Abstract
The preparation of the novel optically active sulfoxides (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(p-tolylsulfinyl)phenyl]propan-2-ol 1, (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(methylsulfinyl)phenyl]propan-2-ol 2 and (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(t-butyl-sulfinyl)phenyl]propan-2-ol 3 according to the Andersen methodology and their spectroscopic characterization is presented. The NMR and CD spectroscopic evidence of the existence of the equilibrium between sulfoxide and hypervalent sulfurane forms of these compounds in solution and attempts at the isolation of corresponding sulfuranes are shown. For compound 3, the unprecedented subsequent irreversible transformation in solution into corresponding cyclic sulfinate ester–sultine 17 was established on the basis of NMR spectroscopy measurements. The mechanism of this transformation was investigated by means of GC-MS analysis and confirmed on the basis of synthesized long alkyl chain analog 23 transformation in solution. Moreover, the oxidation properties of obtained sulfoxides 2 and 3 for the selected compounds are described.
1. Introduction
Sulfuranes, compounds with a central sulfur atom of higher than formal valency and a 3c-4e hypervalent bond, have been known for a relatively long time [1,2]. Their chirality, especially in the case of spirosulfuranes, is also well documented [2,3,4,5,6]. Although there are a few publications presenting stable chiral bicyclic sulfuranes bearing a strongly electronegative halogen atom as an apical substituent [3,7,8,9,10,11], examples of that kind of stable sulfurane with a hydroxyl group in an apical position have not been shown till now. Selected sulfoxides bearing perfluorocumyl moiety seem to be appropriate precursors for obtaining the hydroxysulfuranes mentioned above, because of the presence of two strongly electron-withdrawing perfluoromethyl groups and the phenyl ring, together with the possibility of forming a five-membered ring containing “a half” of a hypervalent bond, the properties of which can stabilize the desired hypervalent structure. Furthermore, those sulfinyl compounds could be useful as bidental chiral ligands and chiral auxiliaries.
2. Results and Discussion
Herein, we present results of studies on the synthesis and behavior in solution of novel optically active sulfoxides bearing a perfluorocumyl moiety: (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(p-tolylsulfinyl)phenyl]propan-2-ol 1, (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(methylsulfinyl)phenyl]propan-2-ol 2 and (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(t-butyl-sulfinyl)phenyl]propan-2-ol 3.
They were obtained according to the Andersen methodology [12] using an ortholithiation reaction of perfluorocumyl alcohol 5 and by treating formed in situ dilithio-derivative 6 with corresponding optically pure sulfinate esters 7, 8 or 9 (Scheme 1). The reactions occur with the inversion of the absolute configuration on a sulfur atom. In the case of sulfoxide 1, the substrate was commercially available O-(-)-(1R,2S,5R)-menthyl (-)-(S)-p-toluenesulfinate 7, and for compounds 2 and 3, O-diacetone-D-glucosyl (-)-(S)-methanesulfinate 8 and O-diacetone-D-glucosyl (-)-(S)-t-butanesulfinate 9, respectively. Compounds 8 and 9 were obtained using a method presented elsewhere [13].
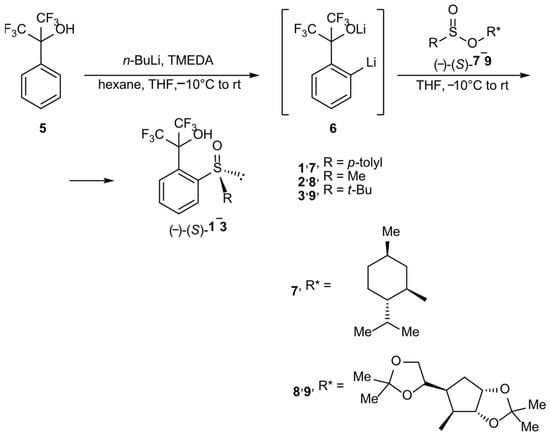
Scheme 1.
Synthesis of sulfoxides 1–3.
The choice of O-diacetone-D-glucosyl sulfinate esters for the synthesis of compounds 2 and 3 instead of corresponding O-(-)-(1R,2S,5R)-menthyl derivatives was dictated by better yields and stereoselectivity. Nevertheless, the yields of the presented processes were rather low; sulfoxide 1 showed the highest yield value, of 40%, but for 2, this was only 16%, probably because of the side reaction of deprotonation of the methyl group bonded to the sulfur atom by dilithio-derivative 6. The lowest yield was for 3, at 7%, which was most likely due to the bulkiness of the t-butyl group. The unsatisfactory yields were compensated by very high stereoselectivity. Product 1 was obtained with 95% ee, and in the case of 2 and 3, the process was stereospecific.
In order to prove the possibility of transformation of sulfoxide 1 form into corresponding hydroxysulfurane 10, compound 1 was crystallized from DCM to give monocrystal. However, its X-ray diffraction analysis allowed us to unambiguously establish that the crystallized compound had a sulfoxide structure with the S absolute configuration on the sulfur atom. On the other hand, the series of 19F NMR spectra of compound 1 solution in CDCl3 was recorded. They showed two quartets characteristic of nonequivalent perfluoromethyl groups as a result of the near presence of the chiral sulfinyl group (Figure 1a). The prolongation of the experiment to 3 h resulted in the appearance of an additional pair of quartets of lower intensity (Figure 1b), which suggested the spontaneous formation of another chiral structure, hydroxysulfurane 10. After 20 days, the intensity of “new” signals increased from 12% to 36% at the expense of the “old” pair (Figure 1c), which, in turn, meant the continuous conversion of sulfoxide 1 to hypervalent structure 10. Attempts to isolate compound 10 from that experiment failed and gave sulfoxide 1 as the only product. These observations proved the existence of the spontaneous and reversible transformation of sulfinyl derivative 1 into hydroxysulfurane structure 10 (Scheme 2). The change in chemical shifts of the quartet signals in Figure 1b,c can be attributed to a change in the chemical composition of the solution and, thus, the change in the intensity of intermolecular interactions between 1 and 10, especially if we take into consideration the presence of hydroxyl groups (hydrogen bond donors) and fluorine atoms (hydrogen bond acceptors), which can interact with each other relatively strongly.
