Death-Associated Protein Kinase 1 Phosphorylation in Neuronal Cell Death and Neurodegenerative Disease

Abstract
1. Introduction
2. Death-associated Protein Kinase Family
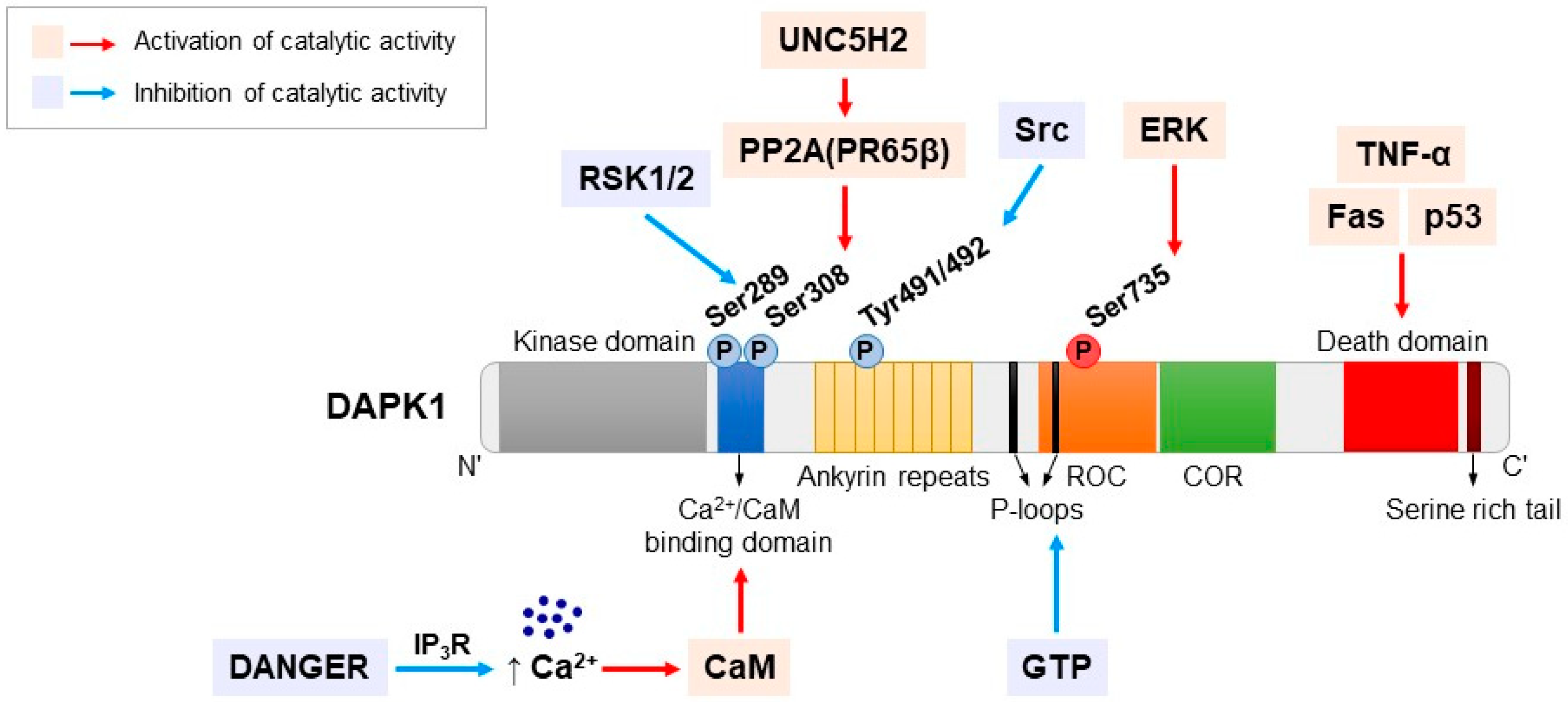
3. DAPK1 Structure
4. Regulation of DAPK1 by Phosphorylation
5. DAPK1 and Neuronal Cell Death
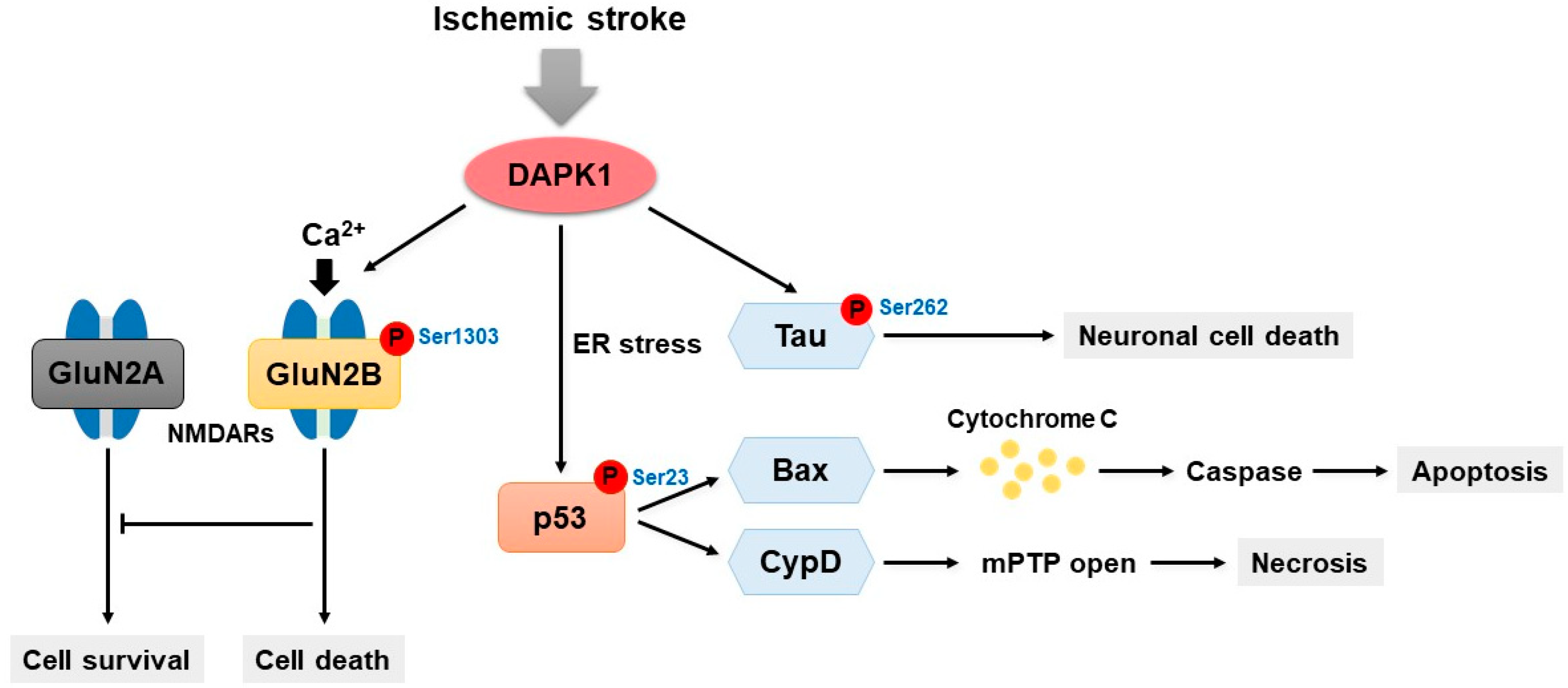
6. DAPK1 and Ischemic Stroke
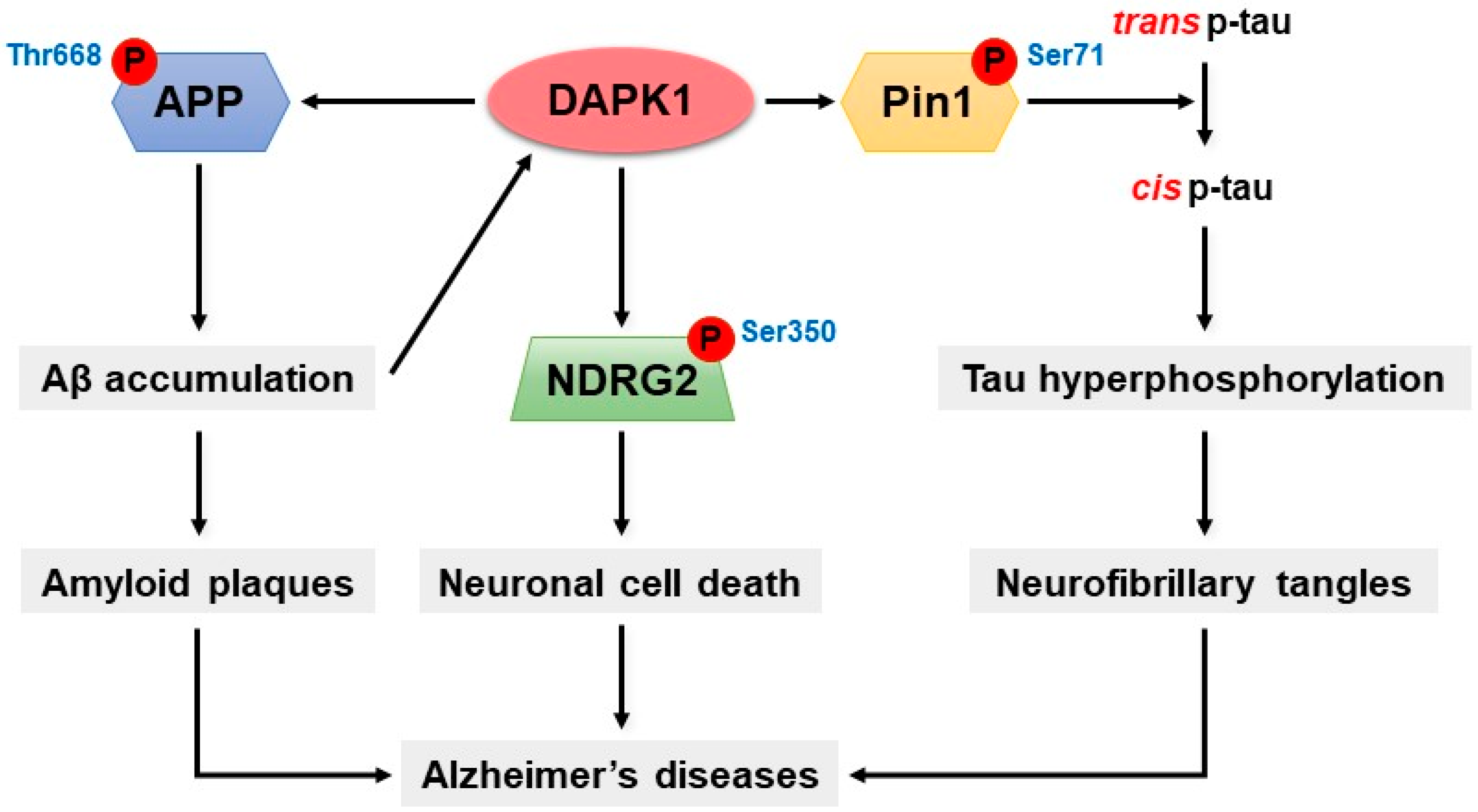
7. DAPK1 and Alzheimer’s Disease
8. DAPK1 as A Potential Target for Neurodegenerative Diseases
9. Conclusions and Perspective
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Chi, H.; Chang, H.Y.; Sang, T.K. Neuronal cell death mechanisms in major neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Peterson, S.E.; Loring, J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 24, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Niemi, N.M.; MacKeigan, J.P. Mitochondrial phosphorylation in apoptosis: Flipping the death switch. Antioxid Redox Signal. 2013, 19, 572–582. [Google Scholar] [CrossRef]
- Singh, P.; Ravanan, P.; Talwar, P. Death associated protein kinase 1 (DAPK1): A regulator of apoptosis and autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. The death-associated protein kinases: Structure, function, and beyond. Annu Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Dapk protein family and cancer. Autophagy 2006, 2, 74–79. [Google Scholar] [CrossRef]
- Chen, R.H.; Wang, W.J.; Kuo, J.C. The tumor suppressor dap-kinase links cell adhesion and cytoskeleton reorganization to cell death regulation. J. Biomed. Sci. 2006, 13, 193–199. [Google Scholar] [CrossRef]
- Sulaiman Alsaadi, M. Role of dapk1 in neuronal cell death, survival and diseases in the nervous system. Int. J. Dev. Neurosci. 2019, 74, 11–17. [Google Scholar] [CrossRef]
- Lai, M.Z.; Chen, R.H. Regulation of inflammation by DAPK. Apoptosis 2014, 19, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Michie, A.M.; McCaig, A.M.; Nakagawa, R.; Vukovic, M. Death-associated protein kinase (dapk) and signal transduction: Regulation in cancer. FEBS J. 2010, 277, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Deiss, L.P.; Feinstein, E.; Berissi, H.; Cohen, O.; Kimchi, A. Identification of a novel serine/threonine kinase and a novel 15-kd protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995, 9, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, R.; Bialik, S.; Kimchi, A. The dapk family: A structure-function analysis. Apoptosis 2014, 19, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Matsumoto, M.; Takeda, K.; Sanjo, H.; Akira, S. Zip kinase, a novel serine/threonine kinase which mediates apoptosis. Mol. Cell. Biol. 1998, 18, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Kogel, D.; Plottner, O.; Landsberg, G.; Christian, S.; Scheidtmann, K.H. Cloning and characterization of dlk, a novel serine/threonine kinase that is tightly associated with chromatin and phosphorylates core histones. Oncogene 1998, 17, 2645–2654. [Google Scholar] [CrossRef]
- Inbal, B.; Shani, G.; Cohen, O.; Kissil, J.L.; Kimchi, A. Death-associated protein kinase-related protein 1, a novel serine/threonine kinase involved in apoptosis. Mol. Cell. Biol. 2000, 20, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Nomura, F.; Hoshino, K.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Akira, S. Death-associated protein kinase 2 is a new calcium/calmodulin-dependent protein kinase that signals apoptosis through its catalytic activity. Oncogene 1999, 18, 3471–3480. [Google Scholar] [CrossRef]
- Sanjo, H.; Kawai, T.; Akira, S. Draks, novel serine/threonine kinases related to death-associated protein kinase that trigger apoptosis. J. Biol. Chem. 1998, 273, 29066–29071. [Google Scholar] [CrossRef]
- Cohen, O.; Feinstein, E.; Kimchi, A. Dap-kinase is a ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J. 1997, 16, 998–1008. [Google Scholar] [CrossRef]
