
Synthesis and Characterization of a Two-Station Two-Gate Calix[6]arene-Based [2]Catenane



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Synthesis of Axle 10
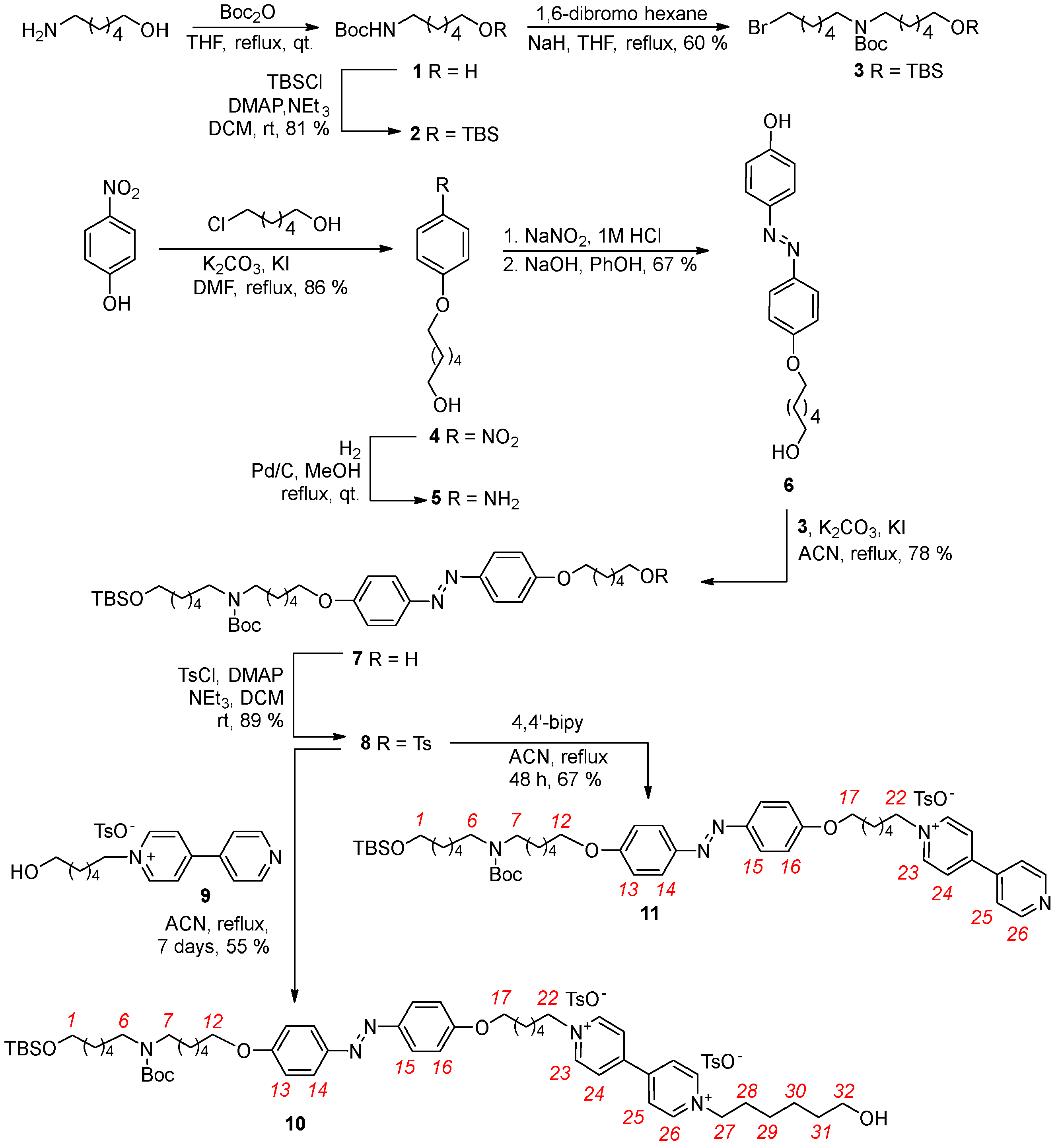
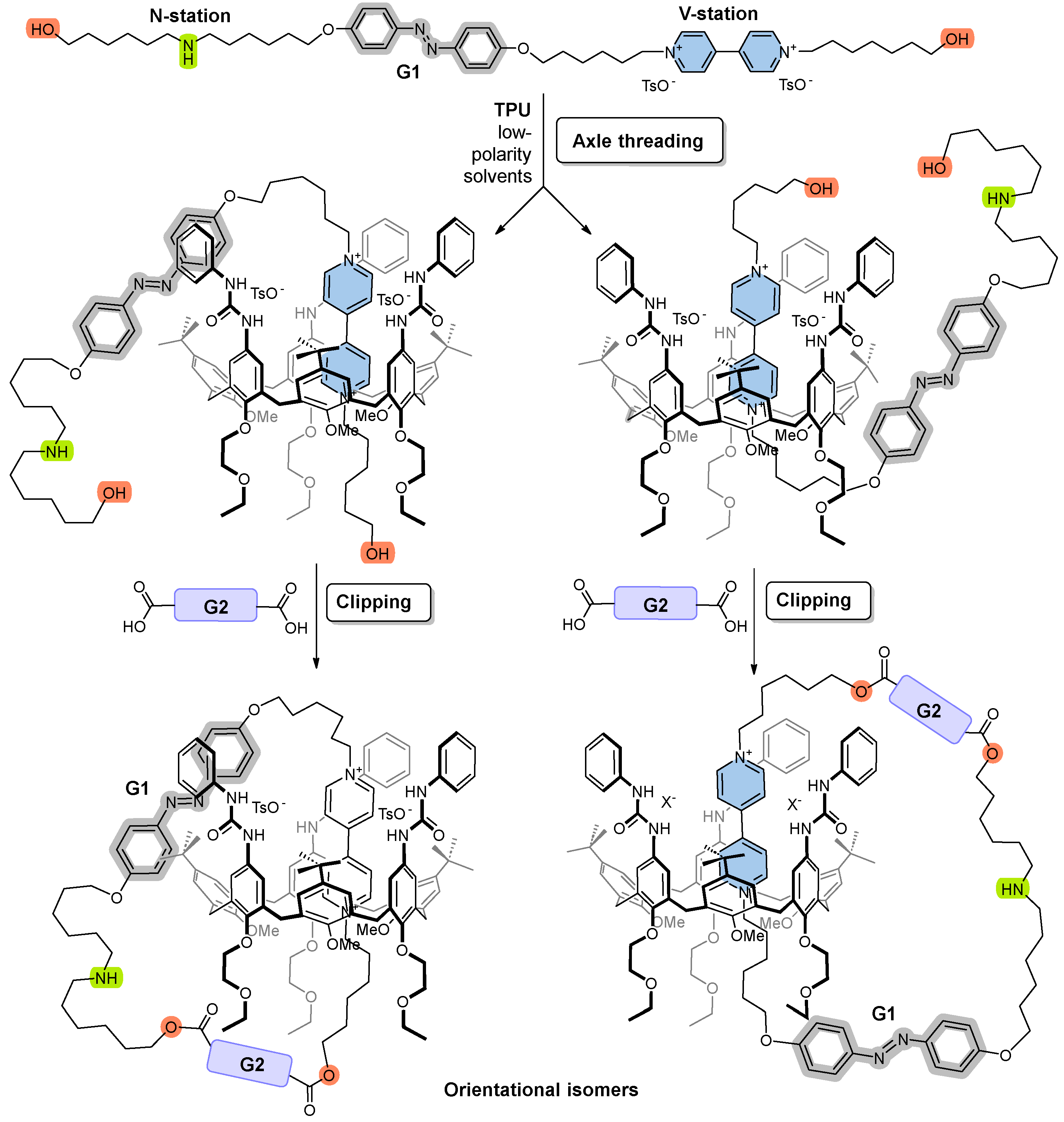
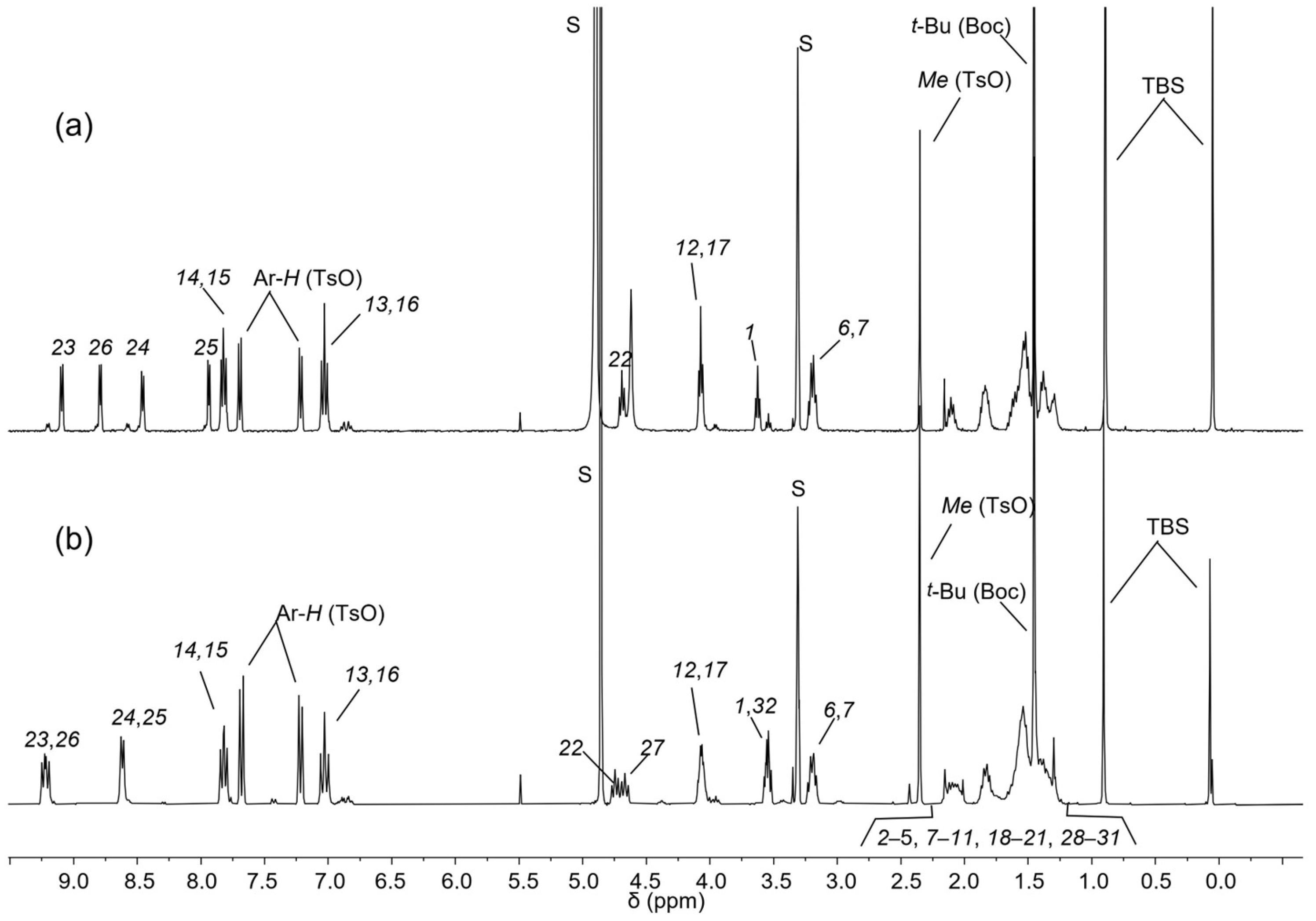
2.2. Synthesis of Rotaxane 12(Azo-Up)
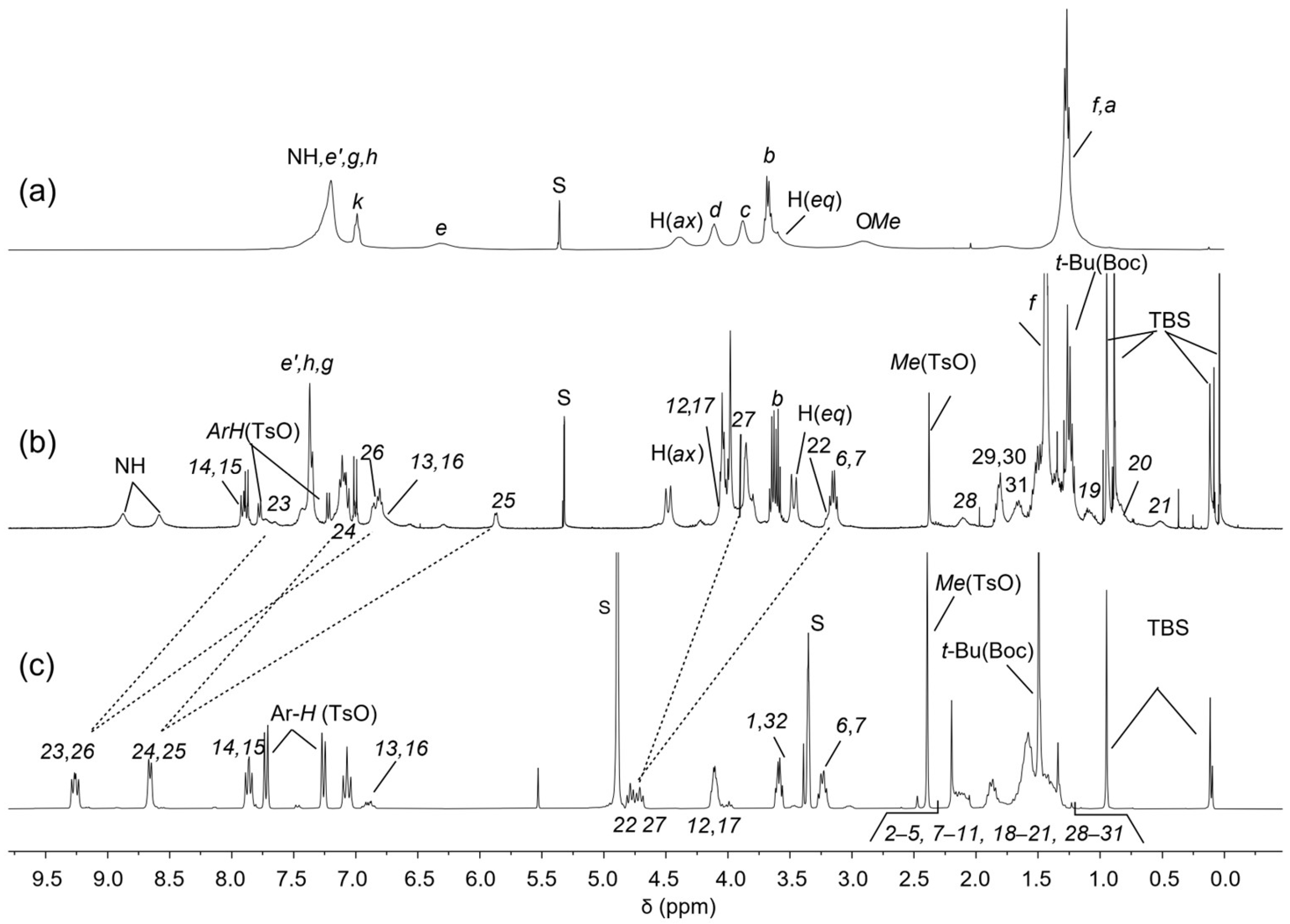
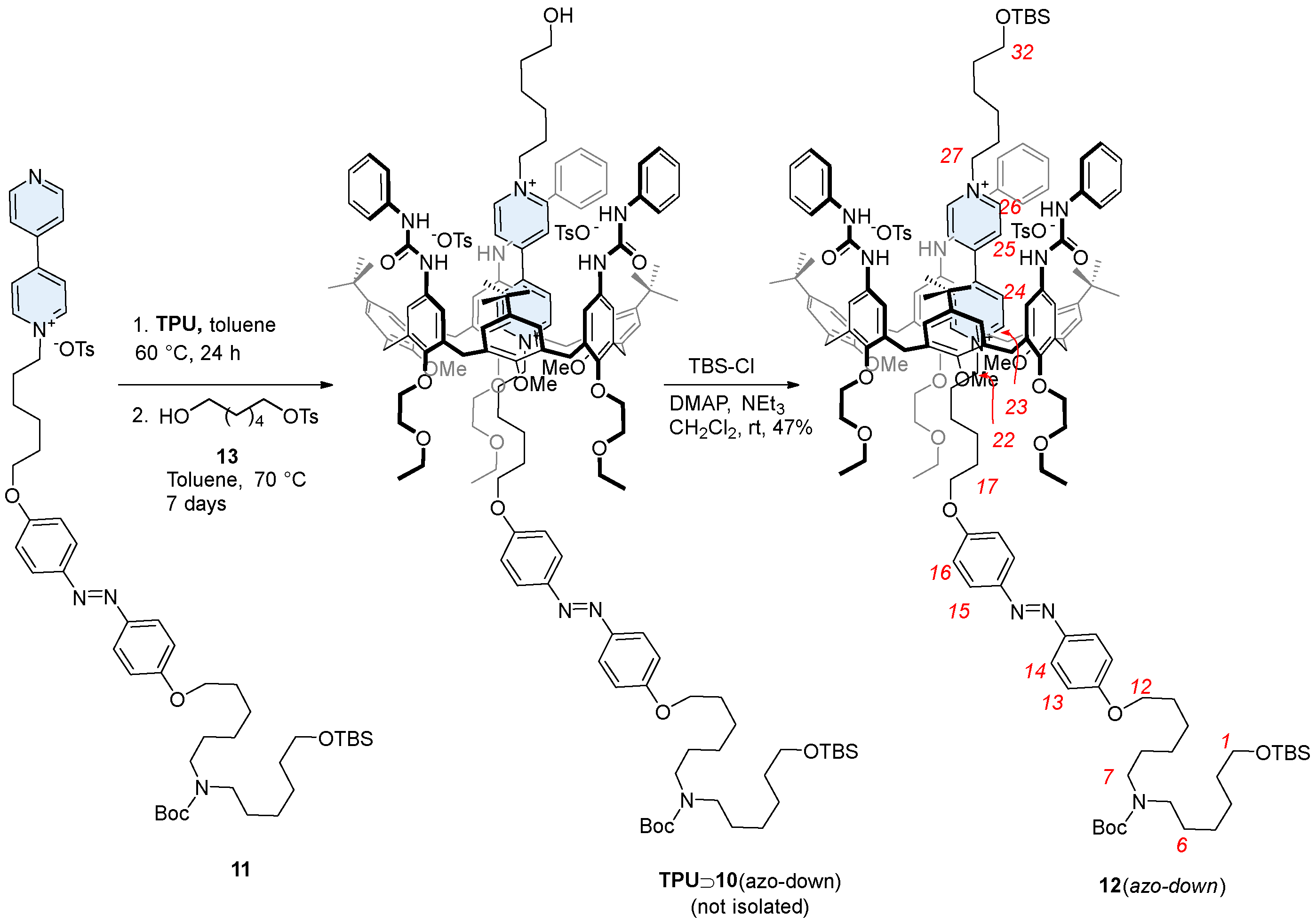
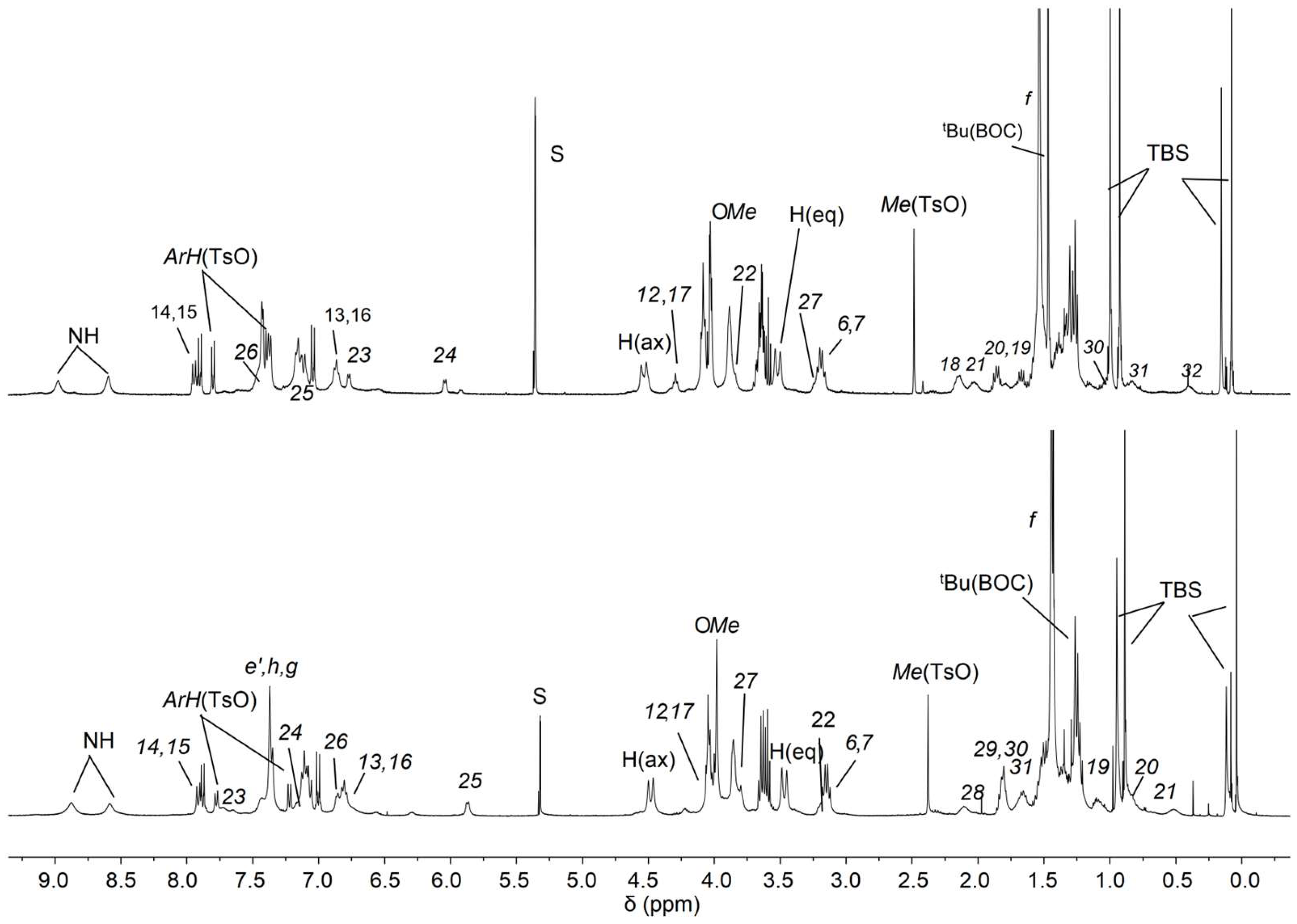
2.3. Synthesis of Rotaxane 12(Azo-Down)
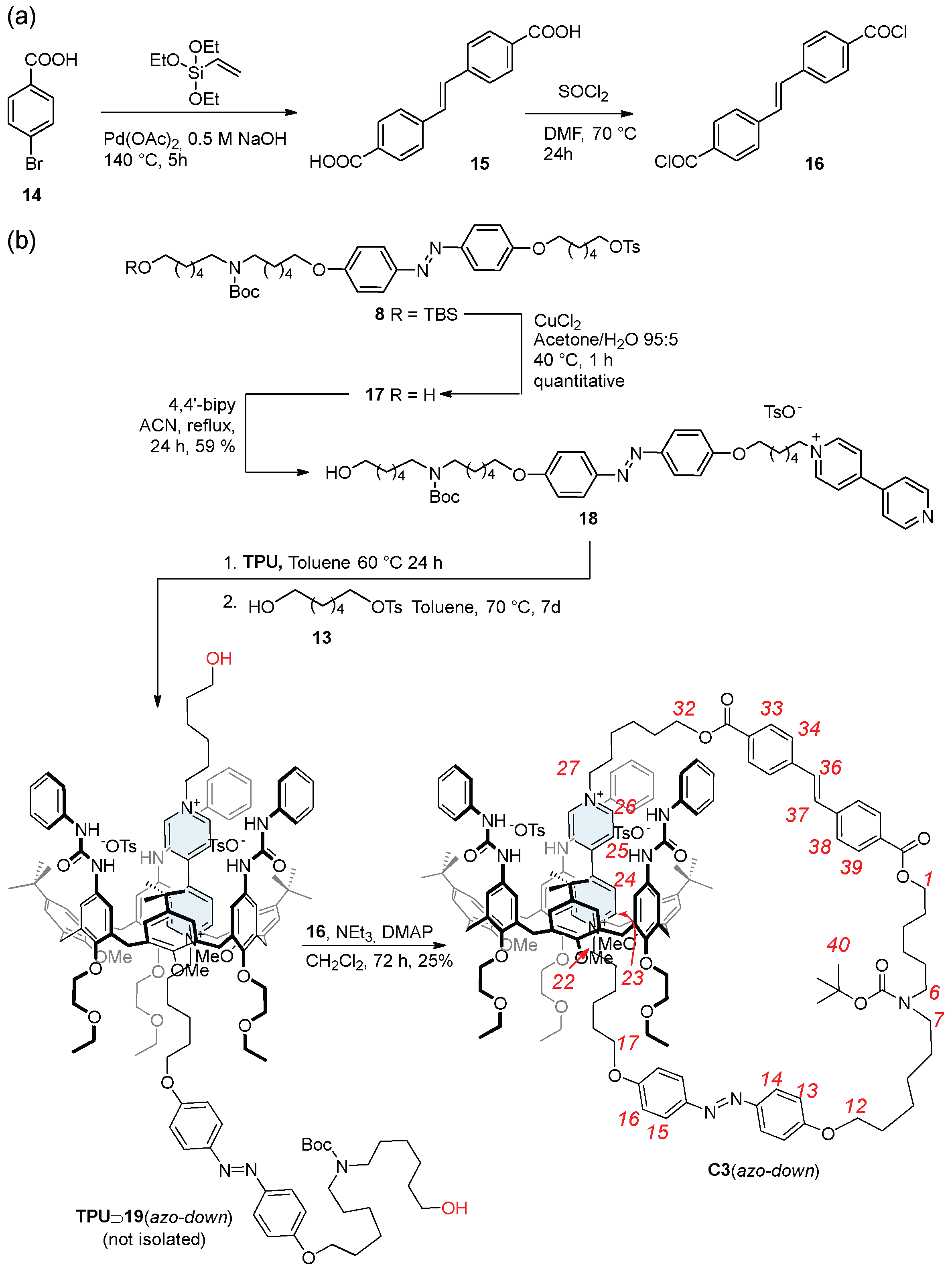
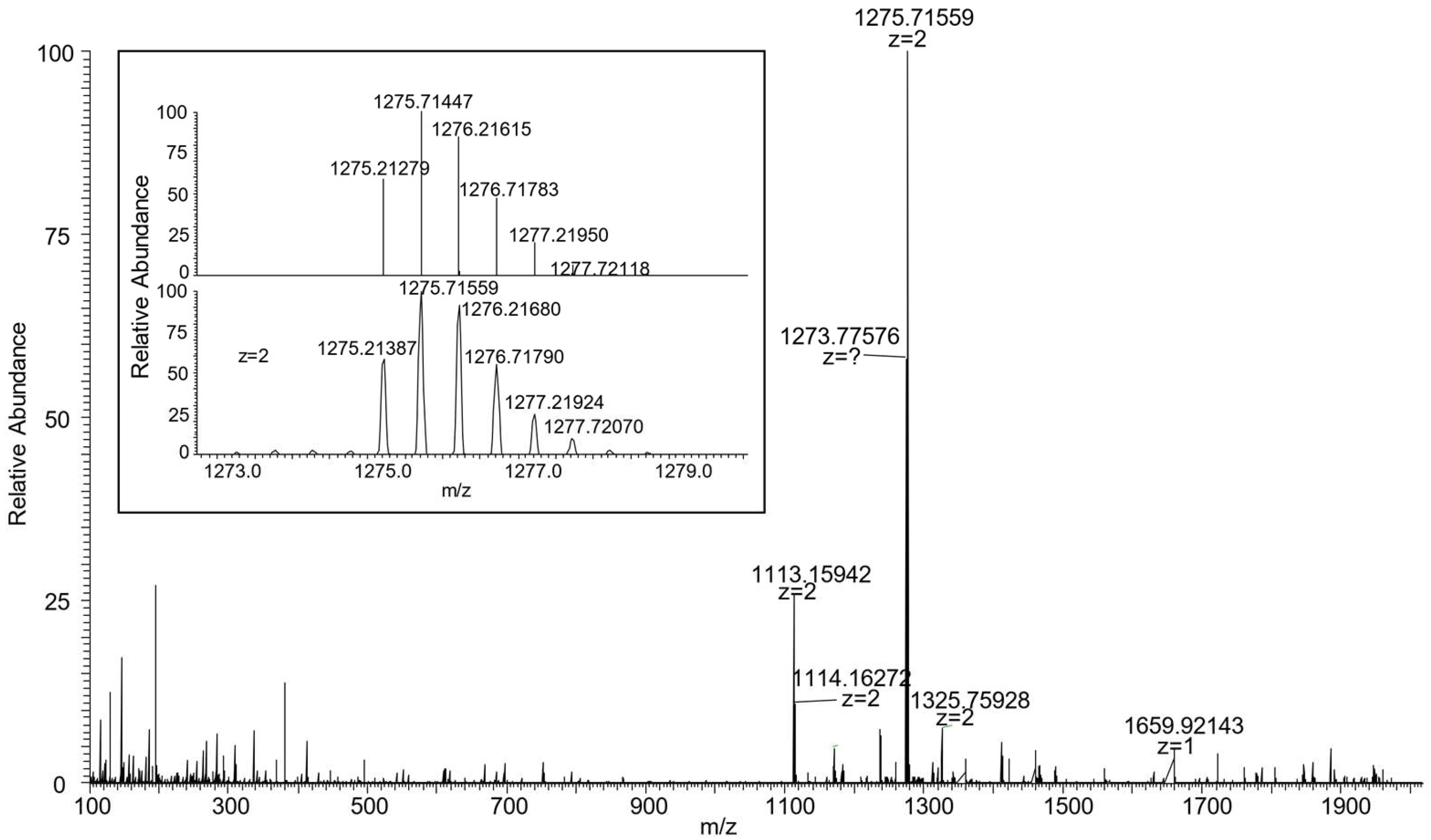
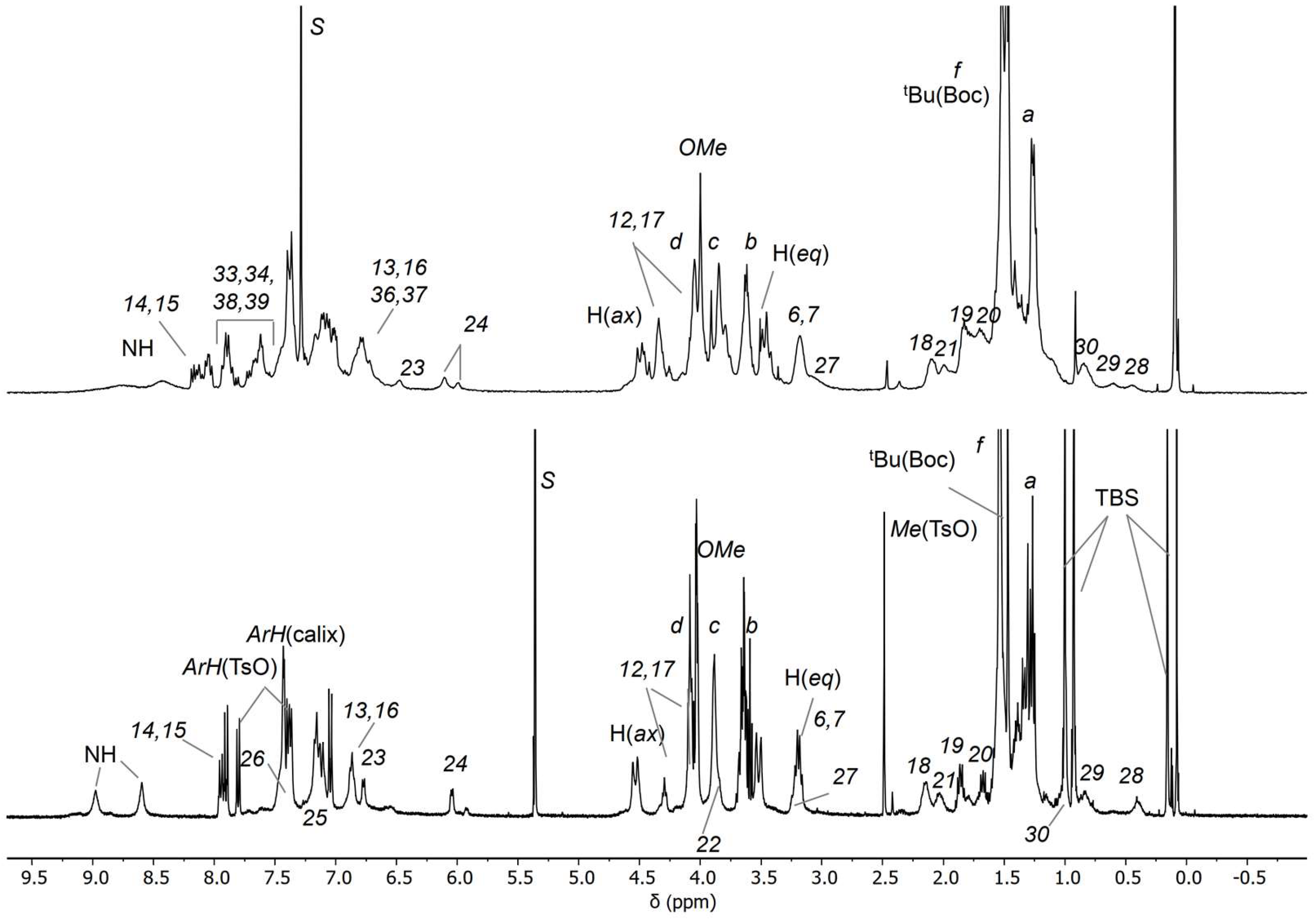
2.4. Synthesis of the Catenane
3. Materials and Methods
3.1. General
3.2. Synthetic Procedures
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stoddart, J.F. Mechanically Interlocked Molecules (MIMs)—Molecular Shuttles, Switches, and Machines (Nobel Lecture). Angew. Chem. Int. Ed. 2017, 56, 11094–11125. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, J.-P. From Chemical Topology to Molecular Machines (Nobel Lecture). Angew. Chem. Int. Ed. 2017, 56, 11080–11093. [Google Scholar] [CrossRef] [PubMed]
- Sluysmans, D.; Stoddart, J.F. The Burgeoning of Mechanically Interlocked Molecules in Chemistry. Trends Chem. 2019, 1, 185–197. [Google Scholar] [CrossRef]
- Heard, A.W.; Goldup, S.M. Simplicity in the Design, Operation, and Applications of Mechanically Interlocked Molecular Machines. ACS Cent. Sci. 2020, 6, 117–128. [Google Scholar] [CrossRef]
- Mena-Hernando, S.; Pérez, E.M. Mechanically Interlocked Materials. Rotaxanes and Catenanes beyond the Small Molecule. Chem. Soc. Rev. 2019, 48, 5016–5032. [Google Scholar] [CrossRef]