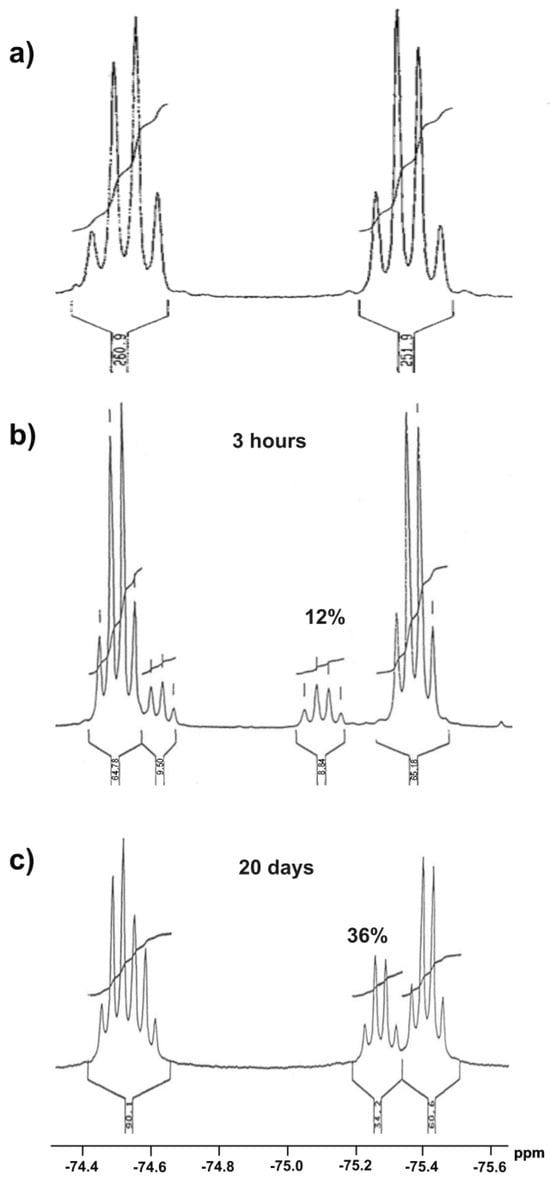
Figure 1.
Series of 19F NMR spectra of sulfoxide 1 in CDCl3: (a) after isolation, (b) after 3 h in solution, (c) after 20 days in solution.
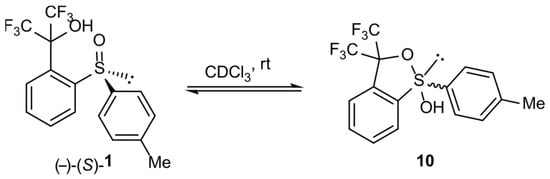
Scheme 2.
Reversible transformation of sulfinyl derivative 1 into hydroxysulfurane structure 10 in solution.
We repeated the above synthesis, and, during purification using column chromatography, two groups of fractions were isolated based on TLC analysis. The 19F NMR spectra in CDCl3 for each group were recorded at time intervals (Figure 2). The experiment clearly showed intense dynamics in both systems, manifested by the cyclic coalescence and differentiation of the quartet signals mentioned above. This result is further evidence of the existence of structures 1 and 10 in each fraction and their dynamic reversible transformation into one another. The formation of two unidentified products (indicated by the appearance of two singlet signals) was also observed. Moreover, for both groups of fractions, CD spectra in DCM were recorded (Figure 3) and showed opposite Cotton effects with shifted maxima relative to each other, and after isolation, their melting points were significantly different (146–150 °C and 80–85 °C), which confirmed the presence of two different chiral structures: sulfoxide 1 and sulfurane 10. Unfortunately, all attempts to crystallize either form did not furnish a signle crystal of sufficient quality for X-ray diffraction analysis. The differences between Figure 1 and Figure 2 can be explained in terms of the significantly different sample histories. In Figure 1, the sample is pure sulfoxide 1 after its preparation, while in Figure 2, two groups of fractions with not precisely established composition are analyzed, which can affect parameters such as chemical shifts. We assume that the driving forces of the transformation of compound 1 into 10 are as follows: (a) intramolecular coordination of the hydroxyl to sulfur; (b) formation of a stabilizing 3c–4e hypervalent bond, which likely requires stabilization by the solvent, as we were unable to isolate this species; (c) the strong electron-withdrawing effect of the perfluorocumyl moiety stabilizing the hypervalent bond; (d) formation of a five-membered ring, which reduces the entropic penalty. On the other hand, the reverse transformation, from 10 to 1, is most probably driven by reaching a certain concentration limit of 10 in solution, followed by destabilizing intermolecular interactions.
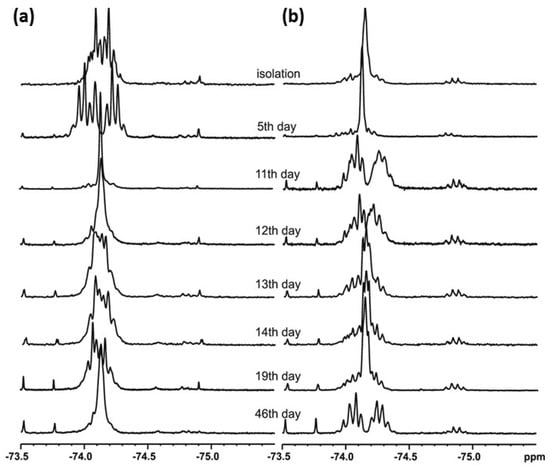
Figure 2.
The 19F NMR spectra in CDCl3 of: (a) fraction 1, (b) fraction 2.
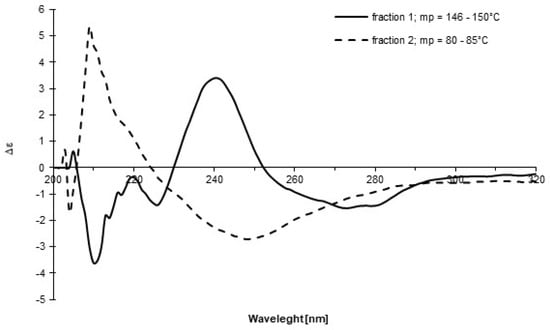
Figure 3.
