
The Role of Noncovalent Interactions in the Infrared Spectra of Lignin Model Compounds: A DFT Study
 , , , , and
, , , , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Lignin Model Compounds
2.2. Geometries and Thermochemical Analysis
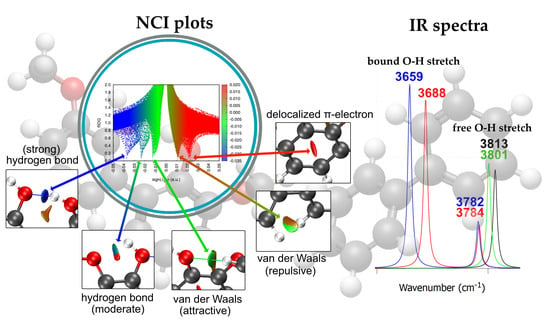
2.3. Noncovalent Interaction (NCI) Analysis of Dimers
- Attractive interactions (λ2 < 0, blue)
- Weak dispersion interactions (λ2 ≈ 0, green)
- Repulsive or steric regions (λ2 > 0, red)
2.4. IR Spectra of Dimers
2.5. Steric Repulsion-Dependent Dihedral Angle of 3,3′-Dimethoxy-1,1′-Biphenyl-2,2′-Diol
- In its most stable conformations (ct-ct-60 and ct-ct-131), the guaiacyl moiety exhibits distinct C–C (linkage) stretching bands in the 1300–1400 cm−1 region of the IR spectrum.
- In ct-at and ct-ag conformations, two types of O–H stretching modes—free and bound hydrogen —appear between 3600 and 3800 cm−1.
- The tt-tt, tt-tg, and tg-tg conformations lack observable C–C or O–H stretching bands but display intense, narrow C–OCH3 stretching peaks in the 1200–1400 cm−1 range.
- At 2900–3300 cm−1, the IR peaks observed in 3,3′-dimethoxy-1,1′-biphenyl-2,2′-diol correspond to C–H stretching modes of the methoxy groups and the aromatic rings, whereas in 2,2′-biphenol and biphenyl they correspond to C–H stretching modes of the aromatic rings.
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riseh, R.S.; Vazvani, M.G.; Hassanisaadi, M.; Thakur, V.K. Agricultural Wastes: A Practical and Potential Source for the Isolation and Preparation of Cellulose and Application in Agriculture and Different Industries. Ind. Crops Prod. 2024, 208, 117904. [Google Scholar] [CrossRef]
- Rodionova, M.V.; Bozieva, A.M.; Zharmukhamedov, S.K.; Leong, Y.K.; Chi-Wei Lan, J.; Veziroglu, A.; Veziroglu, T.N.; Tomo, T.; Chang, J.S.; Allakhverdiev, S.I. A Comprehensive Review on Lignocellulosic Biomass Biorefinery for Sustainable Biofuel Production. Int. J. Hydrog. Energy 2022, 47, 1481–1498. [Google Scholar] [CrossRef]
- Kumar, A.; Anushree; Kumar, J.; Bhaskar, T. Utilization of Lignin: A Sustainable and Eco-Friendly Approach. J. Energy Inst. 2020, 93, 235–271. [Google Scholar] [CrossRef]
- Mujtaba, M.; Fernandes Fraceto, L.; Fazeli, M.; Mukherjee, S.; Savassa, S.M.; Araujo de Medeiros, G.; do Espírito Santo Pereira, A.; Mancini, S.D.; Lipponen, J.; Vilaplana, F. Lignocellulosic Biomass from Agricultural Waste to the Circular Economy: A Review with Focus on Biofuels, Biocomposites and Bioplastics. J. Clean. Prod. 2023, 402, 136815. [Google Scholar] [CrossRef]
- Wang, H.; Pu, Y.; Ragauskas, A.; Yang, B. From Lignin to Valuable Products–Strategies, Challenges, and Prospects. Bioresour. Technol. 2019, 271, 449–461. [Google Scholar] [CrossRef]
- Yu, X.; Yang, B.; Zhu, W.; Deng, T.; Pu, Y.; Ragauskas, A.; Wang, H. Towards Functionalized Lignin and Its Derivatives for High-Value Material Applications. Ind. Crops Prod. 2023, 200, 116824. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Ye, M.; Tang, Y.; Wen, Z.; Liu, X.; Li, C.C. Renewable Lignin and Its Macromolecule Derivatives: An Emerging Platform toward Sustainable Electrochemical Energy Storage. Green Chem. 2023, 25, 4154–4179. [Google Scholar] [CrossRef]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of Lignin-Derived Bio-Oils by Catalytic Hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Adilina, I.B.; Rinaldi, N.; Simanungkalit, S.P.; Aulia, F.; Oemry, F.; Stenning, G.B.G.; Silverwood, I.P.; Parker, S.F. Hydrodeoxygenation of Guaiacol as a Bio-Oil Model Compound over Pillared Clay-Supported Nickel-Molybdenum Catalysts. J. Phys. Chem. C 2019, 123, 21429–21439. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as Renewable Raw Material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Brown, R.C.; Shanks, B.H. Understanding the Fast Pyrolysis of Lignin. ChemSusChem 2011, 4, 1629–1636. [Google Scholar] [CrossRef]
