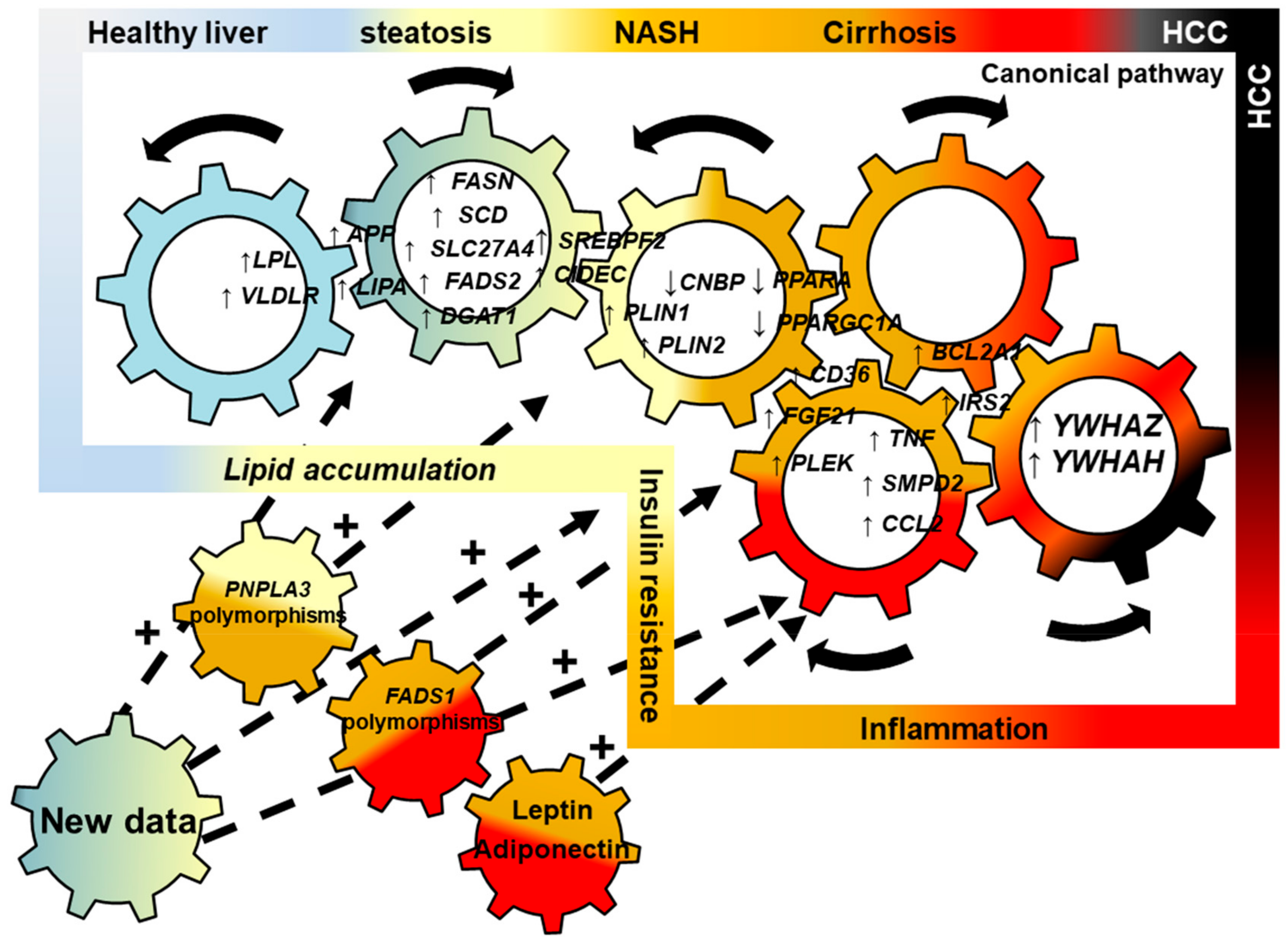
Lipid Related Genes Altered in NASH Connect Inflammation in Liver Pathogenesis Progression to HCC: A Canonical Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. A Subset of Lipid-Related Genes is Differentially Expressed in NASH vs Healthy Obese Human Liver Samples
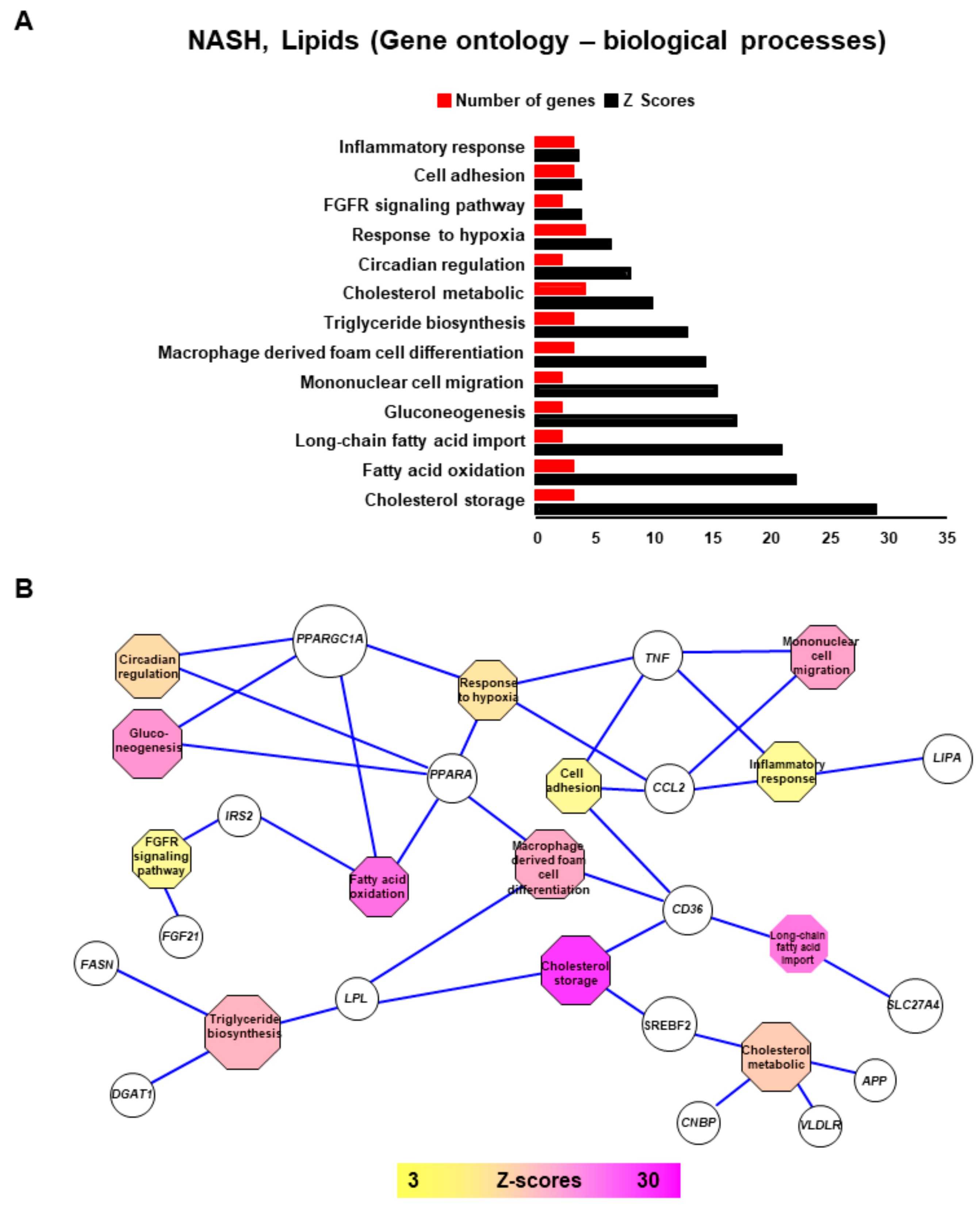
2.2. NASH is Associated with Impaired Function of Genes Implicated in Lipid Metabolism, Insulin-Resistance, Inflammation and Tumorigenesis
2.3. Connecting Lipid Related Genes Altered in NASH to Immunity, Inflammation and Liver Pathogenesis Progression
2.4. The 14-3-3 Protein Family is the Cornerstone between Dysregulated Lipid Metabolism, Inflammatory and Insulin Pathways during NASH Progression to HCC
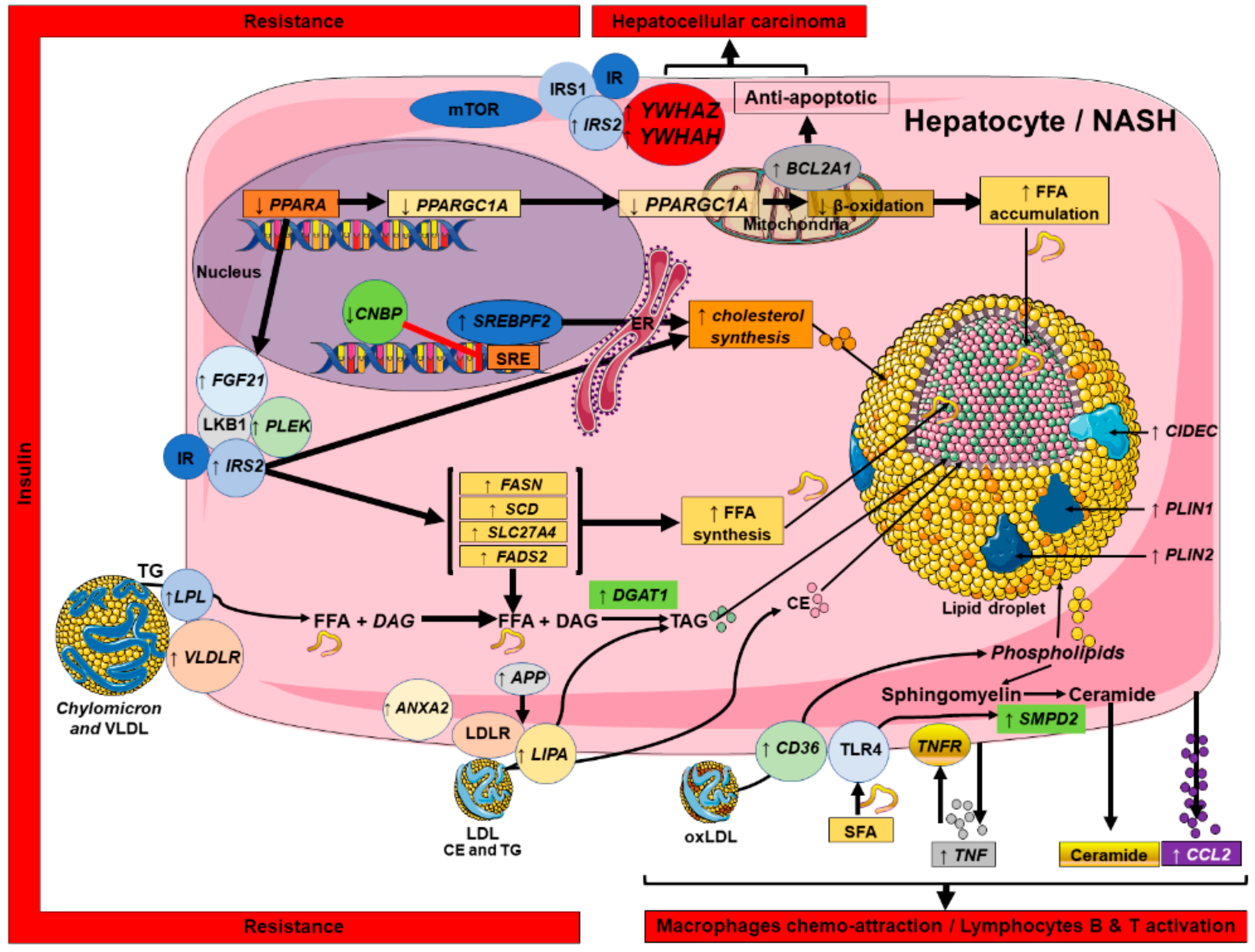
2.5. Connecting Lipid Dysregulation, Insulin Resistance, Inflammatory Processes and HCC: a NASH Canonical Pathway
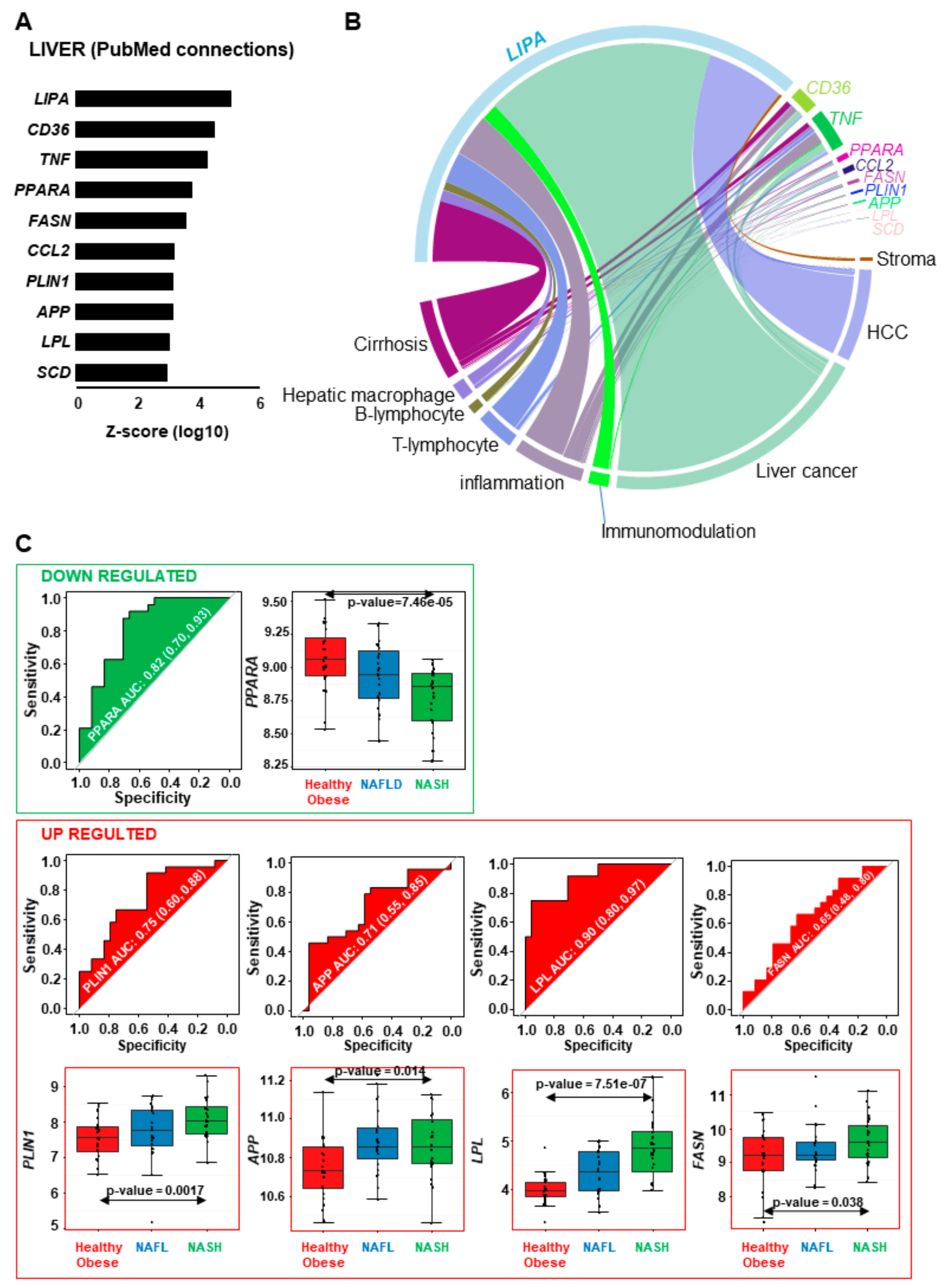
2.6. Confirmation of the NASH Canonical Pathway Using Independent Illumina Gene Expression Beadchip
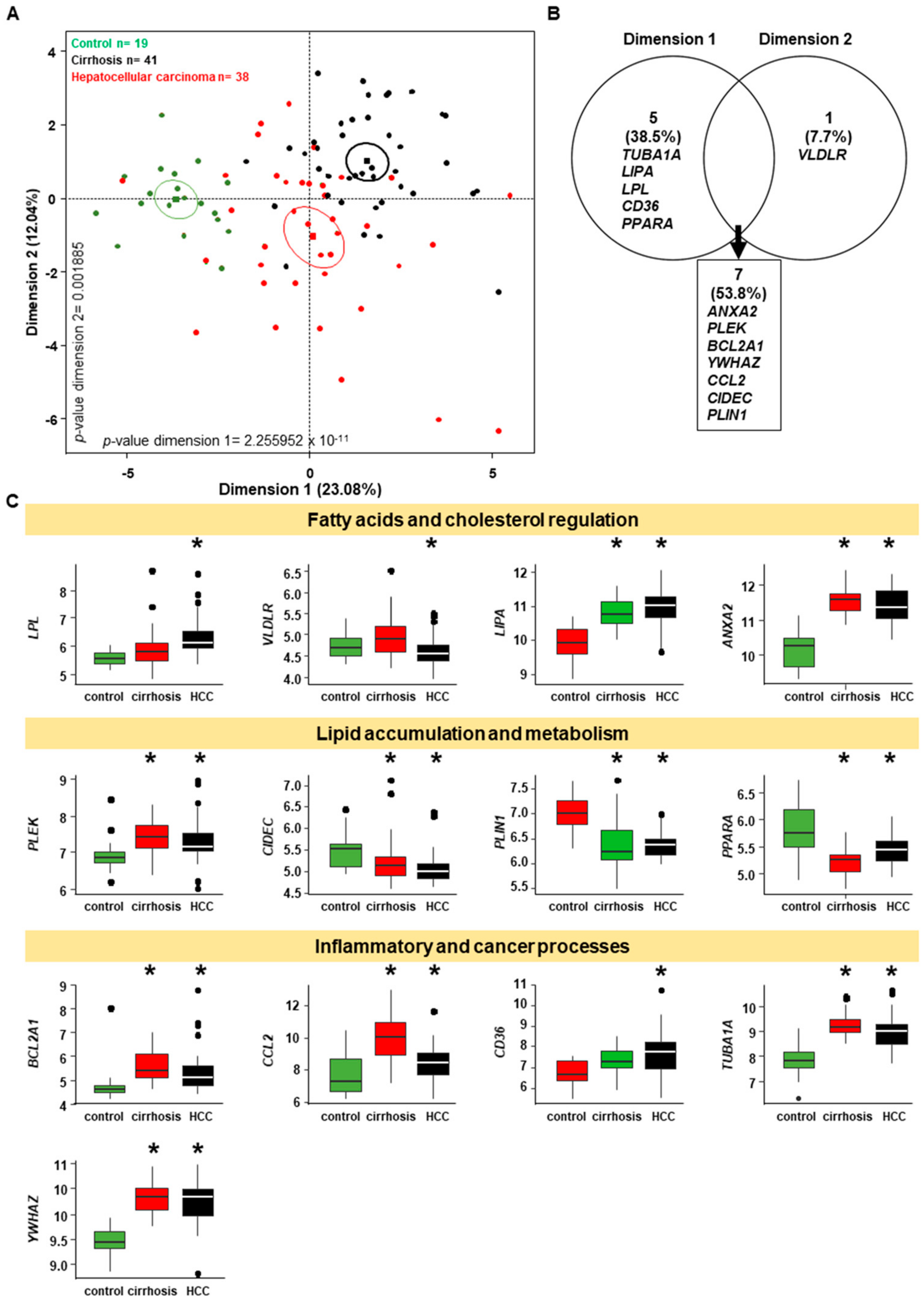
2.7. Genes Implicated in Progression of NASH are Part of the Progression to Hepatocellular Carcinoma
3. Discussion
4. Material and Methods
4.1. Bioinformatics and Statistical Analyses
Text-Mining Approach
4.2. Transcriptome Dataset Analysis Narrowing Text-Mining Discovery
4.3. Confirmation of NASH Canonical Pathway by Using an Independent Validation Cohort and a Different Transcriptome Technology Analysis
4.4. Genes Implicated in NASH Progression Involved in Progression to Hepatocellular Carcinoma: Liver Cancer Genomic Data
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perla, F.M.; Prelati, M.; Lavorato, M.; Visicchio, D.; Anania, C. The role of lipid and lipoprotein metabolism in non-alcoholic fatty liver disease. Children 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B. Fatty acid regulation of hepatic lipid metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.G. Hepatic glucose and lipid metabolism. Diabetologia 2016, 59, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef] [PubMed]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, S.; Farrell, G.C. Etiopathogenesis of nonalcoholic steatohepatitis. Semin. Liver Dis. 2001, 21, 27–41. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Rodriguez-Gallego, E.; Guirro, M.; Riera-Borrull, M.; Hernandez-Aguilera, A.; Marine-Casado, R.; Fernandez-Arroyo, S.; Beltran-Debon, R.; Sabench, F.; Hernandez, M.; Del Castillo, D.; et al. Mapping of the circulating metabolome reveals alpha-ketoglutarate as a predictor of morbid obesity-associated non-alcoholic fatty liver disease. Int. J. Obes. 2014, 279–287. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Snehalatha, C. Rising burden of obesity in Asia. J. Obes 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Nontriglyceride hepatic lipotoxicity: The new paradigm for the pathogenesis of NASH. Curr. Gastroenterol. Rep. 2010, 12, 49–56. [Google Scholar] [CrossRef]
- Calzadilla Bertot, L.; Adams, L.A. The natural course of non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Chiappini, F.; Barrier, A.; Saffroy, R.; Domart, M.C.; Dagues, N.; Azoulay, D.; Sebagh, M.; Franc, B.; Chevalier, S.; Debuire, B.; et al. Exploration of global gene expression in human liver steatosis by high-density oligonucleotide microarray. Lab. Invest. 2006, 86, 154–165. [Google Scholar] [CrossRef]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallee, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef]
- Chiappini, F.; Desterke, C.; Bertrand-Michel, J.; Guettier, C.; Le Naour, F. Hepatic and serum lipid signatures specific to nonalcoholic steatohepatitis in murine models. Sci. Rep. 2016, 6, 31587. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Senturk, H. From fatty liver to fibrosis: A tale of “second hit”. World J. Gastroenterol. 2013, 19, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef]
- Muir, K.; Hazim, A.; He, Y.; Peyressatre, M.; Kim, D.Y.; Song, X.; Beretta, L. Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma. Cancer Res. 2013, 73, 4722–4731. [Google Scholar] [CrossRef]
- Huang, J.; Iqbal, J.; Saha, P.K.; Liu, J.; Chan, L.; Hussain, M.M.; Moore, D.D.; Wang, L. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology 2007, 46, 147–157. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Y.; Fu, Y.; Guo, G.; Zhang, X. Liver fatty acid composition in mice with or without nonalcoholic fatty liver disease. Lipids Health Dis. 2011, 10, 234. [Google Scholar] [CrossRef]