- Kojima, H.; Nemoto, A.; Uemura, T.; Honma, R.; Ogura, M.; Liu, Y. Rdrak1, a novel kinase related to apoptosis, is strongly expressed in active osteoclasts and induces apoptosis. J. Biol. Chem. 2001, 276, 19238–19243. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Hagberg, H.; Krishnamurthy, R.; Thornton, C.; Mallard, C. Death associated protein kinases: Molecular structure and brain injury. Int. J. Mol. Sci. 2013, 14, 13858–13872. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhou, X.Z.; Lee, T.H. Death-associated protein kinase 1 as a promising drug target in cancer and alzheimer’s disease. Recent Pat. Anticancer Drug Discov. 2019, 14, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.K.; Roh, E.J. Death-associated protein kinase (dapk) family modulators: Current and future therapeutic outcomes. Med. Res. Rev. 2019, 39, 349–385. [Google Scholar] [CrossRef] [PubMed]
- Tereshko, V.; Teplova, M.; Brunzelle, J.; Watterson, D.M.; Egli, M. Crystal structures of the catalytic domain of human protein kinase associated with apoptosis and tumor suppression. Nat. Struct. Biol. 2001, 8, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. Dap-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin Cancer Biol. 2004, 14, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; You, M.H.; Chen, C.H.; Suh, J.; Tanzi, R.E.; Ho Lee, T. Inhibition of death-associated protein kinase 1 attenuates the phosphorylation and amyloidogenic processing of amyloid precursor protein. Hum. Mol. Genet. 2016, 25, 2498–2513. [Google Scholar] [CrossRef] [PubMed]
- Velentza, A.V.; Schumacher, A.M.; Weiss, C.; Egli, M.; Watterson, D.M. A protein kinase associated with apoptosis and tumor suppression: Structure, activity, and discovery of peptide substrates. J. Biol. Chem. 2001, 276, 38956–38965. [Google Scholar] [CrossRef]
- Li, J.; Mahajan, A.; Tsai, M.D. Ankyrin repeat: A unique motif mediating protein-protein interactions. Biochemistry 2006, 45, 15168–15178. [Google Scholar] [CrossRef]
- Lin, Y.; Stevens, C.; Harrison, B.; Pathuri, S.; Amin, E.; Hupp, T.R. The alternative splice variant of dapk-1, s-dapk-1, induces proteasome-independent dapk-1 destabilization. Mol. Cell. Biochem. 2009, 328, 101–107. [Google Scholar] [CrossRef]
- Carlessi, R.; Levin-Salomon, V.; Ciprut, S.; Bialik, S.; Berissi, H.; Albeck, S.; Peleg, Y.; Kimchi, A. Gtp binding to the roc domain of dap-kinase regulates its function through intramolecular signalling. EMBO Rep. 2011, 12, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chen, C.H.; Suizu, F.; Huang, P.; Schiene-Fischer, C.; Daum, S.; Zhang, Y.J.; Goate, A.; Chen, R.H.; Zhou, X.Z.; et al. Death-associated protein kinase 1 phosphorylates pin1 and inhibits its prolyl isomerase activity and cellular function. Mol. Cell. 2011, 42, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase pin1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, E.; Kimchi, A.; Wallach, D.; Boldin, M.; Varfolomeev, E. The death domain: A module shared by proteins with diverse cellular functions. Trends Biochem. Sci. 1995, 20, 342–344. [Google Scholar] [CrossRef]
- Cohen, O.; Kimchi, A. Dap-kinase: From functional gene cloning to establishment of its role in apoptosis and cancer. Cell Death Differ. 2001, 8, 6–15. [Google Scholar] [CrossRef]
- Itoh, N.; Nagata, S. A novel protein domain required for apoptosis. Mutational analysis of human fas antigen. J. Biol. Chem. 1993, 268, 10932–10937. [Google Scholar] [PubMed]
- Shohat, G.; Spivak-Kroizman, T.; Cohen, O.; Bialik, S.; Shani, G.; Berrisi, H.; Eisenstein, M.; Kimchi, A. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J. Biol. Chem. 2001, 276, 47460–47467. [Google Scholar] [CrossRef]
- Shani, G.; Henis-Korenblit, S.; Jona, G.; Gileadi, O.; Eisenstein, M.; Ziv, T.; Admon, A.; Kimchi, A. Autophosphorylation restrains the apoptotic activity of drp-1 kinase by controlling dimerization and calmodulin binding. EMBO J. 2001, 20, 1099–1113. [Google Scholar] [CrossRef]