The CD spectra of two fractions obtained during purification process of 1 recorded in DCM.
A similar approach was applied to the other two sulfoxides, 2 and 3. Due to difficulties in crystallizing these compounds, the absolute configuration was determined using an indirect comparative method. CD spectra of these compounds were recorded in DCM (Figure 4) and compared with the spectra of analogous sulfoxides, (R)-p-tolyl-methylsulfoxide and (R)-p-tolyl-t-butylsulfoxide, respectively, as reported by Mislow et al. [14]. The opposite signs of the Cotton effect at similar wavelengths together with the assumption that the reaction gives a product of inverted configuration with respect to precursors 8 and 9 allowed for the conclusion that sulfoxides 2 and 3 possess the SS absolute configuration.
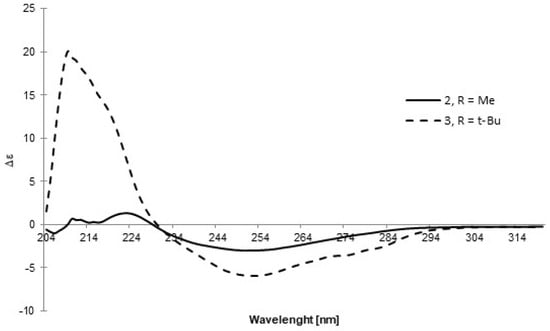
Figure 4.
The CD spectra of sulfoxides 2 and 3 recorded in DCM.
As previously, the enantiomeric excess was determined via the HPLC technique using a column with a chiral stationary phase and racemic mixtures of compounds 2 and 3 as a reference. It is noteworthy that racemization attempts with gaseous HCl in DCM at rt for (-)-(S)-t-butylsulfoxide 3 resulted in the corresponding racemate but only in a 10% yield, whereas the main product was sulfide 11, the result of the reduction of starting sulfoxide 3, obtained in a 90% yield (Equation (1)). In the case of compound 2, the corresponding sulfide 12 was the only product (Scheme 3). These conclusions were confirmed by 19F NMR spectra. These are examples of sulfoxide reduction by gaseous HCl. Moreover, attempts to determine the ee of methylsulfoxide 2 by the use of the chiral solvating agent—t-butylphenylthiophosphinic acid 13—in CDCl3 resulted in the oxidation of compound 13 to disulfide 14, with the simultaneous reduction of 2 to 12 (Scheme 3) according to 31P and 19F NMR spectra, which indicates that a sulfoxide bearing the perfluorocumyl moiety is a stronger oxidant than analogous aryl methyl sulfoxides.

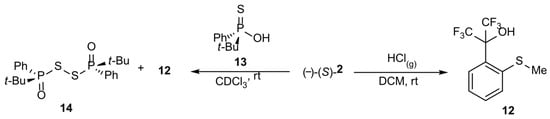
Scheme 3.
Oxidizing effect of sulfoxide 2.
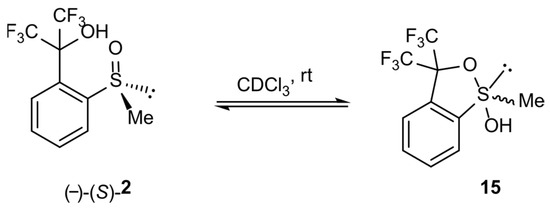
In order to check the possibility of the transformation of alkyl derivatives 2 and 3 into the corresponding hypervalent structures 15 and 16, respectively, similar long-lasting NMR experiments were conducted. The series of 19F NMR spectra (Figure 5a) confirmed the existence of two different chiral structures in a CDCl3 solution. They showed two multiplet signals, which are combinations of two quartet resonance signals each, as in the case of compound 1. Moreover, the 1H NMR spectrum (Figure 5b) recorded for the same solution contained doubled resonance signals related to the protons of the methyl group bonded to the sulfur atom (2.88 and 2.93 ppm, Figure 5b, lower image) and to the aromatic proton at the meta position of the benzene ring (8.2–8.3 ppm region, Figure 5b, upper image). The spectrum remained unchanged until the end of the experiment. The analysis of the spectra indicated an equilibrium between the sulfinyl 2 and sulfurane 15 structures in solution (Scheme 4).
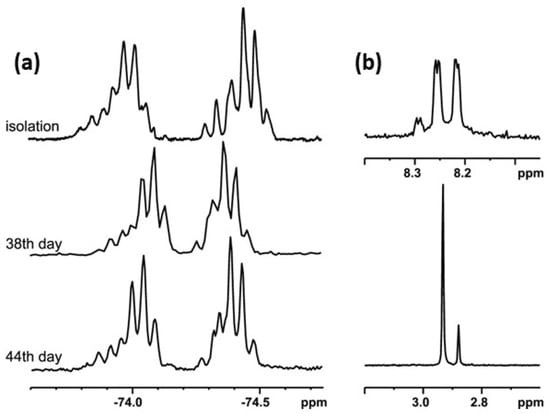
Figure 5.
The NMR spectra of compound 2: (a) 19F NMR, (b) 1H NMR (selected regions).
Scheme 4.
Equilibrium between sulfinyl 2 and sulfurane 15 structures in solution.
t-Butylsulfoxide 3 turned out to be the most interesting among the presented compounds. Three sets of spectra at time intervals were recorded: 19F NMR in CDCl3 (Figure 6a) and in DCM (Figure 6b), and 1H NMR in CDCl3 (Figure 7). The existence of both sulfoxide 3 and hydroxysulfurane 16 (Scheme 5) in solutions was confirmed by the broadening of the quartet resonance signals in the 19F NMR spectrum in DCM after 6 days (Figure 6b), and the presence of a doubled singlet signal referred to the protons of the t-butyl group in the 1H NMR spectrum after isolation (1.32 and 1.56 ppm, Figure 7). Interestingly, compound 3 underwent another irreversible transformation into a structure with a stereogenic sulfur atom. It was evidenced by the appearance of a new pair of quartet signals at −74.80 and −74.04 ppm in the 19F NMR spectra (Figure 6), both in CDCl3 and DCM, which was accompanied by the disappearance of the signal of the t-butyl group protons. These observations, supported by the mass spectrometry analysis [MS EI, m/z = 290.0 (100%)] and comparisons with the data acquired for the product obtained in an independent experiment, allowed for the identification of the product formed from sulfoxide 3 as the cyclic sulfinate ester 17 (Scheme 5). The formation of 17 was preceded by the production of two unidentified achiral products, which was indicated by the appearance of two singlet signals (−75.33 and −73.79 ppm) in the 19F NMR spectra (Figure 6a). Due to the decrease in the intensity of one of them (−75.33 ppm), it was probably assigned to the precursor of compound 17.
