
System Design in CO2 Electrolysis: Integrating Value-Added Anode Reactions with Cathodic Reduction


Abstract
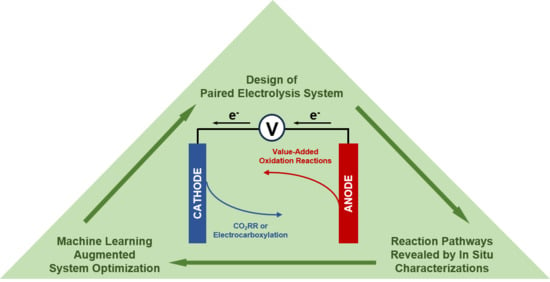
1. Introduction
2. Paired Electrolysis: Fundamentals and Principles
- The product of the counter-electrode reaction can be recovered and repurposed as feedstock for subsequent synthesis;
- The cathodic and anodic processes cooperatively participate in constructing the same target molecule;
- Each electrode generates products with independent practical value.
- Parallel paired electrolysis: Two different substrates are independently transformed at the cathode and anode into valuable products (Figure 1a);
- Divergent paired electrolysis: The same substrate undergoes different reaction path-ways at two electrodes, yielding two distinct valuable products (Figure 1b);
- Convergent paired electrolysis: Reactive intermediates (e.g., radicals or ions) generated at the cathode and anode diffuse into the solution and couple to form a single target molecule (Figure 1c);
- Sequential paired electrolysis: A substrate undergoes sequential electron-transfer reactions, first at one electrode and then at the other, to form a single product (Figure 1d);
- Catalyzed/mediated indirect paired electrolysis: The electrode reactions indirectly drive the target reaction by regenerating a catalyst/mediator in its active oxidation or reduction state (Figure 1e);
- Linear paired electrolysis: A single starting material is transformed into a single target product at both electrodes. This is achieved through direct or indirect electron transfer at one electrode, while the counter electrode reaction, typically consuming a sacrificial starting material, provides a reactive species that drives the transformation toward the same product (Figure 1f).
3. Paired Electrolysis: Engineering Synergy Across Electrolyzers, Electrodes, and Electrolytes
3.1. Configuring Electrolyzers for Kinetic and Mass Transport Synergy
- Beaker-type cell: The workhorse for initial reaction feasibility screening. Its simple, single-compartment design minimizes cost and internal resistance but necessitates exceptional selectivity at both electrodes to prevent cross-talk between reactants and products; its mass transport relies primarily on slow natural convection and diffusion, strictly limiting its performance ceiling.
- H-type cell: Features a physical separator (e.g., an ion-exchange membrane) for compartmentalized studies, essential for mechanistic investigation. However, stagnant electrolytes and large electrode spacing impose severe mass transport limitations, restricting its use to low current densities. Despite its performance being incomparable to high-throughput systems, the H-cell is crucial under controlled mass-transfer conditions for assessing the intrinsic activity and selectivity of catalysts.
- Microfluidic electrolyzer: Employs co-laminar flow in a narrow channel (<1 mm) without a membrane, enabling enhanced mass transport and continuous operation. The key drawback is significant product crossover, which leads to parasitic reactions and low Faradaic efficiency, as key intermediates can be swept away by the flow. Nevertheless, its highly controllable laminar flow characteristics allow for precise tuning of mass transfer, offering a unique platform for mechanistic studies.
- Flow cell: A paradigm for high-rate synthesis. It integrates a membrane into a three-chamber architecture, striking a critical balance between suppressing crossover and enabling efficient ion transport. When coupled with porous gas diffusion electrodes, it can sustain industrially relevant current densities, benefiting from the extreme mass transport enabled by forced convection, albeit at the cost of increased system complexity and challenges like electrode flooding.
- Zero-gap electrolyzer: The state-of-the-art for minimizing ohmic losses. It uses a membrane electrode assembly (MEA) to eliminate the inter-electrode gap. The membrane also defines distinct chemical microenvironments, making it ideal for gas-phase reactions. Persistent challenges include managing steep pH and water gradients, as well as the complexity of MEA fabrication. Its mass transport efficiency ranks at the top among various configurations, making it the preferred platform for high-performance system comparison and assessment of industrialization potential.
3.2. Electrode Design: Multi-Scale Engineering from Atomic Activity to System Integration
3.2.1. Electrode Configurations: Architectures Defined by Mass Transport
- Conventional two-dimensional (2D) planar electrodes (e.g., metal foils, glassy carbon): Serve as foundational model systems for fundamental electrochemical research. Operating under mass-transport-limited liquid-phase diffusion, they typically sustain current densities below 100 mA·cm−2. While unsuitable for high-throughput synthesis, their value resides in providing a well-defined platform with minimal confounding factors for assessing intrinsic catalyst activity and elucidating reaction mechanisms.
- Three-dimensional (3D) porous electrodes (e.g., foams, felts, meshes): Offer superior performance for reactions involving dissolved reactants, such as organic electrosynthesis or metal ion reduction. Their design centers on constructing an interconnected, hierarchical pore network to synergistically achieve a high specific surface area for catalyst loading and efficient electrolyte permeation for enhanced mass transfer.
- Gas diffusion electrodes (GDEs): Are the cornerstone of high-rate gaseous reactant conversion (e.g., CO2, CO, O2 reduction). They are engineered to establish a stable gas–liquid–solid three-phase interface, enabling operation at industrially relevant current densities exceeding 1 A·cm−2. Their design lies in maintaining this interface’s dynamic stability under high-rate conditions through meticulous gradational control from a macroporous, hydrophobic gas diffusion layer (GDL) to a mesoporous, hydrophilic catalyst layer (CL).
- Surface-engineered nanostructured electrodes (e.g., nanowire arrays, nanotube forests): Extend the design paradigm to the nanoscale. They aim to provide directional charge transport pathways and, through precise interface engineering, actively tailor the local chemical microenvironment (e.g., pH, reactant concentration). This creates opportunities to explore and optimize demanding reaction pathways with high energy barriers that are inaccessible to conventional morphologies.
3.2.2. Overarching Design Principles: A Multi-Scale Philosophy
- Atomic-scale electronic structure modulation: The foundational principle. Strategies such as inducing strain, engineering coordination environments (e.g., M-N4 sites), and incorporating dopants are employed to optimize the adsorption energy of key reaction intermediates. This allows for precise steering of the reaction pathway towards desired products while suppressing competing reactions (e.g., HER). This principle applies broadly, from CO2 reduction (tuning Cu-based catalysts for multi-carbon products) to the selective oxidation of organic molecules.