- Molecular Machines and Motors: Recent Advances and Perspectives; Credi, A., Silvi, S., Venturi, M., Eds.; Topics in Current Chemistry; Springer International Publishing: Heidelberg, Germany, 2014. [Google Scholar] [CrossRef]
- Erbas-Cakmak, S.; Leigh, D.A.; McTernan, C.T.; Nussbaumer, A.L. Artificial Molecular Machines. Chem. Rev. 2015, 115, 10081–10206. [Google Scholar] [CrossRef]
- Bruns, C.J.; Stoddart, J.F. The Nature of the Mechanical Bond: From Molecules to Machines; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Feng, Y.; Ovalle, M.; Seale, J.S.W.; Lee, C.K.; Kim, D.J.; Astumian, R.D.; Stoddart, J.F. Molecular Pumps and Motors. J. Am. Chem. Soc. 2021, 143, 5569–5591. [Google Scholar] [CrossRef]
- Aprahamian, I. The Future of Molecular Machines. ACS Cent. Sci. 2020, 6, 347–358. [Google Scholar] [CrossRef]
- Forgan, R.S.; Sauvage, J.-P.; Stoddart, J.F. Chemical Topology: Complex Molecular Knots, Links, and Entanglements. Chem. Rev. 2011, 111, 5434–5464. [Google Scholar] [CrossRef]
- De Bo, G.; Dolphijn, G.; McTernan, C.T.; Leigh, D.A. [2]Rotaxane Formation by Transition State Stabilization. J. Am. Chem. Soc. 2017, 139, 8455–8457. [Google Scholar] [CrossRef]
- Jiao, Y.; Đorđević, L.; Mao, H.; Young, R.M.; Jaynes, T.; Chen, H.; Qiu, Y.; Cai, K.; Zhang, L.; Chen, X.-Y.; et al. A Donor–Acceptor [2]Catenane for Visible Light Photocatalysis. J. Am. Chem. Soc. 2021, 143, 8000–8010. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Tan, Z.; Hu, Y.; Lu, Z.; Yuan, J.; Li, X.; Xie, J.; Zhang, J.; Zhu, K. Precise Control of Radial Catenane Synthesis via Clipping and Pumping. J. Am. Chem. Soc. 2022, 144, 2085–2089. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.V.; Kay, E.R.; Leigh, D.A. A Reversible Synthetic Rotary Molecular Motor. Science 2004, 306, 1532–1537. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-H.; Cecconello, A.; Elbaz, J.; Credi, A.; Willner, I. A Three-Station DNA Catenane Rotary Motor with Controlled Directionality. Nano Lett. 2013, 13, 2303–2308. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Du, Z.; Zhang, S.; Xie, J.; Li, X.; Chen, Q.; Tang, Y.; Chen, J.; Zhu, K. A Compact Chemically Driven [2]Catenane Rotary Motor Operated through Alternate Pumping and Discharging. Chem. Sci. 2024, 15, 14721–14725. [Google Scholar] [CrossRef]
- Pooler, D.R.S.; Lubbe, A.S.; Crespi, S.; Feringa, B.L. Designing Light-Driven Rotary Molecular Motors. Chem. Sci. 2021, 12, 14964–14986. [Google Scholar] [CrossRef]
- Pfeifer, L.; Crespi, S.; van der Meulen, P.; Kemmink, J.; Scheek, R.M.; Hilbers, M.F.; Buma, W.J.; Feringa, B.L. Controlling Forward and Backward Rotary Molecular Motion on Demand. Nat. Commun. 2022, 13, 2124. [Google Scholar] [CrossRef]
- Van Beek, C.L.F.; Feringa, B.L. Coupled Rotary Motion in Molecular Motors. J. Am. Chem. Soc. 2024, 146, 5634–5642. [Google Scholar] [CrossRef]
- Leigh, D.A.; Wong, J.K.; Dehez, F.; Zerbetto, F. Unidirectional Rotation in a Mechanically Interlocked Molecular Rotor. Nature 2003, 424, 174–179. [Google Scholar] [CrossRef]
