Abstract
Angiogenesis, the formation of new blood vessels, is critical for survival and in the regenerative response to tissue injury or ischemia. However, in diseases such as cancer and atherosclerosis, inflammation can cause unregulated angiogenesis leading to excessive neovascularization, which exacerbates disease. Current anti-angiogenic therapies cause complete inhibition of both inflammatory and ischemia driven angiogenesis causing a range of side effects in patients. Specific inhibition of inflammation-driven angiogenesis would therefore be immensely valuable. Increasing evidence suggests that the CC-chemokine class promotes inflammation-driven angiogenesis, whilst there is little evidence for a role in ischemia-mediated angiogenesis. The differential regulation of angiogenesis by CC-chemokines suggests it may provide an alternate strategy to treat angiogenesis associated pathological diseases. The focus of this review is to highlight the significant role of the CC-chemokine class in inflammation, versus ischemia driven angiogenesis, and to discuss the related pathologies including atherosclerosis, cancer, and rheumatoid arthritis. We examine the pros and cons of anti-angiogenic therapies currently in clinical trials. We also reveal novel therapeutic strategies that cause broad-spectrum inhibition of the CC-chemokine class that may have future potential for the specific inhibition of inflammatory angiogenesis.
1. Introduction
Angiogenesis is the formation of new blood vessels from pre-existing blood vessels [1]. It is an essential process for development, growth, and repair. Under postnatal physiological conditions, angiogenesis is important for tissue neovascularization in response to ischemia (e.g., following myocardial infarction and in peripheral vascular disease) and in repair during wound healing. In contrast, uncontrolled regulation of angiogenesis that occurs with excessive or prolonged inflammation can lead to the promotion of pathological angiogenesis that is associated with the acceleration of diseases such as cancer and atherosclerosis. Increasing evidence implicates chemokines as key regulators of angiogenesis. Several members of the CC-chemokine class are implicated in a number of diseases in which inflammation-induced pathological angiogenesis plays an important role. Inflammatory angiogenesis is not, however, exclusive to the CC-chemokine class and members of the CXC and CX3C classes also play a role. Some CXC-chemokines, including those which bind to CXCR2 (and CXCR1 in humans), are also known to promote inflammation-driven angiogenesis. For example, CXCL5 and CXCL8 which bind to CXCR2 promote angiogenesis [2], while CXCL4 and CXCL10, which bind to CXCR3 inhibit angiogenesis in the context of inflammation [3]. Furthermore, in vitro studies have shown that the CX3C chemokine CX3CL1 (fractalkine) is a stimulator of angiogenic functions such as endothelial cell migration, proliferation and tubulogenesis [4].
Inflammation-driven diseases associated with angiogenesis are highly prevalent globally, with cardiovascular related disorders and cancer being the leading causes of mortality worldwide [5]. Current anti-angiogenic therapies suppress angiogenesis in all pathophysiological conditions, and while many are successful in suppressing disease progression, they often cause severe side effects including high blood pressure and gastrointestinal disorders [6,7]. This highlights the need for novel therapeutic targets and/or strategies that can specifically inhibit inflammation-driven angiogenesis while having no effect on important physiological ischemia-mediated angiogenic processes.
2. Conditional Regulation of Angiogenesis
The regulation of angiogenesis involves a number of critical growth factors, cytokines, signaling cascades and cellular processes that are triggered in response to either an inflammatory or ischemic stimulus. Inflammatory and ischemia-driven angiogenesis are regulated via two distinctly different, yet slightly overlapping pathways [8]. Inflammation-driven pathological angiogenesis accelerates a number of disease processes, in particular tumor growth in cancer and plaque expansion in atherosclerosis through intra-plaque neovascularization [8,9,10]. Inflammatory angiogenesis also exacerbates rheumatoid arthritis [11]. A key promoter in the inflammatory angiogenic pathway is the transcription factor nuclear factor-κB (NF-κB). Whilst, in ischemia-induced physiological angiogenesis that is critical for wound healing or following a myocardial infarction to promote re-oxygenation of the heart tissues, the transcription factor, hypoxia inducible transcription factor-1α (HIF-1α), is the key promoter. Interestingly, vascular endothelial growth factor (VEGF) is a key promoter of both inflammation-driven pathological angiogenesis and ischemia-induced physiological angiogenesis. This is possible because VEGF has response elements for both NF-κB and HIF-1α in its promoter region (Figure 1), allowing for activation of its production from both signaling pathways [12,13,14,15]. HIF-1α also possesses a response element for NF-κB signaling [16]. By having both NF-κB and hypoxia response elements (HREs) in the promoter regions of HIF-1α and VEGF, it enables conditional regulation of angiogenesis. This implies for example that specific suppression of inflammatory angiogenesis will not prevent the induction of physiological ischemia-driven angiogenesis via the HREs. Whilst inflammatory and ischemia-driven angiogenesis are regulated by distinct pathways, these two forms of angiogenesis are likely to co-exist. In atherosclerosis and in tumors, as the plaque/tumor grow, cells become distant to nearby vessels creating hypoxic regions that are the initial triggers for neovascularization. Plaque/tumor neovessels then deliver inflammatory cells/cytokines to the site and inflammation becomes the predominate driving force in the progression of the disease [17].
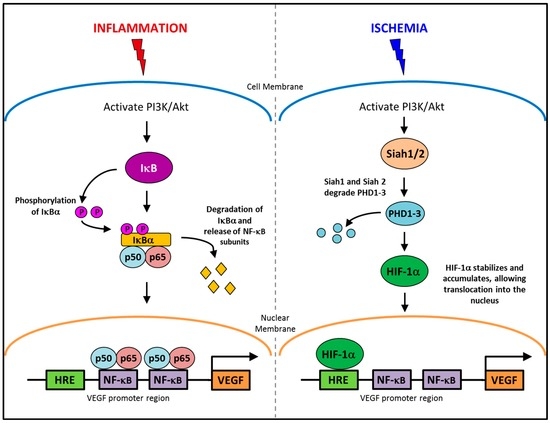
Figure 1.
The conditional regulation of vascular endothelial growth factor (VEGF) in inflammation and ischemia. An inflammatory stimulus activates the PI3K/Akt pathway, leading to the phosphorylation of IκBα. IκBα is degraded allowing NF-κB subunits, p50 and p65 to translocate into the nucleus and activate the production of VEGF. Whereas, ischemia activates the PI3K/Akt pathway to increase the production of Siah1 and Siah2, causing the degradation of PHD1-3 preventing the targeted degradation of HIF-1α, allowing it to accumulate and then translocate to the nucleus where it upregulates VEGF production.
2.1. Pathological Inflammatory Angiogenesis
Pathological angiogenesis is induced at sites of vascular inflammation where there has been increased recruitment of monocytes/macrophages. Macrophages are potent promoters of angiogenesis as they release pro-angiogenic factors, including VEGF, basic fibroblast growth factor (bFGF), platelet derived growth factor (PDGF), tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) that then stimulate excessive neovessel formation [18,19,20]. In plaques, tumors and in RA, neovessels facilitate increased transportation of more inflammatory cells, cytokines and growth factors to the site and exacerbate the disease further [9,10,21]. Neovessels in atherosclerotic plaques are particularly undesirable as they are thin walled and prone to hemorrhage making the plaque unstable [22]. Tumor neovessels also have undesirable effects and lead to metastasis as the local lymphatic systems are unable to support the tumor expansion and shunt tumor cells to neighboring lymph nodes.