- Microscale mass transport and mesoscale charge transfer optimization: The electrode architecture must facilitate efficient transport highways for reactants and products. This is achieved by designing hierarchical pore structures that align with the reactant’s physical state (gas/liquid) and ensure continuous accessibility of active sites, accounting for timely product (especially gas) removal to prevent pore blocking. Concurrently, the electrode must ensure low-resistance electronic pathways to all active sites.
- System-level chemical and mechanical compatibility: Electrodes cannot be designed in isolation. They must be co-optimized with the membrane and electrolyte to mitigate detrimental crossover processes (e.g., product oxidation, salt precipitation) and ensure long-term operational durability under harsh conditions, including extreme potentials, pH gradients, and mechanical stress.
- Scalable manufacturing and sustainability-by-design: For meaningful industrial translation, electrode design must incorporate forward-looking considerations for scalable fabrication processes (e.g., roll-to-roll manufacturing), the use of Earth-abundant materials, and end-of-life recyclability based on green design principles, ensuring both economic and environmental viability.
3.3. Electrolyte Engineering: Tailoring the Ionic and Chemical Microenvironment
3.3.1. Cation and Anion Effects: The Ionic Helmholtz Layer
- Cations (e.g., Li+, Na+, K+, Cs+): Their size and hydration energy affect the electric field strength at the electrode surface. Larger, less hydrated cations (e.g., Cs+) can more effectively stabilize anionic reaction intermediates (e.g., CO2·− in CO2RR) and lower the activation barrier for C–C coupling steps, following the Hofmeister series.
- Anions and pH regulation: The bulk and local pH, dictated by the buffering capacity of the electrolyte, is a master variable. Alkaline media (e.g., KOH) thermodynamically favor certain reactions like C–C coupling but can cause carbonate precipitation and catalyst degradation. Acidic media (e.g., H2SO4/KCl mixtures) prevent carbonate formation but require the in situ generation of a local high-pH environment at the cathode to suppress the HER. Neutral buffers (e.g., KHCO3) offer a compromise but with limited buffering capacity at high current densities. Anions can also specifically adsorb onto catalyst surfaces, altering the electronic structure of active sites and the adsorption energy of key intermediates.
3.3.2. Solvent and Additive Engineering
- Solvent selection: Moving beyond aqueous systems, organic solvents (e.g., acetonitrile) and ionic liquids can drastically increase the solubility of non-polar reactants (e.g., CO2, organic substrates) and provide a different dielectric environment, opening alternative reaction pathways and selectivity.
- Functional additives: Small amounts of halide ions (Cl−, Br−, I−) or organic molecules can act as promoters or surface modifiers. They can selectively block sites for parasitic reactions (e.g., HER), enhance local CO2 concentration by modifying surface hydrophobicity, or directly participate in stabilizing critical reaction intermediates.
3.3.3. System Integration and Trade-Offs
- Synergy with electrodes: The electrolyte’s wetting behavior and viscosity must be compatible with the electrode’s pore structure—flooding of GDEs must be prevented, and mass transport within 3D electrodes must be ensured.
- Compatibility with membranes: The electrolyte’s pH and composition must be compatible with the membrane’s operational stability window, and vice versa. For instance, AEMs require alkaline conditions, while CEMs are suited for acidic environments. BPMs allow for the independent optimization of anolyte and catholyte, a powerful yet complex strategy.
- Serving the paired reactions: The ultimate goal is to formulate an electrolyte (or anolyte/catholyte pair) that sustains optimal microenvironments for both the cathode and anode reactions simultaneously, balancing the needs for reactant supply, product removal, and inhibition of cross-reactions.
4. Paired Electrolysis in Electrocarboxylation
5. Valorized Anode Reactions with Electrochemical CO2 Reduction Reaction (CO2RR)
5.1. Paired Electrolysis via OOR
5.2. Paired Electrolysis via IOR
6. In Situ/Operando Characterizations and Machine Learning of CO2 Electrolysis
7. Summarization and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| OER | Oxygen Evolution Reaction |
| CO2RR | Carbon Dioxide Reduction Reaction |
| FE | Faradaic Efficiency |
| EE | Energy Efficiency |
| STY | Space–Time Yield |
| TEA | Techno-Economic Analysis |
| HER | Hydrogen Evolution Reaction |
| CEM | Cation Exchange Membrane |
| AEM | Anion Exchange Membrane |
| BPM | Bipolar Membrane |
| GDE | Gas Diffusion Electrode |
| GDL | Gas Diffusion Layer |
| MEA | Membrane Electrode Assembly |
| HAT | Hydrogen Atom Transfer |
| OOR | Organic Oxidation Reaction |
| IOR | Inorganic Oxidation Reaction |
| PCET | Proton-Coupled Electron Transfer |
| MOR | Methanol Oxidation Reaction |
| MF | Methyl Formate |
| FDCA | 2,5-Furandicarboxylic Acid |
| HMF | 5-Hydroxymethylfurfural |
| EO | Ethylene Oxide |
| IOR | Iodide Oxidation Reaction |
| DMC | Dimethyl Carbonate |
| HOR | Hydrogen Oxidation Reaction |