- Yamada, K.; Mizukoshi, E.; Sunagozaka, H.; Arai, K.; Yamashita, T.; Takeshita, Y.; Misu, H.; Takamura, T.; Kitamura, S.; Zen, Y.; et al. Characteristics of hepatic fatty acid compositions in patients with nonalcoholic steatohepatitis. Liver Int. 2014. [Google Scholar] [CrossRef]
- Araseki, M.; Kobayashi, H.; Hosokawa, M.; Miyashita, K. Lipid peroxidation of a human hepatoma cell line (HepG2) after incorporation of linoleic acid, arachidonic acid, and docosahexaenoic acid. Biosci. Biotechnol. Biochem. 2005, 69, 483–490. [Google Scholar] [CrossRef]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.L.; Gaudin, F.; Naveau, S.; Prevot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Pusl, T.; Wild, N.; Vennegeerts, T.; Wimmer, R.; Goke, B.; Brand, S.; Rust, C. Free fatty acids sensitize hepatocytes to bile acid-induced apoptosis. Biochem. Biophys. Res. Commun. 2008, 371, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Matsubara, T.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 2012, 56, 118–129. [Google Scholar] [CrossRef]
- Wang, L.; Athinarayanan, S.; Jiang, G.; Chalasani, N.; Zhang, M.; Liu, W. Fatty acid desaturase 1 gene polymorphisms control human hepatic lipid composition. Hepatology 2015, 61, 119–128. [Google Scholar] [CrossRef]
- Wang, Y.; Torres-Gonzalez, M.; Tripathy, S.; Botolin, D.; Christian, B.; Jump, D.B. Elevated hepatic fatty acid elongase-5 activity affects multiple pathways controlling hepatic lipid and carbohydrate composition. J. Lipid Res. 2008, 49, 1538–1552. [Google Scholar] [CrossRef]
- Warensjo, E.; Ohrvall, M.; Vessby, B. Fatty acid composition and estimated desaturase activities are associated with obesity and lifestyle variables in men and women. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 128–136. [Google Scholar] [CrossRef]
- Araya, J.; Rodrigo, R.; Pettinelli, P.; Araya, A.V.; Poniachik, J.; Videla, L.A. Decreased liver fatty acid delta-6 and delta-5 desaturase activity in obese patients. Obesity 2010, 18, 1460–1463. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Monk, J.M.; Hou, T.Y.; Callway, E.; Vincent, L.; Weeks, B.; Yang, P.; Chapkin, R.S. Characterization of an arachidonic acid-deficient (Fads1 knockout) mouse model. J. Lipid Res. 2012, 53, 1287–1295. [Google Scholar] [CrossRef]
