Abstract
Given the known biological activity of both natural and synthetic substituted 1H-phenalen-1-ones, the generation of a small chemically and structurally diverse 1H-phenalen-1-one-based library of compounds was warranted. Herein, we have synthesized several groups of compounds to broaden and improve the chemical diversity of our 1H-phenalen-1-one collection. Additionally, we have also introduced hydroxyl, amides or carboxylic groups onto the core, or nitrogen atoms into the core, to increase the chemical diversity while also lowering the ClogP values to aid their water solubility. Notably, we have also improved the synthetic routes to several compounds of interest and have observed the unexpected formation of phenoxazine and acridin-7-one systems during cross-coupling reactions with polyfunctional anilines. Combining the compounds generated in this work with previous ones has enabled us to create a library of chemically diverse 1H-phenalen-1-one available for screening assays. Evaluation of anti-plasmodial activity against the chloroquine-resistant Plasmodium falciparum strain FCR3 revealed four compounds with notable activity, three of which exhibited IC50 values below 1 µM, while none displayed significant cytotoxicity at 10 µM.
1. Introduction
1H-Phenalen-1-one-based compounds have been isolated from both plants and fungi, where they are believed to play a key role in the organisms’ defense mechanisms [1]. Interestingly, substituted 1H-phenalen-1-ones, whether natural or synthetic, have been shown to display a broad range of biological activities.
Several phenalenones are known to be phototoxins [2,3]. For example, it has been shown that exposure to UV light (355 nm) increased the antifungal activity against Fusarium oxysporum of some natural and synthetic phenylphenalenones, and their increased activity correlated with their quantum yields (1a–b) (Figure 1) [4]. Likewise, Otálvaro and colleagues [5], demonstrated that a collection of phenalenones had improved antifungal activities against Mycosphaerella fijiensis when exposed to light. M. fijiensis and F. oxysporum are both known to reduce the production of banana plants by up to 20%.
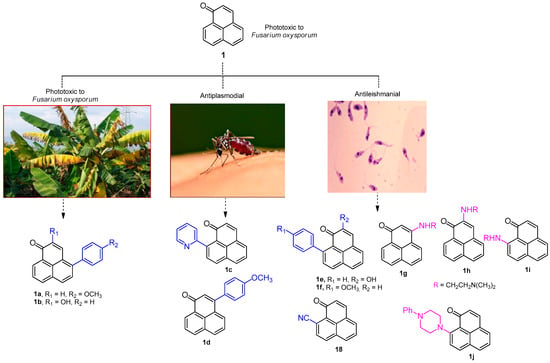
Figure 1.
A representative collection of 1H-phenalen-1-one-based bioactive compounds.
Phenalenone-based compounds have also shown activity against Leishmania species, including compounds 1e, 1f and 18 (Figure 1) [6]. Of note, some amine-functionalized phenalenones, such as 1g–j (Figure 1), have demonstrated 3- to 5-fold greater potency against intracellular amastigotes of L. amazonensis compared to miltefosine, a clinically approved anti-leishmanial drug [7].
Further research has shown that 9- and 3-phenylphenalenone derivatives, as well as 9-heteroaryl-phenalenones, also possess antiplasmodial effects against both chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum [8]. Malaria, which is caused by various Plasmodium species, remains the leading parasitic disease globally in terms of morbidity and mortality, particularly in developing countries, where it imposes substantial social and economic burdens. Although effective treatments exist, they face significant limitations such as resistance, high costs, and complex treatment regimens [9]. Compounds 1c and 1d (Figure 1) represent a new structural class of antiplasmodial agents, offering a new scaffold for antimalarial drug development [8].
In general, the chemical synthesis of substituted phenalenones can be achieved by functionalizing pre-formed phenalenones [6,7,10,11,12,13,14,15] or by constructing the phenalenone core from pre-designed building blocks such as functionalized naphthalenes [16,17,18].
Given their interesting range of biological activities, we wished to supplement and broaden the chemical diversity and novelty of our 1H-phenalen-1-one collection, with the goal of generating a small phenalenone-based library ready for biological screening (Figure 2). As the phenalenone core itself has a relatively high ClogP value of 2.84 and a low predicted water solubility, we also wanted to investigate whether we could generate analogs with lower ClogP values, either by adding polar groups (-OH, -COOH, -CONH2) to the core or by incorporating a nitrogen atom directly into the core. Additionally, where possible, we also wanted to improve the synthetic routes to these phenalenones.
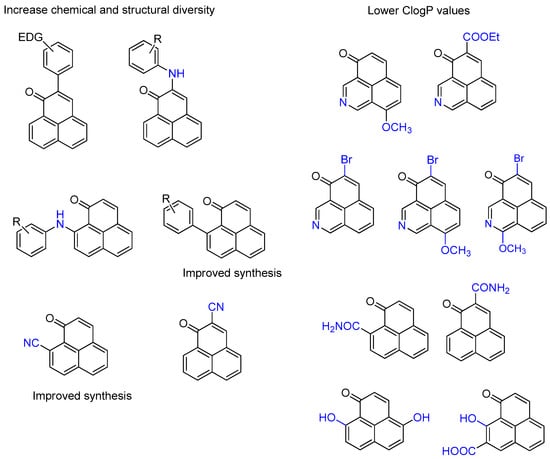
Figure 2.
Compounds of interest described in this work.
2. Results and Discussion
- Preparation of 2-aryl/heteroaryl phenalenones
Previously we had only prepared 2-aryl-phenalenones with electron-donating groups (-OMe) on the aryl entity [19], so to improve the diversity of this sub-series we also needed to incorporate electron-withdrawing groups. It has been demonstrated that the Suzuki–Miyaura coupling works with 2-iodocycloenones, Pd/C, and arylboronic acids. Interestingly, when the reaction was carried out with 2-bromocyclohex-2-en-1-one, no reaction occurred [20]. Based on these results, compounds (4–7) (Scheme 1) were rapidly and efficiently synthesized using a combination of 2-iodo-1H-phenalen-1-one 3 as the phenalenone source and ligand-free palladium on carbon (Pd/C) as the cross-coupling agent.
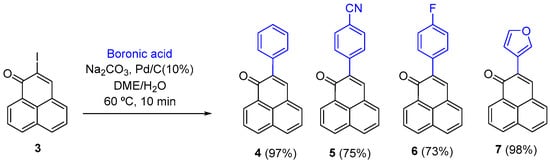
Scheme 1.
Synthesis of 2-substituted 1H-phenalen-1-ones (4–7).
- Preparation of 9-aryl/heteroaryl phenalenones.
Compounds of this type have been prepared using a two-step process [6]. Firstly, a nucleophilic addition using a Grignard reagent followed by oxidation with DDQ. This process, although viable, had certain drawbacks, notably the limitations of functional groups in the Grignard reagent, and often the reagent added to the carbonyl group as well as the 9-position, even at low temperatures. Given the success of the Suzuki–Miyaura coupling observed for the 2-aryl derivatives (4–7), we investigated its use to generate derivatives at the 9-position. Iodides, bromides, and triflates are commonly used in these cross-coupling reactions. Because triflate 8 can be prepared directly from the 9-OH precursor (Scheme 2)—and the corresponding 9-iodide can be generated from 8 [21]—the 9-triflate phenalenone (8) was selected first, thereby avoiding the additional step required to convert 8 into the iodide. The conditions used in Scheme 1 were not compatible with the triflate (8) as it was rapidly hydrolyzed. Fortunately, more standard conditions (K3PO4, Pd(PPh3)4, in 1,4-dioxane at 60 °C), provided the 9-aryl derivatives (9–14) in one-step and with yields typically 10–30% higher than the previous two-step process [12,13,14] (Scheme 2).
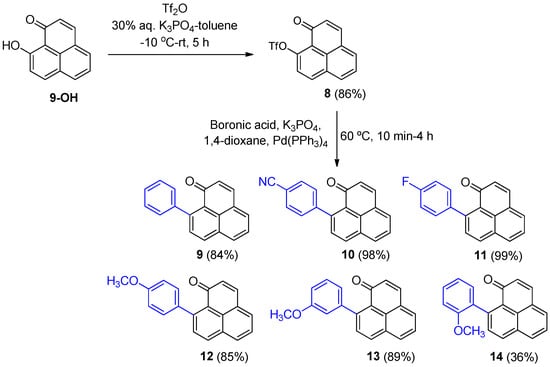
Scheme 2.
Synthesis of 9-substituted aryl 1H-phenalen-1-ones (9–14).
- Preparation of 9-amino-aryl-1H-phenalenones.
The novel 9-aminoaryl-phenalenone derivatives, compounds 15–17 (Scheme 3), were synthesized from compound 8 and an excess of the corresponding anilines via a nucleophilic substitution reaction. For the synthesis of 16, neat 2,6-dimethylaniline was used, with no THF added as solvent Although they required long reaction times (up to 4 days in the case of 16) the products were all obtained in high yields.
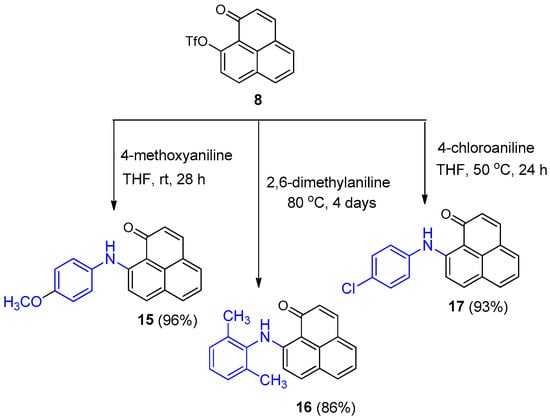
Scheme 3.
Synthesis of 9-arylamino 1H-phenalen-1-ones (15–17).
- Preparation of 2- and 9-cyano/amido 1H-phenalenone derivatives
Compound 18 possesses interesting antiprotozoal activities, especially against L. amazonesis, and L. donovani. Unfortunately, our previous synthesis of 18 from 1 using sodium cyanide in aqueous DMF followed by air oxidation only afforded a 19% yield [6]. We therefore investigated whether other synthetic routes to 18 were more viable (Scheme 4). An initial attempt using a palladium/copper-catalyzed cyanation of triflate 8 in acetonitrile under reflux afforded 18 in a 50% yield. The product was accompanied by a significant amount of 9-hydroxy-1H-phenalen-1-one revealing that triflate hydrolysis was an issue. Changing the solvent to dichloromethane eliminated this side reaction and increased the yield of 18 to 95% after 24 h at reflux. Under the same reaction conditions 2-iodo-1H-phenalenone 3 was transformed cleanly into the 2-cyano analog (20). The difference in yields between isomers 18 (95%) and 20 (67%) is difficult to ascertain as a direct comparison is difficult since the starting materials are different. Interestingly, no 2-cyano 20 was formed when 2-bromo-1H-phenalenone 2 was used as the phenalenone source.
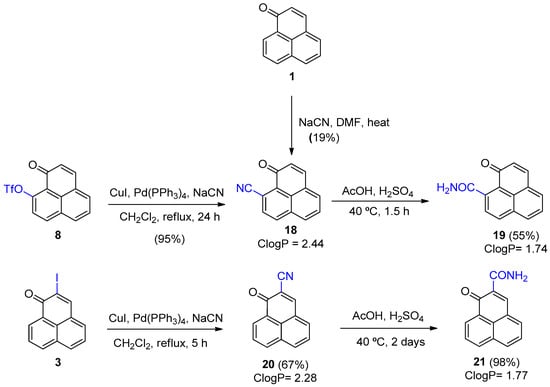
Scheme 4.
Synthesis of 9- and 2-cyano 1H-phenalen-1-ones (18 and 20) and amides (19 and 21).
As one of our goals was also to obtain 1H-phenalenone compounds with lower ClogP values, we converted the cyano compounds 18 and 20 into their respective amido analogs 19 and 21 using acidic conditions. A notable difference in the yields of 19 and 21 was observed. Both reactions proceeded to completion, although 21 took considerably longer. We observed that immediately upon addition of concentrated H2SO4 to a solution of 18 in AcOH, the solution turned dark. Moreover, after extraction of the organics and neutralization of the acid, a significant amount of a dark, insoluble material (char) remained suspended in the aqueous layer. In contrast, no darkening or formation of insoluble material was observed when using 20, even after heating at 40 °C for two days.
- Preparation of 2-amino-aryl/heteroaryl 1H-phenalenone derivatives and the unexpected formation of phenoxarine and acridine derivatives.
Cross-coupling of 2-bromo-1H-phenalenone 2 with weakly nucleophilic amines using Büchwald-Hartwig conditions, generally provides the desired 2-aryl/heteroaryl products as illustrated by the generation of the novel analogs (22–25, Scheme 5). Even the sterically hindered 2,6-dimethylaniline and the delocalized pyrimidin-2-amine afforded their expected products.
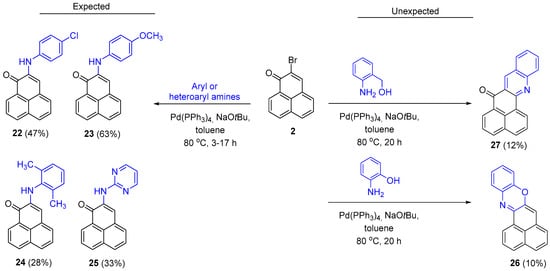
Scheme 5.
Synthesis of 2-substituted 1H-phenalen-1-ones (22–25), the phenoxazine 26 and the acridin-7-one 27.
Interestingly, however, using either 2-aminophenol or (2-aminophenyl)methanol as the aniline did not lead to the generation of their predicted coupled products. Instead, the only products that could be isolated and characterized were the novel naphtho[1,8-ab]phenoxazine (26) and 7H-naphtho[1,8-bc]acridin-7-one (27), respectively, albeit in low yields (Scheme 5).
To confirm the structures of product 26 and 27, 1H NMR, 13C NMR, mass and IR spectra were performed, in addition to bi-dimensional COSY, HMBC, and HSQC NMR experiments. In the 1H NMR spectrum of 26, we observed 11 signals corresponding to aromatic protons (Figure 3), one of which appeared as a singlet at 6.46 ppm, in contrast to the singlet at 7.19 ppm for 2-anilino-1H-phenalen-1-one, the product with the structure closest to what we expected in this reaction [7]. In the 13C NMR spectrum, the quaternary carbon signal corresponding to the enone carbonyl group around 180 ppm was absent. Instead, a quaternary carbon signal was present, shifted around 152 ppm (Figure 3), likely due to a C=N group.