An increase in inflammation can also directly cause the stimulation of angiogenesis in endothelial cells. Under inflammatory conditions, vessels form dense and highly disorganized networks [23]. These new vessels develop from existing vessels in which endothelial cells are recruited from sites adjacent to the injury [24]. These recruited endothelial cells accelerate angiogenesis either by forming part of the new vessel or by stimulating vessel growth in a paracrine fashion. At inflammatory sites in tumors, fibroblasts aid in the remodeling of the extracellular matrix (ECM) to make way for new vessels and have also been shown to have pro-angiogenic properties and promote rapid tumor growth and metastasis [21,25,26]. Due to the imbalance between the pro- and anti-angiogenic switch, pathological angiogenesis persists and exacerbates the disease, preventing its resolution.
2.2. Physiological Ischemia-Driven Angiogenesis
Physiological angiogenesis is primarily triggered when there is a chronic imbalance in tissue oxygen supply versus demand following injury or vessel occlusions, which restricts the supply of blood. Neovessel growth is triggered via signaling through HIF-1α. HIF-1α is regulated post-translationally by the prolyl hydroxylase domain protein family (PHD1, PHD2, PHD3). Under high oxygen conditions, PHDs are upregulated causing ubiquitination and degradation of HIF-1α [27,28]. Whilst in low oxygen conditions, E3 ubiquitin ligases Siah1 and Siah2 are upregulated resulting in the degradation of PHD1-3 leading to stabilization and accumulation of HIF-1α [29,30]. This enables the translocation of HIF-1α into the nucleus, where it complexes with the HIF-1β subunit to bind to the hypoxia response element (HRE). HIF-1α upregulates pro-angiogenic growth factors including a 30-fold increase in VEGF [29,30]. HIF-1α is essential in physiological angiogenesis. For example, inactivation of HIF-1α in mice results in abnormal vascular developmental and embryonic lethality [31,32,33,34].
Endothelial nitric oxide synthase (eNOS) is a key enzyme responsible for the promotion of angiogenesis in ischemia and is downstream of VEGF Receptor 2 (VEGFR2), the main receptor for angiogenesis, following activation by VEGF. Upon ischemic induction, eNOS stimulates the production of nitric oxide (NO), which directly promotes angiogenic processes. NO also controls the expression of growth factors such as VEGF, angiopoietins, and fibroblast growth factors and genes involved in matrix metabolism, including matrix metalloproteinases [35].
The important angiogenic functions are cell migration, proliferation and tubulogenesis, which are induced by both hypoxic and inflammatory stimuli [36,37]. Under each stimulus, endothelial cells undergo increased proliferation and migration prior to assembling into tubular structures to form new blood vessels. During angiogenesis, circulating endothelial progenitor cells (EPCs) from the bone marrow promote angiogenesis in a paracrine fashion by secreting growth factors and cytokines to activate local endothelial cells for angiogenesis [38,39,40]. EPCs have also been shown to incorporate into tubules and participate in angiogenesis directly [41,42].
3. Chemokines
Increasing evidence suggests that small (8–12 kDa) inflammatory cytokines known as chemokines are key regulators of angiogenesis. The main role of chemokines is to direct the recruitment and migration of cells to sites of inflammation or injury. They are divided into four classes according to the placement and number of the cysteine residues at the amino terminus. The CC chemokine group has two cysteine residues adjacent to each other, the CXC chemokine group has two cysteine residues separated by an amino acid. The CX3C chemokine group has three amino acids between two cysteine residues and the C chemokine group has only one cysteine residue at the amino terminus [43,44,45]. Of the four chemokine groups, the largest are the CC-chemokines consisting of 28 members in total across all species. This is followed by the CXC-chemokines (17 members), with the CX3C- and XC-chemokines having one and two members respectively.
Under homeostatic conditions, some chemokines such as CCL19 and CCL21, are constitutively expressed in cells to assist with basal leukocyte migration [46,47]. Other chemokines such as CCL2, CCL3, CCL4 and CCL5 are expressed when induced by an inflammatory stimulus [47]. Endothelial cells are able to generate and release chemokines under normal and inflammatory conditions and this is likely the major source of chemokines found on the luminal surface of the endothelium [48]. Once at the luminal surface, chemokines will first bind to glycosaminoglycans (GAGs) such as heparin and heparin sulfate, present on the surface of endothelial cells. Chemokines will then start to accumulate at the site of inflammation/injury via GAG attachment. This is essential, as many chemokines cannot recruit cells unless they are bound to GAGs [49]. GAGs are highly negatively charged and form an electrostatic interaction with the basic chemokine [50], tethering the chemokines to the cell surface ready for receptor interaction. GAG binding also causes oligomerization which is thought to be important in establishing chemokine activity but not receptor interaction [49,51]. Chemokine retention on the cell surface configures a concentration gradient which attracts leukocytes and monocytes to the area of inflammation or injury [52]. CC-chemokines bind to and activate inflammatory cells (e.g., monocytes, neutrophils) via binding to their cognate receptors. Receptor attachment causes a conformation change of the CC-chemokine, leading to an essential receptor and CC-chemokine N-terminal interaction that activates the inflammatory cell [44,51].
Interestingly, there is promiscuity in chemokine signaling, where chemokines in one class can bind to several receptors from the same class [53,54,55]. Conversely, receptors can have multiple ligands within a chemokine class [53,54,55]. For example in humans, CCR2 can bind CCL2, CCL7, CCL8, CCL13 and CCL16, whilst CCR5 can bind CCL3, CCL4, CCL5, CCL8, CCL11, CCL13, CCL14 and CCL16 [53,54,55]. Chemokine promiscuity is essential for the chemokine network as it allows for the robust control of the inflammatory system, such that if one chemokine is inhibited another may replace its function.
4. Chemokines in Angiogenesis Associated Diseases
In addition to their role in maintaining immune cell homeostasis, some chemokines have also been shown to have roles in the development of and, in particular, the promotion of angiogenesis. Several members of the CC-chemokine class including CCL1, CCL2, CCL5, CCL11, CCL15 and CCL16 [56,57,58,59,60] are involved in stimulating pathological inflammation-driven angiogenesis, with little evidence for a role in ischemia-mediated physiological angiogenesis. In contrast, a member of the CXC-chemokine class, CXCL12, uniquely plays an important role in physiological hypoxia-mediated angiogenesis.
4.1. CC-Chemokines in Pathological Inflammation-Driven Angiogenesis
CC-chemokines are increasingly implicated in disease pathologies in which inflammation-driven angiogenesis plays a key role (Figure 2). They are primarily known to promote angiogenesis indirectly by first recruiting macrophages to the site of inflammation or injury where they then release pro-inflammatory cytokines and growth factors that lead to neovessel formation. For example in liver fibrosis, angiogenesis is caused by macrophage recruitment directed by CCL2 [61]. In inflammatory conditions, CC-chemokines are secreted by a wide variety of cells including endothelial cells, smooth muscle cells and inflammatory cells.