| HzOR | Hydrazine Oxidation Reaction |
| SOR | Sulfide Oxidation Reaction |
| XAS | X-ray Absorption Spectroscopy |
| TEM | Transmission Electron Microscopy |
| XRD | X-ray Diffraction |
| ML | Machine Learning |
References
- Zhang, X.M.; Feng, D.; Wang, J.C.; Sui, A.N. Integrating renewable energy systems: Assessing financial innovation, renewable energy generation intensity, energy transition and environmental regulation with renewable energy sources. Energy Strategy Rev. 2024, 56, 101567. [Google Scholar] [CrossRef]
- Fang, Y.C.; Han, J.P.; Du, E.S.; Jiang, H.Y.; Fang, Y.J.; Zhang, N.; Kang, C.Q. Electric energy system planning considering chronological renewable generation variability and uncertainty. Appl. Energy 2024, 373, 123961. [Google Scholar] [CrossRef]
- Sari, K.; Balamane, W. Reducing intermittency using distributed wind energy: Are wind patterns sufficiently diversified within France. Energy 2024, 313, 133516. [Google Scholar] [CrossRef]
- Palmer, C.; Saadi, F.; McFarland, E.W. Technoeconomics of commodity chemical production using sunlight. ACS Sustain. Chem. Eng. 2018, 6, 7003–7009. [Google Scholar] [CrossRef]
- Herron, J.A.; Kim, J.; Upadhye, A.A.; Huber, G.W.; Maravelias, C.T. A general framework for the assessment of solar fuel technologies. Energy Environ. Sci. 2015, 8, 126–157. [Google Scholar] [CrossRef]
- Kilicaslan, A.F.; Dincer, I. Design and performance assessment of an integrated energy system with compressed air and pumped hydro storage. J. Energy Storage 2025, 116, 116039. [Google Scholar] [CrossRef]
- Boretti, A. Advancing sustainable mobility: Integrating flywheel kinetic energy recovery systems with high-efficiency hydrogen internal combustion engines. Int. J. Hydrogen Energy 2025, 125, 354–363. [Google Scholar] [CrossRef]
- Matos, C.R.; Silva, P.P.; Carneiro, J.F. Overview of compressed air energy storage projects and regulatory framework for energy storage. J. Energy Storage 2022, 55, 105862. [Google Scholar] [CrossRef]
- Zhang, L.L.; Zhou, Z. Electrosynthesis of value-added chemicals: Challenges from laboratory research to industrial application. Chin. J. Catal. 2025, 73, 1–7. [Google Scholar] [CrossRef]
- Wu, C.J.; Zheng, S.S.; Wang, Z.L.; Chen, R.; Hu, X.H.; Chen, J. Discussion on ammonia as one of the energy storage media of solar energy in China. Chin. J. Catal. 2021, 38, 100697. [Google Scholar] [CrossRef]
- Ishaq, H.; Shehzad, M.F.; Crawford, C. Transient modeling of a green ammonia production system to support sustainable development. Int. J. Hydrogen Energy 2023, 48, 39254–39270. [Google Scholar] [CrossRef]
- Liu, J.W.; Li, Z.Y.; Lv, C.D.; Tan, X.Y.; Lee, C.R.; Loh, X.J.; Chua, M.H.; Li, Z.B.; Pan, H.G.; Chen, J.; et al. Electrocatalytic upgrading of nitrogenous wastes into value-added chemicals: A review. Mater. Today 2024, 73, 208–259. [Google Scholar] [CrossRef]
- Xia, R.L.; Zhou, Y.X.; Li, S.J.; Liu, Z.Y.; Liu, Y.P.; Yuan, Z.Y.; Li, W. Metal–nitrogen batteries: Emerging and promising models for energy conversion/storage system and simultaneous NH3 synthesis. ChemSusChem 2025, 18, e202500247. [Google Scholar] [CrossRef]
- Cai, P.W.; Hu, X.; Chen, K.; Lu, Z.W.; Wen, Z.H. The emerging hybrid electrochemical energy technologies. Sci. Bull. 2024, 69, 3571–3589. [Google Scholar] [CrossRef] [PubMed]
- Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S.R. Modern electrochemical aspects for the synthesis of value-added organic products. Angew. Chem. Int. Ed. 2018, 57, 6018–6041. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.X.; Chen, A.; Chen, Y.; Qiao, F.; Wang, J.F.; Yang, N.J.; Zhang, H.; Wen, Z.H. Advancements in electrochemical synthesis: Expanding from water electrolysis to dual-value-added products. eScience 2025, 5, 100333. [Google Scholar] [CrossRef]
- Orlic, M.; Hochenauer, C.; Nagpal, R.; Subotic, V. Electrochemical reduction of CO2: A roadmap to formic and acetic acid synthesis for efficient hydrogen storage. Energy Convers. Manag. 2024, 314, 118601. [Google Scholar] [CrossRef]
- Vega, L.F.; Bahamon, D.; Alkhatib, I.I.I. Perspectives on advancing sustainable CO2 conversion processes: Trinomial technology, environment, and economy. ACS Sustain. Chem. Eng. 2024, 12, 5357–5382. [Google Scholar] [CrossRef]
- Khodadadpour, M.; Bahmanzadegan, F.; Rossi, L.M.; Thonemann, N.; Beale, A.M.; Wojcieszak, R. Hydrogenation of CO2 for sustainable fuel and chemical production. Science 2025, 387, eadn9388. [Google Scholar] [CrossRef]
- Ye, J.Y.; Dimitratos, N.; Ghaemi, A.; Bazyari, A. A review of the performance of zeolitic compounds catalysts for CO2 conversion into valuable hydrocarbons. J. CO2 Util. 2025, 96, 103093. [Google Scholar] [CrossRef]
- Li, J.J.; Qin, X.R.; Wang, X.R.; Wang, L.L.; Yu, Z.Y.; Lu, T.B. Direct electroreduction of low-concentration CO2: Progress and perspective. ACS Nano 2025, 19, 10620–10629. [Google Scholar] [CrossRef]
- O’Carroll, T.; Yang, X.X.; Gordon, K.J.; Fei, L.; Wu, G. Ethylene electrosynthesis via selective CO2 reduction: Fundamental considerations, strategies, and challenges. Adv. Energy Mater. 2024, 14, 2401558. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Xing, G.S.; Sun, C.H.; Chen, Z.K.; Li, S.Y.; Chen, M.; Zhang, P.; Feng, C.R.; Abudula, A.; Guan, G.Q. Electrosynthesis of urea from carbon dioxide and waste nitrates: History, recent progress, and future prospects. Adv. Funct. Mater. 2025, e15635. [Google Scholar] [CrossRef]
