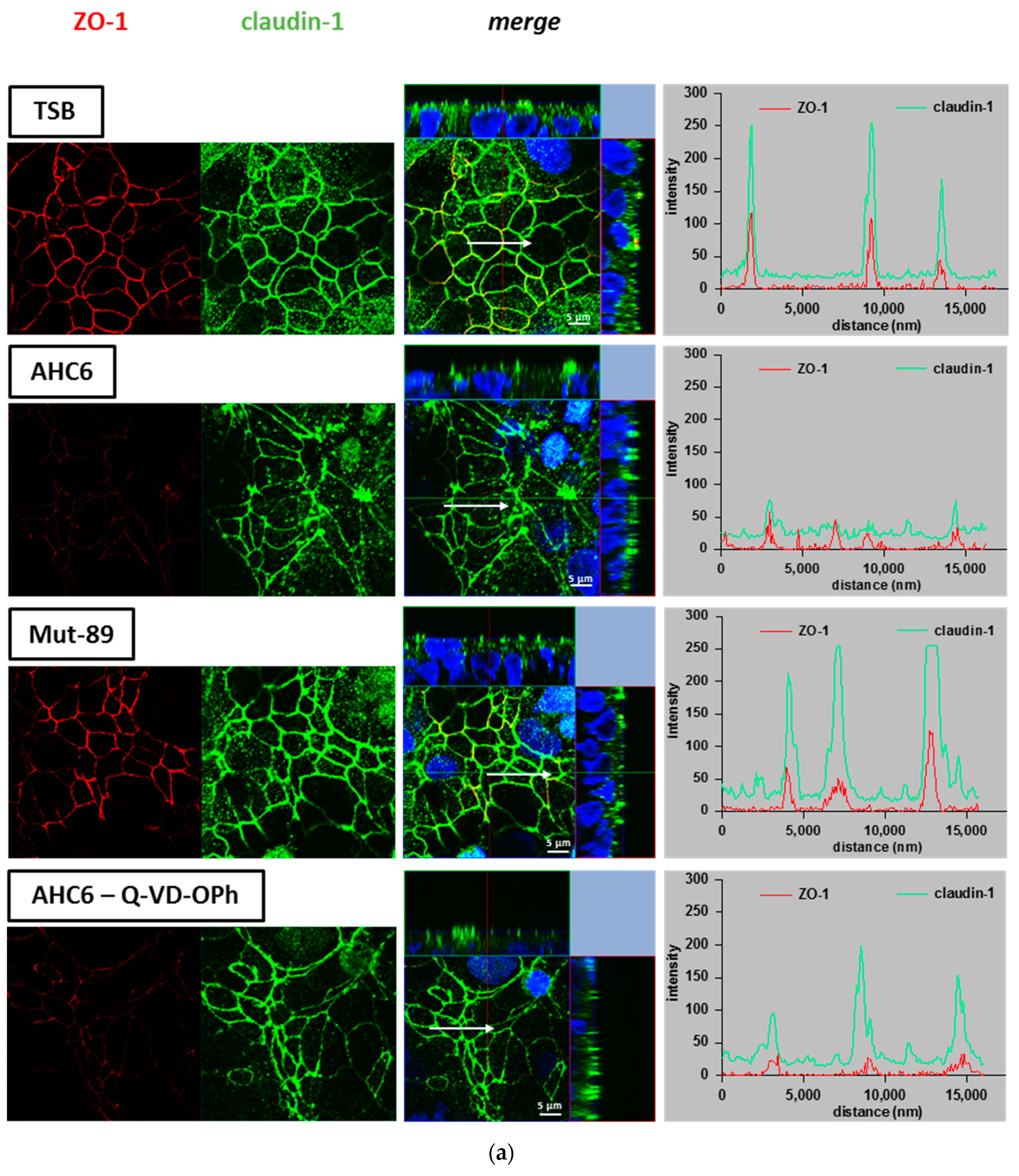
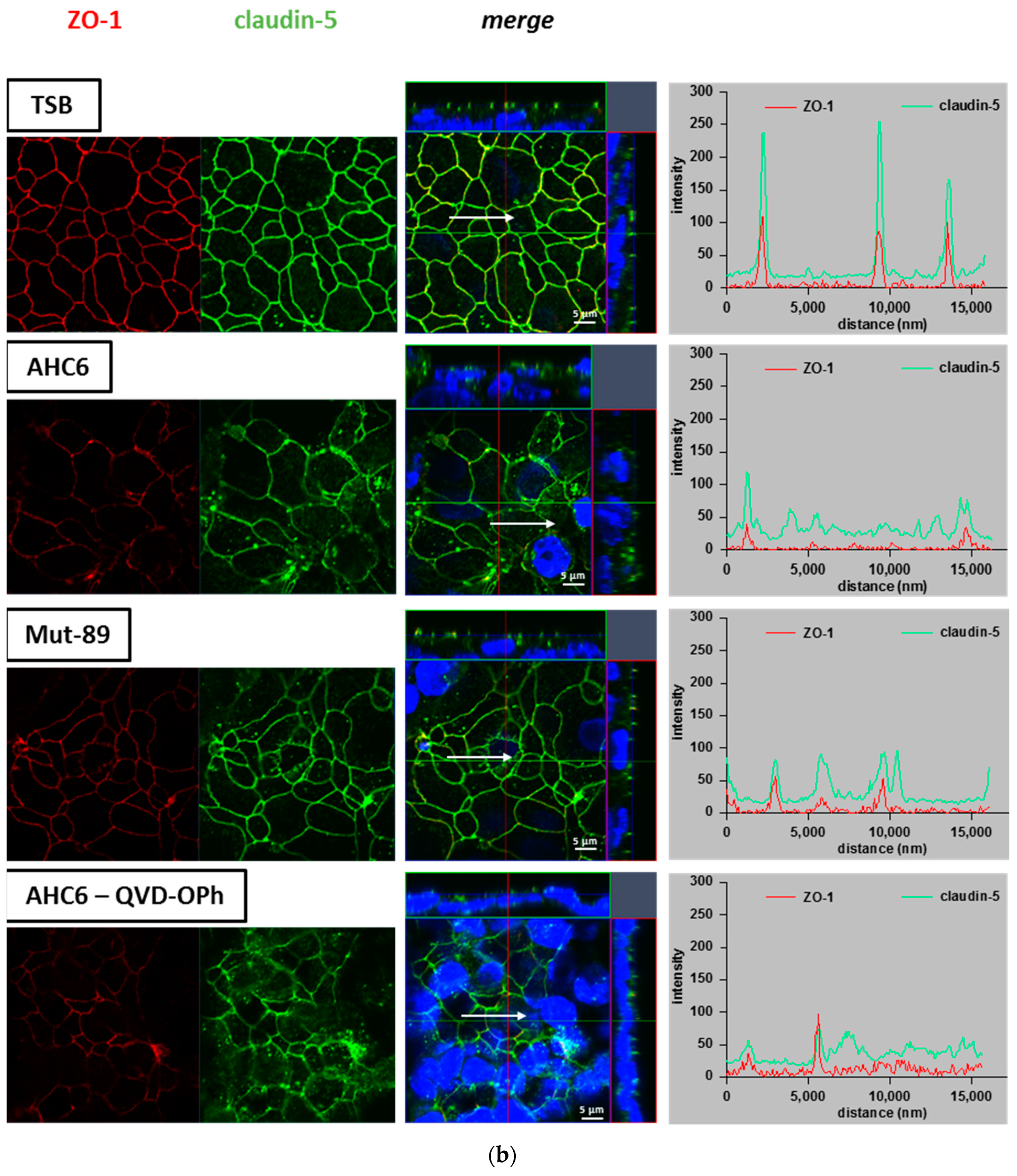
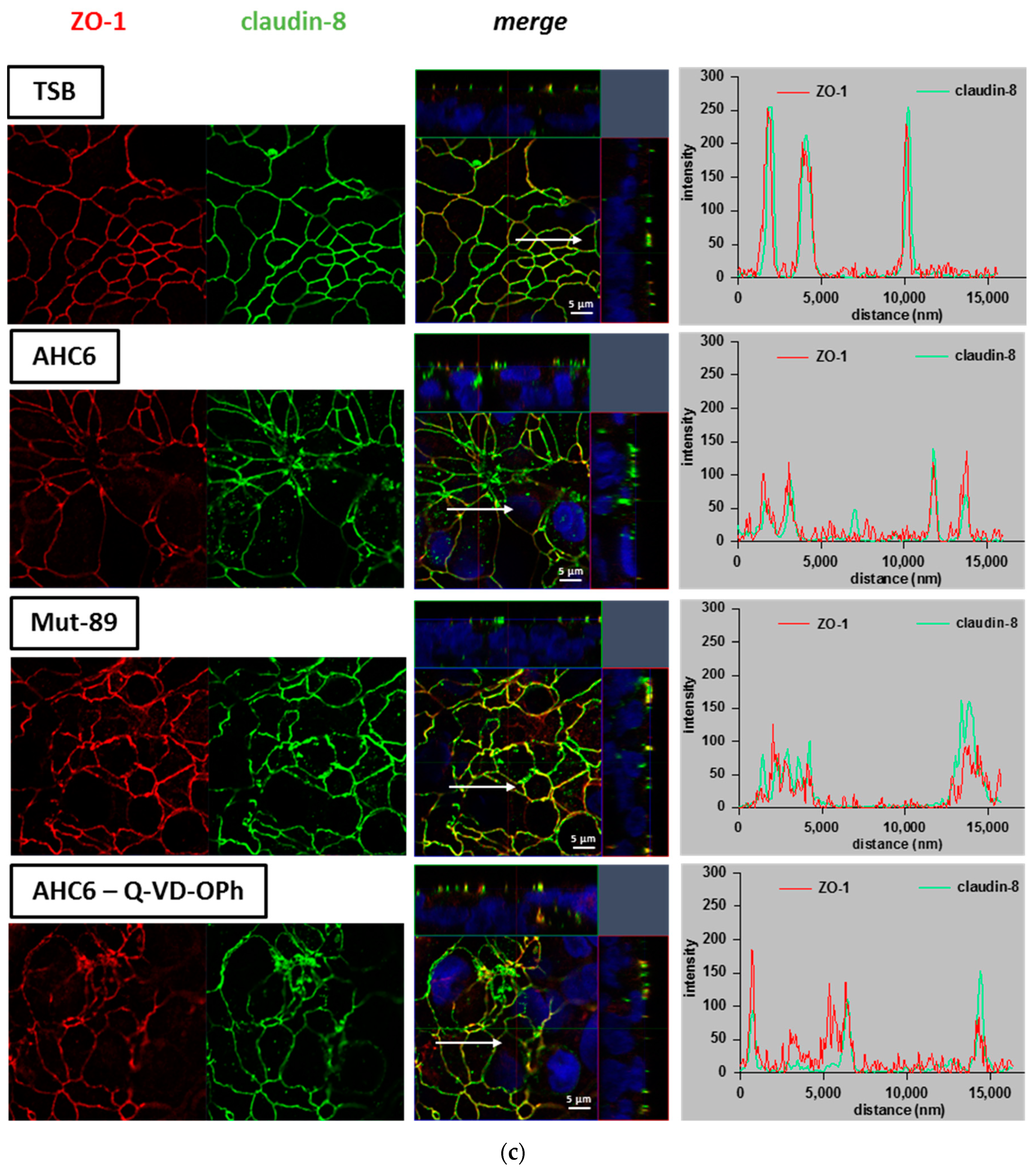
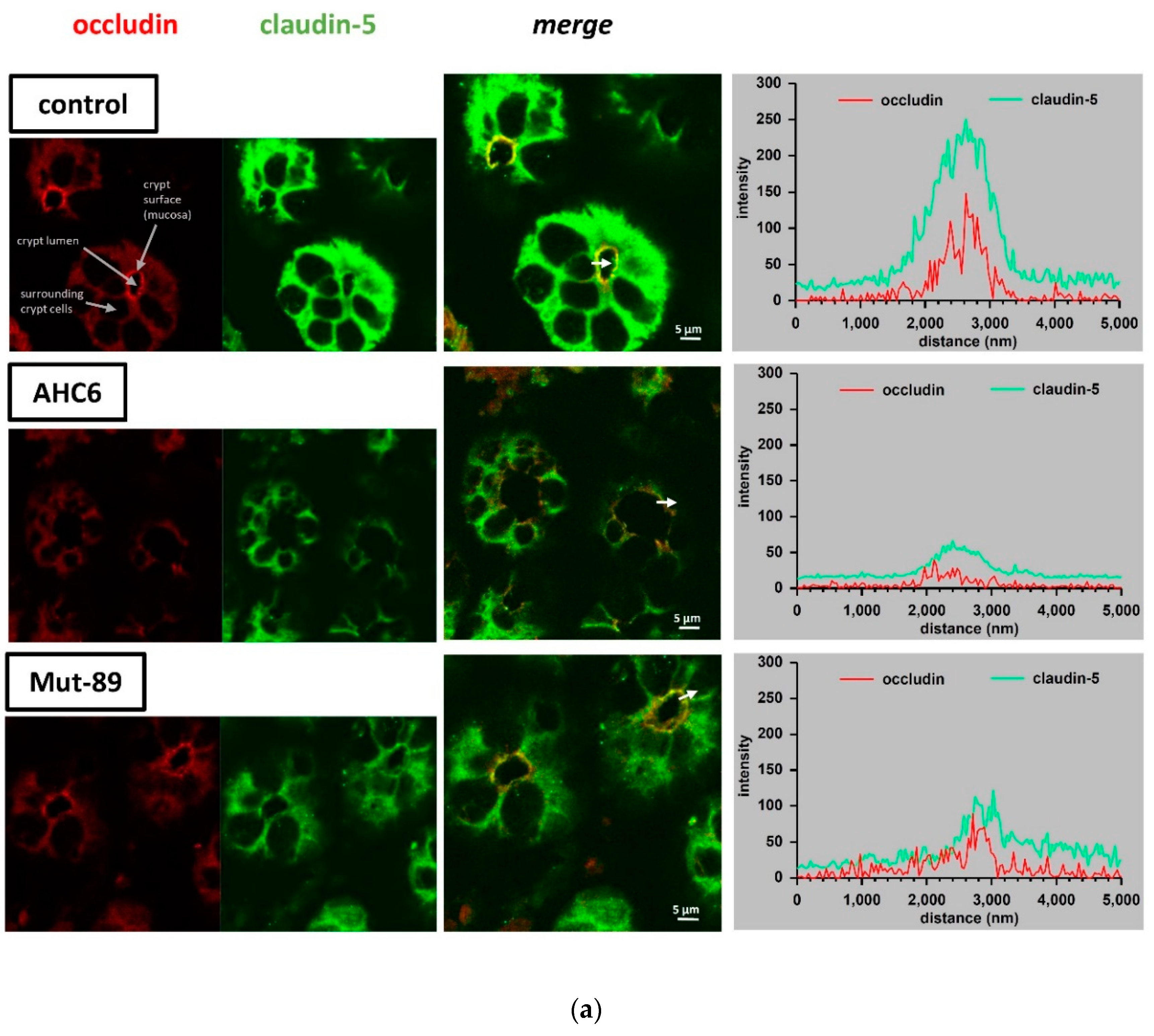
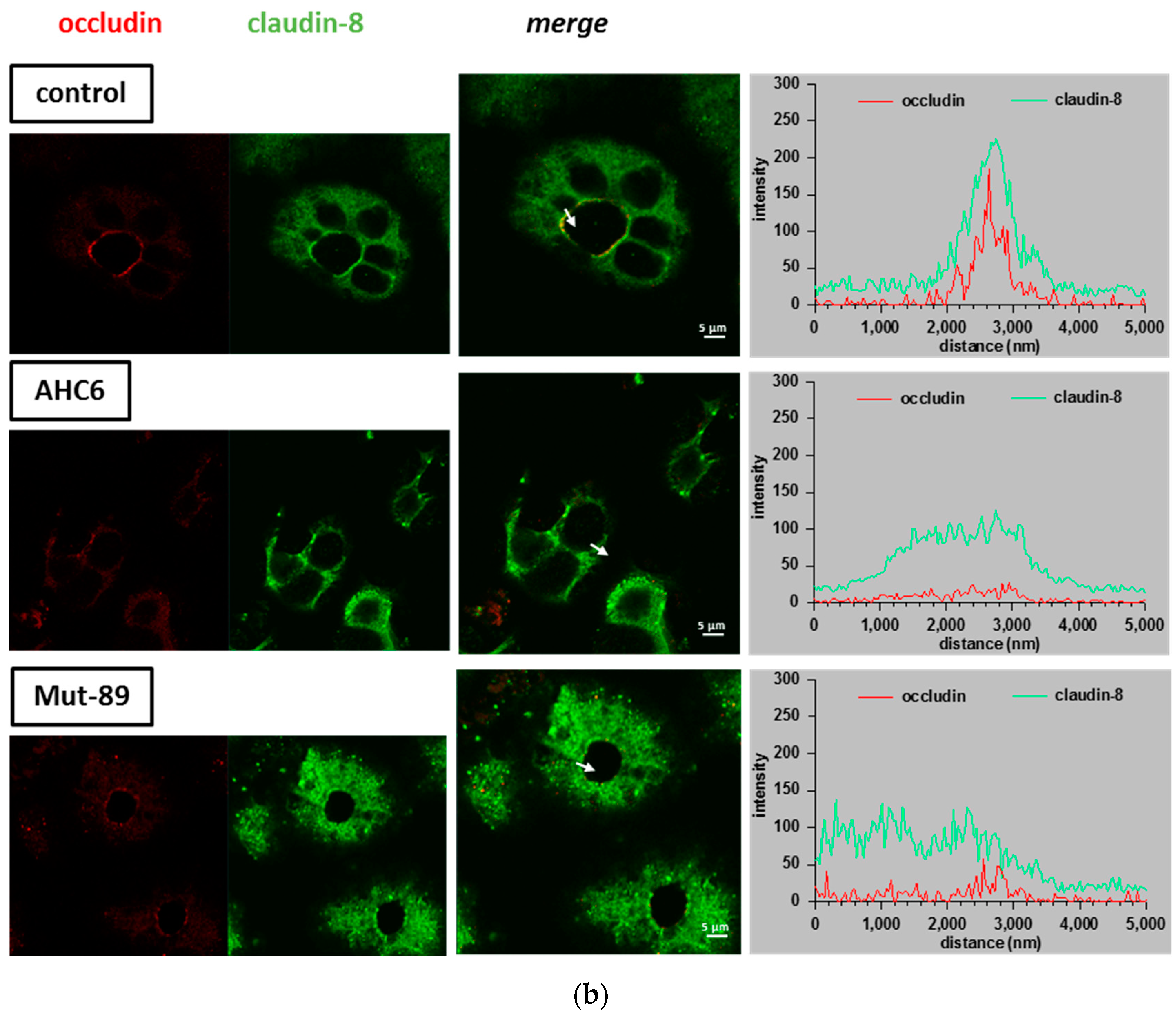
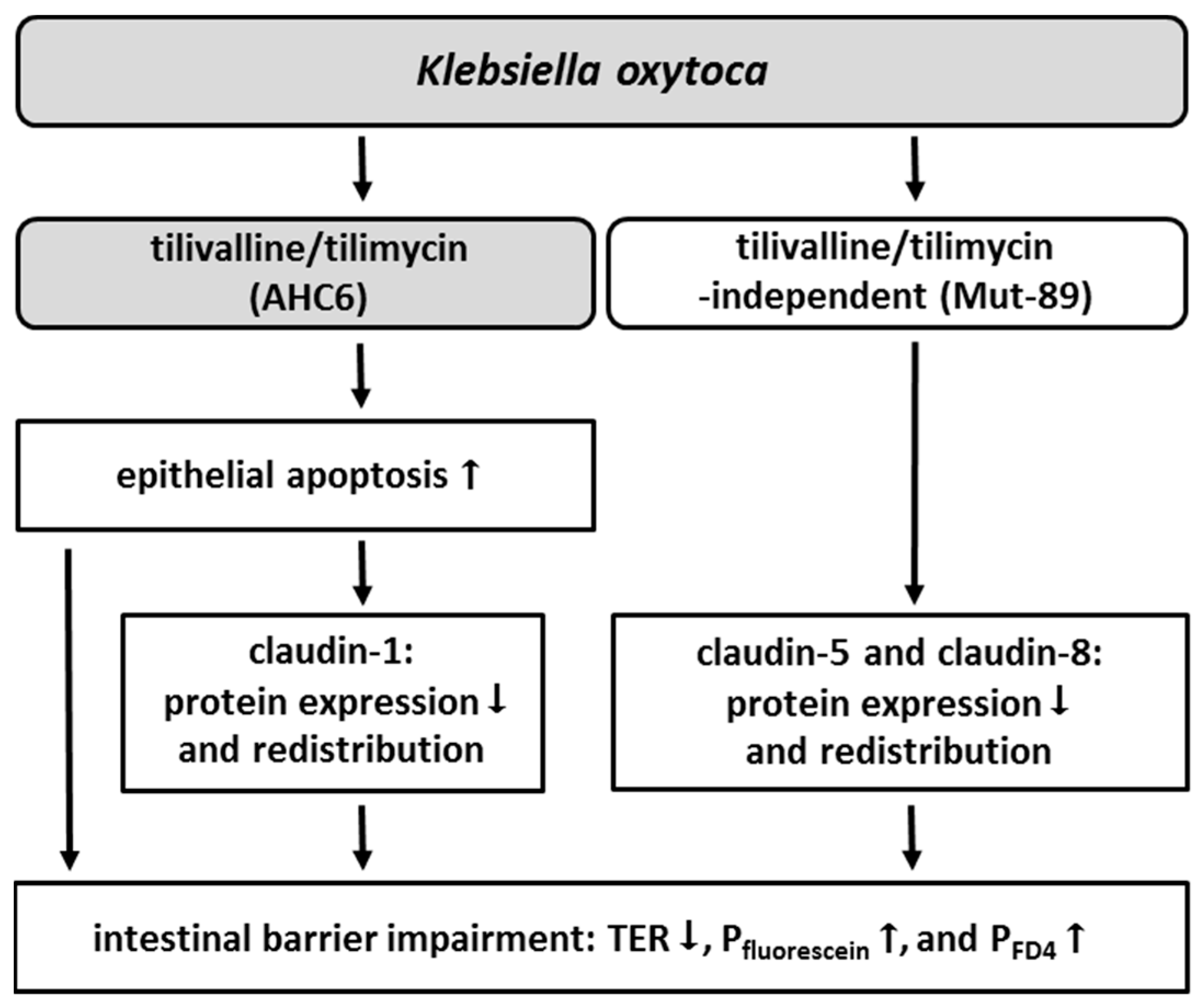
2. Results
3. Discussion
4. Material and Methods
4.1. Cell Culture and Challenge
4.2. TER Measurement
4.3. Permeability Measurements
4.4. Measurement of Para- and Transcellular Resistance
4.5. Western Blot
4.6. Immunofluorescence Staining and Confocal Laser scanning Microscopy
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAHC | antibiotic-associated hemorrhagic colitis |
| FD4 | fluorescein-iso-thio-cyanate-dextran-4000 |
| LSM | laser scanning microscopy |
| K. oxytoca | Klebsiella oxytoca |
| QV | Q-VD-Oph |
| Repi | epithelial resistance |
| Rpara | paracellular resistance |
| Rtrans | transcellular resistance |
| TER | transepithelial resistance |
| TSB | tryptic soy broth |
| ZO-1 | zonula occludens-1 |
References
- Bartlett, J.G. Clostridium difficile-associated Enteric Disease. Curr. Infect. Dis. Rep. 2002, 4, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Hgenauer, C.; Langner, C.; Beubler, E.; Lippe, I.T.; Schicho, R.; Gorkiewicz, G.; Krause, R.; Gerstgrasser, N.; Krejs, G.J.; Hinterleitner, T.A. Klebsiella oxytoca as a causative organism of antibiotic-associated hemorrhagic colitis. N. Engl. J. Med. 2006, 355, 2418–2426. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Metz, M.; Barbut, F.; Bellaiche, G.; Bouhnik, Y.; Raskine, L.; Nicolas, J.C.; Chatelet, F.P.; Lehn, N.; Petit, J.C.; et al. Klebsiella oxytoca as an agent of antibiotic-associated hemorrhagic colitis. Clin. Gastroenterol. Hepatol. 2003, 1, 370–376. [Google Scholar] [CrossRef]