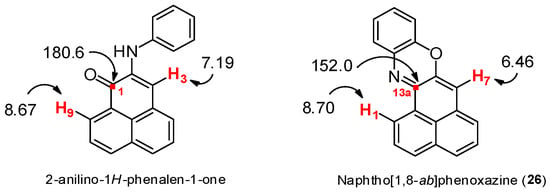
Figure 3.
Comparative analysis of NMR chemical shifts between the anilino derivative and 26.
Based on the couplings observed in the COSY experiment, we assigned the protons H-1 to H-6 of the 1H-phenalen-1-one nucleus. The corresponding CH carbons were assigned using HSQC. To assign quaternary carbons we used HMBC. The IR bands of compound 26 were in agreement with its functional groups (Figure 4).
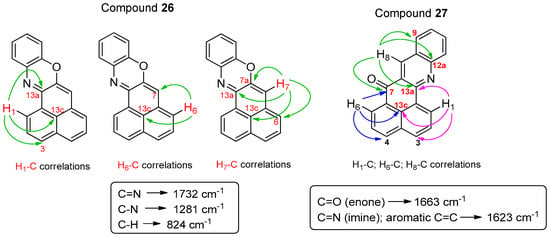
Figure 4.
Key HMBC correlations at two or three bonds and IR bands of compounds 26 and 27.
Although less well known, Buchwald–Hartwig reaction conditions can also generate diaryl ethers, when phenolic groups are present [22,23]. Under the reaction conditions used the phenoxy group in 2-aminophenol can be deprotonated by the base forming the corresponding phenoxide ion. The phenoxide ion, being more nucleophilic than the aniline group, will be preferentially incorporated into the palladium catalytic cycle and will generate the corresponding diaryl ether intermediate 26a (Scheme 6). The aniline group is now ideally positioned to undergo an intramolecular imine reaction resulting in compound 26. The use of more electron-rich and bulkier ligands than triphenylphosphine has been reported to improve the yields of these types of diaryl ethers, and may explain the relatively low yield observed [24].
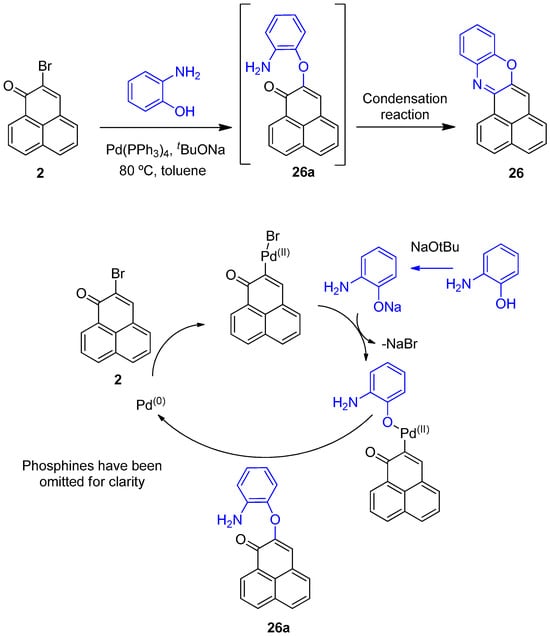
Scheme 6.
Proposed mechanism for the cross-coupling of 2-aminophenol with 2-bromo-1H-phenalen-1-one (2) to give 26a.
For compound 27, all proton and CH carbon signals were assigned using two-dimensional NMR experiments. High-resolution mass spectrometry established its molecular formula as C20H11NO, consistent with a highly unsaturated structure, and the IR spectrum showed bands in agreement with the proposed functional groups (Figure 4).
Mechanistically, the formation of 27 under these reaction conditions is intriguing. An initial Buchwald-Hartwig coupling would attach the nitrogen to position 2, from where it would seem improbable to generate the characterized product 27. Of note, the synthesis of compound 27 has been previously described by Kuroki and co-workers [25] albeit using relatively harsh conditions. Although they also used 2-(aminophenyl)methanol as the amine source, they used 3-hydroxyphenalenone as the phenalenone source. Apart from solvents, no other additives were employed. Boiling in DMSO only generated a trace amount of 27, while heating in boiling HMPT (195 °C) increased the yield of 27 to 33%.
Further experimental studies will be needed to assess the role of each reagent (Pd(Ph3P)4, NaOtBu) in our reaction conditions, and to try and elucidate a plausible mechanism for the formation of 27 under these milder reaction conditions.
- Preparation of hydroxyl and carboxylic acid containing 1H-phenalen-1-ones
As several naturally occurring phenalenones with antifungal activities are hydroxylated, or even poly-hydroxylated [4], we wanted to have more hydroxylated examples in our collection.
The desired derivatives 30 and 33 were synthesized by constructing the phenalenone core using hydroxylated naphthalene’s as building blocks (Scheme 7). Using the same methodology reported for the synthesize 9-hydroxy-1H-phenalen-1-one [17,18,21], but with 29 as the starting material, a complex mixture of products was obtained from which 30 was isolated in yields < 30%. However, substitution of conventional heating for microwave irradiation in a sealed tube afforded the fully demethylated product 30 in 73% yield. Ethyl 3-hydroxynaphthalene-2-carboxylate (32) was converted into its corresponding 1H-phenalenone derivative 33 in a similar fashion; however, the microwave conditions were too harsh for this substrate and so the original conditions were preferred. Subsequent hydrolysis of 33 furnished compound 34, which combines the 1H-phenalen-1-one core with a salicylic acid moiety.
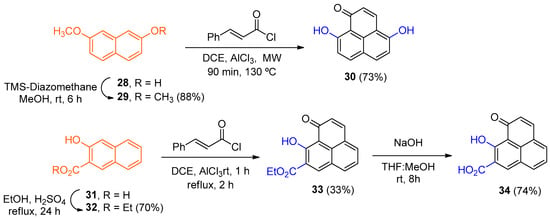
Scheme 7.
Synthesis of hydroxylated analogs (30, 33 and 34).
- Preparation of nitrogen containing 1H-phenalen-1-ones
The 4H-benzo[de]isoquinolin-4-one 35 derivatives 36 and 37 were synthesized from naphthalenes 29 and 32 using 1,3,5-triazine and PPA (polyphosphoric acid) [26] (Scheme 8). The poorer solubility of 32 compared to 29 in PPA was probably accountable for the observed lower yield of 37 compared to 36.
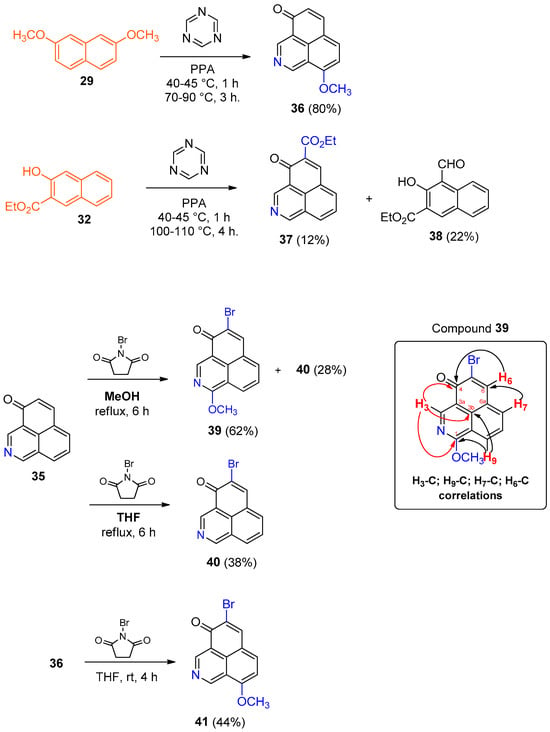
Scheme 8.
Synthesis of heterocyclic 1H-phenalen-1-one analogs (36–37 and 39–41) and key HMBC correlations at three bonds of compound 39.
To enable further functionalization of these heterocycles in a manner similar to the phenalenone core, we explored the introduction of a suitable leaving group at the C-2 position. Bromination of compound 35 with NBS in MeOH afforded compound 39 (62% yield), along with compound 40 (28% yield). Compound 39 was likely formed via activation of the pyridine nitrogen by NBS, followed by regioselective addition of methanol (Scheme 7). Two-dimensional COSY, HMBC, and HSQC NMR experiments were performed to determine which of the positional isomers [either 5-bromo-3-methoxy- or 5-bromo-1-methoxy-4H-benzo[de]isoquinolin-4-one (39)], had been obtained. Based on the couplings observed in the COSY spectrum, we assigned protons H-6 and H-8. The corresponding CH carbons were identified using HSQC. For the assignment of quaternary carbons, we relied on HMBC correlations (Scheme 8), confirming 39 as the isomer formed.
When THF was used instead of MeOH, compounds 40 and 41 were obtained from compounds 35 and 36, respectively (Scheme 8).
- In Vitro Anti-plasmodium Activity and Cytotoxicity
To date, thirty phenalenone-based compounds of the total synthetized, including the 1H-phenalen-1-one (1) as a starting material and compounds 2, 3, 9-hydroxy-1H-phenalen-1-one (9-OH) and 35 as intermediates, have been investigated for their activity against the chloroquine-resistant P. falciparum strain FCR3, and the results are summarized in Table 1. The cytotoxicity of these compounds was also evaluated by measuring their ability to reduce the viability of MCF-7 cells at 10 µM.

Table 1.
In vitro antiplasmodial activity and cytotoxicity of 1H-phenalen-1-one derivatives.
Four compounds exhibited antiplasmodium activity, with three having IC50 values < 1 µM. The most potent compound was the 9-cyano derivative (18). Interestingly, neither the 2-cyano compound (20) nor the 9-amido compound (19) were active, even at 10 µM. Extending the cyano group by introducing a phenyl spacer at the 9-position, compound (10), was also shown to be detrimental to activity. The 9-hydroxy compound also possessed sub-micromolar activity which was unfortunately lost upon the addition of either an ethyl ester (33) or carboxylic acid (34) in the adjacent 8-position. Notably, the introduction of another hydroxyl group at position 4, compound (30), also diminished antiplasmodium activity. While the 2-bromo derivative (2) was active (IC50 = 0.7 µM), substituting it for the larger iodo (3), phenyl (4) or furan (7) groups again caused a significant loss in antiplasmodium activity. Finally, the introduction of a nitrogen atom into the 8-position (compound 35) of phenaleonone (1) resulted in a complete loss of activity relative to the parent structure. On a more positive note, none of the tested compounds produced significant levels of cytotoxicity at 10 µM.
Overall, these active phenalenone derivatives exhibited improved antiplasmodial activity compared to previously tested compounds, such as 1c (IC50 = 21.0 µM) and 1d (IC50 = 2.1 µM) [8] (Figure 1). Importantly, the active compounds showed no evidence of cytotoxicity with IC50 > 10 μM and possess favorable predicted physicochemical properties (Table 2).
Table 2.
Calculated physicochemical properties for the most active compounds a,b.
3. Materials and Methods
3.1. General Methods
All of the reagents and solvents were obtained from Aldrich Chemical Co. (Merck KGaA, Darmstadt, Germany) and used without further purification. Reactions with sensitive reagents were performed under an inert atmosphere (argon or nitrogen), and organic solvents were dried using standard methods. Unless otherwise stated, solvents were removed under reduced pressure using a rotary evaporator at 40–60 °C. All of the reactions were monitored via analytical thin layer chromatography (TLC) on POLIGRAM® SIL G/UV254 silica gel-coated plates (0.20 mm) from MACHEREY-NAGEL (Düren, Germany). Column chromatography was performed on silica gel 60 (0.063–0.20 mm) from MERCK (Merck KGaA, Darmstadt, Germany). Preparative thin layer chromatography was carried out with GF silica gel plates (1 mm) with fluorescent indicator 254 nm (UNIPLATE) (Merck KGaA, Darmstadt, Germany). The compounds were visualized using ultraviolet light (254 nm). 1H and 13C NMR spectra were recorded at room temperature (rt) on a Bruker Avance 500 or 600 MHz NMR spectrometer in the solvent indicated (Bruker Española S.A., Madrid, Spain). Data for 1H NMR are reported as follows: chemical shift (δ ppm), integration, multiplicity and coupling constant (Hz). 13C NMR analyses are reported in terms of chemical shift. IR spectra were recorded as a thin film on NaCl plates with a BRUKER IFS 66 spectrophotometer. UV spectra were recorded on a JASCO modelo V-560 spectrophotometer (Jasco Analítica Spain, S.L., Madrid, Spain). Melting points were determined using a Stuart Scientific SMP11 instrument. Low-resolution (EIMS) and high-resolution mass spectrometry (HRMS) were performed on a Micromass VG AutoSpec magnetic tri-sector (EBE geometry) mass spectrometer (Manchester, UK) via electrospray ionization (ESI) or electron impact (EI) (Agilent Technologies Spain, S.L., Barcelona, Spain). Microwave reactions were performed using a Biotage® Initiator (software version 2.5). Lyophilization was performed using a CHRIST ALPHA 2–4 lyophilizer (TELSTAR S.L.U., Terrassa, Spain).
3.2. Synthesis
Compounds 2 [28], 3 [29], 8 [21] and 35 [26] were prepared as described in the references.
3.2.1. Synthesis of 2-Bromo-1H-phenalen-1-one (2)
1H-Phenalen-1-one (1) (3.0 g, 16.6 mmol), neutral alumina (22.6 g), and NBS (5.0 g, 28.3 mmol) were mixed in a mortar until a uniformly yellow mixture was obtained. This mixture was then heated at 45 °C for 22 h. The crude product was filtered, washing with CH2Cl2, and the solvent was evaporated to dryness under reduced pressure using a rotary evaporator. The resulting mixture was washed with hot water and filtered while hot to afford compound 2 (4.0 g, 93%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ: 8.64 (1H, dd, J = 0.9, 7.4 Hz, H-9), 8.19 (1H, d, J = 8.0 Hz, H-7), 8.14 (1H, s, H-3), 8.02 (1H, d, J = 8.4, H-6), 7.74 (1H, dd, J = 8.0, 7.6 Hz, H-8), 7.67 (1H, d, J = 7.0 Hz, H-4), 7.56 (1H, dd, J = 8.0, 7.2 Hz, H-5). 13C NMR (100 MHz, CDCl3) δ: 178.7 (C=O), 143.1 (CH, C-3), 135.6 (CH), 132.5 (CH), 132.2 (CH), 132.2 (C), 131.5 (CH), 128.6 (C), 127.9 (C), 127.5 (CH), 126.9 (CH), 126.6 (C), 125.9 (C). UV-Vis (EtOH) λmax: 403, 362, 328 nm. Mp: 150–152 °C.