- Jiang, M.H.; Wang, H.Z.; Zhu, M.F.; Luo, X.J.; He, Y.; Wang, M.J.; Wu, C.J.; Zhang, L.Y.; Li, X.; Liao, X.M.; et al. Review on strategies for improving the added value and expanding the scope of CO2 electroreduction products. Chem. Soc. Rev. 2024, 53, 5149–5189. [Google Scholar] [CrossRef]
- Sun, G.Q.; Liao, L.L.; Ran, C.K.; Ye, J.H.; Yu, D.G. Recent advances in electrochemical carboxylation with CO2. Acc. Chem. Res. 2024, 57, 2728–2745. [Google Scholar] [CrossRef] [PubMed]
- Ballard-Kyle, P.; Hsieh, I.; Zhu, H.Y.; Ye, J.H.; Yu, D.G. Electrocatalytic C-N coupling: Advances in urea synthesis and opportunities for alternative products. ChemSusChem 2025, 18, e202402566. [Google Scholar] [CrossRef] [PubMed]
- Mena, S.; Peral, J.; Guirado, G. Use of CO2 for electrosynthesis. Curr. Opin. Electrochem. 2023, 42, 101392. [Google Scholar] [CrossRef]
- Sun, D.L.; Xu, X.M.; Qin, Y.L.; Jiang, S.; Shao, Z.P. Rational design of Ag-based catalysts for the electrochemical CO2 reduction to CO: A review. ChemSusChem 2020, 13, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.F.; Xu, W.C.; Liu, C.; Wang, Y.K.; Huang, X.Z.; Xiao, R.; Xu, X.M.; Li, T.; Shao, Z.P. Innovative electrode designs for low-temperature electrochemical CO2 reduction with ampere-level performance. Energy Environ. Sci. 2025, 18, 7792–7858. [Google Scholar] [CrossRef]
- Verma, S.; Lu, S.; Kenis, P.J.A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nat. Energy 2019, 4, 466–474. [Google Scholar] [CrossRef]
- Na, J.; Seo, B.; Kim, J.; Lee, C.W.; Lee, H.; Hwang, Y.J.; Min, B.K.; Lee, D.K.; Oh, H.S.; Lee, U. General technoeconomic analysis for electrochemical coproduction coupling carbon dioxide reduction with organic oxidation. Nat. Commun. 2019, 10, 5193. [Google Scholar] [CrossRef]
- Li, R.; Xiang, K.; Peng, Z.K.; Zou, Y.Q.; Wang, S.Y. Recent advances on electrolysis for simultaneous generation of valuable chemicals at both anode and cathode. Adv. Energy Mater. 2021, 11, 2102292. [Google Scholar] [CrossRef]
- Huang, B.B.; Sun, Z.M.; Sun, G.B. Recent progress in cathodic reduction-enabled organic electrosynthesis: Trends, challenges, and opportunities. eScience 2022, 2, 243–277. [Google Scholar] [CrossRef]
- Meyer, T.H.; Choi, I.; Tian, C.; Ackermann, L. Powering the future: How can electrochemistry make a difference in organic synthesis. Chem 2020, 6, 2484–2496. [Google Scholar] [CrossRef]
- Xue, H.; Zhao, Z.H.; Yuan, M.L.; Zhang, G.J. Microfluidic reactors for paired electrosynthesis: Fundamentals, applications and future prospects. Green Energy Environ. 2025, 10, 471–499. [Google Scholar] [CrossRef]
- MariaJoseph, A.; Nangan, S.; Verma, D.; Gnanasekaran, L.; Rajendran, S.; Natesan, T.; Pattananuwat, P.; Okhawilai, M. Rational coupling of selective electrochemical oxidation and reduction reactions for in-situ value-added chemical generation. Fuel 2024, 367, 131408. [Google Scholar] [CrossRef]
- Francke, R.; Gonzalez, L.; Little, R.D.; Moeller, K.D. Electrons, electrodes, and the transformation of organic molecules. In Surface and Interface Science: Applications II; Wandelt, K., Ed.; Wiley-VCN: Berlin, Germany, 2020; Volume 10, pp. 827–891. ISBN 978-3527413836. [Google Scholar]
- Baizer, M.; Hallcher, R.C. Paired electro-organic syntheses: I. Cathodic adipate with anodic bimalonate. J. Electrochem. Soc. 1976, 123, 809–813. [Google Scholar] [CrossRef]
- Hilt, G. Recent advances in paired electrolysis and their application in organic electrosynthesis. Curr. Opin. Electrochem. 2024, 43, 101425. [Google Scholar] [CrossRef]
- Duan, F.Y.; Yuan, M.L.; Zhang, J. Paired electrolysis for inorganic small molecules reduction coupled with alternative oxidation reactions. Chin. J. Org. Chem. 2024, 44, 809–824. [Google Scholar] [CrossRef]
- Strehl, J.; Abraham, M.L.; Hilt, G. Linear paired electrolysis-realising 200% current efficiency for stoichiometric transformations-the electrochemical bromination of alkenes. Angew. Chem. Int. Edit. 2021, 60, 9996–10000. [Google Scholar] [CrossRef]
- Ma, C.; Fang, P.; Liu, D.; Jiao, K.J.; Gao, P.S.; Qiu, H.; Mei, T.S. Transition metal-catalyzed organic reactions in undivided electrochemical cells. Chem. Sci. 2021, 12, 12866–12873. [Google Scholar] [CrossRef] [PubMed]
- Appleby, A.J.; Zagal, J.H. Free energy relationships in electrochemistry: A history that started in 1935. J. Solid State Electrchem. 2011, 15, 1811–1832. [Google Scholar] [CrossRef]
- Moeller, K.D. Using physical organic chemistry to shape the course of electrochemical reactions. Chem. Rev. 2018, 118, 4817–4833. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.L.; Yang, P.; Jiang, M.K.; Wang, X.L.; Zhou, H.X.; Duan, J.Y.; Ratova, M.; Wu, D. Electrochemical CO2 reduction coupled with alternative oxidation reactions: Electrocatalysts, electrolytes, and electrolyzers. Appl. Catal. B-Environ. Energy 2024, 341, 123291. [Google Scholar] [CrossRef]
- Zhang, S.J.; Tang, W.; Yin, J.Y.; Wang, S.K.; Yu, Y.J.; Huang, R.; Huo, E.G. Feasibility and prospects of electrocatalytic conversion of CO2 for chemical feedstock production and renewable energy storage. ACS Sustain. Chem. Eng. 2025, 13, 9841–9858. [Google Scholar] [CrossRef]