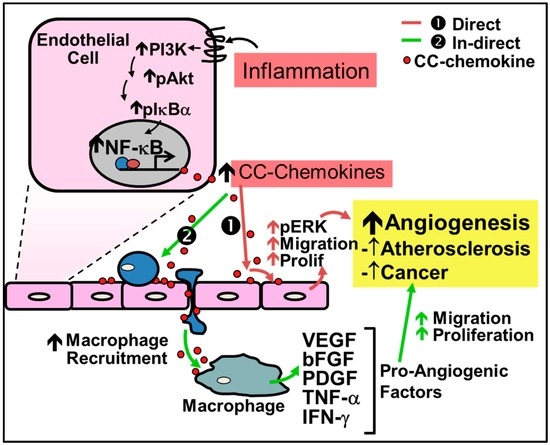
Figure 2.
Regulation of inflammation-driven pathological angiogenesis by CC-chemokines. CC-chemokines regulate angiogenesis through two pathways: (1) the direct pathway; and (2) the indirect pathway. (1) Red Arrows - Direct stimulation by CC-chemokines promotes signaling pathways such as PI3K, MAPK, and ERK1/2 to increase nitric oxide production and promote endothelial cell proliferation and migration leading to neovessel formation; (2) Green Arrows - Indirect stimulation involves the initial recruitment of macrophages to the site of inflammation by CC-chemokines, which secrete pro-angiogenic factors such as VEGF, bFGF, PDGF, TNF-α and IFN-γ that promote angiogenesis.
Recent evidence demonstrates that CC-chemokines can also directly regulate inflammation-driven angiogenesis. For example, CC-chemokines have been shown to promote signaling pathways which induce inflammation-driven angiogenesis including the signaling proteins phosphatidylinositol 3-kinase (PI3K), Akt and mitogen-activated protein kinase (MAPK) and ERK1/2 to increase nitric oxide production, and endothelial cell proliferation and migration, ultimately leading to increased angiogenesis [56,62]. Stimulation of these pathways subsequently increases the production of VEGF to further augment neovascularization [56,62]. Additionally, CCL2 is associated with the increase of MMP14, essential for endothelial cell migration and neovessel formation [62]. CCL2 has also been shown to accelerate endothelialization through the recruitment of EPCs [63]. Endothelial tubule formation in vitro is increased following incubation with CCL1, CCL2, CCL11, CCL15 and CCL16 [56,57,58,59,60]. Furthermore, all CC-chemokines contain NF-κB binding motifs and their expression is dramatically increased under inflammatory conditions [54,64,65].
4.1.1. CC-Chemokines in Atherosclerosis
Atherosclerosis is a chronic inflammatory disease associated with the recruitment of circulating leukocytes into the vascular wall. Inflammation-driven angiogenesis accelerates plaque formation as intra-plaque neovessels deliver more inflammatory cells to the plaque. Plaque neovessels are also fragile and prone to rupture, thereby significantly contributing to plaque instability [9]. Atherosclerosis develops over several decades and encompasses a host of cellular processes involved at various stages of disease from the initiation of plaque formation to plaque rupture, with NF-κB shown to be the key transcription factor involved throughout the entire disease progression. The initial stage of atherosclerosis involves damage to the endothelium and the activation of endothelial cells via the NF-κB signaling pathway. This results in the expression of adhesion molecules and various chemokines such as CCL2, CCL5, macrophage inflammatory factor (MIF), and CX3CL1 [66,67,68], that promote the recruitment of leukocytes to the vascular intima.
A number of CC-chemokines have been identified in human atherosclerotic plaques, including: CCL2, CCL3, CCL5, CCL11, CCL13, CCL18, CCL19, thereby suggesting a role for them in plaque neovessel formation [69,70,71,72]. Furthermore, targeted CC-chemokine receptor or CC-chemokine deletion studies in atherosclerosis-prone ApoE−/− mice have found they develop significantly less plaque [73,74,75]. For example, Met-RANTES (an antagonist to CCL5) causes reductions in the size of atherosclerotic lesions in hypercholesterolemic LDLR−/− mice [73,74,75]. Mice deficient in CCR5, the receptor for CCL5, is associated with reduced macrophage accumulation and fewer more stable plaques [76]. Conversely, overexpression of CCL2 induces macrophage infiltration and enhances atherosclerosis in ApoE−/− mice [77]. Recently, a phase II clinical trial using a CCR2 neutralizing antibody MLN1202 found a decrease in C-reactive protein in patients with high risk of atherosclerosis, suggesting the potential for the suppression of inflammation, plaque angiogenesis and ultimately plaque progression [78].
4.1.2. CC-Chemokines in Rheumatoid Arthritis
Studies have shown that in rheumatoid arthritis (RA), another inflammation-driven pathology, new blood vessels are abundant in the synovial tissue. Neovessels promote the infiltration of leukocytes that increase the progression of RA [11]. Previous studies have found that production of CCL2, CCL3, CCL5, and CCL20 are associated with increased leukocyte infiltration and angiogenesis in models of RA [11,79,80]. Despite the high levels of expression of CC-chemokines in RA, two separate clinical trials with MK-0812 and MLN1202, both CCR2 antagonists, were found to be ineffective [81,82]. Similarly targeting single CC-chemokine receptors CCR1 with Pfizer’s CP-481,715, and CCR5 with Maraviroc were also ineffective [83,84]. Some promising findings have, however, been demonstrated with CCX354-C, a CCR1 antagonist, that improved RA by 20% in 56% of patients after receiving a daily 200 mg dose [85].
4.2. Chemokines in Physiological Ischemia-Mediated Angiogenesis
In contrast to their critical role in inflammation-driven angiogenesis, the CC-chemokines have little involvement in ischemia-mediated angiogenesis (Figure 3). It is the CXC-chemokines, and in particular CXCL12 (also known as stromal-cell derived factor-1α, SDF-1α), that are involved in important developmental processes including hematopoiesis, organogenesis and tissue repair. This is highlighted by the fact that CXCL12 knockout mice are not viable due to the absence of vessel development [86]. HIF-1α augments the increase in expression of CXCL12 in ischemia. Consistent with this, CXCL12 expression in ischemic sites directly correlates with the degree of hypoxia [87]. Furthermore, increased CXCL12 levels have been reported in infarcted myocardium in both human and rodent studies, with increased CXCL12 levels detected as early as 1 h following ischemic induction in the myocardium or in hindlimbs, indicative of a role in the initiation of tissue repair and revascularization [88,89,90]. CXCL12 is the single natural ligand for the chemokine receptor CXCR4 [91] and the CXCL12/CXCR4 axis is important in the mobilization, migration and recruitment of EPCs to sites of ischemia [86,92,93,94,95]. Mechanistically, CXCL12 mobilizes bone marrow-derived EPCs by enhancing PI3K/Akt/eNOS activation [96]. Upregulation of eNOS further promotes EPC mobilization by increasing the production of VEGF [97].
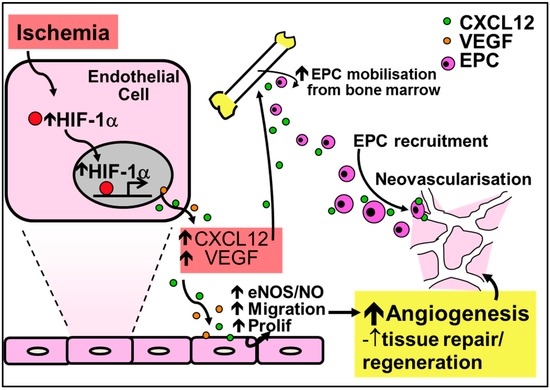
Figure 3.