- Bialik, S.; Kimchi, A. The dap-kinase interactome. Apoptosis 2014, 19, 316–328. [Google Scholar] [CrossRef]
- Widau, R.C.; Jin, Y.; Dixon, S.A.; Wadzinski, B.E.; Gallagher, P.J. Protein phosphatase 2a (pp2a) holoenzymes regulate death-associated protein kinase (dapk) in ceramide-induced anoikis. J. Biol. Chem. 2010, 285, 13827–13838. [Google Scholar] [CrossRef]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2a regulatory subunits and cancer. Biochim. Biophys. Acta 2009, 1795, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Luo, W.; Zeng, C.; Zhang, Y.; Wang, L.; Yao, W.; Nie, C. Pp2a mediates apoptosis or autophagic cell death in multiple myeloma cell lines. Oncotarget 2017, 8, 80770–80789. [Google Scholar] [CrossRef] [PubMed]
- Truttmann, A.C.; Ashraf, Q.; Mishra, O.P.; Delivoria-Papadopoulos, M. Effect of hypoxia on protein phosphatase 2a activity, subcellular distribution and expression in cerebral cortex of newborn piglets. Neuroscience 2004, 127, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Usui, T.; Ohama, T.; Sato, K. Regulation of beclin 1 protein phosphorylation and autophagy by protein phosphatase 2a (pp2a) and death-associated protein kinase 3 (dapk3). J. Biol. Chem. 2016, 291, 10858–10866. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, Y. Novel ser/thr protein phosphatases in cell death regulation. Physiology (Bethesda) 2012, 27, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Guenebeaud, C.; Goldschneider, D.; Castets, M.; Guix, C.; Chazot, G.; Delloye-Bourgeois, C.; Eisenberg-Lerner, A.; Shohat, G.; Zhang, M.; Laudet, V.; et al. The dependence receptor unc5h2/b triggers apoptosis via pp2a-mediated dephosphorylation of dap kinase. Mol. Cell. 2010, 40, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Wang, W.J.; Kuo, J.C.; Tsai, H.C.; Lin, J.R.; Chang, Z.F.; Chen, R.H. Bidirectional signals transduced by dapk-erk interaction promote the apoptotic effect of dapk. EMBO J. 2005, 24, 294–304. [Google Scholar] [CrossRef]
- Dioletis, E.; Dingley, A.J.; Driscoll, P.C. Structural and functional characterization of the recombinant death domain from death-associated protein kinase. PLoS ONE 2013, 8, e70095. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. Erk and cell death: Mechanisms of erk-induced cell death—apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Subramaniam, S.; Zirrgiebel, U.; von Bohlen Und Halbach, O.; Strelau, J.; Laliberte, C.; Kaplan, D.R.; Unsicker, K. Erk activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J. Cell Biol. 2004, 165, 357–369. [Google Scholar] [CrossRef]
- Xiong, W.; Wu, Y.; Xian, W.; Song, L.; Hu, L.; Pan, S.; Liu, M.; Yao, S.; Pei, L.; Shang, Y. Dapk1-erk signal mediates oxygen glucose deprivation reperfusion induced apoptosis in mouse n2a cells. J. Neurol. Sci. 2018, 387, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Novel functions of death-associated protein kinases through mitogen-activated protein kinase-related signals. Int. J. Mol. Sci. 2018, 19, 3031. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.A.; Blenis, J. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (mapk) kinase (mek)-mapk cell survival signals. Cell Growth Differ. 2001, 12, 397–408. [Google Scholar] [PubMed]