- Zollner-Schwetz, I.; Hogenauer, C.; Joainig, M.; Weberhofer, P.; Gorkiewicz, G.; Valentin, T.; Hinterleitner, T.A.; Krause, R. Role of Klebsiella oxytoca in antibiotic-associated diarrhea. Clin. Infect. Dis. 2008, 47, e74–e78. [Google Scholar] [CrossRef] [PubMed]
- Podschun, R.; Ullmann, U. Klebsiella spp. as nosocomial pathogens: Epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. beta-Lactamases in laboratory and clinical resistance. Clin. Microbiol. Rev. 1995, 8, 557–584. [Google Scholar] [CrossRef] [PubMed]
- Joainig, M.M.; Gorkiewicz, G.; Leitner, E.; Weberhofer, P.; Zollner-Schwetz, I.; Lippe, I.; Feierl, G.; Krause, R.; Hinterleitner, T.; Zechner, E.L.; et al. Cytotoxic effects of Klebsiella oxytoca strains isolated from patients with antibiotic-associated hemorrhagic colitis or other diseases caused by infections and from healthy subjects. J. Clin. Microbiol. 2010, 48, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Schneditz, G.; Rentner, J.; Roier, S.; Pletz, J.; Herzog, K.A.; Bucker, R.; Troeger, H.; Schild, S.; Weber, H.; Breinbauer, R.; et al. Enterotoxicity of a nonribosomal peptide causes antibiotic-associated colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13181–13186. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.; Gu, Q.; Sze, K.H.; Chu, I.K.; Kao, R.Y.; Lee, K.C.; Lam, C.W.; Yang, D.; Tai, S.S.; Ke, Y.; et al. A tricyclic pyrrolobenzodiazepine produced by Klebsiella oxytoca is associated with cytotoxicity in antibiotic-associated hemorrhagic colitis. J. Biol. Chem. 2017, 292, 19503–19520. [Google Scholar] [CrossRef] [PubMed]
