
Polystyrene Upcycling via Photocatalytic and Non-Photocatalytic Degradation


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
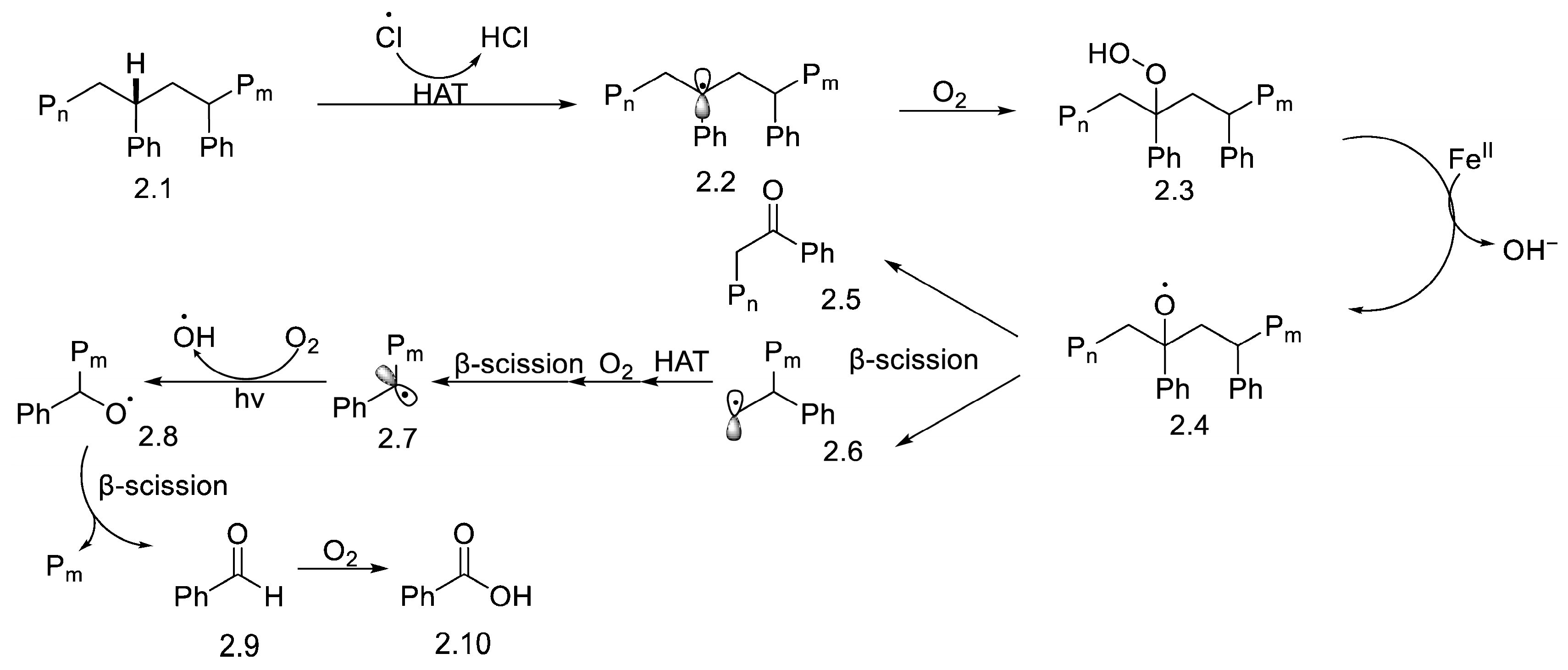
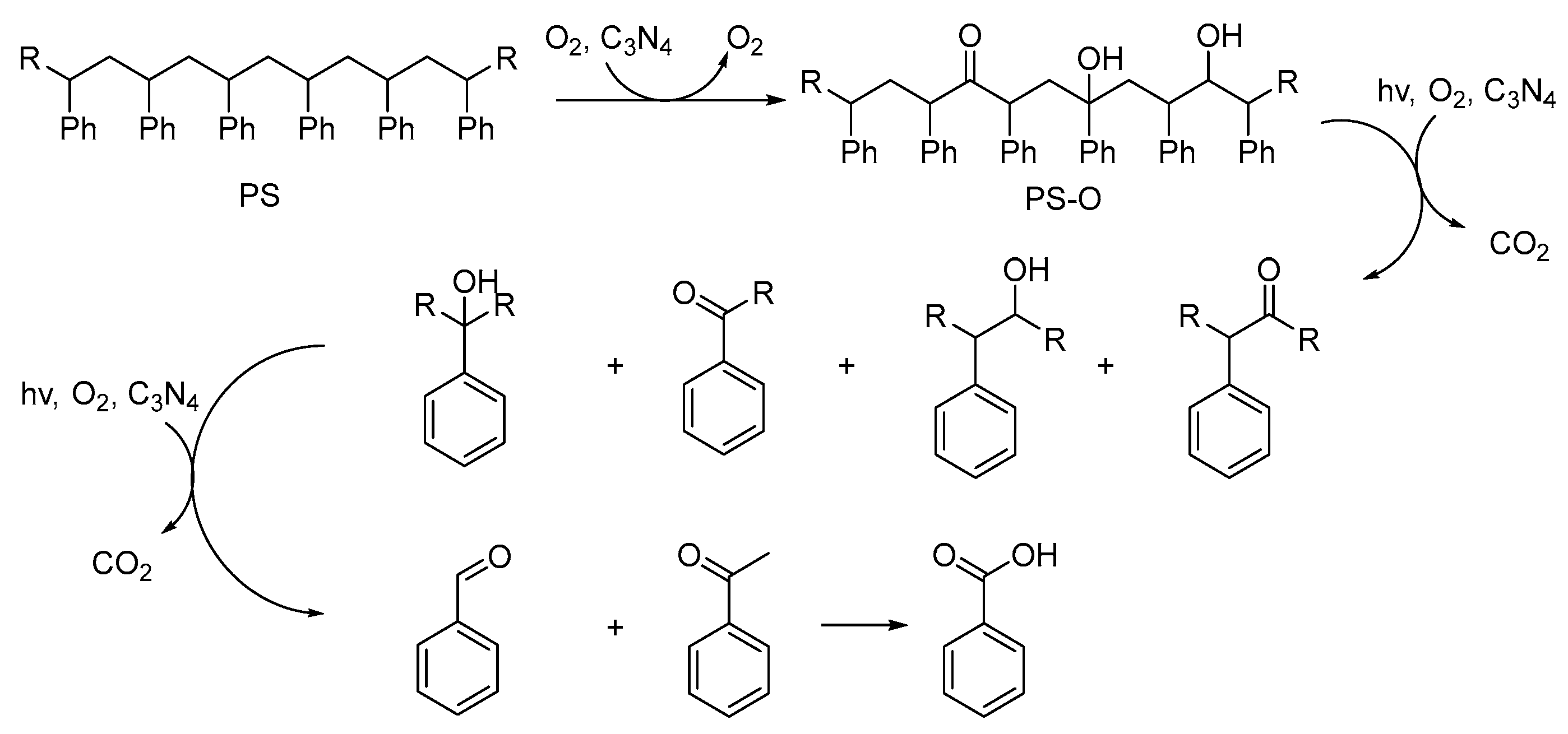
2. Photocatalytic Upcycling of PS