3.2.2. Synthesis of 2-Iodo-1H-phenalen-1-one (3)
A solution of iodine (6.0 g, 20 mmol) in CCl4:Py (20 mL, 1:1) was added dropwise to a solution of 1H-phenalen-1-one (1) (2.0 g, 11.1 mmol) in CCl4:Py (20 mL, 1:1) at 0 °C under an argon atmosphere. The mixture was stirred at room temperature for 48 h. It was then diluted with ether (100 mL) and washed successively with water (50 mL), 1 N HCl solution (2 × 50 mL), water (50 mL), and an aqueous 20% Na2S2O3 solution (50 mL). The organic layer was dried over Na2SO4, filtered, and the solvent was evaporated to dryness under reduced pressure. The crude product was purified by recrystallization from ether to afford compound 3 (1.56 g, 46%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ: 8.55 (1H, dd, J = 1.2, 7.6 Hz, H-9), 8.37 (1H, s, H-3), 8.12 (1H, d, J = 7.2 Hz, H-7), 7.96 (1H, d, J = 8.4 Hz, H-6), 7.66 (1H, dd, J = 7.6, 8.0 Hz, H-8), 7.57 (1H, d, J = 6.8 Hz, H-4), 7.50 (1H, dd, J = 8, 7.2 Hz, H-5). 13C NMR (100 MHz, CDCl3) δ: 179.5 (C=O), 150.5 (CH, C-3), 135.4 (CH), 132.6 (CH), 132.4 (CH), 132.0 (C), 131.2 (C), 128.7 (C), 127.4 (CH), 127.0 (C), 126.9 (C), 126.8 (CH), 106.0 (C, C-2). EIMS: m/z 306 (M+, 15), 305 (M-1, 100), 179 (M-I+, 22), 151 (47), 111 (26), 97 (37), 57 (59). HRMS (EI): calcd. for C13H7IO (M+) 305.9542, found 305.9540.
3.2.3. Synthesis of 1-Oxo-1H-phenalen-9-trifluoromethanesulfonate (8)
To a solution of 9-hydroxy-1H-phenalen-1-one (1) (400 mg, 2.04 mmol) in toluene (20 mL) was added 20 mL of a 30% K3PO4 solution (8.6 g, 40.8 mmol). The mixture was cooled to −10 °C, then triflic anhydride (0.42 mL, 2.45 mmol) was slowly added, and the reaction was stirred at room temperature for 5 h. The aqueous phase was extracted with toluene (3 × 15 mL), the combined organic layers were dried (MgSO4), filtered, and the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography (hexane/EtOAc, 10–30%) to afford compound 8 (575 mg, 86%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ: 8.29 (1H, d, J = 8.8 Hz), 8.07 (1H, d, J = 8.2 Hz), 7.87 (1H, d, J = 7 Hz), 7.76 (1H, d, J = 9.8 Hz), 7.70 (1H, dd, J = 7.8, 7.5 Hz), 7.57 (1H, d, J = 8.8, Hz), 6.74 (1H, d, J = 9.8 Hz). EIMS: m/z 139 (100), 166 (31), 195 (35), 196 (16). 236 (32) 327 (73), 328 (M+, 14). HRMS (EI): calcd. for C14H7F3O4S (M+) 328.0017, found 327.0019. UV-Vis (EtOH) λmax: 307, 340, 355, 382 nm.
3.2.4. Synthesis of 2-Aryl-1H-phenalen-1-one Derivatives: General Procedure
To a solution of 2-iodo-1H-phenalen-1-one (3) in a 1:1 DME/H2O mixture, Na2CO3, boronic acid, and 10% Pd/C were added. The reaction mixture was heated at 60 °C. After this time, EtOAc (10 mL) was added and the mixture was filtered over Celite®. H2O (20 mL) was then added, the mixture was extracted with EtOAc (3 × 8 mL), and dried (MgSO4). After filtration and concentration under vacuum, the crude product was purified by column chromatography or on a preparative silica gel plate.
- Synthesis of 2-phenyl-1H-phenalen-1-one (4).
From 2-iodo-1H-phenalen-1-one (3) (51.8 mg, 0.169 mmol), DME/H2O (1 mL), Na2CO3 (41.6 mg, 0.392 mmol), phenylboronic acid (46.1 mg, 0.378 mmol), and 10% Pd/C (15.6 mg), the mixture was heated for 10 min; column chromatography (CH2Cl2); product 4 (42.3 mg, 97%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ: 8.71 (1H, d, J = 7.4 Hz, H-9), 8.20 (1H, d, J = 8.0 Hz), 8.01 (1H, d, J = 8.3 Hz), 7.85 (1H, s, H-3), 7.85–7.78 (2H, m), 7.69 (2H, d, J = 8.0 Hz), 7.61 (1H, t, J = 7.6), 7.49–7.45 (2H, m), 7.42–7.38 (1H, m). 13C NMR (100 MHz, CDCl3) δ: 184.1 (C=O), 139.7 (CH), 139.4 (C), 136.7 (C), 134.7 (CH), 132.1 (C), 131.5 (CH), 131.1 (CH), 130.0 (C), 129.2 (2 × CH), 128.3 (2 × CH), 128.3 (C), 128.2 (CH), 127.4 (CH), 127.3 (C), 126.9 (CH, C-3). EIMS: m/z 301 (40), 280 (23), 279 (M+ + Na, 100). HRMS (ESI): calcd. for C19H12ONa (M+ + Na) 279.0787, found 279.0787. Mp: 147–149 °C.
- Synthesis of 4-(1-oxo-1H-phenalen-2-yl)benzonitrile (5).
Starting with 2-iodo-1H-phenalen-1-one (3) (70.0 mg, 0.228 mmol), DME/H2O (1.4 mL), Na2CO3 (56.0 mg, 0.524 mmol), (4-cyanophenyl)boronic acid (77.0 mg, 0.524 mmol), and 10% Pd/C (15.6 mg), the mixture was heated for 1 h and 20 min; column chromatography (hexane/EtOAc, 4:1 to 3:2) yielded product 5 (48.0 mg, 75%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 8.73 (1H, dd, J = 1.1, 7.4 Hz, H-9), 8.27 (1H, dd, J = 0.9, 8.0 Hz, H-7), 8.09 (1H, d, J = 8.2 Hz), 7.91 (1H, s, H-3), 7.88–7.85 (2H, m), 7.81 (2H, dd, J = 1.9, 6.0 Hz, H-2’, H-6’,), 7.74 (2H, dd, J = 2.0, 6.6 Hz, H-3’, H-5’), 7.66 (1H, dd, J = 7.6, 8.2 Hz, H-5). 13C NMR (125 MHz, CDCl3) δ: 183.4 (C=O), 141.4 (C, C-2), 140.9 (CH), 137.6 (C), 135.3 (CH), 132.5 (CH), 132.4 (CH), 132.2 (C), 132.1 (2 × CH), 131.5 (CH), 129.9 (2 × CH), 129.7 (C), 127.7 (CH), 127.6 (C), 127.4 (C), 127.1 (CH), 119.1 (CN), 111.7 (C, C-1’). EIMS: m/z 281 (M+, 56), 280 (10), 251 (11), 140 (11). HRMS (EI): calcd. for C20H11NO (M+) 281.0841, found 281.0834. UV-Vis (EtOH) λmax: 400, 363, 282, 249 nm. IR (film) νmax: 2852, 1731, 1634, 1115, 837 cm−1. Mp: 232–234 °C.
- Synthesis of 2-(4-fluorophenyl)-1H-phenalen-1-one (6).
Starting with 2-iodo-1H-phenalen-1-one (3) (70.0 mg, 0.228 mmol), DME/H2O (1.4 mL), Na2CO3 (56.0 mg, 0.524 mmol), (4-fluorophenyl)boronic acid (87.0 mg, 0.524 mmol), and 10% Pd/C (15.6 mg), the mixture was heated for 10 min; column chromatography (hexane/AcOEt, 4:1) gave product 6 (46.0 mg, 73%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 8.70 (1H, dd, J = 1.1, 7.3 Hz, H-9), 8.23 (1H, dd, J = 0.8, 8.0 Hz, H-7), 8.04 (1H, d, J = 8.1 Hz), 7.83 (1H, s, H-3), 7.81 (2H, t, J = 7.6 Hz), 7.66 (1H, dd, J = 2.1, 5.5 Hz), 7.62 (1H, dd, J = 7.1, 8.2 Hz, H-8), 7.14 (1H, dd, J = 2.1, 5.5 Hz). 13C NMR (125 MHz, CDCl3) δ: 184.1 (C=O), 162.9 (d, JC-F = 245.7 Hz), 139.6 (CH), 138.4 (C), 134.9 (CH), 132.6 (d, JC-F = 3.5 Hz), 132.2 (C), 131.7 (CH), 131.6 (CH), 131.2 (CH), 131.0 (d, JCH-F = 8.0 Hz, 2 × CHF), 129.9 (C), 128.1 (C), 127.5 (CH), 127.3 (C), 127.0 (CH), 115.3 (d, JCH-F = 21.2 Hz, 2 × CHF). 19F NMR (470 MHz, CDCl3) δ: −113.8. EIMS: m/z 274 (M+, 68), 273 (100), 244 (17), 136 (11). HRMS (EI): calcd. for C19H11FO (M+) 274.0794, found 274.0786. UV-Vis (EtOH) λmax: 405, 361, 254 nm. IR (film) νmax: 2850, 1631, 1577, 1288, 1220, 833cm−1. Mp: 200–202 °C.
- Synthesis of 2-(3-furyl)-1H-phenalen-1-one (7).
From 2-iodo-1H-phenalen-1-one (3) (50.0 mg, 0.163 mmol), DME/H2O (1 mL), Na2CO3 (34.6 mg, 0.326 mmol), 3-furylboronic acid (35.4 mg, 0.316 mmol), and 10% Pd/C (15.6 mg), the mixture was heated for 10 min; column chromatography (CH2Cl2) gave product 7 (39.5 mg, 98%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ: 8.68 (1H, d, J = 7.3 Hz, H-9), 8.56 (1H, s, H-2’), 8.18 (1H, d, J = 7.9 Hz, H-7), 7.97 (1H, d, J = 8.2 Hz), 7.87 (1H, s, H-3), 7.80–7.76 (2H, m), 7.58 (1H, t, J = 7.6 Hz, H-5), 7.50 (1H, s, H-5’), 6.84 (1H, s, H-4’). 13C NMR (100 MHz, CDCl3) δ: 183.8 (C=O), 143.8 (CH), 142.6 (CH), 135.9 (CH), 134.9 (CH), 132.0 (C), 131.3 (CH), 131.2 (CH), 131.0 (CH), 130.8 (C), 129.7 (C), 128.1 (C), 127.3 (CH), 126.9 (CH), 126.5 (s), 120.3 (C), 108.2 (CH). EIMS: m/z 270 (17), 269 (M+ + Na+,100). HRMS (ESI): calcd. for C17H10O2Na (M+ + Na+) 269.0578, found 269.0572. UV-Vis (EtOH) λmax: 407, 362, 246 nm. IR (film) νmax: 3393, 1754, 1636, 1573, 936 cm−1. Mp: 133–135 °C.
3.2.5. Synthesis of 9-Phenyl-1H-phenalen-1-one (9)
To a solution of 1-oxo-1H-phenalen-9-trifluoromethylsulfonate (8) (46.0 mg, 0.140 mmol), K3PO4 (44.0 mg, 0.210 mmol), and Pd(PPh3)4 (10.5 mg, 0.009 mmol) in 1,4-dioxane (1.2 mL), 95% phenylboronic acid (28 mg, 0.229 mmol) was added, and the mixture was heated for 3 h at 60 °C. The reaction crude was concentrated on a rotary evaporator, H2O (10 mL) was added, extracted with EtOAc (3 × 8 mL), dried (Na2SO4), filtered and the solvent was removed on a rotary evaporator. The crude was purified by preparative silica gel plate chromatography (hexane/EtOAc, 3:2) to obtain product 9 (30 mg, 84%) as a red oil. 1H NMR (500 MHz, CDCl3) δ: 8.16 (1H, d, J = 8.3 Hz, H-7), 8.03 (1H, dd, J = 0.8, 8.2 Hz), 7.77 (1H, dd, J = 0.7, 7.0 Hz), 7.69 (1H, d, J = 9.7 Hz), 7.61 (1H, dd, J = 7.1, 8.2 Hz), 7.59 (1H, d, J = 8.2 Hz), 7.49–7.37 (5H, m), 6.60 (1H, d, J = 9.7 Hz).
- Synthesis of 4-(1-oxo-1H-phenalen-9-yl)benzonitrile (10).
To a solution of 1-oxo-1H-phenalen-9-trifluoromethylsulfonate (8) (50.0 mg, 0.152 mmol), K3PO4 (48 mg, 0.228 mmol), and Pd(PPh3)4 (10 mg, 0.009 mmol) in 1,4-dioxane (1.3 mL), (4-cyanophenyl)boronic acid (36 mg, 0.243 mmol) was added, and the mixture was heated for 2 h and 30 min at 60 °C. The reaction crude was concentrated on a rotary evaporator, H2O (10 mL) was added, the mixture was extracted with EtOAc (3 × 5 mL), dried (Na2SO4), filtered, and the solvent was removed on a rotary evaporator. The crude was purified by silica gel column chromatography (hexane/EtOAc, 4:1 to 3:2) to give product 10 (42 mg, 98%) as an orange solid. 1H NMR (500 MHz, CDCl3) δ: 8.22 (1H, d, J = 8.2 Hz, H-7), 8.07 (1H, dd, J = 0.7, 8.2 Hz), 7.82 (1H, d, J = 6.5 Hz), 7.74–7.72 (3H, m), 7.56 (1H, dd, J = 7.1, 8.2 Hz, H-5), 7.51 (1H, d, J = 8.2 Hz), 7.45 (2H, dd, J = 8.4, 1.9 Hz, H-2’, H-6’), 6.58 (1H, d, J = 9.7 Hz, H-2). 13C NMR (125 MHz, CDCl3) δ: 185.6 (C=O), 148.2 (C, C-9), 145.3 (C), 141.1 (CH), 134.3 (CH), 132.3 (C), 132.2 (2 × CH, C-2’, C-6’), 132.1 (CH), 132.0 (CH), 130.6 (CH), 130.2 (CH), 128.8 (2 × CH, C-3’, C-5’), 128.6 (C), 128.3 (C), 127.1 (CH), 126.1 (C), 119.2 (C, CN), 111.0 (C, C-1’). EIMS: m/z 281 (M+, 40), 280 (100), 251 (7), 140 (8) HRMS (EI): calcd. for C20H11NO (M+) 281.0841, found 281.0833. UV-Vis (EtOH) λmax: 361, 257 nm. IR (film) νmax: 2222, 1623, 1546, 832 cm−1. Mp: 191–193 °C.