Regulation of ischemia-mediated physiological angiogenesis by the CXC-chemokine CXCL12. Increased HIF-1α in response to ischemia causes the release of CXCL12 and VEGF, leading to increased nitric oxide production and augmented endothelial migration and proliferation to promote angiogenesis. Furthermore, CXCL12 upregulates the recruitment and mobilization of EPCs to sites of ischemia. Increased nitric oxide from the induction of CXCL12 further mobilizes EPCs and increases the production of VEGF for neovessel formation.
5. CC-Chemokines in Cancer
Tumor angiogenesis is triggered initially by inflammation and then promoted further by hypoxia. Tumor neovessels facilitate the recruitment of inflammatory cells that express pro-angiogenic mediators. This promotes a positive angiogenic feed-forward loop, similar to that of plaque neovessels. Tumor angiogenesis is regarded as a negative prognostic variable for several malignancies including breast and prostate cancer [98,99,100]. CC-chemokines have been associated with a number of malignancies. Studies reveal that CCL2 and CCL5 expression are elevated in breast and prostate cancer [101,102]. CCL1 and CCL3 are shown to promote development of leukemia and CCL19, CCL21, and CCL25 direct tumor metastasis [103,104]. Murine cancer models have demonstrated the importance of CC-chemokines in tumor development in vivo. Inhibition of CCL2 reduced hemangioma size in nude mice inoculated with cancer cells, while infusion of Met-RANTES inhibited tumor development in murine models of breast cancer [105].
In the progression of tumor development, tumors expand to become solid tumors, causing the microenvironment to become progressively more hypoxic as cells become distant to nearby vessels. This forces cancer cells to adapt promoting tumorigenesis so oxygen and other nutrients can be delivered, expanding the tumor further. This also enhances tumor malignancy and aggressiveness, ultimately resulting in increased resistance to therapy and poor long-term prognosis. This response is induced by a series of hypoxia-mediated proteomic and genomic changes that facilitate cell survival [106]. CC-chemokines are not the only chemokines found in tumors. The CXC-chemokine class have also been shown to play a role in tumor neovascularization. For example, IL-8 (CXCL8) promotes tumor growth, angiogenesis and metastasis [107]. Furthermore, CXCL12 production is triggered in hypoxic environments in tumor cells [108] and is implicated in the promotion of tumor growth in mouse prostate tumors following overexpression of its receptor CXCR4 [109].
6. Anti-Angiogenic Therapies
The development of anti-angiogenic agents is of great interest due to the high prevalence of diseases associated with angiogenesis. Current anti-angiogenic agents such as VEGF inhibitors or VEGF receptor inhibitors are found to reduce atherosclerotic plaque size and tumor growth in a variety of malignancies including breast, lung and prostate cancers [10,43,110,111,112]. However, the VEGF signaling pathway is critically important in physiological ischemia-driven angiogenesis as well as other biological functions (e.g., regulation of blood pressure, activation of the coagulation cascade, and regulation of myelopoiesis [113,114,115,116]) so non-specific inhibition of VEGF in all pathophysiological contexts leads to side effects including: bleeding, vessel clots (leading to stroke or heart attack), proteinuria, severe weight loss, hypertension, diarrhea, nausea gastrointestinal perforations, and reduced wound healing [6,7,117,118]. Furthermore, the clinically available Bevacizumab (an anti-vascular endothelial growth factor, VEGF, antibody) [119] and multi-targeted tyrosine kinase inhibitors (TKIs, target the VEGF receptor), such as Sorafenib and Sunitinib [120], cause endothelial dysfunction and capillary rarefaction [6,7,117]. Both classes of drug cause clinically significant elevations in blood pressure (12%–15%), bleeding and hemorrhage—all of which are associated with impaired regenerative angiogenic responses [6,7,117]. These studies suggest that complete inhibition of angiogenesis may not be the ideal strategy to suppress pathological angiogenic diseases. This highlights the need for the development of novel therapies specifically targeting only pathological angiogenesis whilst preserving physiological angiogenesis.
Other than VEGF inhibitors, on-going clinical trials that target individual CC-chemokine receptors have shown some success, including: (1) MLN1202 that targets CCR2 and causes reductions in circulating C-reactive protein in patients with high risk of atherosclerosis; and (2) Mogamulizumab that targets CCR4 and causes reductions in regulatory T cells in patients with adult T-cell leukemia-lymphoma (ATL) and peripheral T-cell lymphoma [81,121]. However due to the promiscuity in chemokine signaling the inhibition of a single chemokine receptor may not be entirely efficacious [122,123]. An agent capable of simultaneously targeting multiple chemokines is likely to provide a more effective approach. Due to their specific role in inflammation-driven angiogenesis, targeting of the CC-chemokine class as a whole may provide a unique means of inhibiting only pathological angiogenesis without affecting critical physiological angiogenic functions.
Broad-Spectrum CC-Chemokine Inhibition as a Strategy to Inhibit Pathological Angiogenesis
Broad-spectrum chemokine inhibitors, also called chemokine binding proteins, have been found to be expressed from a range of species including the Schistosoma mansoni helminth worm, the Rhipicephalus sanguineus tick and several viruses. Chemokine binding proteins are thought to have evolved as a way to bypass the host inflammatory response to propagate their infection. These chemokine binding proteins target multiple chemokines. For example, the S. mansoni chemokine binding protein (smCKBP), secreted from schistosome eggs, targets CXCL8, CCL2, CCL3, CCL5 and CX3CL1 [124]. Chemokine binding proteins from R. sanguineus tick, called Evasins, bind to (Evasin-1) CCL3, CCL4, CCL5, (Evasin-3) CXCL1, CXCL2, CXCL3, CXL5, CXCL6, CXCL8, (Evasin-4) CCL5 and CCL11 [125]. Viral protein M3 from the mouse herpes virus binds to several inflammatory chemokines across all four classes by binding to GAGs and chemokines such as XCL1, CCL2, CCL5, CXCL8 and CX3CL1 [126,127,128]. Vaccinia viral protein “35K” is another chemokine binding protein that uniquely binds with high affinity to the CC-chemokine class [129,130,131,132,133]. 35K has been shown to suppress inflammation-driven diseases in pre-clinical models including atherosclerosis [134,135,136], liver fibrosis and acute peritonitis [137,138]. Additionally, 35K attached to a Fc fusion protein (35K-Fc) improved pulmonary function and reduced inflammation in the lung [139]. 35K will therefore not inhibit CXCL12 which is essential for development and for tissue repair following ischemia. Broad-spectrum inhibition of the CC-chemokine class by 35K may be an alternate therapeutic strategy to specifically reduce pathological angiogenesis-associated diseases such as atherosclerosis and cancer, without the severe side effects of current non-selective angiogenic inhibitors that block angiogenesis in all pathophysiological contexts, including in ischemia.