- Hubin, T.J.; Busch, D.H. Template Routes to Interlocked Molecular Structures and Orderly Molecular Entanglements. Coord. Chem. Rev. 2000, 200–202, 5–52. [Google Scholar] [CrossRef]
- Evans, N.H.; Beer, P.D. Progress in the Synthesis and Exploitation of Catenanes since the Millennium. Chem. Soc. Rev. 2014, 43, 4658–4683. [Google Scholar] [CrossRef] [PubMed]
- Crowley, J.D.; Goldup, S.M.; Lee, A.-L.; Leigh, D.A.; Mc Burney, R.T. Active Metal Template Synthesis of Rotaxanes, Catenanes and Molecular Shuttles. Chem. Soc. Rev. 2009, 38, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.E.M.; Modicom, F.; Goldup, S.M. Efficient Multicomponent Active Template Synthesis of Catenanes. J. Am. Chem. Soc. 2018, 140, 4787–4791. [Google Scholar] [CrossRef] [PubMed]
- Calixarenes and Beyond; Neri, P., Sessler, J.L., Wang, M.-X., Neri, P., Sessler, J.L., Wang, M.-X., Eds.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Talotta, C.; Gaeta, C.; Neri, P. Large Calixarenes: Synthesis and Properties. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar] [CrossRef]
- Cera, G.; Arduini, A.; Secchi, A.; Credi, A.; Silvi, S. Heteroditopic Calix[6]Arene Based Intervowen and Interlocked Molecular Devices. Chem. Rec. 2021, 21, 1161–1181. [Google Scholar] [CrossRef]
- Credi, A.; Dumas, S.; Silvi, S.; Venturi, M.; Arduini, A.; Pochini, A.; Secchi, A. Viologen-Calix[6]Arene Pseudorotaxanes. Ion-Pair Recognition and Threading/Dethreading Molecular Motions. J. Org. Chem. 2004, 69, 5881–5887. [Google Scholar] [CrossRef]