- Ma, Z.; Custodis, V.; Hemberger, P.; Bährle, C.; Vogel, F.; Jeschke, G.; Van Bokhoven, J.A. Chemicals from Lignin by Catalytic Fast Pyrolysis, from Product Control to Reaction Mechanism. Chimia 2015, 69, 597–602. [Google Scholar] [CrossRef]
- Kawamoto, H.; Horigoshi, S.; Saka, S. Pyrolysis Reactions of Various Lignin Model Dimers. J. Wood Sci. 2007, 53, 168–174. [Google Scholar] [CrossRef]
- Lahive, C.W.; Kamer, P.C.J.; Lancefield, C.S.; Deuss, P.J. An Introduction to Model Compounds of Lignin Linking Motifs; Synthesis and Selection Considerations for Reactivity Studies. ChemSusChem 2020, 13, 4238–4265. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Kim, K.H.; Brown, R.C.; Dalluge, E.; Hutchinson, C.; Lee, Y.J.; Dalluge, D. Formation of Phenolic Oligomers during Fast Pyrolysis of Lignin. Fuel 2014, 128, 170–179. [Google Scholar] [CrossRef]
- Wang, Y.; Kalscheur, J.; Ebikade, E.; Li, Q.; Vlachos, D.G. LigninGraphs: Lignin Structure Determination with Multiscale Graph Modeling. J. Cheminform 2022, 14, 43. [Google Scholar] [CrossRef]
- Karlsson, M.; Romson, J.; Elder, T.; Emmer, Å.; Lawoko, M. Lignin Structure and Reactivity in the Organosolv Process Studied by NMR Spectroscopy, Mass Spectrometry, and Density Functional Theory. Biomacromolecules 2023, 24, 2314–2326. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-González, Á.; Martín-Martínez, F.J.; Dobado, J.A. The Role of Weak Interactions in Lignin Polymerization. J. Mol. Model. 2017, 23, 80. [Google Scholar] [CrossRef]
- Saragi, R.T.; Calabrese, C.; Juanes, M.; Pinacho, R.; Rubio, J.E.; Pérez, C.; Lesarri, A. π-Stacking Isomerism in Polycyclic Aromatic Hydrocarbons: The 2-Naphthalenethiol Dimer. J. Phys. Chem. Lett. 2023, 14, 207–213. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Palos, R.; Hita, I.; Arandes, J.M.; Rodríguez-Mirasol, J.; Cordero, T.; Bilbao, J.; Castaño, P. Revealing the Pathways of Catalyst Deactivation by Coke during the Hydrodeoxygenation of Raw Bio-Oil. Appl. Catal. B 2018, 239, 513–524. [Google Scholar] [CrossRef]
- Erlekam, U.; Frankowski, M.; Meijer, G.; Von Helden, G. An Experimental Value for the B1u C-H Stretch Mode in Benzene. J. Chem. Phys. 2006, 124, 171101. [Google Scholar] [CrossRef]
- Anouar, E.; Calliste, C.A.; Košinová, P.; Di Meo, F.; Duroux, J.L.; Champavier, Y.; Marakchi, K.; Trouillas, P. Free Radical Scavenging Properties of Guaiacol Oligomers: A Combined Experimental and Quantum Study of the Guaiacyl-Moiety Role. J. Phys. Chem. A 2009, 113, 13881–13891. [Google Scholar] [CrossRef]
- Fujii, A.; Iwasaki, A.; Yoshida, K.; Ebata, T.; Mikami, N. Infrared Spectroscopy of (Phenol) n+ (N=2-4) and (Phenol-Benzene)+ Cluster Ions. J. Phys. Chem. A 1997, 101, 1798–1803. [Google Scholar] [CrossRef]
- Longarte, A.; Redondo, C.; Fernández, J.A.; Castaño, F. IR/UV and UV/UV Double-Resonance Study of Guaiacol and Eugenol Dimers. J. Chem. Phys. 2005, 122, 164304. [Google Scholar] [CrossRef]
- Pietraperzia, G.; Pasquini, M.; Schiccheri, N.; Piani, G.; Becucci, M.; Castellucci, E.; Biczysko, M.; Bloino, J.; Barone, V. The Gas Phase Anisole Dimer: A Combined High-Resolution Spectroscopy and Computational Study of a Stacked Molecular System. J. Phys. Chem. A 2009, 113, 14343–14351. [Google Scholar] [CrossRef] [PubMed]