In support of this concept, administration of CCL2 increases inflammatory angiogenesis in mice [140] and induces inflammatory angiogenesis in rabbit cornea [141]. In contrast, CCL2−/− mice show no difference in capillary density compared to wild type mice in a model of myocardial ischemia [142]. Furthermore, CCR2−/− mice have comparatively normal revascularization in the hindlimb ischemia model, compared to wild type mice [143]. Unpublished studies from our laboratory have found that mice injected with adenovirus overexpressing 35K (Ad35K) have reduced neovessel formation in the peri-arterial femoral cuff model of inflammation-driven angiogenesis, when compared to mice injected with control GFP adenovirus (AdGFP). In contrast, we found angiogenesis was preserved in the hindlimb model of ischemia-mediated angiogenesis following CC-chemokine class inhibition with 35K. In vitro functional assays of angiogenesis supported these findings. 35K inhibited endothelial cell proliferation, migration and tubule formation in inflammatory conditions but, conversely, had minimal effects on these angiogenic functions in hypoxia. Mechanistically, we revealed that VEGF was conditionally regulated by 35K, where VEGF is suppressed in inflammation but preserved in hypoxia (Unpublished data). The ability of 35K to inhibit inflammation-driven angiogenesis whilst preserving ischemia-mediated angiogenesis suggests it may present as an alternate therapeutic strategy for the specific inhibition of diseases associated with inflammatory driven angiogenesis.
7. Conclusions
In conclusion, CC-chemokines play an important role in inflammation-driven pathological angiogenesis whilst having minimal effects in ischemia-mediated physiological angiogenesis. This differential regulation of angiogenesis by CC-chemokines highlights the possibility that targeted inhibition of CC-chemokines will prevent inflammation-driven angiogenesis, whilst preserving important ischemia-mediated angiogenic processes. Current strategies targeting VEGF or single CC-chemokines/receptors have caused issues including severe side effects or ineffectiveness. This is likely due to the complete inhibition of VEGF across all pathophysiological contexts and also the promiscuity in chemokine signaling, respectively. A new approach for a more targeted strategy is broad-spectrum CC-chemokine class inhibition and there is recent evidence that it specifically inhibits pathological angiogenesis, whilst leaving the physiological angiogenesis unchanged. This may have immense benefits over current anti-angiogenic therapies.
Acknowledgments
This work was supported by the National Health and Medical Research Council (NHMRC) of Australia Project Grant (#632512 to Christina A. Bursill) and the National Heart Foundation Career Development Fellowship (#CR07S3331 to Christina A. Bursill).
Author Contributions
Christina A. Bursill conceived the idea for the review article. Anisyah Ridiandries, Joanne T. M. Tan and Christina A. Bursill wrote and edited the review article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [PubMed]
- Strieter, R.M.; Polverini, P.J.; Kunkel, S.L.; Arenberg, D.A.; Burdick, M.D.; Kasper, J.; Dzuiba, J.; van Damme, J.; Walz, A.; Marriott, D.; et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J. Biol. Chem. 1995, 270, 27348–27357. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Lasagni, L.; Annunziato, F.; Serio, M.; Romagnani, S. CXC chemokines: The regulatory link between inflammation and angiogenesis. Trends Immunol. 2004, 25, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Namkoong, S.; Kim, Y.-M.; Kim, C.-K.; Lee, H.; Ha, K.-S.; Chung, H.-T.; Kwon, Y.-G.; Kim, Y.-M. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2836–H2846. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Status Report on Noncommunicable Diseases 2014; World Health Organization: Geneva, Switzerland, 2014; p. 298. [Google Scholar]
- Mourad, J.J.; des Guetz, G.; Debbabi, H.; Levy, B.I. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann. Oncol. 2008, 19, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Cabebe, E.; Wakelee, H. Role of anti-angiogenesis agents in treating NSCLC: Focus on bevacizumab and VEGFR tyrosine kinase inhibitors. Curr. Treat. Opt. Oncol. 2007, 8, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Vakili, K.; Zurakowski, D.; Soliman, M.; Butterfield, C.; Sylvin, E.; Lo, K.M.; Gillies, S.; Javaherian, K.; Folkman, J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 4736–4741. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Pakozdi, A.; Szentpetery, A.; Besenyei, T.; Koch, A.E. Chemokines and angiogenesis in rheumatoid arthritis. Front. Biosci. 2009, 1, 44–51. [Google Scholar]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.P.; Levy, N.S.; Wegner, S.; Goldberg, M.A. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J. Biol. Chem. 1995, 270, 13333–13340. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive oxygen species activate the HIF-1α promoter via a functional NFκB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Marsch, E.; Sluimer, J.C.; Daemen, M.J. Hypoxia in atherosclerosis and inflammation. Curr. Opin. Lipidol. 2013, 24, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.R.; Seed, M.P.; Kircher, C.H.; Willoughby, D.A.; Winkler, J.D. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997, 11, 457–465. [Google Scholar] [PubMed]
- Sunderkotter, C.; Steinbrink, K.; Goebeler, M.; Bhardwaj, R.; Sorg, C. Macrophages and angiogenesis. J. Leukoc. Biol. 1994, 55, 410–422. [Google Scholar] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R.; Purushothaman, K.R.; Fuster, V.; Echeverri, D.; Truszczynska, H.; Sharma, S.K.; Badimon, J.J.; O’Connor, W.N. Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: Implications for plaque vulnerability. Circulation 2004, 110, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. Oxygen sensing in the hypoxic response pathway: Regulation of the hypoxia-inducible transcription factor. Genes Dev. 2003, 17, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Frew, I.J.; Hagensen, M.; Skals, M.; Habelhah, H.; Bhoumik, A.; Kadoya, T.; Erdjument-Bromage, H.; Tempst, P.; Frappell, P.B.; et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell 2004, 117, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Qi, J.; Ronai, Z. The ubiquitin ligase Siah2 and the hypoxia response. Mol. Cancer Res. 2009, 7, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 α. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Kotch, L.E.; Iyer, N.V.; Laughner, E.; Semenza, G.L. Defective vascularization of HIF-1α-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev. Biol. 1999, 209, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Licht, A.H.; Müller-Holtkamp, F.; Flamme, I.; Breier, G. Inhibition of hypoxia-inducible factor activity in endothelial cells disrupts embryonic cardiovascular development. Blood 2006, 107, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Munk, V.C.; Miguel, L.S.d.; Humar, R.; Kiefer, F.N.; Butz, N.; Battegay, E.J. INOS is required for in vitro angiogenesis of hypoxic mouse hearts. Semin. Cardiol. 2006, 12, 21–26. [Google Scholar]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar] [PubMed]