- Orlandini, G.; Ragazzon, G.; Zanichelli, V.; Secchi, A.; Silvi, S.; Venturi, M.; Arduini, A.; Credi, A. Covalent Capture of Oriented Calix[6]Arene Rotaxanes by a Metal-Free Active Template Approach. Chem. Commun. 2017, 53, 6172–6174. [Google Scholar] [CrossRef]
- Arduini, A.; Orlandini, G.; Secchi, A.; Credi, A.; Silvi, S.; Venturi, M. Calix-Based Molecular Machines and Devices. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Waltham, MA, USA, 2014; pp. 1–26. [Google Scholar] [CrossRef]
- Bazzoni, M.; Andreoni, L.; Silvi, S.; Credi, A.; Cera, G.; Secchi, A.; Arduini, A. Selective Access to Constitutionally Identical, Orientationally Isomeric Calix[6]Arene-Based [3]Rotaxanes by an Active Template Approach. Chem. Sci. 2021, 12, 6419–6428. [Google Scholar] [CrossRef]
- Cester Bonati, F.; Bazzoni, M.; Baccini, C.; Zanichelli, V.; Orlandini, G.; Arduini, A.; Cera, G.; Secchi, A. Calix[6]Arene-Based [3]Rotaxanes as Prototypes for the Template Synthesis of Molecular Capsules. Molecules 2023, 28, 595. [Google Scholar] [CrossRef]
- Gaeta, C.; Talotta, C.; Mirra, S.; Margarucci, L.; Casapullo, A.; Neri, P. Catenation of Calixarene Annulus. Org. Lett. 2013, 15, 116–119. [Google Scholar] [CrossRef]
- Orlandini, G.; Zanichelli, V.; Secchi, A.; Arduini, A.; Ragazzon, G.; Credi, A.; Venturi, M.; Silvi, S. Synthesis by Ring Closing Metathesis and Properties of an Electroactive Calix[6]Arene [2]Catenane. Supramol. Chem. 2016, 28, 427–435. [Google Scholar] [CrossRef]
- Zanichelli, V.; Dallacasagrande, L.; Arduini, A.; Secchi, A.; Ragazzon, G.; Silvi, S.; Credi, A. Electrochemically Triggered Co-Conformational Switching in a [2]Catenane Comprising a Non-Symmetric Calix[6]Arene Wheel and a Two-Station Oriented Macrocycle. Molecules 2018, 23, 1156. [Google Scholar] [CrossRef] [PubMed]