2.1. Visible-Light-Induced Photocatalytic Upcycling
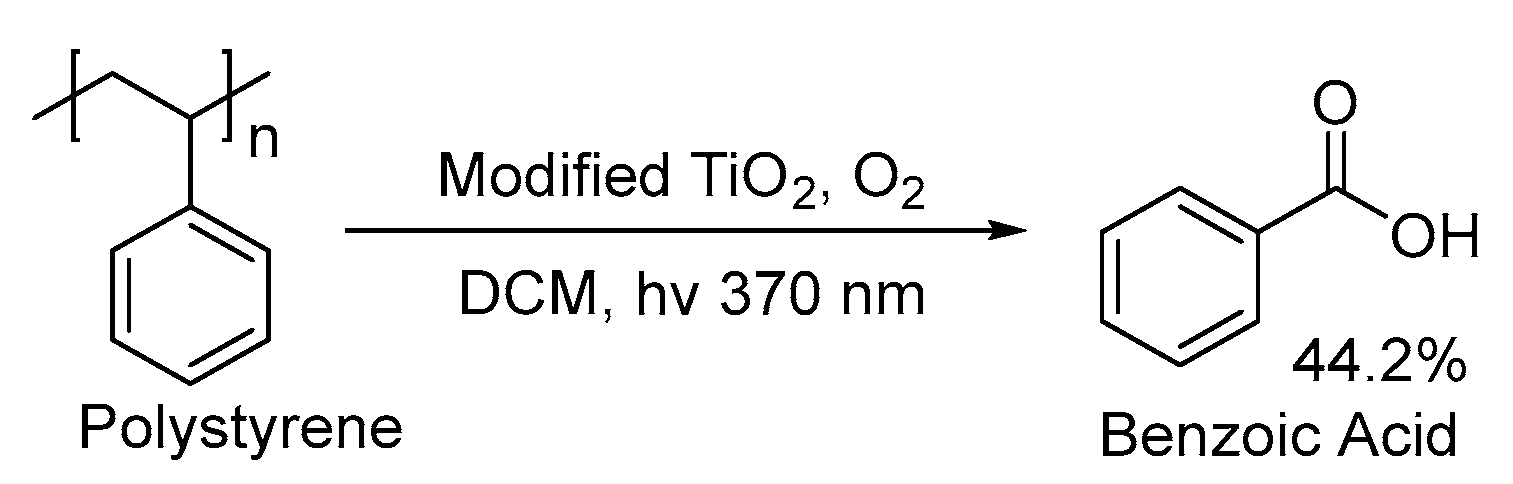