- Synthesis of 9-(4-fluorophenyl)-1H-phenalen-1-one (11).
To a solution of 1-oxo-1H-phenalene-9-trifluoromethylsulfonate (8) (50.0 mg, 0.152 mmol), K3PO4 (48 mg, 0.228 mmol), and Pd(PPh3)4 (10 mg, 0.009 mmol) in 1,4-dioxane (1.3 mL), 95% (4-fluorophenyl)boronic acid (42 mg, 0.253 mmol) was added, and the mixture was heated for 1 h and 30 min at 60 °C. The reaction crude was concentrated on a rotary evaporator, H2O (8 mL) was added, the mixture was extracted with EtOAc (3 × 5 mL), dried (Na2SO4), filtered, and the solvent was removed on a rotary evaporator. The crude was purified by silica gel column chromatography (hexane/EtOAc, 4:1 to 3:2) to give product 11 (41.5 mg, 99%) as an orange solid. 1H NMR (500 MHz, CDCl3) δ: 8.16 (1H, d, J = 8.3 Hz, H-7), 8.03 (1H, dd, J = 0.95, 8.2 Hz), 7.76 (1H, dd, J = 0.8, 7.0 Hz), 7.68 (1H, d, J = 9.7 Hz, H-3), 7.61 (1H, dd, J = 7.1, 8.1 Hz, H-5), 7.55 (1H, d, J = 8.3, H-8), 7.35 (1H, d, J = 8.7 Hz, H-3’, H-5’), 7.34 (1H, d, J = 8.8 Hz), 7.16 (1H, d, J = 8.7, H-2’, H-6’), 7.14 (1H, d, J = 8.8 Hz), 6.59 (1H, d, J = 9.7 Hz, H-2). 13C NMR (125 MHz, CDCl3) δ: 185.9 (C=O), 163.3 (d, JC-F = 244.5 Hz), 146.6 (C), 140.6 (CH), 138.8 (d, JC-F = 3.6 Hz), 133.9 (CH), 131.9 (C), 131.9 (CH), 131.7 (2 × CH), 130.6 (CH), 129.8 (d, JCH-F = 8.0 Hz, 2 × CHF), 128.5 (C), 128.4 (C), 126.6 (CH), 126.2 (C), 115.4 (d, JCH-F = 21.7 Hz, 2 × CHF). 19F NMR (470 MHz, CDCl3) δ: −115.4. EIMS: m/z 274 (M+, 33), 273 (100), 136 (7), 113 (4). HRMS (EI): calcd. for C19H11FO (M+) 274.0794, found 274.0802. UV-Vis (EtOH) λmax: 398, 362, 252 nm. IR (film) νmax: 1624, 1552, 1350, 1211, 829 cm−1. Mp: 136–138 °C.
- Synthesis of 9-(4-methoxyphenyl)-1H-phenalen-1-one (12).
To a solution of 1-oxo-1H-phenalene-9-trifluoromethylsulfonate (8) (28.0 mg, 0.085 mmol), K3PO4 (27 mg, 0.128 mmol), and Pd(PPh3)4 (6 mg, 0.005 mmol) in 1,4-dioxane (0.7 mL) was added 95% (4-methoxyphenyl)boronic acid (33 mg, 0.217 mmol) and heated for 1 h and 30 min at 60 °C under an inert atmosphere. The reaction crude was concentrated on a rotary evaporator, H2O (10 mL) was added, the mixture was extracted with EtOAc (3 × 5 mL), dried (Na2SO4), filtered, and the solvent was removed on a rotary evaporator. The crude was purified by silica gel column chromatography (hexane/EtOAc, 10–30%) to give product 12 (20.7 mg, 85%) as an orange solid. 1H NMR (400 MHz, CDCl3) δ: 8.14 (1H, d, J = 8.3 Hz, H-7), 8.07 (1H, d, J = 8.2 Hz, H-6), 7.77 (1H, d, J = 7 Hz, H-4), 7.68 (1H, d, J = 9.7 Hz, H-3), 7.68 (2H, m, H-5, H-8), 7.34 (2H, d, J = 8.6, Hz, H-2’, H-6’), 7.0 (2H, d, J = 8.6 Hz, H-3’, H-5’), 6.60 (1H, d, J = 9.7 Hz, H-2), 3.88 (3H, s, OCH3). Mp: 175–178 °C.
- Synthesis of 9-(3-methoxyphenyl)-1H-phenalen-1-one (13).
To a solution of 1-oxo-1H-phenalene-9-trifluoromethylsulfonate (8) (80.0 mg, 0.244 mmol), K3PO4 (76 mg, 0.366 mmol), and Pd(PPh3)4 (17 mg, 0.015 mmol) in 1,4-dioxane (2 mL) was added (3-methoxyphenyl)boronic acid (59 mg, 0.390 mmol) and heated for 4 h at 60 °C. The reaction crude was concentrated on a rotary evaporator, H2O (8 mL) was added, the mixture was extracted with EtOAc (3 × 5 mL), dried (Na2SO4), filtered, and the solvent was removed on a rotary evaporator. The crude oil was purified by silica gel column chromatography (hexane/EtOAc, 19:1 to 17:3) to give product 13 (62 m, 89%) as an orange oil. 1H NMR (500 MHz, CDCl3) δ: 8.16 (1H, d, J = 8.3 Hz, H-7), 8.04 (1H, dd, J = 0.9, 8.2 Hz), 7.78 (1H, d, J = 6.7 Hz), 7.68 (1H, d, J = 9.7 Hz, H-3), 7.61 (1H, dd, J = 7.1, 8.2 Hz, H-5), 7.60 (1H, d, J = 8.3, H-8), 7.37 (1H, t, J = 7.8 Hz, H-5), 6.95 (2H, dd, J = 2.1, 7.9 Hz, H-4’, H-6’), 6.90 (1H, m, H-2’), 6.59 (1H, d, J = 9.7 Hz, H-2), 3.83 (3H, s, CH3). 13C NMR (125 MHz, CDCl3) δ: 185.8 (C=O), 159.8 (C, C-3’), 147.5 (C), 144.5 (C), 140.5 (CH), 138.8 (CH), 131.9 (C), 131.8 (CH), 131.6 (CH), 131.5 (CH), 130.6 (CH), 129.4 (CH), 128.7 (C), 128.5 (C), 126.5 (CH), 126.3 (C), 120.4 (CH), 113.7 (CH), 112.7 (CH), 56.4 (CH3, OCH3). EIMS: m/z 286 (M+, 74), 285 (100), 271 (28), 256 (19), 255 (95), 242 (30), 213 (19). HRMS (EI): calcd. for C20H14O2 (M+) 286.0994, found 286.1003. UV-Vis (EtOH) λmax: 362, 253 nm. IR (film) νmax: 1640, 1558, 1240, 850 cm−1.
- Synthesis of 9-(2-methoxyphenyl)-1H-phenalen-1-one (14).
To a solution of 1-oxo-1H-phenalene-9-trifluoromethylsulfonate (8) (20.0 mg, 0.058 mmol), K3PO4 (18 mg, 0.086 mmol), and Pd(PPh3)4 (4 mg, 0.003 mmol) in 1,4-dioxane (0.5 mL) was added (2-methoxyphenyl)boronic acid (14 mg, 0.093 mmol) and heated for 4 h at 60 °C. The reaction crude was concentrated on a rotary evaporator, H2O (8 mL) was added, the mixture was extracted with EtOAc (3 × 5 mL), dried (Na2SO4), filtered, and the solvent was removed on a rotary evaporator. The crude oil was purified by preparative silica gel plate chromatography (CH2Cl2) to give product 14 (6 mg, 36%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 8.18 (1H, d, J = 8.2 Hz, H-7), 8.03 (1H, dd, J = 0.9, 8.2 Hz), 7.75 (1H, dd, J = 0.9, 7.0 Hz), 7.67 (1H, d, J = 9.7 Hz, H-3), 7.60 (1H, dd, J = 7.1, 8.2 Hz, H-5), 7.58 (1H, d, J = 8.2 Hz, H-8), 7.39 (1H, ddd, J = 1.7, 7.4, 8.2 Hz), 7.16 (1H, dd, J = 1.8, 7.5 Hz), 7.07 (1H, ddd, J = 0.9, 7.3, 8.3 Hz), 6.99 (1H, dd, J = 0.6, 8.2 Hz), 6.57 (1H, d, J = 9.7 Hz, H-2), 3.70 (3H, s, CH3). 13C NMR (125 MHz, CDCl3) δ: 185.8 (C=O), 156.6 (C, C-2’), 143.9 (C), 140.4 (CH), 133.9 (CH), 132.4 (C), 132.0 (C), 131.8 (CH), 131.7 (CH), 131.2 (CH), 130.4 (CH), 128.9 (CH), 128.8 (CH), 128.7 (C), 128.4 (C), 127.1 (C), 126.3 (CH), 121.1 (CH), 110.9 (CH), 56.2 (CH3, OCH3). Mp: 123–125 °C.
3.2.6. Synthesis of 9-Arylamino-1H-phenalen-1-ones Derivatives
- Synthesis of 9-[(4-methoxyphenyl)amino]-1H-phenalen-1-one (15).
To a solution of 1-oxo-1H-phenalene-9-trifluromethylsulfonate (8) (25 mg, 0.076 mmol) in THF (0.65 mL) was added (4-methoxyphenyl)amine (21 mg, 0.173 mmol) and stirred at room temperature for 28 h. After that time, the THF was removed under high vacuum. The crude product was purified by silica gel column chromatography (hexane/EtOAc, 4:1, 7:3) to give product 15 (22 mg, 96%) as a red solid. 1H NMR (500 MHz, CDCl3) δ: 13.64 (1H, br s, NH), 7.88–7.85 (4H, m), 7.44 (1H, t, J = 7.6 Hz, H-5), 7.35 (1H, d, J = 9.3 Hz, H-8), 7.30 (2H, d, J = 8.5 Hz, H-3’, H-5’), 7.03 (1H, d, J = 9.4 Hz, H-2), 6.98 (2H, d, J = 8.6 Hz, H-2’, H-6’), 3.86 (3H, s, OCH3). Mp: 128–130 °C.
- Synthesis of 9-[(2,6-dimethylphenyl)amino]-1H-phenalen-1-one (16).
A solution of 1-oxo-1H-phenalene-9-trifluromethylsulfonate (8) (50 mg, 0.152 mmol) and 2,6-dimethylaniline (0.6 mL) was heated at 80 °C for 4 days. After that time, the amine was removed under high vacuum. The crude product was purified by silica gel column chromatography (hexane/EtOAc, 1:1 to 1:9) to obtain product 16 (39 mg, 86%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 13.38 (1H, br s, NH), 7.94 (4H, m), 7.51 (1H, t, J = 7.5 Hz, H-5), 7.26 (3H, m), 7.13 (1H, d, J = 9.4 Hz), 6.77 (1H, d, J = 9.2 Hz, H-2), 2.29 (6H, s, 2 × CH3). 13C NMR (125 MHz, CDCl3) δ: 185.1 (C=O), 155.5 (C, C-9), 138.9 (CH), 138.4 (CH), 136.2 (CH), 135.9 (C), 131.9 (CH), 131.5 (CH), 128.9 (CH), 128.7 (2 × CH), 128.5 (C), 127.6 (2 × C), 125.4 (C), 125.0 (C), 122.2 (CH), 115.7 (CH), 108.2 (C), 18.4 (2 × CH3). EIMS: m/z 299 (M+, 80), 298 (100), 141 (11). HRMS (EI): calcd. for C21H17NO (M+) 299.1310, found 299.1315. UV-Vis (EtOH) λmax: 468, 441, 355, 250 nm. IR (film) νmax: 3397, 1632, 1509, 1242, 852 cm−1. Mp: 205–207 °C.
- Synthesis of 9-[(4-chlorophenyl)amino]-1H-phenalen-1-one (17).
To a solution of 1-oxo-1H-phenalene-9-trifluoromethylsulfonate (8) (50 mg, 0.152 mmol) in THF (1 mL) was added (4-chlorophenyl)amino (54.5 mg, 0.427 mmol) and heated at 50 °C for 24 h. After that time, THF was removed under high vacuum. The crude product was purified by silica gel column chromatography (hexane/EtOAc, 9:1 to 4:1) to give product 17 (43 mg, 93%) as an orange solid. 1H NMR (500 MHz, CDCl3) δ: 13.73 (1H, br s, NH), 7.92 (1H, d, J = 9.3 Hz, H-7), 7.91–7.86 (3H, m), 7.46 (1H, t, J = 7.6 Hz, H-5), 7.42 (1H, d, J = 9.2, H-3), 7.40 (2H, d, J = 8.6 Hz, H-2’, H-6’), 7.31 (2H, d, J = 8.6 Hz, H-3’, H-5’), 7.00 (1H, d, J = 9.4 Hz, H-2). 13C NMR (125 MHz, CDCl3) δ: 185.1 (C=O), 153.6 (C, C-9), 139.3 (CH), 138.2 (CH), 137.2 (C), 132.2 (CH), 131.6 (CH), 131.5 (CH), 131.8 (C), 129.8 (2 × CH, C-2’, C-6’), 128.8 (CH), 128.3 (C), 126.2 (2 × CH, C-3’, C-5’), 125.6 (C), 125.4 (C), 122.7 (CH), 115.5 (CH), 109.1 (C). EIMS: m/z 307 (34), 305 (M+, 100), 270 (18), 241 (12), 120 (23), HRMS (EI): calcd. for C19H12ClNO (M+) 305.0607, found 305.0611. UV-Vis (EtOH) λmax: 469, 358, 283, 244 nm. IR (film) νmax: 3092, 1630, 1581, 1510, 1139, 830cm−1. Mp: 133–135 °C.