- Merino, E.; Ribagorda, M. Control over Molecular Motion Using the Cis–Trans Photoisomerization of the Azo Group. Beilstein J. Org. Chem. 2012, 8, 1071–1090. [Google Scholar] [CrossRef] [PubMed]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Yoshino, T.; Mamiya, J.; Kinoshita, M.; Ikeda, T.; Yu, Y. Preparation and Characterization of Crosslinked Azobenzene Liquid-Crystalline Polymer Fibers. Mol. Cryst. Liq. Cryst. 2007, 478, 233/[989]–243/[999]. [Google Scholar] [CrossRef]
- Arduini, A.; Bussolati, R.; Credi, A.; Faimani, G.; Garaudée, S.; Pochini, A.; Secchi, A.; Semeraro, M.; Silvi, S.; Venturi, M. Towards Controlling the Threading Direction of a Calix[6]Arene Wheel by Using Nonsymmetric Axles. Chem. Eur. J. 2009, 15, 3230–3242. [Google Scholar] [CrossRef]
- Zanichelli, V.; Ragazzon, G.; Orlandini, G.; Venturi, M.; Credi, A.; Silvi, S.; Arduini, A.; Secchi, A. Efficient Active-Template Synthesis of Calix[6]Arene-Based Oriented Pseudorotaxanes and Rotaxanes. Org. Biomol. Chem. 2017, 15, 6753–6763. [Google Scholar] [CrossRef]
- Zanichelli, V.; Bazzoni, M.; Arduini, A.; Franchi, P.; Lucarini, M.; Ragazzon, G.; Secchi, A.; Silvi, S. Redox-Switchable Calix[6]Arene-Based Isomeric Rotaxanes. Chem. Eur. J. 2018, 24, 12370–12382. [Google Scholar] [CrossRef]