- Phillips, P.G.; Birnby, L.M.; Narendran, A. Hypoxia induces capillary network formation in cultured bovine pulmonary microvessel endothelial cells. Am. J. Physiol. 1995, 268, L789–L800. [Google Scholar] [PubMed]
- Rehman, J.; Li, J.; Orschell, C.M.; March, K.L. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 2003, 107, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; von Ballmoos, M.W.; Faessler, D.; Voelzmann, J.; Ortmann, J.; Diehm, N.; Kalka-Moll, W.; Baumgartner, I.; di Santo, S.; Kalka, C. Paracrine factors secreted by endothelial progenitor cells prevent oxidative stress-induced apoptosis of mature endothelial cells. Atherosclerosis 2010, 211, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Chang, S.-J.; Chueh, Y.-N.; Huang, T.-S.; Huang, P.-H.; Cheng, S.-M.; Tsai, T.-N.; Chen, J.-W.; Wang, H.-W. Distinct angiogenesis roles and surface markers of early and late endothelial progenitor cells revealed by functional group analyses. BMC Genom. 2013, 14, 182. [Google Scholar] [CrossRef] [PubMed]
- Sieveking, D.P.; Buckle, A.; Celermajer, D.S.; Ng, M.K. Strikingly different angiogenic properties of endothelial progenitor cell subpopulations: Insights from a novel human angiogenesis assay. J. Am. Coll. Cardiol. 2008, 51, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Mukai, N.; Akahori, T.; Komaki, M.; Li, Q.; Kanayasu-Toyoda, T.; Ishii-Watabe, A.; Kobayashi, A.; Yamaguchi, T.; Abe, M.; Amagasa, T.; et al. A comparison of the tube forming potentials of early and late endothelial progenitor cells. Exp. Cell Res. 2008, 314, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Chan, A. Antiangiogenic therapy for metastatic breast cancer: Current status and future directions. Drugs 2009, 69, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.J.; Lolis, E. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 469–499. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.D.J. Location, movement and survival: The role of chemokines in haematopoiesis and malignancy. Br. J. Haematol. 2006, 132, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Øynebråten, I.; Bakke, O.; Brandtzaeg, P.; Johansen, F.-E.; Haraldsen, G. Rapid chemokine secretion from endothelial cells originates from 2 distinct compartments. Blood 2004, 104, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.C.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Kuschert, G.S.; Coulin, F.; Power, C.A.; Proudfoot, A.E.; Hubbard, R.E.; Hoogewerf, A.J.; Wells, T.N. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 1999, 38, 12959–12968. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Crown, S.E.; Handel, T.M. Chemokine: Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Hub, E.; Rot, A. Binding of RANTES, MCP-1, MCP-3, and MIP-1α to cells in human skin. Am. J. Pathol. 1998, 152, 749–757. [Google Scholar] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Mostarica-Stojkovic, M.; Andjelkovic, A.V. CCL2 regulates angiogenesis via activation of ETS-1 transcription factor. J. Immunol. 2006, 177, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, R.; Young, H.A.; Ponce, M.L.; Ward, J.M.; Kleinman, H.K.; Murphy, W.J.; Oppenheim, J.J. Eotaxin (CCL11) induces in vivo angiogenic responses by human CCR3+ endothelial cells. J. Immunol. 2001, 166, 7571–7578. [Google Scholar] [CrossRef] [PubMed]
- Strasly, M.; Doronzo, G.; Cappello, P.; Valdembri, D.; Arese, M.; Mitola, S.; Moore, P.; Alessandri, G.; Giovarelli, M.; Bussolino, F. CCL16 activates an angiogenic program in vascular endothelial cells. Blood 2004, 103, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Spinetti, G.; Ribatti, D.; Camarda, G.; Morbidelli, L.; Ziche, M.; Santoni, A.; Capogrossi, M.C.; Napolitano, M. I-309 binds to and activates endothelial cell functions and acts as an angiogenic molecule in vivo. Blood 2000, 96, 4039–4045. [Google Scholar] [PubMed]
- Hwang, J.; Kim, C.W.; Son, K.-N.; Han, K.Y.; Lee, K.H.; Kleinman, H.K.; Ko, J.; Na, D.S.; Kwon, B.S.; Gho, Y.S.; et al. Angiogenic activity of human CC chemokine CCL15 in vitro and in vivo. FEBS Lett. 2004, 570, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Möckel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef] [PubMed]
- Galvez, B.G.; Genis, L.; Matias-Roman, S.; Oblander, S.A.; Tryggvason, K.; Apte, S.S.; Arroyo, A.G. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/CCL2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J. Biol. Chem. 2005, 280, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, S.; Amano, K.; Uehira, K.; Yoshida, M.; Nishiwaki, Y.; Nozawa, Y.; Jin, D.; Takai, S.; Miyazaki, M.; Egashira, K.; et al. Bone marrow monocyte lineage cells adhere on injured endothelium in a monocyte chemoattractant protein-1-dependent manner and accelerate reendothelialization as endothelial progenitor cells. Circ. Res. 2003, 93, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.S.; McIntyre, T.M.; McEver, R.P.; Prescott, S.M.; Zimmerman, G.A. Monocyte tethering by p-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-α secretion. Signal integration and NF-κB translocation. J. Clin. Investig. 1995, 95, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Werts, C.; le Bourhis, L.; Liu, J.; Magalhaes, J.G.; Carneiro, L.A.; Fritz, J.H.; Stockinger, S.; Balloy, V.; Chignard, M.; Decker, T.; et al. Nod1 and Nod2 induce CCL5/RANTES through the NF-κB pathway. Eur. J. Immunol. 2007, 37, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Weber, C. Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc. Res. 2010, 86, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Leech, M.; Bernhagen, J. MIF: A new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 2006, 5, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jiang, D. Fractalkine/CX3CR1 and atherosclerosis. Clin. Chim. Acta 2011, 412, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Bursill, C.A.; Cai, S.; Channon, K.M.; Greaves, D.R. Adenoviral-mediated delivery of a viral chemokine binding protein blocks CC-chemokine activity in vitro and in vivo. Immunobiology 2003, 207, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Schober, A.; Zernecke, A. Chemokines: Key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Reape, T.J.; Rayner, K.; Manning, C.D.; Gee, A.N.; Barnette, M.S.; Burnand, K.G.; Groot, P.H. Expression and cellular localization of the CC chemokines PARC and ELC in human atherosclerotic plaques. Am. J. Pathol. 1999, 154, 365–374. [Google Scholar] [CrossRef]
- Berkhout, T.A.; Sarau, H.M.; Moores, K.; White, J.R.; Elshourbagy, N.; Appelbaum, E.; Brawner, T.J.R.; Makwana, J.; Foley, J.J.; Schmidt, D.B.; et al. Cloning, in vitro expression, and functional characterization of a novel human CC chemokine of the monocyte chemotactic protein (MCP) family (MCP-4) that binds and signals through the CC chemokine receptor 2B. J. Biol. Chem. 1997, 272, 16404–16413. [Google Scholar] [CrossRef] [PubMed]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nat. Med. 1998, 394, 894–897. [Google Scholar]