- Simon, B.; Huart, A.S.; Temmerman, K.; Vahokoski, J.; Mertens, H.D.; Komadina, D.; Hoffmann, J.E.; Yumerefendi, H.; Svergun, D.I.; Kursula, P.; et al. Death-associated protein kinase activity is regulated by coupled calcium/calmodulin binding to two distinct sites. Structure 2016, 24, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, S.; Scheibe, K.; Erlenbach-Wuensch, K.; Neufert, C.; Schneider-Stock, R. Death-associated protein kinase: A molecule with functional antagonistic duality and a potential role in inflammatory bowel disease (review). Int. J. Oncol. 2015, 47, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Hupp, T.R.; Stevens, C. Death-associated protein kinase (dapk) and signal transduction: Additional roles beyond cell death. FEBS J. 2010, 277, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Anjum, R.; Roux, P.P.; Ballif, B.A.; Gygi, S.P.; Blenis, J. The tumor suppressor dap kinase is a target of rsk-mediated survival signaling. Curr. Biol. 2005, 15, 1762–1767. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Kuo, J.C.; Ku, W.; Lee, Y.R.; Lin, F.C.; Chang, Y.L.; Lin, Y.M.; Chen, C.H.; Huang, Y.P.; Chiang, M.J.; et al. The tumor suppressor dapk is reciprocally regulated by tyrosine kinase src and phosphatase lar. Mol. Cell 2007, 27, 701–716. [Google Scholar] [CrossRef]
- Tur, M.K.; Daramola, A.K.; Gattenlohner, S.; Herling, M.; Chetty, S.; Barth, S. Restoration of dap kinase tumor suppressor function: A therapeutic strategy to selectively induce apoptosis in cancer cells using immunokinase fusion proteins. Biomedicines 2017, 5. [Google Scholar] [CrossRef]
- Tsai, Y.T.; Chuang, M.J.; Tang, S.H.; Wu, S.T.; Chen, Y.C.; Sun, G.H.; Hsiao, P.W.; Huang, S.M.; Lee, H.J.; Yu, C.P.; et al. Novel cancer therapeutics with allosteric modulation of the mitochondrial c-raf-dapk complex by raf inhibitor combination therapy. Cancer Res. 2015, 75, 3568–3582. [Google Scholar] [CrossRef]
- Gorman, A.M. Neuronal cell death in neurodegenerative diseases: Recurring themes around protein handling. J. Cell Mol. Med. 2008, 12, 2263–2280. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, A.G.; Faden, A.I. Mechanisms of neural cell death: Implications for development of neuroprotective treatment strategies. NeuroRx 2004, 1, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Merighi, A. In vivo cellular and molecular mechanisms of neuronal apoptosis in the mammalian cns. Prog. Neurobiol. 2003, 69, 287–312. [Google Scholar] [CrossRef]
- Yuan, J.; Lipinski, M.; Degterev, A. Diversity in the mechanisms of neuronal cell death. Neuron 2003, 40, 401–413. [Google Scholar] [CrossRef]
- Chan, F.K.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, K.A. Molecular mechanisms of neuronal cell death. Ann. N Y Acad Sci 2003, 991, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Dachsel, J.C.; Wider, C.; Vilarino-Guell, C.; Aasly, J.O.; Rajput, A.; Rajput, A.H.; Lynch, T.; Craig, D.; Krygowska-Wajs, A.; Jasinska-Myga, B.; et al. Death-associated protein kinase 1 variation and parkinson’s disease. Eur. J. Neurol. 2011, 18, 1090–1093. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; You, M.H.; Chen, C.H.; Lee, S.; Hong, Y.; Hong, Y.; Kimchi, A.; Zhou, X.Z.; Lee, T.H. Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis. 2014, 5, e1237. [Google Scholar] [CrossRef]
- Su, Y.; Deng, M.F.; Xiong, W.; Xie, A.J.; Guo, J.; Liang, Z.H.; Hu, B.; Chen, J.G.; Zhu, X.; Man, H.Y.; et al. Microrna-26a/death-associated protein kinase 1 signaling induces synucleinopathy and dopaminergic neuron degeneration in parkinson’s disease. Biol. Psychiatry 2019, 85, 769–781. [Google Scholar] [CrossRef]
- Wang, S.; Shi, X.; Li, H.; Pang, P.; Pei, L.; Shen, H.; Lu, Y. Dapk1 signaling pathways in stroke: From mechanisms to therapies. Mol. Neurobiol. 2017, 54, 4716–4722. [Google Scholar] [CrossRef]
- Cohen, O.; Inbal, B.; Kissil, J.L.; Raveh, T.; Berissi, H.; Spivak-Kroizaman, T.; Feinstein, E.; Kimchi, A. Dap-kinase participates in tnf-α–and fas-induced apoptosis and its function requires the death domain. J. Cell Biol. 1999, 146, 141–148. [Google Scholar]
- You, M.H.; Kim, B.M.; Chen, C.H.; Begley, M.J.; Cantley, L.C.; Lee, T.H. Death-associated protein kinase 1 phosphorylates ndrg2 and induces neuronal cell death. Cell Death Differ. 2017, 24, 238–250. [Google Scholar] [CrossRef]