- Unterhauser, K.; Poltl, L.; Schneditz, G.; Kienesberger, S.; Glabonjat, R.A.; Kitsera, M.; Pletz, J.; Josa-Prado, F.; Dornisch, E.; Lembacher-Fadum, C.; et al. Klebsiella oxytoca enterotoxins tilimycin and tilivalline have distinct host DNA-damaging and microtubule-stabilizing activities. Proc. Natl. Acad. Sci. USA 2019, 116, 3774–3783. [Google Scholar] [CrossRef] [PubMed]
- Dornisch, E.; Pletz, J.; Glabonjat, R.A.; Martin, F.; Lembacher-Fadum, C.; Neger, M.; Hogenauer, C.; Francesconi, K.; Kroutil, W.; Zangger, K.; et al. Biosynthesis of the Enterotoxic Pyrrolobenzodiazepine Natural Product Tilivalline. Angew. Chem. Int. Ed. Engl. 2017, 56, 14753–14757. [Google Scholar] [CrossRef] [PubMed]
- von Tesmar, A.; Hoffmann, M.; Abou Fayad, A.; Huttel, S.; Schmitt, V.; Herrmann, J.; Muller, R. Biosynthesis of the Klebsiella oxytoca Pathogenicity Factor Tilivalline: Heterologous Expression, in Vitro Biosynthesis, and Inhibitor Development. ACS Chem. Biol. 2018, 13, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Quigley, E.M. Leaky gut-concept or clinical entity? Curr. Opin. Gastroenterol. 2016, 32, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Gitter, A.H.; Bendfeldt, K.; Schulzke, J.D.; Fromm, M. Leaks in the epithelial barrier caused by spontaneous and TNF-alpha-induced single-cell apoptosis. Faseb. J. 2000, 14, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.L.; Nielsen, H.; Ejlertsen, T.; Engberg, J.; Gunzel, D.; Zeitz, M.; Hering, N.A.; Fromm, M.; Schulzke, J.D.; Bucker, R. Oral and fecal Campylobacter concisus strains perturb barrier function by apoptosis induction in HT-29/B6 intestinal epithelial cells. PLoS ONE 2011, 6, e23858. [Google Scholar] [CrossRef] [PubMed]