- Green, C.D.; Ozguden-Akkoc, C.G.; Wang, Y.; Jump, D.B.; Olson, L.K. Role of fatty acid elongases in determination of de novo synthesized monounsaturated fatty acid species. J. Lipid Res. 2010, 51, 1871–1877. [Google Scholar] [CrossRef]
- Portolesi, R.; Powell, B.C.; Gibson, R.A. Competition between 24:5n-3 and ALA for Delta 6 desaturase may limit the accumulation of DHA in HepG2 cell membranes. J. Lipid Res. 2007, 48, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Kumadaki, S.; Matsuzaka, T.; Kato, T.; Yahagi, N.; Yamamoto, T.; Okada, S.; Kobayashi, K.; Takahashi, A.; Yatoh, S.; Suzuki, H.; et al. Mouse elovl-6 promoter is an SREBP target. Biochem. Biophys. Res. Commun. 2008, 368, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Dalen, K.T.; Gustafsson, J.A.; Nebb, H.I. Regulation of hepatic fatty acid elongase 5 by LXRalpha-SREBP-1c. Biochim. Biophys. Acta 2009, 1791, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, S.; Torres-Gonzalez, M.; Jump, D.B. Elevated hepatic fatty acid elongase-5 activity corrects dietary fat-induced hyperglycemia in obese C57BL/6J mice. J. Lipid Res. 2010, 51, 2642–2654. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.C.; Cook-Johnson, R.J.; James, M.J.; Muhlhausler, B.S.; Gibson, R.A. Omega-3 long chain fatty acid synthesis is regulated more by substrate levels than gene expression. Prostaglandins Leukot. Essent. Fatty Acids 2010, 83, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Botolin, D.; Xu, J.; Christian, B.; Mitchell, E.; Jayaprakasam, B.; Nair, M.G.; Peters, J.M.; Busik, J.V.; Olson, L.K.; et al. Regulation of hepatic fatty acid elongase and desaturase expression in diabetes and obesity. J. Lipid Res. 2006, 47, 2028–2041. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Rebholz-Schuhmann, D.; Oellrich, A.; Hoehndorf, R. Text-mining solutions for biomedical research: Enabling integrative biology. Nat. Rev. Genet. 2012, 13, 829–839. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Federhen, S.; et al. Database resources of the national center for biotechnology information. Nucl. Acids Res. 2012, 40, D13–D25. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucl. Acids Res. 2009, 37, D5–D15. [Google Scholar] [CrossRef]