- Inbal, B.; Cohen, O.; Polak-Charcon, S.; Kopolovic, J.; Vadai, E.; Eisenbach, L.; Kimchi, A. Dap kinase links the control of apoptosis to metastasis. Nature 1997, 390, 180–184. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by tgf-beta and potentiates tgf-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef]
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. Tgf-beta induces apoptosis through smad-mediated expression of dap-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. Dap-kinase-mediated phosphorylation on the bh3 domain of beclin 1 promotes dissociation of beclin 1 from bcl-xl and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Gambaro, K.; Aberdam, E.; Virolle, T.; Aberdam, D.; Rouleau, M. Bmp-4 induces a smad-dependent apoptotic cell death of mouse embryonic stem cell-derived neural precursors. Cell Death Differ. 2006, 13, 1075–1087. [Google Scholar] [CrossRef]
- Pei, L.; Shang, Y.; Jin, H.; Wang, S.; Wei, N.; Yan, H.; Wu, Y.; Yao, C.; Wang, X.; Zhu, L.Q.; et al. Dapk1-p53 interaction converges necrotic and apoptotic pathways of ischemic neuronal death. J. Neurosci. 2014, 34, 6546–6556. [Google Scholar] [CrossRef]
- Araki, T.; Shinoda, S.; Schindler, C.K.; Quan-Lan, J.; Meller, R.; Taki, W.; Simon, R.P.; Henshall, D.C. Expression, interaction, and proteolysis of death-associated protein kinase and p53 within vulnerable and resistant hippocampal subfields following seizures. Hippocampus 2004, 14, 326–336. [Google Scholar] [CrossRef]
- Wu, L.; Wang, H.M.; Li, J.L.; Feng, H.X.; Zhao, W.M.; Zhang, H.Y. Dual anti-ischemic effects of rosmarinic acid n-butyl ester via alleviation of dapk-p53-mediated neuronal damage and microglial inflammation. Acta Pharmacol. Sin. 2017, 38, 459–468. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Kimchi, A. Dap kinase regulates jnk signaling by binding and activating protein kinase d under oxidative stress. Cell Death Differ. 2007, 14, 1908–1915. [Google Scholar] [CrossRef]
- Kang, C.; Avery, L. Death-associated protein kinase (dapk) and signal transduction: Fine-tuning of autophagy in caenorhabditis elegans homeostasis. FEBS J. 2010, 277, 66–73. [Google Scholar] [CrossRef]
- Pattingre, S.; Bauvy, C.; Carpentier, S.; Levade, T.; Levine, B.; Codogno, P. Role of jnk1-dependent bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009, 284, 2719–2728. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. Jnk1-mediated phosphorylation of bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamashita, T. Role of dapk in neuronal cell death. Apoptosis 2014, 19, 339–345. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic nmda receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef]
- Martin, H.G.; Wang, Y.T. Blocking the deadly effects of the nmda receptor in stroke. Cell 2010, 140, 174–176. [Google Scholar] [CrossRef]
- Paudice, P.; Gemignani, A.; Raiteri, M. Evidence for functional native nmda receptors activated by glycine or d-serine alone in the absence of glutamatergic coagonist. Eur. J. Neurosci. 1998, 10, 2934–2944. [Google Scholar] [CrossRef]
- Wang, C.C.; Held, R.G.; Chang, S.C.; Yang, L.; Delpire, E.; Ghosh, A.; Hall, B.J. A critical role for glun2b-containing nmda receptors in cortical development and function. Neuron 2011, 72, 789–805. [Google Scholar] [CrossRef]
- Li, S.X.; Han, Y.; Xu, L.Z.; Yuan, K.; Zhang, R.X.; Sun, C.Y.; Xu, D.F.; Yuan, M.; Deng, J.H.; Meng, S.Q.; et al. Uncoupling dapk1 from nmda receptor glun2b subunit exerts rapid antidepressant-like effects. Mol. Psychiatry 2018, 23, 597–608. [Google Scholar] [CrossRef]
- Tu, W.; Xu, X.; Peng, L.; Zhong, X.; Zhang, W.; Soundarapandian, M.M.; Balel, C.; Wang, M.; Jia, N.; Zhang, W.; et al. Dapk1 interaction with nmda receptor nr2b subunits mediates brain damage in stroke. Cell 2010, 140, 222–234. [Google Scholar] [CrossRef]
- Van Rossum, D.B.; Patterson, R.L.; Cheung, K.H.; Barrow, R.K.; Syrovatkina, V.; Gessell, G.S.; Burkholder, S.G.; Watkins, D.N.; Foskett, J.K.; Snyder, S.H. Danger, a novel regulatory protein of inositol 1,4,5-trisphosphate-receptor activity. J. Biol. Chem. 2006, 281, 37111–37116. [Google Scholar] [CrossRef]