- Fedor, A.M.; Toda, M.J. Investigating Hydrogen Bonding in Phenol Using Infrared Spectroscopy and Computational Chemistry. J. Chem. Educ. 2014, 91, 2191–2194. [Google Scholar] [CrossRef]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Otero-De-La-Roza, A.; Johnson, E.R.; Luaña, V. Critic2: A Program for Real-Space Analysis of Quantum Chemical Interactions in Solids. Comput. Phys. Commun. 2014, 185, 1007–1018. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Otero-De-La-Roza, A.; Johnson, E.R.; Contreras-García, J. Revealing Non-Covalent Interactions in Solids: NCI Plots Revisited. Phys. Chem. Chem. Phys. 2012, 14, 12165–12172. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Martinez, C.R.; Iverson, B.L. Rethinking the Term “Pi-Stacking.”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef]
- Lewis, M.; Bagwill, C.; Hardebeck, L.; Wireduaah, S. Modern Computational Approaches to Understanding of Aromatics. In Aromatic Interactions: Frontiers in Knowledge and Application; Johnson, D.W., Hof, F., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 1–15. [Google Scholar]
- Thakuria, R.; Nath, N.K.; Saha, B.K. The Nature and Applications of π-π Interactions: A Perspective. Cryst. Growth Des. 2019, 19, 523–528. [Google Scholar] [CrossRef]
- Rusydi, F.; Aisyah, N.D.; Fadilla, R.N.; Dipojono, H.K.; Ahmad, F.; Mudasir; Puspitasari, I.; Rusydi, A. The Transition State Conformational Effect on the Activation Energy of Ethyl Acetate Neutral Hydrolysis. Heliyon 2019, 5, e02409. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Houk, K.N. Substituent Effects in the Benzene Dimer Are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene. J. Am. Chem. Soc. 2008, 130, 10854–10855. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K.M. The Nature of π-π Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Rezáč, J.; Hobza, P. Benzene Dimer: Dynamic Structure and Thermodynamics Derived from on-the-Fly Ab Initio DFT-D Molecular Dynamic Simulations. J. Chem. Theory Comput. 2008, 4, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, V.; Biennier, L.; Arunan, E.; Talbi, D.; Georges, R. Direct Infrared Absorption Spectroscopy of Benzene Dimer. J. Phys. Chem. A 2011, 115, 11263–11268. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S. Accurate Description of van Der Waals Complexes by Density Functional Theory Including Empirical Corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Linstrom, P.J.; Mallard, W.G. (Eds.) Infrared Spectra. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2025. Available online: https://webbook.nist.gov/chemistry/ (accessed on 29 October 2025). [CrossRef]
- Cappelli, C.; Silva, C.O.; Tomasi, J. Solvent Effects on Vibrational Modes: Ab-Initio Calculations, Scaling and Solvent Functions with Applications to the Carbonyl Stretch of Dialkyl Ketones. J. Mol. Struct. 2001, 544, 191–203. [Google Scholar] [CrossRef]
- Louis, J.; Daniel, R.; Dillet, R.V. Solvent Effects in Infrared Spectroscopy a Computational Approach. Mol. Phys. 1996, 89, 1521–1529. [Google Scholar] [CrossRef]
- Dunkelberger, E.B.; Grechko, M.; Zanni, M.T. Transition Dipoles from 1D and 2D Infrared Spectroscopy Help Reveal the Secondary Structures of Proteins: Application to Amyloids. J. Phys. Chem. B 2015, 119, 14065–14075. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, S.; Ghosh, A. Inconsistent Hydrogen Bond-Mediated Vibrational Coupling of Amide I. RSC Adv. 2023, 13, 1295–1300. [Google Scholar] [CrossRef]
- Smith, B.C. Infrared Spectral Interpretation: A Systematic Approach, 1st ed.; Shapiro, F., Williams, M., Eds.; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Smith, B.C. Quantitative Spectroscopy: Theory and Practice, 1st ed.; Roberts, E., Kinney, C., Coleman, D., Eds.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Baroni, S.; De Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and Related Crystal Properties from Density-Functional Perturbation Theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Simmons, K.E.; Minard, R.D.; Bollag, J.-M. Oxidative Coupling and Polymerization of Guaiacol, a Lignin Derivative. Soil Sci. Soc. Am. J. 1988, 52, 1356–1360. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef]