- Lai, W.C.; Qiao, T.; Zhang, J.W.; Lin, Z.Q.; Huang, H.W. Design strategies for markedly enhancing energy efficiency in the electrocatalytic CO2 reduction reaction. Energy Environ. Sci. 2022, 15, 3603–3629. [Google Scholar] [CrossRef]
- O’Brien, C.P.; Miao, R.K.; Zeraati, A.S.; Lee, G.H.; Sargent, E.H.; Sinton, D. CO2 Electrolyzers. Chem. Rev. 2024, 124, 3648–3693. [Google Scholar] [CrossRef] [PubMed]
- Wakerley, D.; Lamaison, S.; Wicks, J.; Clemens, A.; Feaster, J.; Corral, D.; Jaffer, S.A.; Sarkar, A.; Fontecave, M.; Duoss, E.B.; et al. Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers. Nat. Energy 2022, 7, 130–143. [Google Scholar] [CrossRef]
- Lees, E.W.; Mowbray, B.A.W.; Parlane, F.G.L.; Berlinguette, C.P. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat. Rev. Mater. 2022, 7, 55–64. [Google Scholar] [CrossRef]
- Chen, J.Y.; Peng, X.Y.; Li, Z.J.; Yang, B.; Zhang, Q.H.; Lu, J.G.; Lei, L.C.; Hou, Y. Rational modulation of Interface microenvironment and design of the flow electrolyzer for COx electroreduction to alcohol. Adv. Mater. 2025, 37, 2409106. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.F.; Dong, Y.L.; Zhao, H.P.; Lei, Y. CO2 conversion toward real-world applications: Electrocatalysis versus CO2 batteries. Adv. Funct. Mater. 2023, 33, 2300926. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, K.; Zheng, S.J.; Cheng, X.; He, Y.X.; Qin, W.; Zhang, X.H.; Chang, H.X.; Zhong, N.B.; He, X.F. Advancements in electrochemical CO2 reduction reaction: A review on CO2 mass transport enhancement strategies. Chem. Eng. J. 2024, 486, 150169. [Google Scholar] [CrossRef]
- Han, S.G.; Wang, S.M.; Hu, M.K.; Wei, W.B.; Zhan, C.; Ma, D.D.; Zhu, Q.L. Low-cost, facile, and scalable manufacturing of single-molecule-integrated catalytic electrodes. ACS Nano 2025, 19, 11273–11283. [Google Scholar] [CrossRef]
- Yan, T.X.; Chen, X.Y.; Kumari, L.; Lin, J.L.; Li, M.L.; Fan, Q.; Chi, H.Y.; Meyer, T.J.; Zhang, S.; Ma, X.B. Multiscale CO2 electrocatalysis to C2+ products: Reaction mechanisms, catalyst design, and device fabrication. Chem. Rev. 2023, 123, 10530–10583. [Google Scholar] [CrossRef]
- Zhao, Z.W.; Liu, Y.; Wang, S.Y.; Tang, S.Y.; Ma, D.K.; Zhu, Z.L.; Guo, C.C.; Qiu, Y.A. Site-selective electrochemical C-H carboxylation of arenes with CO2. Angew. Chem. Int. Edit. 2023, 62, e202214710. [Google Scholar] [CrossRef]
- Alkayal, A.; Tabas, V.; Montanaro, S.; Wright, I.A.; Malkov, A.V.; Buckley, B.R. Harnessing applied potential: Selective β-hydrocarboxylation of substituted olefins. J. Am. Chem. Soc. 2020, 142, 1780–1785. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiang, H.F.; Zhang, Y.N.; Tao, Y.Y.; Liu, X.C.; Huo, C.D. Electroreductive deoxygenative carboxylation of alkyl oxalates with CO2. Green Chem. 2024, 26, 10811–10817. [Google Scholar] [CrossRef]
- Ran, C.K.; Qu, Q.; Tao, Y.Y.; Chen, Y.F.; Liao, L.L.; Ye, J.H.; Yu, D.G. Electro-reductive carboxylation of acyclic C(sp3)-C(sp3) bonds in aromatic hydrocarbons with CO2. Sci. China Chem. 2024, 615, 3366–3372. [Google Scholar] [CrossRef]
- Ware, S.D.; Zhang, W.Y.; Guan, W.Y.; Lin, S.; See, K.A. A guide to troubleshooting metal sacrificial anodes for organic electrosynthesis. Chem. Sci. 2024, 15, 5814–5831. [Google Scholar] [CrossRef] [PubMed]
- Chantarojsiri, T.; Soisuwan, T.; Kongkiatkrai, P. Toward green syntheses of carboxylates: Considerations of mechanisms and reactions at the electrodes for electrocarboxylation of organohalides and alkenes. Chin. J. Catal. 2022, 43, 3046–3061. [Google Scholar] [CrossRef]
- Sun, G.Q.; Zhang, W.; Liao, L.L.; Li, L.; Nie, Z.H.; Wu, J.G.; Zhang, Z.; Yu, D.G. Nickel-catalyzed electrochemical carboxylation of unactivated aryl and alkyl halides with CO2. Nat. Commun. 2021, 12, 7086. [Google Scholar] [CrossRef]
- Sun, G.Q.; Yu, P.; Zhang, W.; Zhang, W.; Wang, Y.; Zhang, Z.; Li, L.; Lu, Z.P.; Yu, D.G.; Lin, S. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 2023, 615, 67–72. [Google Scholar] [CrossRef]
- Zhao, R.; Surke, M.; Lin, Z.P.; Alsalme, A.; Ackermann, L. Site-selective electrochemical carboxylation of aromatic C(sp2)–H bonds with CO2. Curr. Res. Green Sustain. Chem. 2023, 7, 100377. [Google Scholar] [CrossRef]
- Zeng, W.M.; Peng, C.Y.; Qiu, Y.A. Electrochemical benzylic C-H carboxylation. J. Am. Chem. Soc. 2025, 147, 13461–13470. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.H.; Chen, H.S.; Shen, C.R.; Wu, H.H.; Dong, K.W. Pairing electrocarboxylation of unsaturated bonds with oxidative transformation of alcohol and amine. ChemSusChem 2023, 16, e202300807. [Google Scholar] [CrossRef]
- Villo, P.; Lill, M.; Alsaman, Z.; Soto Kronberg, A.; Chu, V.; Ahumada, G.; Agarwala, H.; Ahlquist, M.; Lundberg, H. Electroreductive deoxygenative C-H and C-C bond formation from non-derivatized alcohols fueled by anodic borohydride oxidation. ChemElectroChem 2023, 10, e202300420. [Google Scholar] [CrossRef]
- Wu, T.Y. Alternative anode paired to electrocatalytic CO2 reduction. Int. J. Hydrogen Energy 2024, 94, 72–79. [Google Scholar] [CrossRef]
- Chen, H.J.; Shin, H.; Huang, J.E.; Liu, H.Z.; Miao, R.K.; Xia, R.; Ni, W.Y.; Yu, J.Q.; Liang, Y.X.; Peng, B.S.; et al. Electrolysis of ethylene to ethylene glycol paired with acidic CO2-to-CO conversion. ChemElectroChem 2025, 18, 8600–8607. [Google Scholar] [CrossRef]