- Sfakianaki, P.; Koumakis, L.; Sfakianakis, S.; Iatraki, G.; Zacharioudakis, G.; Graf, N.; Marias, K.; Tsiknakis, M. Semantic biomedical resource discovery: A natural language processing framework. BMC Med. Inf. Decis. Mak. 2015, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucl. Acids Res. 2008, 36, D13–D21. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucl. Acids Res. 2006, 34, D173–D180. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucl. Acids Res. 2007, 35, D5–D12. [Google Scholar] [CrossRef]
- Krallinger, M.; Valencia, A.; Hirschman, L. Linking genes to literature: Text mining, information extraction, and retrieval applications for biology. Genome Biol. 2008, 9, S8. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucl. Acids Res. 2011, 39, D38–D51. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; King, B.L.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The comparative toxicogenomics database: Update 2017. Nucl. Acids Res. 2017, 45, D972–D978. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucl. Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Zhao, J.F.; Zhao, Q.; Hu, H.; Liao, J.Z.; Lin, J.S.; Xia, C.; Chang, Y.; Liu, J.; Guo, A.Y.; He, X.X. The ASH1-miR-375-YWHAZ signaling axis regulates tumor properties in hepatocellular carcinoma. Mol. Ther. Nucl. Acids 2018, 11, 538–553. [Google Scholar] [CrossRef]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schonfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl Acad Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.; Zimmer, A.R.; Zimmer, E.R.; de Souza, D.O.; Portela, L.V.; Saraiva-Pereira, M.L. Changes in brain 14-3-3 proteins in response to insulin resistance induced by a high palatable diet. Mol. Neurobiol. 2015, 52, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, N.; Tsunedomi, R.; Tamesa, T.; Okada, T.; Sakamoto, K.; Hamaguchi, T.; Yamada-Okabe, H.; Miyamoto, T.; Uchimura, S.; Hamamoto, Y.; et al. Involvement of c-myc-regulated genes in hepatocellular carcinoma related to genotype-C hepatitis B virus. J. Cancer Res. Clin. Oncol 2006, 132, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Estall, J.L.; Ruas, J.L.; Choi, C.S.; Laznik, D.; Badman, M.; Maratos-Flier, E.; Shulman, G.I.; Spiegelman, B.M. PGC-1alpha negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erb(alpha) axis. Proc. Natl Acad Sci. USA 2009, 106, 22510–22515. [Google Scholar] [CrossRef] [PubMed]