- Gosling, J.; Slaymaker, S.; Gu, L.; Tseng, S.; Zlot, C.H.; Young, S.G.; Rollins, B.J.; Charo, I.F. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J. Clin. Investig. 1999, 103, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Potteaux, S.; Combadiere, C.; Esposito, B.; Lecureuil, C.; Ait-Oufella, H.; Merval, R.; Ardouin, P.; Tedgui, A.; Mallat, Z. Role of bone marrow-derived CC-chemokine receptor 5 in the development of atherosclerosis of low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1858–1863. [Google Scholar] [CrossRef] [PubMed]
- Aiello, R.J.; Bourassa, P.-A.K.; Lindsey, S.; Weng, W.; Natoli, E.; Rollins, B.J.; Milos, P.M. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Koch, A.E. Chemokines and angiogenesis. Curr. Opin. Rheumatol. 2001, 13, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, E.; Struyf, S.; van Damme, J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003, 14, 409–426. [Google Scholar] [CrossRef]
- Horuk, R. Chemokine receptor antagonists: Overcoming developmental hurdles. Nat. Rev. Drug Discov. 2009, 8, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Vergunst, C.E.; Gerlag, D.M.; Lopatinskaya, L.; Klareskog, L.; Smith, M.D.; van den Bosch, F.; Dinant, H.J.; Lee, Y.; Wyant, T.; Jacobson, E.W.; et al. Modulation of CCR2 in rheumatoid arthritis: A double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008, 58, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.F.; Bahnck, K.B.; Blumberg, L.C.; Brissette, W.H.; Burrell, S.A.; Driscoll, J.P.; Fedeles, F.; Fisher, M.B.; Foti, R.S.; Gladue, R.P.; et al. Piperazinyl CCR1 antagonists—Optimization of human liver microsome stability. Bioorg. Med. Chem. Lett. 2007, 17, 3109–3112. [Google Scholar] [CrossRef] [PubMed]
- Fleishaker, D.L.; Garcia Meijide, J.A.; Petrov, A.; Kohen, M.D.; Wang, X.; Menon, S.; Stock, T.C.; Mebus, C.A.; Goodrich, J.M.; Mayer, H.B.; et al. Maraviroc, a chemokine receptor-5 antagonist, fails to demonstrate efficacy in the treatment of patients with rheumatoid arthritis in a randomized, double-blind placebo-controlled trial. Arthritis Res. Ther. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Balanescu, A.; Tseluyko, V.; Bojin, S.; Drescher, E.; Dairaghi, D.; Miao, S.; Marchesin, V.; Jaen, J.; Schall, T.J.; et al. Chemokine receptor CCR1 antagonist CCX354-C treatment for rheumatoid arthritis: Carat-2, a randomised, placebo controlled clinical trial. Ann. Rheum. Dis. 2013, 72, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Karshovska, E.; Zernecke, A.; Weber, C. SDF-1α-mediated tissue repair by stem cells: A promising tool in cardiovascular medicine? Trends Cardiovasc. Med. 2006, 16, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.P.; Youn, S.W.; Cho, H.J.; Li, L.; Kim, T.Y.; Yook, H.S.; Chung, J.W.; Hur, J.; Yoon, C.H.; Park, K.W.; et al. Integrin-linked kinase, a hypoxia-responsive molecule, controls postnatal vasculogenesis by recruitment of endothelial progenitor cells to ischemic tissue. Circulation 2006, 114, 150–159. [Google Scholar] [CrossRef] [PubMed]
- De Falco, E.; Porcelli, D.; Torella, A.R.; Straino, S.; Iachininoto, M.G.; Orlandi, A.; Truffa, S.; Biglioli, P.; Napolitano, M.; Capogrossi, M.C.; et al. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood 2004, 104, 3472–3482. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, Z.H.; Li, Z.J.; Yang, R.C.; Qian, G.Q.; Han, Z.C. Enhancement of neovascularization with cord blood CD133+ cell-derived endothelial progenitor cell transplantation. Thromb. Haemost. 2004, 91, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Liekens, S.; Schols, D.; Hatse, S. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr. Pharm. Des. 2010, 16, 3903–3920. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.T.; Unzek, S.; Popovic, Z.B.; Goldman, C.K.; Forudi, F.; Kiedrowski, M.; Rovner, A.; Ellis, S.G.; Thomas, J.D.; di Corleto, P.E.; et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 2003, 362, 697–703. [Google Scholar] [CrossRef]
- Wang, Y.; Haider, H.; Ahmad, N.; Zhang, D.; Ashraf, M. Evidence for ischemia induced host-derived bone marrow cell mobilization into cardiac allografts. J. Mol. Cell. Cardiol. 2006, 41, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, K.; Gupta, S.K. Cloning and relative expression analysis of rat stromal cell derived factor-1 (SDF-1)1: SDF-1 α mRNA is selectively induced in rat model of myocardial infarction. Inflammation 2001, 25, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ge, J.; Zhang, S.; Sun, A.; Shen, J.; Chen, L.; Wang, K.; Zou, Y. Time course of myocardial stromal cell-derived factor 1 expression and beneficial effects of intravenously administered bone marrow stem cells in rats with experimental myocardial infarction. Basic Res. Cardiol. 2005, 100, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fu, G.; Dai, T.; Huang, H. Migration of endothelial progenitor cells mediated by stromal cell-derived factor-1α/CXCR4 via PI3K/AKT/eNOS signal transduction pathway. J. Cardiovasc. Pharmacol. 2007, 50, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Aicher, A.; Heeschen, C.; Mildner-Rihm, C.; Urbich, C.; Ihling, C.; Technau-Ihling, K.; Zeiher, A.M.; Dimmeler, S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 2003, 9, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Molecular mechanisms of tumor angiogenesis and tumor progression. J. Neuro-Oncol. 2000, 50, 63–70. [Google Scholar] [CrossRef]
- Zachary, I. Vascular endothelial growth factor. Int. J. Biochem. Cell Biol. 1998, 30, 1169–1174. [Google Scholar] [CrossRef]
- Van Hinsbergh, V.W.; Collen, A.; Koolwijk, P. Angiogenesis and anti-angiogenesis: Perspectives for the treatment of solid tumors. Ann. Oncol. 1999, 10, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, Y.; Pienta, K.J. Multiple roles of chemokine (C-C MOTIF) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer Inst. 2010, 102, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Bai, Z.; Srinoulprasert, Y.; Yang, B.; Hayasaka, H.; Miyasaka, M. Chemokines in tumor progression and metastasis. Cancer Sci. 2005, 96, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.C.; Scott, K.A.; Wilson, J.L.; Thompson, R.G.; Proudfoot, A.E.I.; Balkwill, F.R. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003, 63, 8360–8365. [Google Scholar] [PubMed]
- Vaupel, P. The role of hypoxia-induced factors in tumor progression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Lange, A.; Mottram, P.; Alvarez, X.; Cheng, P.; Hogan, M.; Moons, L.; Wei, S.; Zou, L.; Machelon, V.; et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res. 2005, 65, 465–472. [Google Scholar] [PubMed]
- Darash-Yahana, M.; Pikarsky, E.; Abramovitch, R.; Zeira, E.; Pal, B.; Karplus, R.; Beider, K.; Avniel, S.; Kasem, S.; Galun, E.; et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004, 18, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.J. Targeting angiogenesis in cancer: Clinical development of bevacizumab. Nat. Clin. Pract. Oncol. 2004, 1, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, F.; Mazzoni, F.; Micol Mela, M.; Antonuzzo, L.; Checcacci, D.; Saggese, M. Bevacizumab in non-small cell lung cancer. Drugs 2008, 68, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Borgstrom, P.; Bourdon, M.A.; Hillan, K.J.; Sriramarao, P.; Ferrara, N. Neutralizing anti-vascular endothelial growth factor antibody completely inhibits angiogenesis and growth of human prostate carcinoma micro tumors in vivo. Prostate 1998, 35, 1–10. [Google Scholar] [CrossRef]