- Barat-Abtahi, S.; Jafari-Hafshejani, F.; Varmaghani, F.; Karimi, B.; Veisi, H.H. Robust interaction of cobalt phthalocyanine and nitrogen-doped ordered mesoporous carbon for CO2 reduction paired with the electro-oxidative synthesis of sulfonamide derivatives. Green Chem. 2024, 26, 362–374. [Google Scholar] [CrossRef]
- Khaledian, S.; Omidi, S.; Varmaghani, F.; Karimi, B.; Sepehri, Z.; Veisi, H.H.; Vali, H. Two targets with one shot: Cost-efficient electrochemical CO2 reduction coupled with the synthesis of Cu- and Zn-based metal-organic frameworks. ACS Sustainable Chem. Eng. 2025, 13, 17309–17323. [Google Scholar] [CrossRef]
- Baessler, J.; Oliveira, T.; Keller, R.; Wessling, M. Paired electrosynthesis of formic acid from CO2 and formaldehyde from methanol. ACS Sustain. Chem. Eng. 2023, 11, 6822–6828. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Zhao, K.; Feng, H.Z.; Ke, L.; Wang, X.D.; Liu, Y.C.; Li, L.J.; Sun, P.F.; Chen, Z.; Sun, Y.F.; et al. Unraveling side reactions in paired CO2 electrolysis at operando conditions: A case study of ethylene glycol oxidation. J. Am. Chem. Soc. 2025, 147, 13471–13482. [Google Scholar] [CrossRef]
- Junqueira, J.R.C.; Das, D.; Brix, A.C.; Dieckhoefer, S.; Weidner, J.; Wang, X.; Shi, J.L.; Schuhmann, W. Simultaneous anodic and cathodic formate production in a paired electrolyzer by CO2 reduction and glycerol oxidation. ChemSusChem 2023, 16, e202202349. [Google Scholar] [CrossRef]
- Shi, J.Y.; Wang, Z.L.; Huang, G.Y.; Wang, K.A.; Zhu, H.B. CuS nanoparticle/TiO2 nanosheets heterojuncture boosting paired electrosynthesis of formate. J. Electroanal. Chem. 2024, 973, 118665. [Google Scholar] [CrossRef]
- Kormányos, A.; Szirmai, A.; Endrodi, B.; Janáky, C. Pairing electrochemical CO2 reduction with glycerol oxidation: Bottlenecks today, opportunities tomorrow. Joule 2025, 9, 102096. [Google Scholar] [CrossRef]
- Chen, D.W.; Yang, S.H.; Gao, J.; Zheng, X.; Mao, J.W.; Hu, Q.Y.; Sun, X.H.; Ji, L.; Zheng, X.L.; Fu, H.Y.; et al. Electrochemical co-upgrading CO2 and glycerol for selective formate production with 190% overall Faradaic efficiency. Nano Res. 2025, 18, 94907399. [Google Scholar] [CrossRef]
- Chen, T.; Hu, Q.Z.; Wu, C.; Sun, M.D.; Fu, P.; Liu, X.L.; Li, Y.L.; Zhou, Y.; Xi, S.B.; Wang, J. Ultralow-coordinated Ni species boosting paired electrosynthesis of formate from waste plastic and carbon dioxide. J. Energy Chem. 2025, 107, 285–295. [Google Scholar] [CrossRef]
- Yeo, J.B.; Jang, J.H.; Jo, Y.I.; Koo, J.W.; Nam, K.T. Paired electrosynthesis of formaldehyde derivatives from CO2 reduction and methanol oxidation. Angew. Chem. Int. Edit. 2024, 63, e202316020. [Google Scholar] [CrossRef]
- Ye, F.H.; Zhang, S.S.; Cheng, Q.Q.; Long, Y.D.; Liu, D.; Paul, R.; Fang, Y.M.; Su, Y.Q.; Qu, L.T.; Dai, L.M.; et al. The role of oxygen-vacancy in bifunctional indium oxyhydroxide catalysts for electrochemical coupling of biomass valorization with CO2 conversion. Nat. Commun. 2023, 14, 2040. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Dutta, S. Effect of single-metal-atoms in electrovalorization of biomass and paired electrolysis. Chem. Eng. J. 2024, 494, 152950. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.C.; Li, Y.R.; Yan, B.H.; Zhao, X.B. Facile preparation of 2D nanomaterials by chelating agent-assisted electrodeposition for efficient electrochemical oxidation of biomass-derived aldehydes coupled with CO2 reduction. Appl. Catal. B Environ. 2025, 386, 125146. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, X.Y.; Ding, J.J.; Ban, C.G.; Feng, Y.J.; Xu, C.H.; Zhou, X.Y. Realizing ampere-level CO2 electrolysis at low voltage over a woven network of few-atom-layer ultralong silverene nanobelts with ultrahigh aspect ratio by pairing with formaldehyde oxidation. Nanoscale 2024, 16, 7076–7084. [Google Scholar] [CrossRef]
- Li, Z.Y.; Wang, P.; Han, G.Q.; Yang, S.Z.; Roy, S.; Xiang, S.T.; Jimenez, J.D.; Kondapalli, V.K.R.; Lyu, X.; Li, J.L.; et al. Ampere-level co-electrosynthesis of formate from CO2 reduction paired with formaldehyde dehydrogenation reactions. Nat. Commun. 2025, 16, 4850. [Google Scholar] [CrossRef]
- Wang, X.; Li, P.H.; Tam, J.; Howe, J.Y.; O’Brien, C.P.; Rasouli, A.S.; Miao, R.K.; Liu, Y.; Ozden, A.; Xie, K.; et al. Efficient CO and acrolein co-production via paired electrolysis. Nat. Sustain. 2024, 7, 931–937. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Y.S.; Yan, S.; Lv, X.M.; Yang, C.; Kuang, M.; Zheng, G.F. Coupling of electrocatalytic CO2 reduction and CH4 oxidation for efficient methyl formate electrosynthesis. Energy Environ. Sci. 2024, 17, 2309–2314. [Google Scholar] [CrossRef]
- Xue, W.J.; Quan, L.; Liu, H.X.; Yu, B.; Chen, X.Q.; Xia, B.Y.; You, B. Bromine-enhanced generation and epoxidation of ethylene in tandem CO2 electrolysis towards ethylene oxide. Angew. Chem. Int. Edit. 2023, 62, e202311570. [Google Scholar] [CrossRef]
- Li, Q.; Ma, D.D.; Wei, W.B.; Han, S.G.; Zheng, L.R.; Zhu, Q.L. Value-added cascade synthesis mediated by paired-electrolysis using an ultrathin microenvironment-inbuilt metalized covalent organic framework heterojunction. Adv. Energy Mater. 2024, 14, 2401314. [Google Scholar] [CrossRef]