- Kang, B.N.; Ahmad, A.S.; Saleem, S.; Patterson, R.L.; Hester, L.; Dore, S.; Snyder, S.H. Death-associated protein kinase-mediated cell death modulated by interaction with danger. J. Neurosci. 2010, 30, 93–98. [Google Scholar] [CrossRef]
- Del Rosario, J.S.; Feldmann, K.G.; Ahmed, T.; Amjad, U.; Ko, B.; An, J.; Mahmud, T.; Salama, M.; Mei, S.; Asemota, D.; et al. Death associated protein kinase (dapk)-mediated neurodegenerative mechanisms in nematode excitotoxicity. BMC Neurosci. 2015, 16, 25. [Google Scholar] [CrossRef]
- Kwon, T.; Youn, H.; Son, B.; Kim, D.; Seong, K.M.; Park, S.; Kim, W.; Youn, B. Danger is involved in high glucose-induced radioresistance through inhibiting dapk-mediated anoikis in non-small cell lung cancer. Oncotarget 2016, 7, 7193–7206. [Google Scholar] [CrossRef]
- Hou, S.T.; MacManus, J.P. Molecular mechanisms of cerebral ischemia-induced neuronal death. Int. Rev. Cytol. 2002, 221, 93–148. [Google Scholar]
- Shamloo, M.; Soriano, L.; Wieloch, T.; Nikolich, K.; Urfer, R.; Oksenberg, D. Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J. Biol. Chem. 2005, 280, 42290–42299. [Google Scholar] [CrossRef]
- Schumacher, A.M.; Velentza, A.V.; Watterson, D.M.; Wainwright, M.S. Dapk catalytic activity in the hippocampus increases during the recovery phase in an animal model of brain hypoxic-ischemic injury. Biochim. Biophys. Acta 2002, 1600, 128–137. [Google Scholar] [CrossRef]
- Zhao, L.P.; Ji, C.; Lu, P.H.; Li, C.; Xu, B.; Gao, H. Oxygen glucose deprivation (ogd)/re-oxygenation-induced in vitro neuronal cell death involves mitochondrial cyclophilin-d/p53 signaling axis. Neurochem. Res. 2013, 38, 705–713. [Google Scholar] [CrossRef]
- Takarada, T.; Hara, T.; Konishi, S.; Nakazato, R.; Yoneda, Y. Selective downregulation of n-methyl-d-aspartate receptor (nmdar) rather than non-nmdar subunits in ipsilateral cerebral hemispheres in rats with middle cerebral artery occlusion. Nihon Shinkei Seishin Yakurigaku Zasshi 2011, 31, 187–194. [Google Scholar]
- Pei, L.; Wang, S.; Jin, H.; Bi, L.; Wei, N.; Yan, H.; Yang, X.; Yao, C.; Xu, M.; Shu, S.; et al. A novel mechanism of spine damages in stroke via dapk1 and tau. Cereb Cortex 2015, 25, 4559–4571. [Google Scholar] [CrossRef]
- Sidaway, P. Stroke: Tau—a new target in acute brain ischaemia. Nat. Rev. Neurol 2017, 13, 639. [Google Scholar] [CrossRef]
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Muller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473. [Google Scholar] [CrossRef]
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Yankner, B.A. Mechanisms of neuronal degeneration in alzheimer’s disease. Neuron 1996, 16, 921–932. [Google Scholar] [CrossRef]
- Zhou, Z.-d.; Chan, C.H.-s.; Ma, Q.-h.; Xu, X.-h.; Xiao, Z.-c.; Tan, E.-K. The roles of amyloid precursor protein (app) in neurogenesis. Cell Adhes. Migr. 2014, 5, 280–292. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in alzheimer cytoskeletal pathology. Proc. Natl. Acad Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Duan, D.X.; Chai, G.S.; Ni, Z.F.; Hu, Y.; Luo, Y.; Cheng, X.S.; Chen, N.N.; Wang, J.Z.; Liu, G.P. Phosphorylation of tau by death-associated protein kinase 1 antagonizes the kinase-induced cell apoptosis. J. Alzheimers Dis. 2013, 37, 795–808. [Google Scholar] [CrossRef]
- Matsuo, E.S.; Shin, R.W.; Billingsley, M.L.; Van deVoorde, A.; O’Connor, M.; Trojanowski, J.Q.; Lee, V.M. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Adlard, P.A.; Masters, C.L.; Bush, A.I. Tau protein: Relevance to parkinson’s disease. Int. J. Biochem. Cell Biol. 2010, 42, 1775–1778. [Google Scholar] [CrossRef]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef]