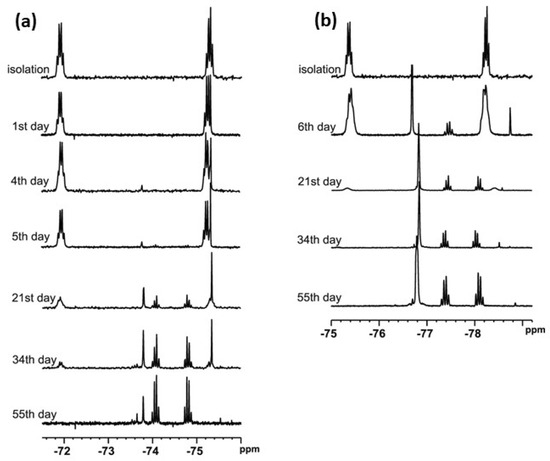
Figure 6.
The 19F NMR spectra of compound 3 recorded at time intervals in: (a) CDCl3, (b) DCM.
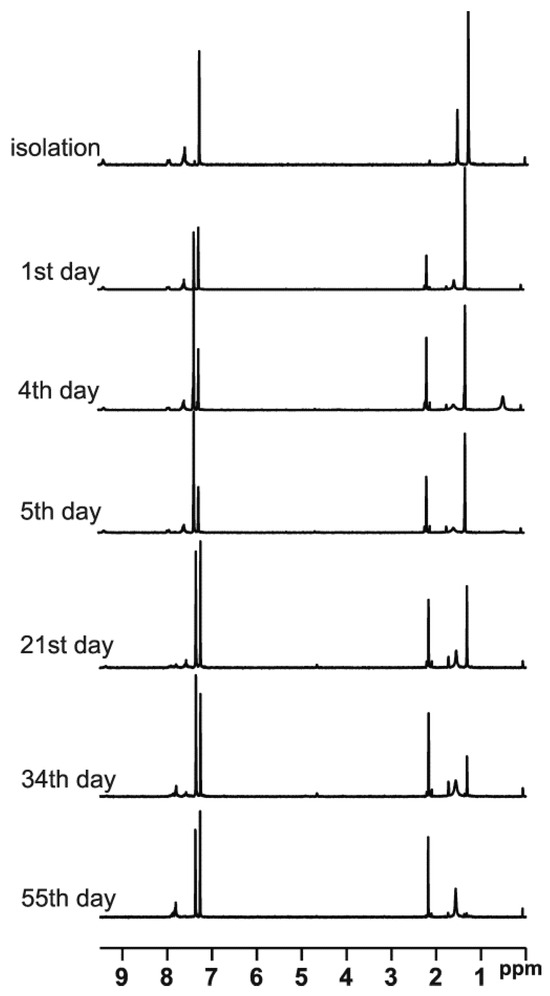
Figure 7.
The 1H NMR spectra of compound 3 recorded at time intervals in CDCl3.
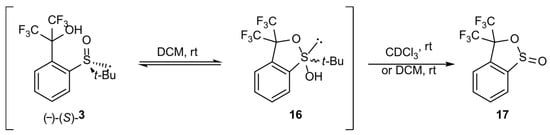
Scheme 5.
Transformations of sulfoxide 3 in solution.
Two hypothetical alternative mechanisms were proposed. According to the first one, existing in equilibrium with the sulfoxide structure 3, sulfurane 16 underwent rearrangement to another sulfurane 18, followed by turnstile rotation (TR) [1,2] of the latter to give structure 19. Sulfurane 19 underwent a ligand coupling reaction to furnish sultine 17 and 2-methylpropane as a side product (Scheme 6). The mechanism would constitute an explanation for the formation of ester 17 in DCM solution.
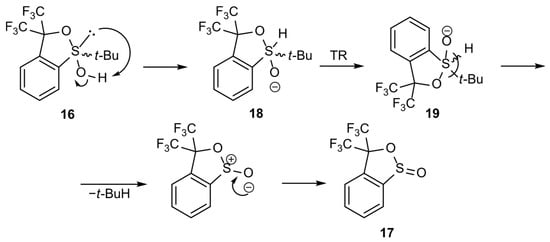
Scheme 6.
Hypothetical mechanism of sulfurane 16 transformation into sultine 17.
The second mechanism could be more appropriate for the CDCl3 solution of 3. It assumes the initial elimination of 1,1-dimethylethylene from sulfoxide 3 and the formation of sulfenic acid 20. Acid 20 furnishes sultine 17 either via the direct elimination of a hydrogen molecule from the tetravalent tautomer 20′ and cyclization (Scheme 7, path a) or its dimerization to thiosulfinate 21 (Scheme 7, path b) and subsequent cyclization as a result of the nucleophilic attack of the oxygen atom in the hydroxyl group on the sulfinyl sulfur atom (Scheme 7). Path b requires the formation of thiol 22, whose existence could explain the appearance of one of the singlet resonance signals in the 19F NMR spectrum.
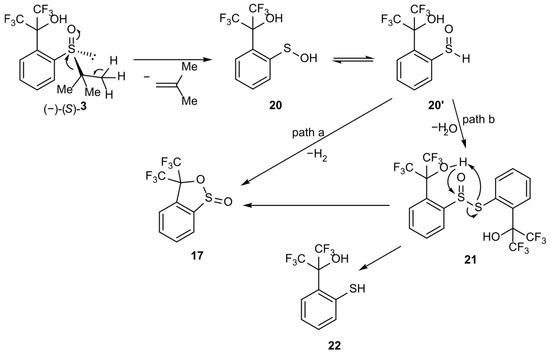
Scheme 7.