- Asakawa, M.; Ashton, P.R.; Balzani, V.; Brown, C.L.; Credi, A.; Matthews, O.A.; Newton, S.P.; Raymo, F.M.; Shipway, A.N.; Spencer, N.; et al. Photoactive Azobenzene-Containing Supramolecular Complexes and Related Interlocked Molecular Compounds. Chem. Eur. J. 1999, 5, 860–875. [Google Scholar] [CrossRef]
- Ikejiri, S.; Takashima, Y.; Osaki, M.; Yamaguchi, H.; Harada, A. Solvent-Free Photoresponsive Artificial Muscles Rapidly Driven by Molecular Machines. J. Am. Chem. Soc. 2018, 140, 17308–17315. [Google Scholar] [CrossRef]
- Gordillo, Á.; de Jesús, E.; López-Mardomingo, C. Consecutive Palladium-Catalyzed Hiyama–Heck Reactions in Aqueous Media under Ligand-Free Conditions. Chem. Commun. 2007, 4056–4058. [Google Scholar] [CrossRef]
- Fisher, B.F.; Gellman, S.H. Impact of γ-Amino Acid Residue Preorganization on α/γ-Peptide Foldamer Helicity in Aqueous Solution. J. Am. Chem. Soc. 2016, 138, 10766–10769. [Google Scholar] [CrossRef] [PubMed]
- Yoritate, M.; Meguro, T.; Matsuo, N.; Shirokane, K.; Sato, T.; Chida, N. Two-Step Synthesis of Multi-Substituted Amines by Using an N-Methoxy Group as a Reactivity Control Element. Chem. Eur. J. 2014, 20, 8210–8216. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, C.N.; Bartlett, R.D.; Esslinger, C.S.; Cybulski, K.A.; Tongcharoensirikul, P.; Bridges, R.J.; Thompson, C.M. Synthesis and in Vitro Pharmacology of Substituted Quinoline-2,4-Dicarboxylic Acids as Inhibitors of Vesicular Glutamate Transport. J. Med. Chem. 2002, 45, 2260–2276. [Google Scholar] [CrossRef]
- Kunitake, T.; Okahata, Y.; Shimomura, M.; Yasunami, S.; Takarabe, K. Formation of Stable Bilayer Assemblies in Water from Single-Chain Amphiphiles. Relationship between the Amphiphile Structure and the Aggregate Morphology. J. Am. Chem. Soc. 1981, 103, 5401–5413. [Google Scholar] [CrossRef]