- Robinson, E.S.; Khankin, E.V.; Karumanchi, S.A.; Humphreys, B.D. Hypertension induced by VEGF signaling pathway inhibition: Mechanisms and potential use as a biomarker. Semin. Nephrol. 2010, 30, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.W.; Hoekman, K.; Lupu, F.; Broxterman, H.J.; van der Valk, P.; Kakkar, A.K.; Pinedo, H.M. Platelet and coagulation activation with vascular endothelial growth factor generation in soft tissue sarcomas. Am. Assoc. Cancer Res. 2000, 6, 166–171. [Google Scholar]
- Verheul, H.M.; van Erp, K.; Homs, M.Y.; Yoon, G.S.; van der Groep, P.; Rogers, C.; Hansel, D.E.; Netto, G.J.; Pili, R. The relationship of vascular endothelial growth factor and coagulation factor (fibrin and fibrinogen) expression in clear cell renal cell carcinoma. Urology 2010, 75, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Larrivee, B.; Pollet, I.; Karsan, A. Activation of vascular endothelial growth factor receptor-2 in bone marrow leads to accumulation of myeloid cells: Role of granulocyte-macrophage colony-stimulating factor. J. Immunol. 2005, 175, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Satchi-Fainaro, R.; Puder, M.; Davies, J.W.; Tran, H.T.; Sampson, D.A.; Greene, A.K.; Corfas, G.; Folkman, J. Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat. Med. 2004, 10, 255–261. [Google Scholar] [CrossRef]
- Chen, H.X.; Cleck, J.N. Adverse effects of anticancer agents that target the VEGF pathway. Nat. Rev. Clin. Oncol. 2009, 6, 465–477. [Google Scholar]
- Shih, T.; Lindley, C. Bevacizumab: An angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 2006, 28, 1779–1802. [Google Scholar] [CrossRef]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vela, M.; Aris, M.; Llorente, M.; Garcia-Sanz, J.A.; Kremer, L. Chemokine receptor-specific antibodies in cancer immunotherapy: Achievements and challenges. Front. Immunol. 2015, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Devalaraja, M.N.; Richmond, A. Multiple chemotactic factors: Fine control or redundancy? Trends Pharmacol. Sci. 1999, 20, 151–156. [Google Scholar] [CrossRef]
- Mantovani, A. The chemokine system: Redundancy for robust outputs. Immunol. Today 1999, 20, 254–257. [Google Scholar] [CrossRef]
- Smith, P.; Fallon, R.E.; Mangan, N.E.; Walsh, C.M.; Saraiva, M.; Sayers, J.R.; McKenzie, A.N.J.; Alcami, A.; Fallon, P.G. Schistosoma mansoni secretes a chemokine binding protein with antiinflammatory activity. J. Exp. Med. 2005, 202, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, P.; Power, C.A.; Proudfoot, A.E.I. Evasins: Therapeutic potential of a new family of chemokine-binding proteins from ticks. Front. Immunol. 2016, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Brett, J.M.; Fremont, D.H. Dual GPCR and GAG mimicry by the M3 chemokine decoy receptor. J. Exp. Med. 2007, 204, 3157–3172. [Google Scholar] [CrossRef] [PubMed]
- Parry, C.M.; Simas, J.P.; Smith, V.P.; Stewart, C.A.; Minson, A.C.; Efstathiou, S.; Alcami, A. A broad-spectrum secreted chemokine binding protein encoded by a herpesvirus. J. Exp. Med. 2000, 191, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Webb, L.M.C.; Smith, V.P.; Alcami, A. The gammaherpesvirus chemokine binding protein can inhibit the interaction of chemokines with glycosaminoglycans. J. Fed. Am. Soc. Exp. Biol. 2004, 18, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Symons, J.A.; Collins, P.D.; Williams, T.J.; Smith, G.L. Blockade of chemokine activity by a soluble chemokine binding protein from vaccinia virus. J. Immunol. 1998, 160, 624–633. [Google Scholar] [PubMed]
- Graham, K.A.; Lalani, A.S.; Macen, J.L.; Ness, T.L.; Barry, M.; Liu, L.Y.; Lucas, A.; Clark-Lewis, I.; Moyer, R.W.; McFadden, G. The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology 1997, 229, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Smith, T.D.; Smolak, P.J.; Friend, D.; Hagen, H.; Gerhart, M.; Park, L.; Pickup, D.J.; Torrance, D.; Mohler, K.; et al. Poxvirus genomes encode a secreted, soluble protein that preferentially inhibits β chemokine activity yet lacks sequence homology to known chemokine receptors. Virology 1997, 236, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Derider, M.; McCornack, M.A.; Jao, S.C.; Isern, N.; Ness, T.; Moyer, R.; LiWang, P.J. Solution structure of the complex between poxvirus-encoded CC chemokine inhibitor vCCI and human MIP-1β. Proc. Natl. Acad. Sci. USA 2006, 103, 13985–13990. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.G.; Studer, C.; Zuber, J.F.; Demange, B.J.; Manning, U.; Urfer, R. The viral CC chemokine-binding protein vCCI inhibits monocyte chemoattractant protein-1 activity by masking its CCR2B-binding site. J. Biol. Chem. 2001, 276, 43270–43276. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.A.; Bursill, C.A.; Hu, Y.; Choudhury, R.P.; Xu, Q.; Greaves, D.R.; Channon, K.M. Gene transfer of a broad-spectrum CC-chemokine inhibitor reduces vein graft atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005, 112, I235–I241. [Google Scholar] [PubMed]
- Bursill, C.A.; Choudhury, R.P.; Ali, Z.; Greaves, D.R.; Channon, K.M. Broad-spectrum CC-chemokine blockade by gene transfer inhibits macrophage recruitment and atherosclerotic plaque formation in apolipoprotein e-knockout mice. Circulation 2004, 110, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Bursill, C.A.; McNeill, E.; Wang, L.; Hibbitt, O.C.; Wade-Martins, R.; Paterson, D.J.; Greaves, D.R.; Channon, K.M. Lentiviral gene transfer to reduce atherosclerosis progression by long-term CC-chemokine inhibition. Gene Ther. 2009, 16, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bursill, C.A.; Cash, J.L.; Channon, K.M.; Greaves, D.R. Membrane-bound CC chemokine inhibitor 35K provides localized inhibition of CC chemokine activity in vitro and in vivo. J. Immunol. 2006, 177, 5567–5573. [Google Scholar] [CrossRef]
- Seki, E.; Minicis, S.D.; Gwak, G.-Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic firbosis in mice. J. Clin. Investig. 2009, 119, 1858–1870. [Google Scholar]
- Dabbagh, K.; Xiao, Y.; Smith, C.; Stepick-Biek, P.; Kim, S.G.; Lamm, W.J.E.; Liggitt, D.H.; Lewis, D.B. Local blockade of allergic airway hyperreactivity and inflammation by the poxvirus-derived pan-CC-chemokine inhibitor vCCI. J. Immunol. 2000, 165, 3418–3422. [Google Scholar]
- Barcelos, L.S.; Talvani, A.; Teixeira, A.S.; Cassali, G.D.; Andrade, S.P.; Teixeira, M.M. Production and in vivo effects of chemokines CXCL1–3/KC and CCL2/JE in a model of inflammatory angiogenesis in mice. Inflamm. Res. 2004, 53, 576–584. [Google Scholar] [PubMed]
- Goede, V.; Brogelli, L.; Ziche, M.; Augustin, H.G. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int. J. Cancer 1999, 82, 765–770. [Google Scholar] [CrossRef]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Charo, D.N.; Wang, R.; Charo, I.F.; Messina, L. CCR2−/− knockout mice revascularize normally in response to severe hindlimb ischemia. J. Vasc. Surg. 2004, 40, 786–795. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).