- Bucker, R.; Krug, S.M.; Moos, V.; Bojarski, C.; Schweiger, M.R.; Kerick, M.; Fromm, A.; Janssen, S.; Fromm, M.; Hering, N.A.; et al. Campylobacter jejuni impairs sodium transport and epithelial barrier function via cytokine release in human colon. Mucosal. Immunol. 2018, 11, 75–577. [Google Scholar]
- Bucker, R.; Troeger, H.; Kleer, J.; Fromm, M.; Schulzke, J.D. Arcobacter butzleri induces barrier dysfunction in intestinal HT-29/B6 cells. J. Infect. Dis. 2009, 200, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Steinhusen, U.; Badock, V.; Bauer, A.; Behrens, J.; Wittman-Liebold, B.; Dorken, B.; Bommert, K. Apoptosis-induced cleavage of beta-catenin by caspase-3 results in proteolytic fragments with reduced transactivation potential. J. Biol. Chem. 2000, 275, 16345–16353. [Google Scholar] [CrossRef] [PubMed]
- Steinhusen, U.; Weiske, J.; Badock, V.; Tauber, R.; Bommert, K.; Huber, O. Cleavage and shedding of E-cadherin after induction of apoptosis. J. Biol. Chem. 2001, 276, 4972–4980. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, C.; Weiske, J.; Schoneberg, T.; Schroder, W.; Mankertz, J.; Schulzke, J.D.; Florian, P.; Fromm, M.; Tauber, R.; Huber, O. The specific fates of tight junction proteins in apoptotic epithelial cells. J. Cell. Sci. 2004, 117, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Mineta, K.; Yamamoto, Y.; Yamazaki, Y.; Tanaka, H.; Tada, Y.; Saito, K.; Tamura, A.; Igarashi, M.; Endo, T.; Takeuchi, K.; et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, D.R.; Marchiando, A.M.; Zhang, Y.; Shen, L.; Sasaki, H.; Wang, Y.; Long, M.; Turner, J.R. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol. Biol. Cell. 2010, 21, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Schulzke, J.D.; Fromm, M. Tight junction, selective permeability, and related diseases. Semin. Cell. Dev. Biol. 2014, 36, 166–176. [Google Scholar] [CrossRef] [PubMed]