Hypothetical mechanism of sulfoxide 3 transformation into sultine 17.
In order to confirm one of the proposed mechanisms, recordable products of the described transformation were required. That goal was achieved through the synthesis of racemic analog 23 bearing a long alkyl substituent with the use of hexadecanesulfinyl chloride 24 (Scheme 8). The reaction was carried out by ortho-lithiation of alcohol 5, followed by treatment of the resulting mixture with an equimolar amount of chloride 24 in THF at room temperature for 22 h. The obtained crude product was then subjected to column chromatography followed by preparative thin-layer chromatography to give pure product 23 in a low yield (10%).
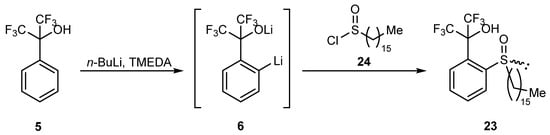
Scheme 8.
Synthesis of sulfoxide 23.
The sample of the obtained compound was kept in a CDCl3 solution at room temperature for 72 days (until the complete disappearance of the initial quartet signals in the 19F NMR spectrum), and the resulting mixture was then subjected to GC-MS analysis. According to both proposed mechanisms, the main product of the discussed transformation would be sultine 17, but in the first case (Scheme 6), it would be accompanied by the corresponding saturated hydrocarbon and, in the second case (Scheme 7), by the corresponding unsaturated hydrocarbon and probably thiol 22. In view of the above, based on initial GC-MS analyses of commercially available references, 1-hexadecene and hexadecane were recorded (Figure 8c,e, respectively). As expected, the retention times of these two compounds were similar (23.54 and 23.57 min., correspondingly), but the results of MS analysis differed significantly, excluding the possibility of a mistake. Next, GC analysis of the sample mixture was conducted to give the chromatogram shown in Figure 8a. The obtained peak MS profiles are shown in Figure 8b,c,f. Figure 8b presents the MS profile of the peak with a retention time equal to 19.97 min and clearly indicates the presence of sultine 17 in the mixture (m/z 290). In Figure 8c, the MS profile of the peak at 23.54 min. is shown. Both the retention time with m/z values and MS peak intensities are almost identical to those recorded for the 1-hexadecene reference, which indicates the presence of this compound in the analyzed mixture. Moreover, MS analysis of the peak with a retention time of 38.44 min. revealed the presence of thiol 22 (m/z 276).
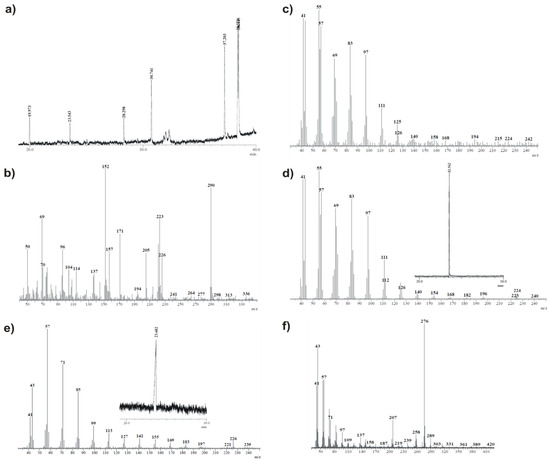
Figure 8.
The GC-MS analysis results obtained for compound 23 kept in CDCl3 solution for 72 days and corresponding reference materials: (a) chromatogram of 23 solution after 72 days, (b) MS profile of 19.97 min. GC peak, (c) MS profile of 23.54 min GC peak, (d) MS profile of 23.56 min. GC peak recorded for commercial 1-hexadecene, serving as reference, (e) MS profile of 23.57 min. GC peak recorded for commercial hexadecane, serving as reference, (f) MS profile of 38.44 min. GC peak.
On the basis of the above results, it can be claimed that the presented transformation occurs according to the mechanism shown in Scheme 7, path b, as follows:
- Elimination of a substituent at the sulfur atom with formation of the corresponding alk-1-ene and sulfenic acid 20. The presence of a hydrogen atom at the β-carbon atom and its aliphatic nature is necessary. In the absence of these features, the described process does not occur, as can be observed for compounds 1 (methyl group at the sulfur atom—no β-hydrogen atom) and 2 (p-tolyl group at the sulfur atom—aromatic β-carbon atom).
- Reversible rearrangement of sulfenic acid 20 into sulfoxide 20′.
- Spontaneous condensation of molecules 20 and 20′ into the unstable thiosulfinate 21 with the release of a water molecule.
- Sultine ring closure in structure 21 leading to the formation of sultine 17 with simultaneous elimination of thiol 22.
The above-described process seems to be general for every S-substituted o-sulfinyl derivative of 1,1,1,3,3,3-hexafluoro-2-phenylpropan-2-ol after the fulfilment of the structural restrictions mentioned above. This aspect will be further investigated.
The above results also explain the changes in the NMR spectra of sulfoxide 3. In Figure 6a, the broadened singlet signal at ~−75.5 ppm can be attributed to unstable thiosulfinate 21 due to the fact that it disappears at the end of the process, although this assignment is not certain. In turn, the sharp singlet signal at ~−73.9 ppm, appearing on day 4–5 of the process and increasing its intensity from that point, can be attributed to the emerging thiol 22 (the chemical shift is in acceptable agreement with the literature data [15]). Obviously, the initial disappearance of the first pair of quartet signals in favor of the second pair of quartets is related to initial sulfoxide decomposition and the formation of the final sultine 17. An analogous situation can be observed in Figure 6b, though with different chemical shifts due to the lack of lock during recording of the spectra. In the 1H NMR spectra evolution, some newly formed signals can also be observed. New signals in the aliphatic region can be attributed to the formation of the alkene—1,1-dimethylethylene—partially soluble in CDCl3, while two new groups of signals in the aromatic region can be attributed to sultine 17 and thiol 22.