- Madinah, R.; Rusydi, F.; Fadilla, R.N.; Khoirunisa, V.; Boli, L.S.P.; Saputro, A.G.; Hassan, N.H.; Ahmad, A. First-Principles Study of the Dispersion Effects in the Structures and Keto-Enol Tautomerization of Curcumin. ACS Omega 2023, 8, 34022–34033. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1; Gaussian Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; Wiley: Chichester, UK, 2001. [Google Scholar]
- Shimanouchi, T. Tables of Molecular Vibrational Frequencies; Consolidated Volume I; National Bureau of Standards: Washington, DC, USA, 1972; pp. 1–160. [Google Scholar]
- Balfour, W.J. The vibrational spectrum of anisole. Spectrochim. Acta A 1983, 39, 1155–1162. [Google Scholar] [CrossRef]
- Scoullos, E.V.; Hofman, M.S.; Zheng, Y.; Potapenko, D.V.; Tang, Z.; Podkolzin, S.G.; Koel, B.E. Guaiacol Adsorption and Decomposition on Platinum: Insights from IR Spectroscopy and DFT Calculations. J. Phys. Chem. C 2018, 122, 24576–24588. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Labels | Interaction Geometry | Interaction Description | Type of Interaction |
|---|---|---|---|
| A | Single aromatic ring (self-interaction) | Opposing C···C atoms (in para position) | delocalized π-electron |
| B | Parallel offset/Edge-to-edge | C···π, CH···π and π···π | σ···π and π···π staking interactions |
| C | Parallel offset/Edge-to-edge | C···C and/or C···H | attractive van der Waals |
| D | Parallel offset/Edge-to-edge | O···H (aromatic–hydroxyl) | H-bond |
| E | Edge-to-edge | O···H (hydroxyl–hydroxyl) | strong H-bond |
| F | Parallel offset | O···H (hydroxyl–hydroxyl) | H-bond |
| G | Single aromatic ring (self-interaction) | Opposing C···C atoms (aromatic–methoxy) | steric repulsion |
| H | Parallel offset | O···H (methoxy–methoxy) | H-bond |
| I | Single aromatic ring (self-interaction) | C···H (aromatic–methoxy) | attractive van der Waals |
| J | Single aromatic ring (self-interaction) | Opposing O···O atoms (hydroxyl–methoxy) | steric repulsion |
| K | Parallel offset | C···O and/or C···H (aromatic–methoxy) | attractive van der Waals |
| L | Single aromatic ring (self-interaction) | O···H (hydroxyl–methoxy) | strong H-bond |
| M | Parallel offset | O···H (hydroxyl–methoxy) | H-bond |
| N | Parallel offset | C···O (aromatic–hydroxyl) | attractive van der Waals |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusydi, F.; Boli, L.S.P.; Adilina, I.B.; Cahyanto, W.T.; Parker, S.F.; Oemry, F. The Role of Noncovalent Interactions in the Infrared Spectra of Lignin Model Compounds: A DFT Study. Molecules 2025, 30, 4694. https://doi.org/10.3390/molecules30244694
Rusydi F, Boli LSP, Adilina IB, Cahyanto WT, Parker SF, Oemry F. The Role of Noncovalent Interactions in the Infrared Spectra of Lignin Model Compounds: A DFT Study. Molecules. 2025; 30(24):4694. https://doi.org/10.3390/molecules30244694
Chicago/Turabian StyleRusydi, Febdian, Lusia Silfia Pulo Boli, Indri Badria Adilina, Wahyu Tri Cahyanto, Stewart F. Parker, and Ferensa Oemry. 2025. "The Role of Noncovalent Interactions in the Infrared Spectra of Lignin Model Compounds: A DFT Study" Molecules 30, no. 24: 4694. https://doi.org/10.3390/molecules30244694
APA StyleRusydi, F., Boli, L. S. P., Adilina, I. B., Cahyanto, W. T., Parker, S. F., & Oemry, F. (2025). The Role of Noncovalent Interactions in the Infrared Spectra of Lignin Model Compounds: A DFT Study. Molecules, 30(24), 4694. https://doi.org/10.3390/molecules30244694