- Cai, M.L.; Dai, S.Y.; Xuan, J.; Mo, Y.M. Bromide-mediated membraneless electrosynthesis of ethylene carbonate from CO2 and ethylene. Nat. Commun. 2025, 16, 3285. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Han, S.G.; Wu, W.M.; Zhang, K.X.; Chen, B.; Zhou, S.H.; Ma, D.D.; Wei, W.B.; Wu, X.T.; Zou, R.Q.; et al. Convergent paired electrosynthesis of dimethyl carbonate from carbon dioxide enabled by designing the superstructure of axial oxygen coordinated nickel single-atom catalysts. Energy Environ. Sci. 2023, 16, 502–512. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Ke, L.; Zhao, K.; Yan, X.Y.; Wang, H.B.; Cao, X.J.; Liu, Y.C.; Li, L.J.; Sun, Y.F.; Wang, Z.P.; et al. Integrating hydrogen utilization in CO2 electrolysis with reduced energy loss. Nat. Commun. 2024, 15, 1427. [Google Scholar] [CrossRef]
- Hu, X.; Mei, G.L.; Chen, X.X.; Liu, J.L.; Xia, B.Y.; You, B. Simultaneous generation of H2O2 and formate by Co-electrolysis of water and CO2 over bifunctional Zn/SnO2 nanodots. Angew. Chem. Int. Edit. 2023, 62, e202304050. [Google Scholar] [CrossRef] [PubMed]
- Mavrikis, S.; Nieuwoudt, M.; Göltz, M.; Ehles, S.; Körner, A.; Hutzler, A.; Fossy, E.; Zervas, A.; Brai, O.; Wegener, M.; et al. Continuous production of ethylene and hydrogen peroxide from paired electrochemical carbon dioxide reduction and water oxidation. Adv. Energy Mater. 2024, 14, 2304247. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wei, K.X.; Li, Z.X.; Gong, H.Z.; Deng, J.N.; Yuan, H.Z.; Lian, H.Y.; Zheng, H.; Zhao, H.H.; Zhang, X.; et al. Engineering the coordination of Cu-Ni dual-atom catalysts to enhance the electrochemical CO2 overall splitting. J. Energy Chem. 2025, 103, 333–343. [Google Scholar] [CrossRef]
- Pan, W.F.; Yuan, J.; Wang, P.; Wang, J.; Zhao, Y.; Wang, G.X.; Yu, H.; Wen, Z.H. Efficient ultra-low voltage electrolysis of CO2 coupling with hydrazine oxidation degradation. Appl. Catal. B-Environ. Energy 2024, 351, 124011. [Google Scholar] [CrossRef]
- Yang, K.X.; Zhang, N.; Yang, J.F.; Xu, Z.; Yan, J.Q.; Li, D.; Liu, S.Z. Synergistic marriage of CO2 reduction and sulfide oxidation towards a sustainable co-electrolysis process. Appl. Catal. B-Environ. Energy 2023, 332, 122718. [Google Scholar] [CrossRef]
- Liu, X.Z.; Zhao, P.C.; Liu, F.F.; Lin, R.C.; Yao, H.F.; Zhu, S.Q. Attenuated total reflection infrared spectroscopy for studying electrochemical cycling of hydrogen, carbon, and nitrogen-containing molecules. J. Energy Chem. 2024, 99, 495–511. [Google Scholar] [CrossRef]
- Xie, Z.Z.; Liu, Y.K.; He, L.Q.; Chen, J.; Wu, X.; Li, M.Y.; Wang, K.; Tong, Y.X. In situ/operando characterization techniques for reaction interface in electrocatalytic CO2 reduction. Small 2025, e2502083. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.S.; Wang, J.L.; Chu, Y.C.; Chen, J.H.; Chien, C.Y.; Lin, K.H.; Tsai, L.D.; Chen, H.C.; Liao, Y.F.; Hiraoka, N.; et al. Activating dynamic atomic-configuration for single-site electrocatalyst in electrochemical CO2 reduction. Nat. Commun. 2023, 14, 5245. [Google Scholar] [CrossRef]
- Hou, X.Y.; Li, S.W.; Frey, J.; Hong, X.; Ackermann, L. Machine learning-guided yield optimization for palladaelectro-catalyzed annulation reaction. Chem 2024, 10, 2283–2294. [Google Scholar] [CrossRef]
- Martini, A.; Hursán, D.; Timoshenko, J.; Rüscher, M.; Haase, F.; Rettenmaier, C.; Ortega, E.; Etxebarria, A.; Cuenya, B.R. Tracking the evolution of single-atom catalysts for the CO2 electrocatalytic reduction using operando X-ray absorption spectroscopy and machine learning. J. Am. Chem. Soc. 2023, 145, 17351–17366. [Google Scholar] [CrossRef]
- Shi, J.J.; Prslja, P.; Jin, B.J.; Suominen, M.; Sainio, J.; Jiang, H.; Han, N.; Robertson, D.; Kosir, J.; Caro, M.; et al. Experimental and computational study toward identifying active sites of supported SnOx nanoparticles for electrochemical CO2 reduction using machine-learned interatomic potentials. Small 2024, 20, 2402190. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Definition * | Role in System Design * |
|---|---|---|
| (Total) Current Density (j) | The electric current flowing per unit geometric area (or electrochemical active surface area) of the electrode. | Determines the reaction rate and reactor’s scale; a primary driver of the capital expenditure. |
| Faradaic Efficiency (FE) | The fraction of the total electric charge used to produce a specific desired product. | Evaluates the selectivity of each half-reaction; the product of the FE at both electrodes defines the upper limit of the system’s atom economy and revenue potential. |
| Cell Voltage (Vcell) | The total voltage applied between the anode and cathode required to drive the stable operation of the entire electrolyzer. | Directly determines the system’s energy consumption; the primary determinant of operating expenses. |
| Energy efficiency (EE) | The ratio of the total chemical energy stored in the products (anodic and cathodic) to the total electrical energy input into the system. | A comprehensive metric of overall energy utilization, highlighting the synergy from co-producing valuable products at both electrodes. |
| Electrode Stability | The rate of performance decay over time for a single electrode under operational conditions. | Ensures the longevity operational reliability of each half-cell; a prerequisite for system-level stability. |