- Zanichelli, V.; Ragazzon, G.; Arduini, A.; Credi, A.; Franchi, P.; Orlandini, G.; Venturi, M.; Lucarini, M.; Secchi, A.; Silvi, S. Synthesis and Characterization of Constitutionally Isomeric Oriented Calix[6]Arene-Based Rotaxanes. Eur. J. Org. Chem. 2016, 2016, 1033–1042. [Google Scholar] [CrossRef]
- Kluger, R.; Grant, A.S.; Bearne, S.L.; Trachsel, M.R. Dicarboxylic Acid Bis(Methyl Phosphates): Anionic Biomimetic Crosslinking Reagents. J. Org. Chem. 1990, 55, 2864–2868. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzoni, M.; Rispoli, F.; Venturelli, S.; Cera, G.; Secchi, A. Synthesis and Characterization of a Two-Station Two-Gate Calix[6]arene-Based [2]Catenane. Molecules 2025, 30, 732. https://doi.org/10.3390/molecules30030732
Bazzoni M, Rispoli F, Venturelli S, Cera G, Secchi A. Synthesis and Characterization of a Two-Station Two-Gate Calix[6]arene-Based [2]Catenane. Molecules. 2025; 30(3):732. https://doi.org/10.3390/molecules30030732
Chicago/Turabian StyleBazzoni, Margherita, Francesco Rispoli, Sara Venturelli, Gianpiero Cera, and Andrea Secchi. 2025. "Synthesis and Characterization of a Two-Station Two-Gate Calix[6]arene-Based [2]Catenane" Molecules 30, no. 3: 732. https://doi.org/10.3390/molecules30030732
APA StyleBazzoni, M., Rispoli, F., Venturelli, S., Cera, G., & Secchi, A. (2025). Synthesis and Characterization of a Two-Station Two-Gate Calix[6]arene-Based [2]Catenane. Molecules, 30(3), 732. https://doi.org/10.3390/molecules30030732