3. Materials and Methods
For column chromatography, silica gel 60 (70–230 mesh) (Merck, Darmstadt, Germany) was used. The NMR spectra were obtained using 200 MHz and 600 MHz spectrometers (Bruker, Billerica, MA, USA). The IR spectra were recorded using FTIR spectrometer (Bruker, Billerica, MA, USA) via the KBr tablet method. The mass spectra and HRMS were measured using double-focusing (BE geometry) mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) utilizing chemical ionization (CI) method with isobutane as an ionizing agent or electron ionization (EI) technique. The melting point was measured using capillary apparatus (Electrothermal, Rochford, UK). The specific optical rotation values were measured with the use of a polarimeter (Anton Paar, Graz, Austria) at wavelength of 589 nm and room temperature. The enantiomeric excesses were determined using HPLC technique (Varian, Palo Alto, CA, USA) on the analytical column with a chiral stationary phase and corresponding racemic mixtures as a reference. The CD spectra were recorded with circular dichroism spectrometer (JASCO, Tokyo, Japan). The GC-MS analysis was conducted at injection temperature of 200 °C in split mode (split ratio 1:40), and the analyte separation was achieved at column gas flow equal to 1.30 mL/min and the following oven temperature program: initially, 35 °C for 5 min; then, the temperature was increased to 300 °C within 8 min. and kept at this level for next 10 min. The MS analysis of separated fractions was realized using electrospray ionization (ESI) technique.
- (-)-(S)-1,1,1,3,3,3-Hexafluoro-2-[o-(p-tolylsulfinyl)phenyl]propan-2-ol (1); General procedure: [15]
Into a stirred-in-argon-atmosphere solution of n-BuLi in hexane (2.5 M, 10 mL, 25.0 mmol), TMEDA was added dropwise (0.57 mL, 3.75 mmol, 15%mol with respect to n-BuLi) at rt, and the solution was stirred for further 30 min. After this time, the mixture was cooled to −10 °C and a solution of perfluorocumyl alcohol 5 (2.08 g, 8.50 mmol) in dry THF (5 mL) was added dropwise for 5 min. Next, the mixture was allowed to warm up to rt and stirred for 24 h. After that, the solution was cooled to −10 °C and a solution of (-)-(S)-menthyl p-toluenesulfinate 7 (2.35 g, 8.00 mmol) in dry THF (10 mL) was added dropwise for 10 min. The obtained solution was allowed to warm up to rt and stirred for 24 h. Next, the reaction mixture was quenched with H2SO4 aqueous solution (5%; 5 mL) and H2O (20 mL), and the obtained two-phase system was extracted with DCM (4 × 30 mL); organic layers were combined, dried over MgSO4, and the solvent was evaporated. The obtained crude reddish oil was purified using column chromatography (SiO; Et2O/petroleum ether 1:1, v/v) to furnish pale-yellow solid (1.154 g, 40%).
- = −95.8 (c 1.22; DCM); ee = 95% [HPLC; Chiralpak AS; hexane/(iPrOH/EtOH 4:1, v/v) 9:1, v/v; flow rate 0.5 mL/min]; mp = 148–150 °C; Rf = 0.6 (Et2O).
- 1H NMR (200 MHz, CDCl3): δ = 8.44 (bs, 1H), 8.16 (d, 1H, 3JH-H = 6.7 Hz), 7.82–7.37 (m, 5H), 7.19 (d, 2H, 3JH-H = 1.7 Hz), 2.34 (s, 3H).
- 13C NMR (50 MHz, CDCl3): δ = 145.1, 142.6, 141.0, 130.8, 129.6, 128.3, 127.7, 127.3, 126.0, 79.9 (h, 2JF-13C = 119.3 Hz), 21.2.
- 19F NMR (188 MHz, CDCl3): δ = −74.11 (q, 3F, 4JF-F = 9.1 Hz), −74.39 (q, 3F, 4JF-F = 9.1 Hz).
- IR (KBr): 3434 (O-H stretch), 1400 (O-H bend), 1153 (S=O sulfoxide/O-S-O sulfurane), 1130 (C-F stretch), 707 (C-S stretch) cm−1.
- MS (CI): m/z (%) = 383.0 (100) [M + H]+.
- HRMS (EI): m/z calcd for C16H12SO2F6 382.04622; found 382.04547 (Δ 1.96380).
- (-)-(S)-1,1,1,3,3,3-Hexafluoro-2-[o-(methylsulfinyl)phenyl]propan-2-ol (2)
The synthesis was carried out according to the general procedure with use of the following: n-BuLi solution in hexane (2.5 M, 8.40 mL, 21.00 mmol), TMEDA (0.47 mL, 3.15 mmol, 15%mol with respect to n-BuLi), perfluorocumyl alcohol 5 (1.71 g, 7 mmol) solution in dry THF (1.5 mL), (-)-(S)-O-diaceton-D-glucosyl methanesulfinate 8 (0.99 g, 3.1 mmol) solution in dry THF (5 mL), aqueous solution of H2SO4 (5%, 5 mL) and H2O (20 mL). The reaction mixture after quenching was extracted with Et2O (8 × 20 mL); organic layers were combined, washed with H2O, and dried over MgSO4. The solvent evaporation furnished crude product as a orange oil (2.43 g). After purification with two subsequent rounds of column chromatography, [SiO2; Et2O (24 × 20 mL), acetone (17 × 20 mL)] followed by [SiO2; acetone/petroleum ether 3:1, v/v (30 × 20 mL), acetone (6 × 20 mL)], a white solid was obtained (153 mg; 16%).
- = −178.1 (c 0.53; CDCl3); ee > 99% (HPLC; Chiralpak AS; hexane/iPrOH 4:1, v/v; flow rate 0.5 mL/min), mp = 125 °C; Rf = 0.3 (acetone/Et2O 1:1, v/v).
- 1H NMR (200 MHz, CDCl3): δ = 8.68 (bs, 1H), 8.28 (d, 1H, 3JH-H = 8.0 Hz), 7.53–7.74 (m, 3H), 2.94 (s, 3H).
- 13C NMR (50 MHz, CDCl3): δ = 144.7, 136.6, 131.1, 131.0, 130.7, 128.4, 123.8 (q, 1JC-F = 288.3 Hz), 79.1 (h, 2JF-C = 30.0 Hz), 44.7.
- 19F NMR (188 MHz, CDCl3): δ = −74.11–−73.85 (m), −74.53–−74.29 (m), −74.81 (q, 4JF-F = 9.8 Hz),
- IR (KBr): 3487 (O-H stretch), 1421 (O-H bend), 1130 (C-F stretch), 1066 (S=O sulfoxide), 704 (C-S stretch) cm−1.