| System Stability | The duration over which the entire paired electrolysis system maintains its target performance | The critical metric for commercial viability, assessing the compatibility and mutual degradation of all integrated components under paired conditions; often the key gap between lab-scale demonstration and industrial application. |
| Space-Time Yield (STY) | The mass of product produced per unit time per unit reactor volume. | Measures the productivity and compactness of the reactor; key for process intensification. |
| Product Concentration/Putative | The concentration of the target product in the outlet electrolyte stream, or the purity of a gaseous product. | Directly determines the energy and cost intensity of downstream separation and purification. |
| Techno-economic Analysis (TEA) | A systematic framework for quantifying the technical and economic feasibility of a technology. | Translates the technical synergies of paired electrolysis into a clear assessment of its commercial viability. |
| Cathode System | Cathode Substrate | Cathode Electrode/ Electrolyte | Cathode Product and FE* | Cathode Potential | Paired Anode Substrate | Anode Electrode/ Electrolyte | Anode Product and FE * | Anode Potential | Full Cell Voltage |
|---|---|---|---|---|---|---|---|---|---|
| Ketone | 2-Acetyl-6-methoxynaphthalene | Electrode: Pb sheet Electrolyte: Bu4NClO4 (0.1 M) Solvent: MeCN | 2-Aryl lactic acid FE: 95% | ~−2.23 V vs. Fc/Fc+ | 4-Methoxybenzyl alcohol | Electrode: Pt sheet Electrolyte: Bu4NBr (0.1 M) Solvent: MeCN Additives: TEMPO, 2,6-lutidine | 4-Methoxybenzaldehyde FE: 73% | ~0.242 V vs. Fc/Fc+ | ~10.55 V |
| Cell Type | Anodic Reaction | Type and Role of Membrane | Proton Flux & Charge Compensation | Ref. |
|---|---|---|---|---|
| H-cell (no GDE) | CH3OH + 4OH− → HCOO− + 3H2O + 3e− | BPM interface enables water dissociation to balance charge and prevent pH crossover, critical for stabilizing environments in paired electrolysis. | Cathodic H+ modulation via TiO2 nanosheets; anodic H+ effects not addressed. | [75] |
| H-cell (no GDE) | CH3OH → HCHO + 2H+ + 2e− | CEM maintains the directional migration of H+ to balance the charge and prevent crossover between the electrodes. | Cathodic H+ modulation directly determines by the anode reaction | [79] |
| H-cell (no GDE) | HMF + 6OH− → FDCA + 4H2O + 6e− | BPM, water dissociation at the interface; supplies H+ and OH− to balance the charge and prevent crossover between the electrodes. | None H+ flux from the anode; cathode H+ modulation directly determines by the BPM. | [80] |
| Flow cell (with GDE) | CH4 + Cl− → CH3Cl + H+ + 2e− | AEM, guided migration of HCOO− from cathode to anode and prevent crossover between the electrodes. | None H+ flux from the an-ode; H+ generated at the anode undergoes immediate neutralization by the abundant OH− in anolyte. | [86] |
| Tandem flow cell system (with GDE) | [Fe(CN)6]4− → [Fe(CN)6]3− + e− (CO2RR chamber) 2Br− → Br2 + 2e− (C2H4 oxidation chamber) | AEM (CO2RR chamber), selective ion transport (predominantly anions) for environmental charge balance and prevent crossover between the electrodes. | None H+ flux from the an-ode in CO2RR chamber. | [87] |
| Flow cell (with GDE) | 3I− → I3− + 2e− | BPM, water dissociation at the interface; supplies H+ and OH− to balance the charge and prevent crossover between the electrodes. | Negligible H+ flux from the anode; cathode H+ modulation directly determines by the BPM. | [88] |
| Flow cell (with GDE) | Ni(OH)2 + OH− → NiOOH + H2O + e− (Step 1) H2 + 2OH− → 2H2O + 2e− (Step 2) | A Ni(OH)2/NiOOH-mediated temporal separation strategy that suppresses carbon loss & catalyst poisoning while boosting energy efficiency | None H+ flux from the anode in step 1. | [91] |
| Flow cell (with GDE) | 2H2O → H2O2 + 2H+ + 2e− | AEM maintains the directional migration of CO32−/HCO3− to balance the charge and prevent crossover be-tween the electrodes. | Effective anode H+ buffering by K2CO3 aq. for minimal cathode flux impact. | [92] |
| Flow cell (with GDE) | N2H4 + 4OH− → N2 + 4H2O + 4e− | CEM maintains the directional migration of K+ to balance the charge and prevent crossover be-tween the electrodes. | Negligible H+ flux from the anode | [95] |
| Flow cell (with GDE) | HS− + OH− → S + H2O + 2e− | CEM maintains the directional migration of K+ to balance the charge and prevent crossover between the electrodes. | Negligible H+ flux from the anode | [96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, Y.; Wang, C.; Chen, Z. System Design in CO2 Electrolysis: Integrating Value-Added Anode Reactions with Cathodic Reduction. Molecules 2025, 30, 4485. https://doi.org/10.3390/molecules30224485
Zhai Y, Wang C, Chen Z. System Design in CO2 Electrolysis: Integrating Value-Added Anode Reactions with Cathodic Reduction. Molecules. 2025; 30(22):4485. https://doi.org/10.3390/molecules30224485
Chicago/Turabian StyleZhai, Yuehui, Chong Wang, and Zheng Chen. 2025. "System Design in CO2 Electrolysis: Integrating Value-Added Anode Reactions with Cathodic Reduction" Molecules 30, no. 22: 4485. https://doi.org/10.3390/molecules30224485
APA StyleZhai, Y., Wang, C., & Chen, Z. (2025). System Design in CO2 Electrolysis: Integrating Value-Added Anode Reactions with Cathodic Reduction. Molecules, 30(22), 4485. https://doi.org/10.3390/molecules30224485