3.2.7. Synthesis of 1-Oxo-1H-phenalene-9-carbonitrile (18) and Its Derivative (19)
- Synthesis of 1-oxo-1H-phenalene-9-carbonitrile (18).
To a solution of 1-oxo-1H-phenalene-9-trifluoromethylsulfonate (8) (200 mg, 0.609 mmol), CuI (60 mg, 0.318 mmol), and Pd(PPh3)4 (106 mg, 0.092 mmol) in DCM (4 mL), NaCN (60 mg, 1.223 mmol) was added, and the mixture was heated under reflux for 24 h. After this time, the mixture was filtered over Celite®, a saturated solution of NH4Cl (10 mL) was added, the mixture was extracted with CH2Cl2 (3 × 5 mL), dried (Na2SO4), filtered, and the solvent was removed using a rotary evaporator. The crude product was purified by column chromatography on silica gel (hexane/CH2Cl2, 9:1 to 2:3) to give product 18 (118 mg, 95%) as an orange solid. 1H NMR (500 MHz, CDCl3) δ: 8.28 (1H, d, J = 8.3 Hz, H-7), 8.06 (1H, d, J = 8.4 Hz, H-6), 8.02 (1H, d, J = 8.03 Hz, H-3), 7.84 (1H, d, J = 6.6 Hz, H-4), 7.75 (1H, dd, J = 8.7, 10, H-5), 7.73 (1H, d, J = 8.3 Hz, H-8), 6.79 (1H, d, J = 9.8 Hz, H-2). Mp: 172–175 °C.
- Synthesis of 1-oxo-1H-phenalene-9-carboxamide (19).
Concentrated H2SO4 (2 mL) was added to a solution of 1-oxo-1H-phenalene-9-carbonitrile (18) (30.0 mg, 0.146 mmol) in AcOH (2 mL) and heated for 1 h and 30 min at 40 °C. H2O (10 mL) was added to the reaction mixture, which was extracted with EtOAc (3 × 8 mL), washed with 1N NaOH solution until pH = 7–8, dried (Na2SO4), filtered, and the solvent was removed on a rotary evaporator. The crude oil was purified by preparative silica gel plate chromatography (hexane/CH2Cl2, 1:4) to give product 19 (18 mg, 55%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 8.25 (1H, d, J = 8.2 Hz, H-7), 8.04 (1H, d, J = 8.2 Hz, H-6), 7.81 (1H, d, J = 7.0 Hz, H-4), 7.74 (2H, d, J = 9.1 Hz, H-8, H-3), 7.65 (1H, dd, J = 7.1, 8.1 Hz, H-5), 6.71 (1H, d, J = 9.7, H-2), 5.84 (2H, s, NH2). 13C NMR (125 MHz, CDCl3) δ: 185.1 (C=O), 141.9 (C, CONH2), 139.9 (C, C-9), 135.2 (CH), 132.6 (CH), 132.5 (CH), 132.0 (2 × C), 129.0 (CH), 128.3 (C), 127.5 (2 × C), 126.5 (CH), 125.2 (C). EIMS: m/z 223 (M+, 100), 207 (80), 271 (28), 195 (35), 180 (30), 152 (49), 151 (68). HRMS (EI): calcd. for C14H9NO2 (M+) 223.0633, found 223.0638. UV-Vis (EtOH) λmax: 391, 359, 247 nm. IR (film) νmax: 3378, 3262, 1610, 1390, 1264, 847 cm−1. Mp: 242–244 °C.
3.2.8. Synthesis of 1-Oxo-1H-phenalene-2-carbonitrile (20) and Its Derivative (21)
- Synthesis of 1-oxo-1H-phenalene-2-carbonitrile (20).
To a solution of 2-iodo-1H-phenalen-1-one (3) (100.0 mg, 0.327), anhydrous CuI (30 mg, 0.163 mmol), and Pd(PPh3)4 (57.0 mg, 0.049 mmol) in anhydrous DCM (2 mL), finely ground NaCN (32 mg, 0.654 mmol) was added and the mixture was refluxed for 5 h under an inert atmosphere. After this time, the mixture was filtered over Celite®, a saturated NH4Cl solution (10 mL) was added, the mixture was extracted with CH2Cl2 (3 × 5 mL), dried (Na2SO4), filtered, and the solvent was removed on a rotary evaporator. The crude oil was purified by silica gel column chromatography (hexane/CH2Cl2, 9:1 to 7:3) to obtain product 20 (45.0 mg, 67%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 8.73 (1H, dd, J = 1.2, 7.4 Hz, H-9), 8.32 (1H, dd, J = 2.1, 7.0 Hz, H-7), 8.32 (1H, s, H-3), 8.27 (1H, dd, J = 0.8, 8.3 Hz, H-6), 7.96 (1H, dd, J = 0.8, 7.1 Hz, H-4), 7.87 (1H, t, J = 7.2 Hz, H-8), 7.71 (1H, dd, J = 7.1, 8.2 Hz, H-5). 13C NMR (125 MHz, CDCl3) δ: 179.8 (C=O), 150.1 (CH), 136.4 (CH), 135.6 (CH), 134.7 (CH), 132.4 (CH), 132.3 (C), 128.4 (C), 128.1 (CH), 127.8 (C), 127.4 (C), 125.7 (C), 115.4 (C), 114.9 (C). EIMS: m/z 206 (15), 205 (M+, 100), 177 (50), 88 (9). HRMS (EI): calcd. for C14H7NO (M+) 205.0528, found 205.0530. UV-Vis (EtOH) λmax: 397, 365, 253 nm. IR (film) νmax: 2226, 1640, 1582, 1357, 1257, 845 cm−1. Mp: 225–227 °C.
- Synthesis of N-(1-oxo-1H-phenalene-2-yl)carboxamide (21).
To a solution of 1-oxo-1H-phenalene-2-carbonitrile (20) (34.0 mg, 0.165 mmol) in AcOH (2 mL) was added H2SO4 (2 mL) and heated for 2 days at 40 °C. The crude product was poured onto an ice-water mixture, and the precipitate was filtered, washed with water and 5% Na2CO3 solution until the filtrate reached a pH = 6–7, to obtain product 21 (36.0 mg, 98%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 9.24 (1H, br s, NH), 8.97 (1H, s, H-3), 8.71 (1H, d, J = 7.4 Hz, H-9), 8.26 (1H, d, J = 7.9 Hz, H-7), 8.16 (1H, d, J = 8.3 Hz), 8.02 (1H, d, J = 7.0 Hz), 7.83 (1H, dd, J = 7.6, 7.8 Hz, H-5), 7.67 (1H, dd, J = 7.5, 7.8 Hz), 5.98 (1H, br s, NH). 13C NMR (125 MHz, CDCl3) δ: 184.7 (C=O), 165.7 (C), 148.6 (CH), 135.9 (CH), 135.5 (CH), 134.6 (CH), 132.1 (CH), 130.0 (C), 129.9 (C), 128.3 (C), 127.8 (C), 127.7 (CH), 127.3 (CH), 126.4 (C). EIMS: m/z 223 (M+, 100), 207 (52), 180 (61), 151 (50). HRMS (EI): calcd. for C14H9NO2 (M+) 223.0633, found 223.0633. UV-Vis (EtOH) λmax: 390, 252 nm. IR (film) νmax: 3347, 1695, 1574, 697cm−1. Mp: 186–188 °C.
3.2.9. Synthesis of 2-Arylamino-1H-phenalen-1-one Derivatives: General Procedure
Arylamine was added to a solution of 2-bromo-1H-phenalen-1-one (2), NaOtBu, Pd(PPh3)4 in anhydrous toluene. The reaction mixture was heated to 80 °C with stirring for 2 h–4 days in the dark under an argon atmosphere. After this time, CH2Cl2 (10 mL) was added and the mixture was filtered over Celite®. The mixture was then concentrated under vacuum to dryness on a rotary evaporator, and the crude product was purified by column chromatography or preparative plate chromatography on silica gel.
- Synthesis of 2-[(4-chloroanilino]-1H-phenalen-1-one (22).
From 2-bromo-1H-phenalen-1-one (2) (101.2 mg, 0.391 mmol), NaOtBu (53.8 mg, 0.560 mmol), Pd(PPh3)4 (46 mg, 0.04 mmol), anhydrous toluene (1.5 mL) and 4-chloroaniline (63.7 mg, 0.5 mmol); it was heated at 80 °C for 17 h; column chromatography (hexane/EtOAc, 95:5); product 22 (56.4 mg, 47%) was obtained as a dark red solid. 1H NMR (400 MHz, CDCl3) δ: 8.75 (1H, dd, J = 0.8, 7.2 Hz, H-9), 8.24 (1H, d, J = 8 Hz, H-7), 7.85 (1H, d, J = 7.7 Hz), 7.80 (1H, dd, J = 7.6, 8.0 Hz), 7.60–7.53 (2H, m), 7.36 (2H, d, J =8.8 Hz, H-3’, H-5’), 7.28–7.23 (4H, m, H-3, H-2’, H-6’, NH). 13C HMR (100 MHz, CDCl3) δ: 180.28 (C=O), 139.56 (C, C-1’), 137.51 (C, C-2), 136.21 (CH), 132.06 (C), 131.11 (CH), 129.64 (2 × CH, C-3’, C-5’), 129.08 (C), 128.85 (CH), 128.45 (CH), 127.92 (C), 127.46 (C), 127.37 (CH), 126.87 (CH), 124.16 (C), 121.64 (2 × CH, C-2’, C-6’), 109.80 (C, C-3). EIMS: m/z 307 (34), 306 (23), 305 (M+, 100), 135 (25), 68 (20). HRMS (EI): calcd. for C19H12ClNO (M+) 305.0607, found 305.0601. Mp: 182–183 °C.
- Synthesis of 2-(4-methoxyanilino)-1H-phenalen-1-one (23).
From 2-bromo-1H-phenalen-1-one (2) (105.8 mg, 0.408 mmol), NaOtBu (51.9 mg, 0.540 mmol), Pd(PPh3)4 (45.8 mg, 0.039 mmol), anhydrous toluene (1.5 mL) and 4-methoxyaniline (61.3 mg, 0.498 mmol); it was heated at 80 °C for 17 h; column chromatography (toluene/EOAc, 95:5); product 23 (77.0 mg, 63%) was obtained as a dark red solid. 1H NMR (400 MHz, CDCl3) δ: 8.72 (1H, dd, J = 1.0, 7.4 Hz, H-9), 8.19 (1H, d, J = 8 Hz, H-7), 7.76–7.73 (2H, m), 7.49–7.48 (2H, m), 7.24 (2H, d, J = 8.8 Hz, H-3’, H-5’), 7.05 (1H, s, H-3), 7.0 (1H, br s, NH), 6.97 (2H, dd, J =1.9, 8.8 Hz, H-2’, H-6’), 3.83 (3H, s, OCH3). 13C NMR (100 MHz, CDCl3) δ: 180.6 (C=O), 155.9 (C, C-4’), 139.2 (C), 135.9 (CH), 133.7 (C), 132.0 (C), 130.8 (CH), 129.6 (C), 128.1 (CH), 128.0 (C), 127.7 (CH), 127.3 (CH), 126.7 (CH), 123.9 (C), 123.6 (2 × CH, C-3’, C-5’), 114.8 (2 × CH, C-2’, C-6’), 107.7 (CH, C-3), 55.69 (CH3, OCH3). EIMS: m/z 302 (22), 301 (M+, 100), 286 (44). HRMS (EI): calcd. for C20H15NO2 (M+) 301.1103, found 301.1092. Mp: 137–139 °C.
- Synthesis of 2-((2,6-dimethylphenyl)amino)-2,3-dihydro-1H-phenalen-1-one (24).
From 2-bromo-1H-phenalen-1-one (2) (104.9 mg, 0.405 mmol), NaOtBu (53.3 mg, 0.555 mmol), Pd(PPh3)4 (47.5 mg, 0.041 mmol), anhydrous toluene (1.5 mL) and 2,6-dimethylaniline (58 μL, 0.463 mmol); it was heated at 80 °C for 4 days; column chromatography (toluene/EOAc, 95:5); product 24 (32.3 mg, 28%) was obtained as a red oil. 1H NMR (400 MHz, CDCl3) δ: 8.78 (1H, dd, J = 0.7, 7.2 Hz, H-9), 8.21 (1H, d, J = 8 Hz, H-7), 7.79 (1H, t, J = 7.6 Hz, H-5), 7.76 (1H, d, J = 7.5 Hz, H-6), 7.47 (1H, dd, J = 7.3, 8.0 Hz, H-8), 7.41 (1H, d, J = 7.0 Hz, H-4), 7.19 (3H, br s, H-3’, H-4’, H-5’), 6.61 (1H, br s, NH), 6.12 (1H, s, H-3), 2.28 (6H, s, CH3). 13C NMR (100 MHz, CDCl3) δ: 180.5 (C=O), 139.8 (C, C-1’), 136.9 (C, C-2), 136.4 (CH), 135.9 (CH), 132.1 (C), 130.7 (CH), 129.9 (C), 128.7 (CH, 2 × C, C-3’, C-5’), 128.3 (C), 127.9 (CH), 127.5 (CH), 127.3 (CH), 126.7 (C, CH, 2 × C), 123.9 (C), 107.5 (CH, C-3), 18.4 (CH3, 2 × C). EIMS: m/z 299 (M+, 100), 282 (44), 141 (29). HRMS (EI): calcd. for C21H17NO (M+) 299.1310, found 299.1310.
- Synthesis of 2-(pyrimidin-2-ylamino)-1H-phenalen-1-one (25).