- De Ryck, T.; Vanlancker, E.; Grootaert, C.; Roman, B.I.; De Coen, L.M.; Vandenberghe, I.; Stevens, C.V.; Bracke, M.; Van de Wiele, T.; Vanhoecke, B. Microbial inhibition of oral epithelial wound recovery: Potential role for quorum sensing molecules? AMB Express. 2015, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Higaki, M.; Chida, T.; Takano, H.; Nakaya, R. Cytotoxic component(s) of Klebsiella oxytoca on HEp-2 cells. Microbiol. Immunol. 1990, 34, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.M.; Lee, H.J.; Jang, S.E.; Han, M.J.; Kim, D.H. Evidence for interplay among antibacterial-induced gut microbiota disturbance, neuro-inflammation, and anxiety in mice. Mucosal. Immunol. 2018, 11, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Fromm, M. Claudins and other tight junction proteins. Compr. Physiol. 2012, 2, 1819–1852. [Google Scholar] [PubMed]
- Hering, N.A.; Fromm, A.; Kikhney, J.; Lee, I.F.; Moter, A.; Schulzke, J.D.; Bucker, R. Yersinia enterocolitica Affects Intestinal Barrier Function in the Colon. J. Infect. Dis. 2016, 213, 1157–1162. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Bucker, R.; Krug, S.M.; Rosenthal, R.; Gunzel, D.; Fromm, A.; Zeitz, M.; Chakraborty, T.; Fromm, M.; Epple, H.J.; Schulzke, J.D. Aerolysin from Aeromonas hydrophila perturbs tight junction integrity and cell lesion repair in intestinal epithelial HT-29/B6 cells. J. Infect. Dis. 2011, 204, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Veshnyakova, A.; Protze, J.; Rossa, J.; Blasig, I.E.; Krause, G.; Piontek, J. On the interaction of Clostridium perfringens enterotoxin with claudins. Toxins (Basel) 2010, 2, 1336–1356. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.S.; McCarthy, K.M.; Francis, S.A.; McCormack, J.M.; Lai, J.; Rogers, R.A.; Lynch, R.D.; Schneeberger, E.E. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am. J. Physiol. Cell. Physiol. 2005, 288, C1231–C1241. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Marchiando, A.M.; Weber, C.R.; Raleigh, D.R.; Wang, Y.; Shen, L.; Turner, J.R. MLCK-dependent exchange and actin binding region-dependent anchoring of ZO-1 regulate tight junction barrier function. Proc. Natl. Acad. Sci. USA 2010, 107, 8237–8241. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Choi, W.; Buckley, A.; Shashikanth, N.; Joseph, N.E.; Wang, Y.; Warren, M.H.; Buschmann, M.M.; Pavlyuk, R.; Hildebrand, J.; et al. ZO-1 interactions with F-actin and occludin direct epithelial polarization and single lumen specification in 3D culture. J. Cell. Sci. 2017, 130, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Zyrek, A.A.; Cichon, C.; Helms, S.; Enders, C.; Sonnenborn, U.; Schmidt, M.A. Molecular mechanisms underlying the probiotic effects of Escherichia coli Nissle 1917 involve ZO-2 and PKCzeta redistribution resulting in tight junction and epithelial barrier repair. Cell Microbiol. 2007, 9, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Potgens, S.A.; Brossel, H.; Sboarina, M.; Catry, E.; Cani, P.D.; Neyrinck, A.M.; Delzenne, N.M.; Bindels, L.B. Klebsiella oxytoca expands in cancer cachexia and acts as a gut pathobiont contributing to intestinal dysfunction. Sci. Rep. 2018, 8, 12321. [Google Scholar] [CrossRef] [PubMed]