- Starmann, J.; Falth, M.; Spindelbock, W.; Lanz, K.L.; Lackner, C.; Zatloukal, K.; Trauner, M.; Sultmann, H. Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PLoS ONE 2012, 7, e46584. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Seo, D.; Gomez-Quiroz, L.E.; Uchida, K.; Gillen, M.C.; Kitade, M.; Kaposi-Novak, P.; Conner, E.A.; Factor, V.M.; Thorgeirsson, S.S. Loss of c-Met accelerates development of liver fibrosis in response to CCl(4) exposure through deregulation of multiple molecular pathways. Biochim. Biophys. Acta 2012, 1822, 942–951. [Google Scholar] [CrossRef]
- Shi, H.; Xiao, L.; Duan, W.; He, H.; Ma, L.; Da, M.; Duan, Y.; Wang, Q.; Wu, H.; Song, X.; et al. ANXA2 enhances the progression of hepatocellular carcinoma via remodeling the cell motility associated structures. Micron 2016, 85, 26–33. [Google Scholar] [CrossRef]
- Mas, V.R.; Maluf, D.G.; Archer, K.J.; Yanek, K.; Kong, X.; Kulik, L.; Freise, C.E.; Olthoff, K.M.; Ghobrial, R.M.; McIver, P.; et al. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol. Med. 2009, 15, 85–94. [Google Scholar] [CrossRef]
- Li, Z.; Guan, M.; Lin, Y.; Cui, X.; Zhang, Y.; Zhao, Z.; Zhu, J. Aberrant lipid metabolism in hepatocellular carcinoma revealed by liver lipidomics. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Salomao, M.; Remotti, H.; Vaughan, R.; Siegel, A.B.; Lefkowitch, J.H.; Moreira, R.K. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum. Pathol. 2012, 43, 737–746. [Google Scholar] [CrossRef]
- Shibahara, J.; Ando, S.; Sakamoto, Y.; Kokudo, N.; Fukayama, M. Hepatocellular carcinoma with steatohepatitic features: A clinicopathological study of Japanese patients. Histopathology 2014, 64, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Perveen, S.; Shahbaz, M.; Keshavjee, K.; Guergachi, A. A systematic machine learning based approach for the diagnosis of non-alcoholic fatty liver disease risk and progression. Sci. Rep. 2018, 8, 2112. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Tokushige, K.; Hyogo, H.; Aikata, H.; Nakajima, T.; Ono, M.; Kawanaka, M.; Sawada, K.; Imajo, K.; Honda, K.; et al. A data mining-based prognostic algorithm for NAFLD-related hepatoma patients: A nationwide study by the Japan Study Group of NAFLD. Sci. Rep. 2018, 8, 10434. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology 2018, 155, 282–302. [Google Scholar] [CrossRef]
- Davison, E.J.; Pennington, K.; Hung, C.C.; Peng, J.; Rafiq, R.; Ostareck-Lederer, A.; Ostareck, D.H.; Ardley, H.C.; Banks, R.E.; Robinson, P.A. Proteomic analysis of increased Parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics 2009, 9, 4284–4297. [Google Scholar] [CrossRef]
- Sutheesophon, K.; Kobayashi, Y.; Takatoku, M.A.; Ozawa, K.; Kano, Y.; Ishii, H.; Furukawa, Y. Histone deacetylase inhibitor depsipeptide (FK228) induces apoptosis in leukemic cells by facilitating mitochondrial translocation of Bax, which is enhanced by the proteasome inhibitor bortezomib. Acta Haematol 2006, 115, 78–90. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid metabolic reprogramming in hepatocellular carcinoma. Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Pilo, M.G.; Cigliano, A.; Latte, G.; Simile, M.M.; Ribback, S.; Dombrowski, F.; Evert, M.; Chen, X.; Calvisi, D.F. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle 2017, 16, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Song, X.; Che, L.; Li, X.; Pilo, M.G.; Vidili, G.; Porcu, A.; Solinas, A.; Cigliano, A.; Pes, G.M.; et al. Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells. Liver Int. 2017, 37, 80–89. [Google Scholar] [CrossRef]
- Boissan, M.; Beurel, E.; Wendum, D.; Rey, C.; Lecluse, Y.; Housset, C.; Lacombe, M.L.; Desbois-Mouthon, C. Overexpression of insulin receptor substrate-2 in human and murine hepatocellular carcinoma. Am. J. Pathol. 2005, 167, 869–877. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Takamoto, I.; Obata, A.; Iwamoto, M.; Hayashi, T.; Aihara, M.; Kubota, T.; Nishihara, H.; Kadowaki, T. Role of insulin receptor substrates in the progression of hepatocellular carcinoma. Sci. Rep. 2017, 7, 5387. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Schulze, A. Tumours use a metabolic twist to make lipids. Nature 2019, 566, 333–334. [Google Scholar] [CrossRef]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A pratical and powerful approach tomultiple testing. J. Royal Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 9 October 2019).