- MS (CI): m/z (%) = 307.0 (100) [M + H]+.
- HRMS (EI): m/z calcd for C10H8SO2F6 306.014920, found 306.014650 (Δ 0.88277).
- (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(t-butyl-sulfinyl)phenyl]propan-2-ol (3)
The synthesis was carried out according to the general procedure with use of the following: n-BuLi solution in hexane (2.5 M, 3.20 mL, 8.0 mmol), TMEDA (0.24 mL, 1.6 mmol, 20%mol with respect to n-BuLi), perfluorocumyl alcohol 5 (680 mg, 2.8 mmol) solution in dry THF (2 mL), (-)-(S)-O-diaceton-D-glucosyl t-butanesulfinate 9 (505 mg, 1.4 mmol) solution in dry THF (5 mL), aqueous solution of H2SO4 (5%, 10 mL) and H2O (30 mL). After addition of sulfinate, the reaction time was prolonged to 72 h. The reaction mixture after quenching was extracted with Et2O (8 × 20 mL); organic layers were combined, washed with H2O, and dried over MgSO4. The solvent evaporation furnished crude product as an orange oil (903 mg). After purification using column chromatography, [SiO2; Et2O/petroleum ether 1:2, v/v (24 × 20 mL), Et2O/petroleum ether 1:1, v/v (12 × 20 mL), Et2O (5 × 20 mL)] followed by crystallization from Et2O solution, a white solid was obtained (34 mg; 7%).
- = −159.9 (c 0.88, DCM); ee > 99% (HPLC; Chiralpak AS; hexane/iPrOH 9:1, v/v; flow rate 0.5 mL/min), mp = 132 °C; Rf = 0.1 (Et2O/petroleum ether 1:1, v/v).
- 1H NMR (200 MHz, CDCl3): δ = 9.39 (bs, 1H), 7.93 (d, 1H, 3JH-H = 7.7 Hz), 7.58–7.62 (m, 3H), 1.32 (s, 9H).
- 19F NMR (188 MHz, CDCl3): δ = −71.92 (q, 3F, 4JF-F = 8.9 Hz), −75.27 (q, 3F, 4JF-F = 8.9 Hz),
- MS (CI): m/z (%) = 349.0 (100) [M + H]+.
- HRMS (CI): m/z calcd for C13H15SO2F6 349.06969, found 349.06870 (Δ 2.85144).
- 1,1,1,3,3,3-Hexafluoro-2-[o-(hexadecylsulfinyl)phenyl]propan-2-ol (23)
Into a stirred-in-argon-atmosphere solution of n-BuLi in hexane (2.70 M, 9.30 mL, 25.0 mmol), TMEDA was added dropwise (0.57 mL, 3.75 mmol, 15%mol with respect to n-BuLi) at rt, and the solution was stirred for further 30 min. After this time, the mixture was cooled to −10 °C, and a solution of perfluorocumyl alcohol 5 (2.08 g, 8.50 mmol) in dry THF (5 mL) was added dropwise for 5 min. Next, the mixture was allowed to warm up to rt and stirred for 26 h. After that, the solution was cooled to −10 °C, and a solution of hexadecanesulfinyl chloride 24 (2.60 g, 8.50 mmol) in dry THF (10 mL) was added dropwise for 5 min. The obtained solution was allowed to warm up to rt and stirred for 22 h. Next, the reaction mixture was quenched with H2SO4 aqueous solution (5%; 5 mL) and H2O (20 mL), and the obtained two-phase system was extracted with DCM (5 × 50 mL); organic layers were combined, dried over MgSO4, and the solvent was evaporated. The obtained crude brownish oil was purified using column chromatography (SiO; CHCl3/acetone 10:1, v/v), followed by preparative thin-layer chromatography (SiO2; CHCl3/acetone 8:1, v/v), to furnish pale-yellow solid (428 mg, 10%).
- 1H NMR (200 MHz, CDCl3): δ = 9.39 (bs, 1H), 8.18 (d, 1H, 3JH-H = 7.8 Hz), 7.71–7.37 (m, 3H), 3.26–3.11 (m, 1H), 2.89–2.75 (m, 1H), 1.77–1.62 (m, 4H), 1.31–1.16 (m, 24H), 0.85 (t, 3H, 3JH-H = 6.2 Hz).
- 13C NMR (150 MHz, CDCl3): δ = 144.5, 130.6, 129.4, 128.3, 128.2, 126.2, 122.7 (q, 1JC-F = 287.4 Hz), 80.0 (h, 2JF-C = 29.6 Hz), 59.0, 32.0, 29.74, 29.73, 29.64, 29.61, 29.4, 29.1, 28.6, 23.5, 23.0, 22.7, 14.2.
- 19F NMR (188 MHz, CDCl3): δ = −73.79 (q, 3F, 4JF-F = 9.0 Hz), −74.35 (q, 3F, 4JF-F = 9.0 Hz).
- MS (CI): m/z (%) = 517.3 (27) [M+H]+, 499.2 (100) [M + H − H2O]+.
- Reduction and racemization procedure of compound (-)-(S)-3
HCl gas was bubbled through a stirred solution of sulfoxide (-)-(S)-3 (6.3 mg; 0.018 mmol) in DCM (2 mL) for 5 s at rt. The resulting solution was washed with K2CO3 solution (5%), dried over MgSO4, and the solvent was evaporated in vacuo to give a mixture of the reduction product 11 (90%) and rac-3 (10%), as judged via 19F NMR analysis and the lack of optical activity.
- 19F NMR (188 MHz, CDCl3): δ = −74.83 (q, 3F, 4JF-F = 8.9 Hz), −73.79, −72.80 (q, 3F, 4JF-F = 8.9 Hz)
- Reduction procedure of compound (-)-(S)-2
HCl gas was bubbled through a stirred solution of sulfoxide (-)-(S)-2 (1.5 mg; 0.005 mmol) in DCM (2 mL) for 5 s at rt. The resulting solution was washed with K2CO3 solution (5%), dried over MgSO4, and the solvent was evaporated in vacuo to give the reduction product, sulfide 12, as a yellowish oil (1.4 mg, 100%) based on 19F NMR spectrum.