- Herzog, K.A.; Schneditz, G.; Leitner, E.; Feierl, G.; Hoffmann, K.M.; Zollner-Schwetz, I.; Krause, R.; Gorkiewicz, G.; Zechner, E.L.; Hogenauer, C. Genotypes of Klebsiella oxytoca isolates from patients with nosocomial pneumonia are distinct from those of isolates from patients with antibiotic-associated hemorrhagic colitis. J. Clin. Microbiol. 2014, 52, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Kreusel, K.M.; Fromm, M.; Schulzke, J.D.; Hegel, U. Cl- secretion in epithelial monolayers of mucus-forming human colon cells (HT-29/B6). Am. J. Physiol. 1991, 261. [Google Scholar] [CrossRef] [PubMed]
- Hering, N.A.; Luettig, J.; Krug, S.M.; Wiegand, S.; Gross, G.; van Tol, E.A.; Schulzke, J.D.; Rosenthal, R. Lactoferrin protects against intestinal inflammation and bacteria-induced barrier dysfunction in vitro. Ann. N. Y. Acad. Sci. 2017, 1405, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Burgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Fromm, M.; Gunzel, D. Two-path impedance spectroscopy for measuring paracellular and transcellular epithelial resistance. Biophys. J. 2009, 97, 2202–2211. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PFluorescein (10−6 cm⋅s−1) | PFD4 (10−6 cm⋅s−1) | TER (Ω∙cm2) | |
|---|---|---|---|
| TSB | 0.10 ± 0.01 (7) | 0.02 ± 0.00 (8) | 1754 ± 179 (7) |
| AHC6 | 1.46 ± 0.31 (7) ** | 0.09 ± 0.01 (8) *** | 231 ± 53 (7) *** |
| Mut-89 | 0.21 ± 0.02 (6) * | 0.03 ± 0.00 (8) * | 814 ± 174 (6) |
| Tight Junction Protein | Expression (%) | ||||
|---|---|---|---|---|---|
| AHC6 | Mut-89 | TSB QV | AHC6 QV | Mut-89 QV | |
| claudin-1 | 65 ± 6 (9) *** | 88 ± 11 (9) | 81 ± 12 (5) | 87 ± 11 (6) | 95 ± 11 (6) |
| claudin-5 | 47 ± 10 (8) ** | 48 ± 8 (8) *** | 90 ± 6 (5) | 38 ± 12 (5) * | 47 ± 14 (5) * |