From 2-bromo-1H-phenalen-1-one (2) (50.7 mg, 0.196 mmol), NaOtBu (28.1 mg, 0.292 mmol), Pd(PPh3)4 (26.2 mg, 0.023 mmol), anhydrous toluene (0.75 mL) and pyrimidin-2-amine (25.6 mg, 0.269 mmol); it was heated at 80 °C for 3 h; column chromatography (hexane/EtOAc, 3:2); product 25 (17.2 mg, 33%) was obtained as a red solid. 1H NMR (400 MHz, CDCl3) δ: 8.86 (1H, s, H-3), 8.73 (1H, dd, J = 1.0, 7.4 Hz, H-9), 8.58 (1H, br s, NH), 8.53 (2H, d, J = 4.8 Hz, H-4’, H-6’), 8.21 (1H, dd, J = 0.6, 8.0 Hz, H-7), 7.88 (1H, d, J = 8.2 Hz), 7.79–7.74 (2H, m), 7.57 (1H, dd, J = 7.3, 8.0 Hz), 6.79 (1H, t, J = 4.8 Hz, H-5’). 13C NMR (100 MHz, CDCl3) δ: 179.7 (C=O), 159.9 (CH, C-2’), 158.0 (2 × CH, C-4’, C-6’), 135.9 (CH), 134.4 (C, C-2’), 132.0 (C), 131.1 (CH), 130.7 (CH), 129.4 (CH), 128.9 (C), 127.8 (C), 127.3 (CH), 126.8 (CH), 124.7 (C), 119.1 (CH), 113.3 (CH, C-3). EIMS: m/z 274 (18), 273 (M+, 100), 272 (54), 244 (14). HRMS (EI): calcd. for C17H11N3O (M+) 273.0902, found 273.0893. UV-Vis (EtOH) λmax: 457, 365, 282 nm. IR (film) νmax: 3349, 1622, 1564, 1519, 1395, 842 cm−1. Mp: 181–183 °C.
- Synthesis of naphtho[1,8-ab]phenoxazine (26).
From 2-bromo-1H-phenalen-1-one (2) (105.2 mg, 0.406 mmol), NaOtBu (107.6 mg, 1.12 mmol), Pd(PPh3)4 (88 mg, 0.078 mmol), anhydrous toluene (1.5 mL) and 2-aminophenol (56.1 mg, 0.514 mmol); it was heated at 80 °C for 4 days; column chromatography (toluene/EtOAc, 9.5:0.5); product 26 (11.3 mg, 10%) was obtained as a red solid. 1H NMR (500 MHz, CDCl3) δ: 8.71 (1H, d, J = 7.5 Hz, H-1), 7.92 (1H, d, J = 8.0 Hz, H-3), 7.59–7.62 (2H, m, H-2, H-4), 7.38 (1H, dd, J = 1.2, 7.7 Hz), 7.35 (1H, t, J = 7.3 Hz, H-5), 7.24 (1H, m, H-6), 7.12 (1H, m), 7.03 (1H, m), 6.87 (1H, dd, J = 0.9, 7.9 Hz), 6.46 (1H, s, H-7). 13C NMR (125 MHz, CDCl3) δ: 152.7 (C, C-13a), 146.1 (C), 145.1 (C, C-7a), 135.0 (C), 133.3 (C, C-3a), 132.3 (CH, C-3), 131.4 (C, C-6a), 130.4 (C, C-13b), 128.8 (CH), 128.2 (CH), 127.1 (2 × CH), 127.0 (C, C-13c), 126.9 (CH), 125.5 (CH, C-1), 125.2 (CH, C-6), 124.1 (CH), 114.8 (CH), 108.5 (CH, C-7). EIMS: m/z 270 (M++1, 21) 269 (M+, 100), 240 (13), 134 (18). HRMS (EI): calcd. for C19H11NO (M+) 269.0841, found 269.0842. UV-Vis (EtOH) λmax: 524, 491, 269, 237 nm. IR (film) νmax: 1732, 1281, 824 cm−1. Mp: 168–170 °C.
- Synthesis of 7H-naphtho[1,8-bc]acridin-7-one (27).
From 2-bromo-1H-phenalen-1-one (2) (51.5 mg, 0.168 mmol), NaOtBu (23.0 mg, 0.239 mmol), Pd(PPh3)4 (19.6 mg, 0.017 mmol), anhydrous toluene (0.75 mL) and (2-aminophenyl)methanol (24.2 mg, 0.196 mmol); it was heated at 80 °C for 20 h; preparative plate chromatography (toluene/EtOAc, 9.8:0.2); product 27 (6.0 mg, 12%) was obtained as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 9.36 (1H, dd, J = 1.1, 7.3 Hz, H-1), 9.28 (1H, s, H-8), 8.79 (1H, d, J = 1.3, 7.3 Hz, H-6), 8.29 (1H, dd, J = 1.1, 8.1 Hz, H-4), 8.25 (1H, dd, J = 0.7, 8.5 Hz, H-12), 8.14 (1H, dd, J = 0.9, 8.1 Hz, H-3), 8.06 (1H, m, J = 7.0 Hz, H-9), 7.87 (1H, ddd, J = 1.5, 6.8, 8.4 Hz, H-11), 7.83 (1H, dd, J = 7.6, 7.9 Hz, H-2), 7.81 (1H, dd, J = 7.4, 7.9 Hz, H-5), 7.61 (1H, ddd, J = 1.1, 6.8, 8.0 Hz, H-10). 13C NMR (125 MHz, CDCl3) δ: 184.2 (C, C-7), 152.4 (C, C-13a), 150.4 (C, C-12a), 137.8 (CH, C-8), 135.6 (CH, C-4), 133.2 (C, C-6a), 132.4 (CH, C-11), 131.7 (CH, C-3), 129.9 (CH), 129.8 (CH), 129.7 (CH, C-9), 129.6 (C, C-13c), 128.2 (2 × C, C-3a, C-13b), 127.6 (C, 8a), 127.3 (CH, C-1), 127.2 (CH, C-2), 127.2 (CH, C-10), 126.5 (CH, C-5), 125.1 (C, C-7a). EIMS: m/z 311 (15), 305 (30), 304 (M+ + Na, 100), 301 (50). HRMS (ESI): calcd. for C20H11NO (M+ + Na) 304.0738, found 304.0743. Mp: 221–224 °C.
3.2.10. Synthesis of 9-Hydroxy-1H-phenalen-1-one Derivatives
- Synthesis of 2,7-dimethoxynaphthalene (29).
Diazomethyl(trimethyl)silane (2.03 mL, 4.018 mmol) was slowly added to a solution of 7-methoxynaphthalene-2-ol (28) (100.0 mg, 0.574 mmol) in MeOH (2 mL) and stirred for 6 h at room temperature. The resulting precipitate was gravity filtered to give product 29 (95.2 mg, 88%) as a white solid. 1H NMR (400 MHz, CDCl3) δ: 7.68 (2H, d, J = 8.8 Hz, H-4, H-5), 7.09 (2H, s, H-1, H-8), 7.03 (2H, d, J = 2.0, 8.8 Hz, H-3, H-6), 3.92 (6H, s, OCH3). EIMS: m/z 189 (21), 145 (51), 188 (M+, 100). HRMS (EI): calcd. for C12H12O2 (M+) 188.0737, found 188.0754. Mp: 135–137 °C.
- Synthesis of 4,9-dihydroxy-1H-phenalen-1-one (30).
To a solution of 2,7-dimethoxynaphthalene (29) (30.0 mg, 0.159 mmol) and (2E)-3-phenylacryloyl chloride (27.0 g, 159 mmol) in DCE (1 mL) was added AlCl3 (48 mg, 0.318 mmol) portionwise, and the mixture was irradiated with MW for 60 min at 130 °C. Additional DCE (1 mL) and AlCl3 (25 mg, 0.159 mmol) were then added, and the mixture was irradiated with MW for 30 min at 130 °C. The reaction crude was concentrated in a rotary evaporator, cooled in an ice bath, and a cold 1N HCl solution (1 mL) was added. The aqueous phase was extracted with EtOAc (3 × 15 mL). The organic phase was washed with a 1% NaHCO3 solution (3 × 15 mL) and with H2O (10 mL). On the other hand, the aqueous phase was acidified to pH = 2–3 and extracted with EtOAc (3 × 15 mL). The organic phase was dried (MgSO4), filtered, and the solvent was removed in a rotary evaporator. The crude was purified by silica gel column chromatography (hexane/EtOAc, 50–70%, EtOAc, acetone) to obtain product 30 (24.5 mg, 73%) as a yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ: 10.57 (1H, br s, OH), 8.62 (1H, d, J = 9.6 Hz, H-3), 8.19 (1H, d, J = 9.2 Hz), 8.09 (1H, d, J = 8.4 Hz), 7.25 (1H, d, J = 8.4 Hz), 7.08 (1H, d, J = 9.6 Hz, H-2), 6.99 (1H, d, J = 9.2 Hz), 3.20 (br s, OH). EIMS: m/z 212 (M+, 100), 105 (89), 51 (15). HRMS (EI): calcd. for C13H8O3 (M+) 212.0473, found 212.0468. Mp: 240 °C.
- Synthesis of ethyl 3-hydroxynaphthalene-2-carboxylate (32).
Concentrated H2SO4 (0.1 mL) was added to a solution of 3-hydroxy-2-naphthoic acid (31) (1 g, 5.3 mmol) in EtOH (10 mL). The reaction mixture was heated under reflux for 24 h. The solvent was then removed, dissolved in a hexane/EtOAc mixture (10 mL, 2:1), and washed with H2O, saturated NaHCO3 solution, and saturated NaCl solution. The mixture was then dried (MgSO4), filtered, and concentrated under vacuum to give 32 (800 mg, 70%) as a white solid. 1H NMR (400 MHz, CDCl3) δ: 11.92 (1H, br s, OH), 10.87(1H, s, H-1), 9.09 (1H, d, J = 8.6 Hz), 8.58 (1H, s, H-4), 7.76 (2H, dt, J = 0.4, 6.5 Hz), 7.67–7.64 (1H, cd, J = 1.1, 5.5 Hz), 7.41–7.38 (1H, cd, J = 0.8, 5.5 Hz), 4.49 (2H, c, J = 7.1 Hz, CH2), 1.48 (3H, t, J = 7.1 Hz, CH3). EIMS: m/z 216 (M+, 45), 171 (20), 169 (100), 142 (45), 114 (19). HRMS (EI): calcd. for C13H12O3 (M+) 216.0786, found 216. 0782. Mp: 85–88 °C.
- Synthesis of ethyl 9-hydroxy-1-oxo-1H-phenalene-8-carboxylate (33).
To a solution of ethyl 3-hydroxynaphthalene-2-carboxylate (32) (101 mg, 0.463 mmol) and (2E)-3-phenylacryloyl chloride (77.0 g, 463 mmol) in DCE (2 mL) was added AlCl3 (123 mg, 0.926 mmol) slowly, and the mixture was stirred at room temperature for 1 h. After this time, the mixture was heated under reflux for 2 h. The reaction mixture was then cooled and DCE (1 mL) and AlCl3 (62 mg, 0.463 mmol) were added, followed by reflux for 4 h. The crude product was concentrated on a rotary evaporator, cooled in an ice bath, and cold 1N HCl (1 mL) was added. The mixture was then lyophilized, the solid dissolved, and filtered with hot EtOAc. The crude product was purified by silica gel column chromatography (hexane/EtOAc, 10–80%) to yield 3 products: ethyl 4-hydroxy-3-oxo-1-phenyl-2,3-dihydro-1H-phenalene-5-carboxylate (7.1 mg, 4%), Ethyl 9-hydroxy-1-oxo-3-phenyl-1H-phenalene-8-carboxylate (2.1 mg, 1.3%), and product 33 (33 mg, 33%) as orange oils. Ethyl 4-hydroxy-3-oxo-1-phenyl-2,3-dihydro-1H-phenalene-5-carboxylate: 1H NMR (500 MHz, CDCl3) δ: 13.98 (1H, s, OH), 8.65 (1H, s, H-6), 7.77 (1H, d, J = 7.9 Hz), 7.36–7.27 (5H, m), 7.16 (2H, d, J = 7.2 Hz), 4.66 (1H, t, J = 7.2 Hz, H-1), 4.48 (2H, c, J = 7.1, Hz, H-1’), 3.28 (2H, d, J = 6.8 Hz, H-2), 1.46 (3H, t, J = 7.1 Hz, H-2’). 13C NMR (125 MHz, CDCl3) δ: 202.8 (C), 165.1 (C), 161.6 (C), 142.5 (C), 141.6 (CH), 134.11 (C), 133.3 (C), 130.0 (CH), 129.1 (2 × CH), 128.5 (CH), 127.9 (2 × CH), 127.5 (CH), 126.1 (C), 124.9 (CH), 121.4 (C), 111.2 (C), 61.7 (CH2), 44.91 (CH), 44.5 (CH2), 14.5 (CH3). EIMS: m/z 346 (M+, 100), 301 (27), 300 (43), 224 (25), 215 (19), 196 (14), 139 (14). HRMS (EI): calcd. for C22H18O4 (M+) 346.1205, found 346.1203. UV-Vis (EtOH) λmax: 378, 328, 234 nm. IR (film) νmax: 2983, 1729, 1631, 1207, cm−1. Ethyl 9-hydroxy-1-oxo-3-phenyl-1H-phenalene-8-carboxylate: 1H NMR (400 MHz, CDCl3) δ: 8.82 (1H, s, H-7), 8.15 (2H, d, J = 7.9 Hz, H-6, H-4), 7.60 (1H, t, J = 7.8 Hz, H-5), 7.56–7.51 (5H, m, J = 7.24 Hz, phenyl), 4.51 (2H, c, J = 7.12 Hz, H-1’), 1.48 (3H, t, J = 7.12 Hz, H-2’). 13C NMR (125 MHz, CDCl3) δ: 179 0, 176.8, 164.9, 153.9, 145.7, 138.1, 135.0, 134.4, 129.8 (2 × C), 129.0, 128.7 (3 × C), 125.6, 125.2, 124.5, 124.4, 123.7, 111.6, 61.7 (C-1’), 14.6 (C-2’). EIMS: m/z 344 (M+, 60), 299 (33), 272 (100), 270 (20), 215 (20), 213 (21). HRMS (EI): calcd. for C22H16O4 (M+) 344.1049, found 344.1047. UV-Vis (EtOH) λmax: 450, 425, 356, 256 nm. IR (film) νmax: 2921, 1719, 1625, 1200, 1110, 1025 cm−1. Compound 33: 1H NMR (500 MHz, CDCl3) δ: 8.72 (1H, s, H-7), 8.09 (1H, d, J = 9.2 Hz, H-3), 8.09–8.08 (2H, m), 7.62 (1H, t, J = 7.6 Hz, H-5), 7.20 (1H, d, J = 9.2 Hz, H-2), 4.48 (2H, c, J = 7.2 Hz, H-1’), 1.46 (3H, t, J = 7.1 Hz, H-2’). 13C NMR (125 MHz, CDCl3) δ: 178.8 (C), 177.7 (C), 164.8 (C), 145.5 (CH), 140.9 (CH), 135.0 (CH), 134 8 (H), 127.9 (C), 125.5 (2 × C), 124.6 (CH), 124.0 (C), 123.5 (CH), 111.7 (C), 61.6 (CH2), 14.5 (CH3). EIMS: m/z 268 (M+, 64), 223 (52), 222 (71), 196 (100), 194 (29), 139 (36), 138 (15). HRMS (EI): calcd. for C16H12O4 (M+) 268.0736, found 268.0742. UV-Vis (EtOH) λmax: 446, 421, 354, 252 nm. IR (film) νmax: 3333, 1630, 1001, 845 cm−1.