- 19F NMR (188 MHz, CDCl3): δ = −77.17
4. Conclusions
We presented the synthesis and characterization of three novel optically active sulfoxides: (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(p-tolylsulfinyl)phenyl]propan-2-ol 1, (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(methylsulfinyl)phenyl]propan-2-ol 2 and (-)-(S)-1,1,1,3,3,3-hexafluoro-2-[o-(t-butyl-sulfinyl)phenyl]propan-2-ol 3. We identified their interesting properties and confirmed the existence of spontaneous reversible transformations of these compounds into the corresponding hypervalent structures, hydroxysulfuranes 10, 15 and 16, on the basis of the analysis of CD, 1H and 19F NMR spectra. It constitutes the first example of sulfuranes stable in solution having a hydroxyl group at the apical position. Nevertheless, all attempts to isolate these sulfuranes failed, and in the case of 1 and 2, the starting sulfinyl structures were recovered. For t-butyl derivative 3, the unprecedented transformation into the cyclic sulfinate ester 17 in solution was observed. Two speculative mechanisms of the process were proposed, and one of them was confirmed to be correct by means of GC-MS and NMR analyses. Furthermore, during attempts to racemize 2 and 3, we found that they were stronger oxidants in comparison with analogous aryl-alkyl sulfoxides without perfluoromethyl groups, whose property can be useful in stereochemistry, particularly for asymmetric oxidation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules31060969/s1, NMR spectra, crystallographic data of compound (S)-1.
Author Contributions
Conceptualization, J.D. and A.Z.; methodology, A.Z.; validation, J.D., A.Z. and E.W.; investigation, A.Z. and E.W.; resources, J.D.; writing—original draft preparation, A.Z.; writing—review and editing, A.Z.; visualization, A.Z.; supervision, J.D.; funding acquisition, J.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Polish Government Fund, grant number N N204 323137, and National Science Center, grant number UMO-2011/01/B/ST5/06304.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
The data underlying this study are available in the published article and its Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chemistry of Hypervalent Compounds; Akiba, K.-Y., Ed.; Wiley-VCH: New York, NY, USA, 1999. [Google Scholar]
- Drabowicz, J.; Halaba, G. Review on Sulphur-Containing Compounds. Rev. Heteroat. Chem. 2000, 22, 1. [Google Scholar]
- Martin, J.C.; Balthazor, T.M. Sulfuranes. 22. Stereochemical course of an associative displacement at tetracoordinate sulfur(IV) in a sulfurane of known absolute configuration. A proposed system of nomenclature for optically active pentacoordinate species. J. Am. Chem. Soc. 1977, 99, 152. [Google Scholar] [CrossRef]
- Kawashima, T.; Ohno, F.; Okazaki, R. Synthesis, Structure, and Thermolysis of a Tetracoordinate 1,2λ4-Oxathietane. Angew. Chem. Int. Ed. 1994, 33, 2094–2095. [Google Scholar] [CrossRef]
- Kawashima, T. Four-membered heterocyclic compounds containing high coordinate group 16 elements. Coord. Chem. Rev. 2003, 244, 137. [Google Scholar] [CrossRef]
- Allenmark, S. Recent advances in spirochalcogenurane stereochemistry—A mini review. Chirality 2008, 20, 544. [Google Scholar] [CrossRef] [PubMed]
- Balthazor, T.M.; Martin, J.C. Sulfuranes. XVIII. Stereochemical course of an associative nucleophilic displacement at tetracoordinate sulfur(IV). Optically active trigonal bipyramidal molecule, a chlorosulfurane. J. Am. Chem. Soc. 1975, 97, 5634. [Google Scholar] [CrossRef]
- Datta, A.K.; Livant, P.D. Synthesis and reactivity of a cyclopropyloxy sulfurane. J. Org. Chem. 1983, 48, 2447. [Google Scholar] [CrossRef]
- Zhang, J.; Takahashi, T.; Koizumi, T. Optically pure alkoxychlorosulfuranes. Synthesis and transformation to chiral sulfoxides, N-p-tosylsulfilimines, and sulfonium ylides. Heterocycles 1997, 44, 325. [Google Scholar] [CrossRef]
- Zhang, J.; Saito, S.; Koizumi, T. Stereochemical Research on the Hydrolysis of Optically Pure Spirosulfuranes: Efficient Synthesis of Chiral Sulfoxides with Completely Opposite Stereochemistry. J. Org. Chem. 1998, 63, 5265. [Google Scholar] [CrossRef]
- Akiba, K.-Y. Studies on hypervalent compounds and synthetic work using heteroaromatic cations. Heteroat. Chem. 2011, 22, 207. [Google Scholar] [CrossRef]
- Andersen, K.K. Synthesis of (+)-ethyl p-tolyl sulfoxide from (−)-menthyl (−)-p-toluenesulfinate. Tetrahedron Lett. 1962, 3, 93. [Google Scholar] [CrossRef]
- Fernádez, I.; Khiar, N.; Llera, J.M.; Alcudia, F. Asymmetric synthesis of alkane- and arenesulfinates of diacetone-D-glucose (DAG): An improved and general route to both enantiomerically pure sulfoxides. J. Org. Chem. 1992, 57, 6789. [Google Scholar] [CrossRef]
- Mislow, K.; Green, M.M.; Laur, P.; Melillo, J.T.; Simmons, T.; Ternay, A.L., Jr. Absolute Configuration and Optical Rotatory Power of Sulfoxides and Sulfinate Esters. J. Am. Chem. Soc. 1965, 87, 1958–1976. [Google Scholar] [CrossRef]
- Perozzi, E.F.; Michalak, R.S.; Figuly, G.D.; Stevenson, W.H., III; Dess, D.B.; Ross, M.R.; Martin, J.C. Directed dilithiation of hexafluorocumyl alcohol—Formation of a reagent for the facile introduction of a stabilizing bidentate ligand in compounds of hypervalent sulfur (10-S-4), phosphorus (10-P-5), silicon (10-Si-5), and iodine (10-I-3). J. Org. Chem. 1981, 46, 1049. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.