- Zambon, A.C.; Gaj, S.; Ho, I.; Hanspers, K.; Vranizan, K.; Evelo, C.T.; Conklin, B.R.; Pico, A.R.; Salomonis, N. GO-Elite: A flexible solution for pathway and ontology over-representation. Bioinformatics 2012, 28, 2209–2210. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Brancotte, B.; Biton, A.; Bernard-Pierrot, I.; Radvanyi, F.; Reyal, F.; Cohen-Boulakia, S. Gene list significance at-a-glance with gene valorization. Bioinformatics 2011, 27, 1187–1189. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Carstensen, B.; Plummer, M. Using lexis objects for multi-state models in R. J. Stat. Softw. 2011, 38, 1–18. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011, 12, 77. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef]
- Culhane, A.C.; Thioulouse, J.; Perriere, G.; Higgins, D.G. MADE4: An R package for multivariate analysis of gene expression data. Bioinformatics 2005, 21, 2789–2790. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desterke, C.; Chiappini, F. Lipid Related Genes Altered in NASH Connect Inflammation in Liver Pathogenesis Progression to HCC: A Canonical Pathway. Int. J. Mol. Sci. 2019, 20, 5594. https://doi.org/10.3390/ijms20225594
Desterke C, Chiappini F. Lipid Related Genes Altered in NASH Connect Inflammation in Liver Pathogenesis Progression to HCC: A Canonical Pathway. International Journal of Molecular Sciences. 2019; 20(22):5594. https://doi.org/10.3390/ijms20225594
Chicago/Turabian StyleDesterke, Christophe, and Franck Chiappini. 2019. "Lipid Related Genes Altered in NASH Connect Inflammation in Liver Pathogenesis Progression to HCC: A Canonical Pathway" International Journal of Molecular Sciences 20, no. 22: 5594. https://doi.org/10.3390/ijms20225594
APA StyleDesterke, C., & Chiappini, F. (2019). Lipid Related Genes Altered in NASH Connect Inflammation in Liver Pathogenesis Progression to HCC: A Canonical Pathway. International Journal of Molecular Sciences, 20(22), 5594. https://doi.org/10.3390/ijms20225594