- Synthesis of 9-hydroxy-1-oxo-1H-phenalene-8-carboxylic acid (34).
To a solution of the 33 (2.31 g, 7.77 mmol) in MeOH (1mL) and H2O (0.2 mL) was added NaOH (4 mg, 0.1 mmol). The mixture was stirred at rt for 8 h. The mixture was concentrated and the crude was diluted with H2O (5 mL) and acidified with an aq solution of 2M HCl (1 mL). This aqueous mixture was extracted with EtOAc (2 × 5 mL) and the combined organic extracts were washed with brine (10 mL), dried with MgSO4, and concentrated under reduce pressure to provide the product 34 (13 mg, 74%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 14.87 (1H, s, OH), 13.5 (1H, br s, OH), 9.24 (1H, d, J = 1.8 Hz, H-7), 8.37 (1H, d, J = 9.1 Hz, H-3), 8.32 (2H, d, J = 8.0 Hz, H-6, H-4), 7.79 (1H, t, J = 7.7 Hz, H-5), 7.47 (1H, d, J = 9.1 Hz, H-2). 13C NMR (125 MHz, CDCl3) δ: 185.7 (C, COOH), 172.8 (C, C-1), 165.3 (C, C-9), 149.9 (CH), 142.5 (CH), 137.7 (CH), 137.1 (CH), 127.8 (C), 125.9 (C), 125.5 (CH), 124.1 (C), 122.8 (C), 121.2 (CH), 111.1 (C). EIMS: m/z 240 (M+, 49), 222 (22), 196 (100), 194 (25), 168 (25), 83 (13). HRMS (EI): calcd. for C14H8O4 (M+) 240.0423, found 240.0417. UV-Vis (EtOH) λmax: 443, 351, 236 nm. IR (film) νmax: 3452, 2401, 1738, 1628, 1575, 1405 cm−1. Mp: 242–244 °C.
3.2.11. Synthesis of 4H-Benzo[de]isoquinolin-4-ones
- Synthesis of 4H-benzo[de]isoquinolin-4-one (35).
A mixture of 2-methoxynaphthalene (316 mg, 1.98 mmol) and triazine (180 mg, 2.22 mmol) in PPA (6–8 g) was stirred at 40–45 °C for 1 h and then at 100–110 °C for 3 h. The reaction mixture was poured into cold H2O (60 mL) with vigorous stirring. The resulting mixture was extracted with EtOAc (3 × 50 mL), dried (MgSO4), filtered, and the solvent was removed under vacuum. The residue was purified by column chromatography on silica gel (CH2Cl2) to give 2-methoxy-1,6-naphthalenedicarbaldehyde (38 mg, 9%) as a pale yellow solid. The aqueous phase was treated with ammonia until pH = 8–9 and, after cooling, the oil was extracted with EtOAc (3 × 50 mL). The organic phase was dried (MgSO4), filtered and the solvent was removed under vacuum and the residue was purified by column chromatography on silica gel (CH2Cl2) to give product 35 (41 mg, 11%) as a yellow solid. Compound 35: 1H NMR (600 MHz, CDCl3) δ: 9.53 (1H, s), 9.52 (1H, s), 8.15 (1H, d, J = 8.4 Hz), 7.93 (1H, d, J = 7.2 Hz), 7.78 (1H, d, J = 9.6 Hz, H-5), 7.72 (1H, dd, J = 7.6, 7.7 Hz, H-8), 6.74 (1H, d, J = 10.2 Hz, H-6). 13C NMR (150 MHz, CDCl3) δ: 185.6 (C), 157.1 (CH), 147.4 (CH), 141.0 (CH), 134.0 (CH), 130.9 (CH), 130.6 (C), 130.4 (CH), 128.4 (CH), 127.4 (C), 127.0 (C), 122.6 (C). EIMS: m/z 181 (M+, 100), 153 (58), 126 (15), 76 (15), 636 (17). HRMS (EI): calcd. for C12H7NO (M+) 181.0528, found 181.0520. UV-Vis (EtOH) λmax: 383, 340, 324, 252, 245, 232 nm. IR (film) νmax: 1638, 1574, 515, 495, 470 cm−1. Mp: 170–172 °C. 2-Methoxy-1,6-naphthalenedicarbaldehyde: 1H NMR (400 MHz, CDCl3) δ: 10.81 (1H, s), 10.05 (1H, s), 9.30 (1H, d, J = 8.9 Hz, H-8), 8.20 (1H, d, J = 1.2 Hz, H-5), 8.14 (1H, d, J = 9.2 Hz), 8.0 (1H, dd, J = 1.6, 8.9 Hz, H-7), 7.35 (1H, d, J = 9.8 Hz), 4.04 (3H, s). EIMS: m/z 214 (M+, 100), 213 (47), 185 (16), 154 (18). HRMS (EI): calcd. for C13H10O3 (M+) 214.0630, found 214.0620. IR (film) νmax: 2873, 1660, 1179, 1057, 506, 490 cm−1. Mp: 156–159 °C.
- Synthesis of 9-methoxy-4H-benzo[de]isoquinolin-4-one (36).
A mixture of 2,7-dimethoxynaphthalene (29) (100 mg, 0.53 mmol) and triazine (47 mg, 0.58 mmol) in APP (3–4 g) was stirred at 40–45 °C for 1 h and then at 70–90 °C for 3 h. The reaction mixture was poured into cold H2O (20 mL) with vigorous stirring. The resulting mixture was extracted with EtOAc (3 × 25 mL), dried (MgSO4), filtered, and the solvent was removed under vacuum. The aqueous phase was treated with ammonia until pH = 8–9 and, after cooling, the oil was extracted with EtOAc (3 × 25 mL). The organic phase was dried (MgSO4), filtered and the solvent was removed under vacuum and the residue was purified by silica gel column chromatography (CH2Cl2), to give product 36 (90 mg, 80%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ: 9.75 (1H, s), 9.48 (1H, s), 7.75 (1H, d, J = 8.1 Hz), 7.62 (1H, d, J = 9.7 Hz, H-5), 6.87 (1H, d, J = 8.1 Hz), 6.56 (1H, d, J = 9.7 Hz, H-6), 4.09 (3H, s, CH3). 13C NMR (125 MHz, CDCl3) δ: 185.5 (C, C-4), 160.5 (C), 152.1 (CH), 148.1 (CH), 141.1 (CH), 136.2 (CH), 131.4 (C), 127.5 (CH), 122.3 (C), 122.3 (C), 119.8 (C), 119.7 (C), 106.1 (CH), 56.4 (CH3, OCH3). EIMS: m/z 212 (14), 211 (M+, 100), 168 (17), 140 (27). HRMS (EI): calcd. for C13H9NO2 (M+) 211.0633, found 211.0626. UV-Vis (EtOH) λmax: 431, 278, 256, 230 nm. IR (film) νmax: 3336, 1644, 1581, 1236, 837, 489 cm−1. Mp: 202–204 °C.
- Synthesis of ethyl 4-oxo-4H-benzo[de]isoquinoline-5-carboxylate (37).
A mixture of ethyl 3-hydroxy-2-naphthoate (32) (800 mg, 3.7 mmol) and triazine (329 mg, 4.1 mmol) in APP (8–9 g) was stirred at 40–45 °C for 1 h and then at 100–110 °C for 4 h. The reaction mixture was poured into cold H2O (80 mL) with vigorous stirring. The resulting mixture was extracted with EtOAc (3 × 60 mL), dried (MgSO4), filtered, and the solvent was removed under vacuum. The residue was purified by silica gel column chromatography (hexane/EtOAc, 1:2) to give ethyl 4-formyl-3-methoxy-2-naphthoate (38) (196 mg, 22%) as a pale yellow solid. The aqueous phase was treated with ammonia until pH = 8–9 and, after cooling, the oil was extracted with EtOAc (3 × 60 mL). The organic phase was dried (MgSO4), filtered and the solvent was removed under vacuum. The residue was purified by silica gel column chromatography (hexane/EtOAc, 1:2) to give product 37 (100 mg, 12%) as an orange oil. Compound 37: 1H NMR (500 MHz, CDCl3) δ: 9.58 (1H, s), 9.57 (1H, s), 8.45 (1H, s, H-6), 8.26 (1H, d, J = 8.3 Hz), 8.10 (1H, d, J = 7.0 Hz), 7.79 (1H, d, J = 7.6, 7.8 Hz, H-8), 4.45 (1H, c, J = 7.1 Hz), 1.44 (3H, t, J = 7.1 Hz). 13C NMR (125 MHz, CDCl3) δ: 181.2 (C, C-4), 164.9 (C), 157.0 (CH), 148.2 (CH), 144.9 (CH), 136.2 (CH), 132.2 (CH), 132.5 (CH), 131.4 (C), 130.7 (CH), 128.4 (CH), 126.7 (C), 125.7 (C), 123.0 (C), 61.7 (CH2), 14.3 (CH3). EIMS: m/z 209 (42), 208 (54), 181 (M+, 100), 180 (24), 152 (32), 148 (76), 57 (30). HRMS (EI): calcd. for C15H11NO3 (M+) 253.0739, found 253.0729. UV-Vis (EtOH) λmax: 389, 341, 326, 244, 223 nm. IR (film) νmax: 3024, 1218, 748, 672, 476 cm−1. Compound 38: 1H NMR (500 MHz, CDCl3) δ: 11.91 (1H, br s, OH), 10.87 (1H, s, CHO), 9.10 (1H, d, J = 8.6 Hz), 8.58 (1H, s, H-1), 7.76 (1H, d, J = 8.6 Hz), 7.66 (1H, ddd, J = 1.4, 6.9, 8.5 Hz), 7.40 (1H, ddd, J = 1.0, 6.9, 8.0 Hz), 4.49 (2H, c, J = 7.1 Hz), 1.49 (3H, t, J = 7.1Hz). EIMS: m/z 244 (M+, 35), 216 (28), 170 (100), 142 (34). HRMS (EI): calcd. for C14H12O4 (M+) 244.0736, found 244.0726. Mp: 140–142 °C.
- Synthesis of 5-bromo-1-methoxy-4H-benzo[de]isoquinolin-4-one (39).
To a solution of 35 (5 mg, 0.028 mmol) in MeOH (1 mL) was added NBS (5 mg, 0.028 mmol), and the reaction mixture was heated under reflux for 6 h. The solvent was removed under vacuum, and the residue was purified by preparative silica gel plate chromatography (hexane/EtOAc, 3:2) to give the products 40 (2 mg, 28%) as an orange oil and 39 (5 mg, 62%) as an orange solid. 1H NMR (500 MHz, CDCl3) δ: 9.24 (1H, s, H-3), 8.39 (1H, dd, J = 0.7, 8.7 Hz, H-9), 8.21 (1H, s, H-6), 7.15 (1H, d, J = 7.0 Hz, H-7), 7.64 (1H, dd, J = 8.1 Hz, H-8), 4.27 (3H, s, CH3). 13C NMR (125 MHz, CDCl3) δ: 177.8 (C, C-4), 165.1 (C, C-1), 150.1 (CH, C-3), 142.1 (CH, C-6), 133.0 (CH, C-7), 131.8 (C, C-9b), 127.8 (CH, C-9), 127.4 (CH, C-8), 127.3 (C, C-5), 127.0 (C, C-6a), 118.2 (C, C-3a), 117.1 (C, C-9a), 55.0 (CH3). EIMS: m/z 290 (M+, 100), 287 (98), 285 (35). HRMS (EI): calcd. for C13H8NO2Br (M+) 290.9716, found 290.9713. Mp: 220–223 °C.
- Synthesis of 5-bromo-4H-benzo[de]isoquinolin-4-one (40).
To a solution of 35 (6 mg, 0.033 mmol) in THF (0.7 mL) was added NBS (5 mg, 0.028 mmol) and the reaction mixture was heated under reflux for 6 h. The solvent was removed under vacuum and the residue was purified by preparative silica gel plate chromatography (hexane/EtOAc, 3:2) to give product 40 (3.3 mg, 38%) as an orange oil. 1H NMR (500 MHz, CDCl3) δ: 9.62 (1H, s), 9.58 (1H, s), 8.28 (1H, s, H-6), 8.21 (1H, dd, J = 0.7, 8.2 Hz), 7.94 (1H, d, J = 7.1 Hz), 7.75 (1H, dd, J = 8.3, 7.3 Hz, H-8). 13C NMR (125 MHz, CDCl3) δ: 178.7 (s, C-4), 157.5 (d), 149.0 (d), 142.4 (d), 133.9 (d), 131.3 (d), 129.8 (s), 128.7 (d), 127.5 (s), 127.3 (s), 127.0 (s), 122.0 (s). EIMS: m/z 260 (M+, 100), 258 (99), 180 (32), 152 (46), 125 (20). HRMS (EI): calcd. for C12H6NOBr (M+) 258.9633, found 260.9641. UV-Vis (EtOH) λmax: 399, 344, 328, 252, 237 nm. IR (film) νmax: 3019, 1214, 647, 466 cm−1.
- Synthesis of 5-bromo-9-methoxy-4H-benzo[de]isoquinolin-4-one (41).