- Wu, P.R.; Tsai, P.I.; Chen, G.C.; Chou, H.J.; Huang, Y.P.; Chen, Y.H.; Lin, M.Y.; Kimchi, A.; Chien, C.T.; Chen, R.H. Dapk activates mark1/2 to regulate microtubule assembly, neuronal differentiation, and tau toxicity. Cell Death Differ. 2011, 18, 1507–1520. [Google Scholar] [CrossRef]
- Lee, T.H.; Pastorino, L.; Lu, K.P. Peptidyl-prolyl cis-trans isomerase pin1 in ageing, cancer and alzheimer disease. Expert. Rev. Mol. Med. 2011, 13, e21. [Google Scholar] [CrossRef]
- Zhou, X.Z.; Lu, K.P. The isomerase pin1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef]
- Lu, K.P.; Kondo, A.; Albayram, O.; Herbert, M.K.; Liu, H.; Zhou, X.Z. Potential of the antibody against cis-phosphorylated tau in the early diagnosis, treatment, and prevention of alzheimer disease and brain injury. JAMA Neurol. 2016, 73, 1356–1362. [Google Scholar] [CrossRef]
- Balastik, M.; Lim, J.; Pastorino, L.; Lu, K.P. Pin1 in alzheimer’s disease: Multiple substrates, one regulatory mechanism? Biochim. Biophys. Acta 2007, 1772, 422–429. [Google Scholar] [CrossRef]
- Holzer, M.; Gartner, U.; Stobe, A.; Hartig, W.; Gruschka, H.; Bruckner, M.K.; Arendt, T. Inverse association of pin1 and tau accumulation in alzheimer’s disease hippocampus. Acta Neuropathol. 2002, 104, 471–481. [Google Scholar] [PubMed]
- Ramakrishnan, P.; Dickson, D.W.; Davies, P. Pin1 colocalization with phosphorylated tau in Alzheimer’s disease and other tauopathies. Neurobiol. Dis. 2003, 14, 251–264. [Google Scholar] [CrossRef]
- Albayram, O.; Herbert, M.K.; Kondo, A.; Tsai, C.Y.; Baxley, S.; Lian, X.; Hansen, M.; Zhou, X.Z.; Lu, K.P. Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci. 2016, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and alzheimer’s disease. Annu Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kao, S.C.; Lemere, C.A.; Xia, W.; Tseng, H.C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.H. App processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.F.; Sun, Y.N.; Liu, D.M.; Yin, H.J.; Peng, Y.; Xu, S.F.; Wang, L.; Wang, X.L. The pathological roles of ndrg2 in Alzheimer’s disease, a study using animal models and appwt-overexpressed cells. CNS Neurosci. Ther. 2017, 23, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Melotte, V.; Qu, X.; Ongenaert, M.; van Criekinge, W.; de Bruine, A.P.; Baldwin, H.S.; van Engeland, M. The n-myc downstream regulated gene (ndrg) family: Diverse functions, multiple applications. FASEB J. 2010, 24, 4153–4166. [Google Scholar] [CrossRef]
- Mitchelmore, C.; Buchmann-Moller, S.; Rask, L.; West, M.J.; Troncoso, J.C.; Jensen, N.A. Ndrg2: A novel Alzheimer’s disease associated protein. Neurobiol. Dis. 2004, 16, 48–58. [Google Scholar] [CrossRef]
- Velentza, A.V.; Wainwright, M.S.; Zasadzki, M.; Mirzoeva, S.; Schumacher, A.M.; Haiech, J.; Focia, P.J.; Egli, M.; Watterson, D.M. An aminopyridazine-based inhibitor of a pro-apoptotic protein kinase attenuates hypoxia-ischemia induced acute brain injury. Bioorg. Med. Chem. Lett. 2003, 13, 3465–3470. [Google Scholar] [CrossRef]
- Mirzoeva, S.; Sawkar, A.; Zasadzki, M.; Guo, L.; Velentza, A.V.; Dunlap, V.; Bourguignon, J.J.; Ramstrom, H.; Haiech, J.; Van Eldik, L.J.; et al. Discovery of a 3-amino-6-phenyl-pyridazine derivative as a new synthetic antineuroinflammatory compound. J. Med. Chem. 2002, 45, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Takayama, K.; Shimizu, T.; Muroya, A.; Furuya, T. Structure-activity relationship of novel dapk inhibitors identified by structure-based virtual screening. Bioorg. Med. Chem. 2010, 18, 2728–2734. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.A.; Franke, A.S.; Weitzel, D.H.; Speer, B.L.; Hughes, P.F.; Hagerty, L.; Fortner, C.N.; Veal, J.M.; Barta, T.E.; Zieba, B.J.; et al. Fluorescence linked enzyme chemoproteomic strategy for discovery of a potent and selective dapk1 and zipk inhibitor. ACS Chem. Biol. 2013, 8, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Kosaka, Y.; Mizuguchi, M. Structural insight into the interactions between death-associated protein kinase 1 and natural flavonoids. J. Med. Chem. 2015, 58, 7400–7408. [Google Scholar] [CrossRef] [PubMed]