| claudin-8 | 33 ± 5 (9) *** | 43 ± 6 (8) *** | 102 ± 5 (5) | 36 ± 10 (6) ** | 45 ± 10 (6) ** |
| claudin-2 | 91 ± 11 (6) | 78 ± 11 (6) | |||
| claudin-4 | 91 ± 8 (6) | 89 ± 9 (6) | |||
| tricellulin-a | 83 ± 9 (6) | 101 ± 7 (6) | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hering, N.A.; Fromm, A.; Bücker, R.; Gorkiewicz, G.; Zechner, E.; Högenauer, C.; Fromm, M.; Schulzke, J.-D.; Troeger, H. Tilivalline- and Tilimycin-Independent Effects of Klebsiella oxytoca on Tight Junction-Mediated Intestinal Barrier Impairment. Int. J. Mol. Sci. 2019, 20, 5595. https://doi.org/10.3390/ijms20225595
Hering NA, Fromm A, Bücker R, Gorkiewicz G, Zechner E, Högenauer C, Fromm M, Schulzke J-D, Troeger H. Tilivalline- and Tilimycin-Independent Effects of Klebsiella oxytoca on Tight Junction-Mediated Intestinal Barrier Impairment. International Journal of Molecular Sciences. 2019; 20(22):5595. https://doi.org/10.3390/ijms20225595
Chicago/Turabian StyleHering, Nina A., Anja Fromm, Roland Bücker, Gregor Gorkiewicz, Ellen Zechner, Christoph Högenauer, Michael Fromm, Jörg-Dieter Schulzke, and Hanno Troeger. 2019. "Tilivalline- and Tilimycin-Independent Effects of Klebsiella oxytoca on Tight Junction-Mediated Intestinal Barrier Impairment" International Journal of Molecular Sciences 20, no. 22: 5595. https://doi.org/10.3390/ijms20225595
APA StyleHering, N. A., Fromm, A., Bücker, R., Gorkiewicz, G., Zechner, E., Högenauer, C., Fromm, M., Schulzke, J.-D., & Troeger, H. (2019). Tilivalline- and Tilimycin-Independent Effects of Klebsiella oxytoca on Tight Junction-Mediated Intestinal Barrier Impairment. International Journal of Molecular Sciences, 20(22), 5595. https://doi.org/10.3390/ijms20225595