To a solution of 36 (37 mg, 0.175 mmol) in THF (2 mL) was added NBS (31 mg, 0.175 mmol), and the reaction mixture was stirred at room temperature for 4 h. The solvent was removed under vacuum, and the residue was purified by silica gel column chromatography (CH2Cl2) to give product 41 (22 mg, 44%) as an orange solid. 1H NMR (500 MHz, CDCl3) δ: 9.83 (1H, s), 9.60 (1H, s), 8.12 (1H, s, H-6), 7.79 (1H, d, J = 8.1 Hz), 6.94 (1H, d, J = 8.1 Hz), 4.15 (3H, s, CH3). 13C NMR (125 MHz, CDCl3) δ: 178.5 (s, C-4), 161.0 (s), 152.5 (d), 149.6 (d), 142.4 (d), 136.4 (d), 130.6 (s), 123.6 (s), 121.6 (s), 119.9 (s), 119.6 (s), 106.5 (d), 56.5 (c, CH3). EIMS: m/z 290 (M+, 100), 289 (49), 288 (99), 287 (37), 261 (30), 259 (30). HRMS (EI): calcd. for C13H8NO2Br (M+) 290.9718, found 290.9715. UV-Vis (EtOH) λmax: 397, 351, 257, 224 nm. IR (film) νmax: 3016, 1214, 744, 668, 462 cm−1. Mp: 215–217 °C.
3.3. Biological Assays
- In vitro antimalarial assay.
FCR-3 chloroquine-resistant strain of P. falciparum (kindly provided by Dr. Fandeur, Pasteur Institute, Cayenne, France) was cultured [30] on glucose-enriched RPMI 1640 medium, supplemented with 10% human serum at 37 °C. After 24 h of incubation at 37 °C, the medium was replaced by fresh medium supplemented with the compound to be evaluated (initial a screening used a concentration of 10 μg/mL), and incubation was continued for a further 48 h. On the third day of the test, a blood smear was taken from each well, and parasitemia was calculated for each concentration of sample compared to the control [31]. Chloroquine (0.1 μg/mL) was used as a positive control. IC50 values were determined graphically by plotting concentrations (0.01–10 μg/mL) versus percent inhibition. All tests were performed in triplicate.
- Cytotoxicity study by MTT proliferation assay.
MCF-7 cells were purchased from ATCC (Manassas, VA, USA) and cultured in DMEN containing 10% FBS. For 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) (Sigma–Aldrich, St. Louis, MO, USA) analysis, cells were plated in 96-well plates at 10,000 cells/well. Twenty-four hours after plating, vehicle (0.1% DMSO, final concentration) or compound was added to cells at indicated doses. Seventy-two hours following compound addition, MTT was added to each well (0.5 mg/mL, final concentration) and plates were incubated for an additional 3 h at 37 °C. Medium was then aspirated and the formazan product was solubilized in SDS–HCl (20% SDS; HCl 0.02 M). The absorbance of each well was measured at 570 nm using a microplate reader (Sigma–Aldrich, St. Louis, MO, USA).
4. Conclusions
A diverse set of 1H-phenalen-1-one analogs was successfully synthesized using several complementary synthetic strategies. These compounds were designed to expand the chemical diversity of our 1H-phenalen-1-one collection and to assemble a focused library centered on this core scaffold. This library enables evaluation of how electronic and steric modifications influence both the chemical reactivity and biological activity of 1H-phenalen1-one derivatives. The inclusion of benzo[de]isoquinolin-4-one nitrogen analogs further enriches the structural scope. Additionally, the unexpected formation of phenoxazine and acrid-7-one systems was observed when 2-bromo-1H-phenalen-1-one was treated with selected anilines under cross-coupling conditions. Most compounds were obtained in moderate to good yields, with 23 identified as novel. Biological screening against the chloroquine-resistant Plasmodium falciparum strain FCR3 revealed three new antiplasmodial agents (compounds 2, 9-OH, and 18) with IC50 values below 1 µM, representing a significant improvement over previously reported phenalenone derivatives (compounds 1c and 1d). None of the active compounds displayed detectable cytotoxicity. Together with their favorable predicted physicochemical properties, these results support further exploration of structure–activity relationships (SAR) within this promising class and investigation into their mechanism of action. We also highlight that, to date, these constitute the only reports describing antiplasmodial activity for this structural family, offering a new scaffold for antimalarial drug discovery and one of particular interest to medicinal chemists working with phenalenone derivatives.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules30244667/s1, Supplementary Materials-S1: 1H and 13C NMR spectra; 19F NMR spectra of compound 6; 19F NMR spectra of compound 11; 1H NMR, 13C NMR, COSY, HSQC and HMBC spectra of compound 26; 1H NMR, 13C NMR, COSY, HSQC and HMBC spectra of compound 27; 1H NMR, 13C NMR, COSY, HSQC and HMBC spectra of compound 39; Scheme S1. Proposed mechanism for the formation of compounds 36 and 37; Supplementary Materials-S2: High-resolution mass spectra; Supplementary Materials-S3: UV-VIS spectra.
Author Contributions
Conceptualization, T.A.-G. and G.M.-S.; investigation, T.A.-G., G.M.-S. and M.B.F.; writing—original draft preparation, T.A.-G.; writing—review and editing, T.A.-G. and G.M.-S.; evaluation of the anti-plasmodial activity and cytotoxicity, D.G. and N.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Spanish Ministerio de Economía, Industria y Competitividad [Proyect SAF 2012-37344-C03-01] and by MAEC-AECID and project X.5 “Búsqueda, Obtención y Evaluación de Nuevos Agentes Antiparasitarios” CYTED (Iberoamerican Program of Science and Technology for Development), Subprogram.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article and Supplementary Materials.
Acknowledgments
T.A.-G. acknowledges support given by the Departamento de Química Orgánica de la ULL (Universidad de La Laguna).
Conflicts of Interest
Author Grant McNaughton-Smith was employed by the company Centro Atlántico del Medicamento S.A. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Echeverri, F.; Torres, F.; Quiñones, W.; Escobar, G.; Archbold, R. Phenylphenalenone phytoalexins, will they be a new type of fungicide? Phytochem. Rev. 2012, 11, 1–12. [Google Scholar] [CrossRef]
- Downum, K.R. Light Activated Plant Defence. New. Phytol. 1992, 122, 401–420. [Google Scholar] [CrossRef] [PubMed]
- Flors, C.; Nonell, S. Light and Singlet Oxygen in Plant Defense against Pathogens: Phototoxic Phenalenone Phytoalexins. Acc. Chem. Res. 2006, 39, 293–300. [Google Scholar] [CrossRef]
- Lazzaro, A.; Corominas, M.; Martí, C.; Flors, C.; Izquierdo, L.R.; Abad-Grillo, T.; Luis, J.G.; Nonell, S. Light- and singlet oxygen mediated anti-fungal activity of phenylphenalenone phytoalexins. Photochem. Photobiol. Sci. 2004, 3, 706–710. [Google Scholar] [CrossRef]
- Hidalgo, W.; Duque, L.; Saez, J.; Arango, R.; Gil, J.; Benjamin, R.; Schneider, B.; Otálvaro, F. Structure-activity relationship in the interaction of substituted perinaphthenones with Mycosphaerella fijiensis. J. Agric. Food Chem. 2009, 57, 7417–7421. [Google Scholar] [CrossRef]
- Rosquete, L.I.; Cabrera-Serra, M.G.; Piñero, J.E.; Rodríguez, P.M.; Pérez, L.F.; Luis, J.G.; McNaughton-Smith, G.; Grillo, T.A. Synthesis and in vitro antiprotozoal evaluation of substituted phenalenone analogues. Bioorg. Med. Chem. 2010, 18, 4530–4534. [Google Scholar] [CrossRef]
- Freijo, M.B.; López-Arencibia, A.; Piñero, J.E.; McNaughton-Smith, G.; Abad-Grillo, T. Design, synthesis and evaluation of amino-substituted 1H-phenalen-1-ones as anti-leishmanial agents. Eur. J. Med. Chem. 2018, 143, 1312–1324. [Google Scholar] [CrossRef]
- Gutiérrez, G.; Flores, N.; Grillo, T.A.; McNaughton-Smith, G. Evaluation of substituted phenalenone analogues as anti-plasmodial agents. Exp. Parasitol. 2013, 135, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, T.; Lv, K.; Liao, F.; Wang, J.; Tu, Y.; Chen, Q. Malaria: Past, present, and future. Signal Transduct Target Ther. 2025, 10, 188. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quiñones, W.; Escobar, G.; Echeverri, F.; Torres, F.; Rosero, Y.; Arango, V.; Cardona, G.; Gallego, A. Synthesis and Anti-fungal Activity of Musa Phytoalexins and Structural Analogs. Molecules 2000, 5, 974–980. [Google Scholar] [CrossRef]
- Song, X.; Ku, C.-F.; Si, T.-X.; Jaiswal, Y.S.; Williams, L.L.; Lu, D.-Y.; Huang, J.-J.; He, Z.-D.; Wang, M.-Z. Synthesis and Biological Activities Assessment of 4-, 6-, and 9-Phenylphenalenone Derivatives. ChemistrySelect 2022, 7, e202203793. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Zhong, L.; Xu-Sheng, S. Synthesis and insecticidal evaluation of phytoalexin phenalenones derivatives. Chin. Chem. Lett. 2017, 28, 1228–1231. [Google Scholar] [CrossRef]
- Suenaga, M.; Miyahara, Y.; Inazu, T. A novel approach to extended phenalenones. J. Org. Chem. 1993, 58, 5846–5848. [Google Scholar] [CrossRef]
- Ospina, F.; Ramirez, A.; Cano, M.; Hidalgo, W.; Schneider, B.; Otalvaro, F. Synthesis of Positional Isomeric Phenylphenalenones. J. Org. Chem. 2017, 82, 3873–3879. [Google Scholar] [CrossRef]
- Ajvazi, N.; Stavber, S. Alcohols in direct carbon-carbon and carbon-heteroatom bond-forming reactions: Recent advances. Arkivoc 2018, 2018, 288–329. [Google Scholar] [CrossRef]
- Xiaowan, L.; Tongxu, S.; Chuenfai, K.; Hongjie, Z.; Mingzhong, W.; Chan, A.S.C. Synthesis of 2,6-dimethoxy-9-phenyl-1H-phenalen-1-one and structural revision of the benzoindenone from Eichhornia crassipes. Z. Naturforsch. 2019, 74, 183–189. [Google Scholar] [CrossRef]
- Manimaran, T.; Jayachandran, T.; Ramakrishnan, V.T. Reaction of β-naphthyl cinnamate with aluminum chloride. Prec. Indian Acad. Sci. (Chem. Sci.) 1980, 89, 301–307. [Google Scholar] [CrossRef]
- Haddon, R.C.; Rayford, R.; Hirani, A.M. 2-Methyl- and 5-methyl-9- hydroxyphenalenone. J. Org. Chem. 1981, 46, 4587–4588. [Google Scholar] [CrossRef]
- Lopez-Arencibia, A.; Bethencourt-Estrella, C.J.; Freijo, M.B.; Reyes-Batlle, M.; Sifaoui, I.; San Nicolas-Hernández, D.; McNaughton-Smith, G.; Lorenzo-Morales, J.; Abad-Grillo, T.; Piñero, J.E. New phenalenone analogues with improved activity against Leishmania species. Biomed. Pharmacother. 2020, 132, 110814. [Google Scholar] [CrossRef]
- Francüois-Xavier, F. Practical and Efficient Suzuki-Miyaura Cross-Coupling of 2-Iodocycloenones with Arylboronic Acids Catalyzed by Recyclable Pd(0)/C. J. Org. Chem. 2005, 70, 8575–8578. [Google Scholar] [CrossRef]
- Amimoghadam, O.; Long, D.L.; Bucher, G. 9-Iodophenalenone and 9-trifluoromethanesulfonyloxyphenalenone: Convenient entry points to new phenalenones functionalised at the 9-position. Iodine-carbonyl interaction studies by X-ray crystallography. RSC Adv. 2014, 4, 56654–56657. [Google Scholar] [CrossRef]
- Palucki, M.; Wolfe, J.P.; Buchwald, S.L. Palladium-Catalyzed Intermolecular Carbon−Oxygen Bond Formation: A New Synthesis of Aryl Ethers. J. Am. Chem. Soc. 1997, 119, 3395–3396. [Google Scholar] [CrossRef]
- Mann, G.; Incarvito, C.; Rheingold, A.L.; Hartwig, J.F. Palladium-Catalyzed C−O Coupling Involving Unactivated Aryl Halides. Sterically Induced Reductive Elimination To Form the C−O Bond in Diaryl Ethers. J. Am. Chem. Soc. 1999, 121, 3224–3225. [Google Scholar] [CrossRef]
- Aranyos, A.; Old, D.W.; Kiyomori, A.; Wolfe, J.P.; Sadighi, J.P.; Buchwald, S.L. Novel Electron-Rich Bulky Phosphine Ligands Facilitate the Palladium-Catalyzed Preparation of Diaryl Ethers. J. Am. Chem. Soc. 1999, 121, 4369–4378. [Google Scholar] [CrossRef]
- Kuroki, M.; Terachi, Y.; Tsunashima, Y. Phenalenones. IV. Heterocycles from 3-hydroxyphenalenone (I). J. Heterocyclic Chem. 1981, 18, 873–876. [Google Scholar] [CrossRef]
- Aksenov, V.A.; Aksenov, N.A.; Lyakhovnenko, A.S.; Aksenova, I.V. Regioselectivity Change in the Reaction of Naphthalene and 2-Naphthyl Ethers with 1,3,5-Triazines Depending on Reagent Quantities. Synthesis 2009, 20, 3439–3442. [Google Scholar] [CrossRef]
- FAF-Drugs4 (Free ADME-Tox Filtering Tool). Available online: https://fafdrugs4.rpbs.univ-paris-diderot.fr/descriptors.html (accessed on 20 October 2025).
- Imanzadeh, G.; Zamanloo, M.; Eskandari, H.; Shayesteh, K. A new ring bromination method for aromatic compounds under solventfree conditions with NBS/Al2O3. J. Chem. Res. 2006, 2006, 151–153. [Google Scholar] [CrossRef]
- Johnson, C.R.; Adams, J.P.; Braun, M.P.; Senanayake, C.B.W.; Wovkulich, P.M.; Uskoković, M.R. Direct α-iodination of cycloalkenones. Tetrahedron Lett. 1992, 33, 917–918. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Deharo, E.; Gautret, P.; Muñoz, V.; Sauvain, M. Técnicas de Laboratorio Para la Selección de Sustancias Antimaláricas; Cytedird: La Paz, Bolivia, 2000. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).