- Wilbek, T.S.; Skovgaard, T.; Sorrell, F.J.; Knapp, S.; Berthelsen, J.; Stromgaard, K. Identification and characterization of a small-molecule inhibitor of death-associated protein kinase 1. Chembiochem 2015, 16, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Talwar, P. Exploring putative inhibitors of death associated protein kinase 1 (dapk1) via targeting gly-glu-leu (gel) and pro-glu-asn (pen) substrate recognition motifs. J. Mol. Graph. Model. 2017, 77, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.K.; Hassan, A.H.E.; Jeong, H.; Kwon, Y.; Choi, J.G.; Oh, M.S.; Park, K.D.; Kim, Y.K.; Roh, E.J. First-in-class dapk1/csf1r dual inhibitors: Discovery of 3,5-dimethoxy-n-(4-(4-methoxyphenoxy)-2-((6-morpholinopyridin-3-yl)amino)pyrimidi n-5-yl)benzamide as a potential anti-tauopathies agent. Eur. J. Med. Chem. 2019, 162, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.H.; Yao, Y.; Lin, Y.M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis p-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Greenwood, A.; Binder, L.; Bigio, E.H.; Denial, S.; Nicholson, L.; Zhou, X.Z.; Lu, K.P. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in alzheimer’s disease. Cell 2012, 149, 232–244. [Google Scholar] [CrossRef]
- Albayram, O.; Angeli, P.; Bernstein, E.; Baxley, S.; Gao, Z.; Lu, K.P.; Zhou, X.Z. Targeting prion-like cis phosphorylated tau pathology in neurodegenerative diseases. J. Alzheimers Dis. Parkinsonism 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Albayram, O.; Kondo, A.; Mannix, R.; Smith, C.; Tsai, C.Y.; Li, C.; Herbert, M.K.; Qiu, J.; Monuteaux, M.; Driver, J.; et al. Cis p-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 2017, 8, 1000. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Molecule | IC50 | Function of Inhibitor in Neuron | Ref. |
|---|---|---|---|
| Alkylated 3-amino-6-phenylpyridazine | 13 μM | Reduction of brain injury and treatment of neuronal cell death | [130,131] |
| 4-(pyridin-3-ylmethylene)oxazol-5(4H)-one | 69 nM | Potential treatment of ischemic stroke and attenuation of Aβ-induced cell death | [27,72,132] |
| Peptide-based DAPK1 inhibitor targeting GluN2B and CTM | N/A | Protection of neuron in ischemic brain | [133] |
| Pyrazolo[3,4-d]pyrimidinone (HS38) | 200 nM | N/A | [134] |
| Morin | 11 μM | N/A | [135] |
| Imidazo-pyramidazine | 247 nM | N/A | [136] |
| Peptide-based DAPK1 inhibitor targeting GEL and PEN | 30 nM | N/A | [137] |
| 3,5-dimethoxy-N-(4-(4-methoxyphenoxy)-2-((6-morpholinopyridin-3-yl)amino)pyrimidin-5-yl)benzamide | 1.25 μM | Reduction of tau aggregation | [138] |
| Conformational-specific cis p-tau antibody | N/A | Reduce tau pathology and prevent neural degeneration | [33,118,123,139,140,141,142] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, N.; Chen, D.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 Phosphorylation in Neuronal Cell Death and Neurodegenerative Disease. Int. J. Mol. Sci. 2019, 20, 3131. https://doi.org/10.3390/ijms20133131
Kim N, Chen D, Zhou XZ, Lee TH. Death-Associated Protein Kinase 1 Phosphorylation in Neuronal Cell Death and Neurodegenerative Disease. International Journal of Molecular Sciences. 2019; 20(13):3131. https://doi.org/10.3390/ijms20133131
Chicago/Turabian StyleKim, Nami, Dongmei Chen, Xiao Zhen Zhou, and Tae Ho Lee. 2019. "Death-Associated Protein Kinase 1 Phosphorylation in Neuronal Cell Death and Neurodegenerative Disease" International Journal of Molecular Sciences 20, no. 13: 3131. https://doi.org/10.3390/ijms20133131
APA StyleKim, N., Chen, D., Zhou, X. Z., & Lee, T. H. (2019). Death-Associated Protein Kinase 1 Phosphorylation in Neuronal Cell Death and Neurodegenerative Disease. International Journal of Molecular Sciences, 20(13), 3131. https://doi.org/10.3390/ijms20133131