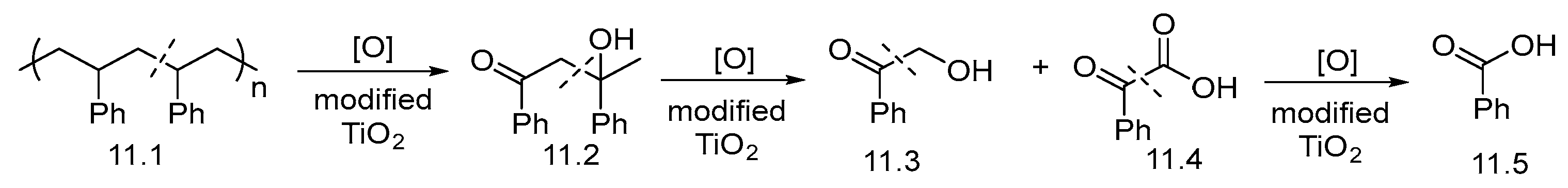
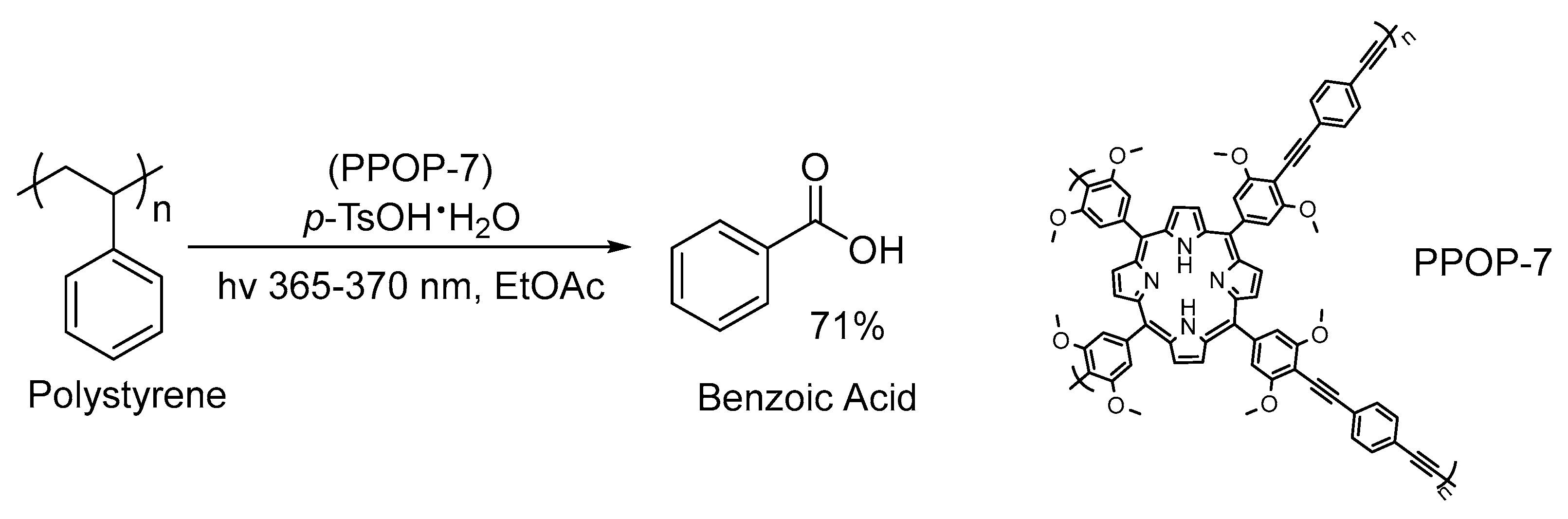
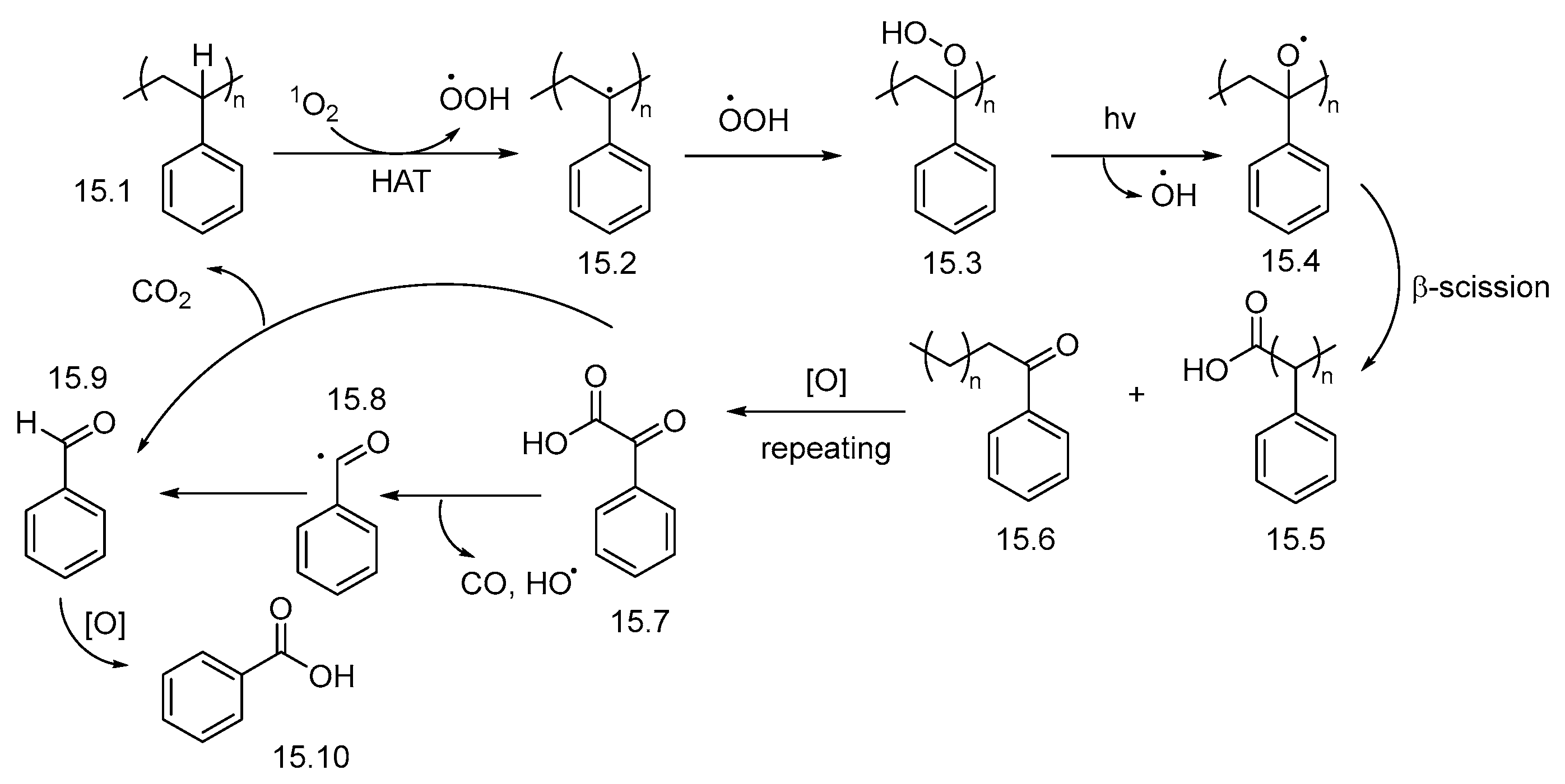
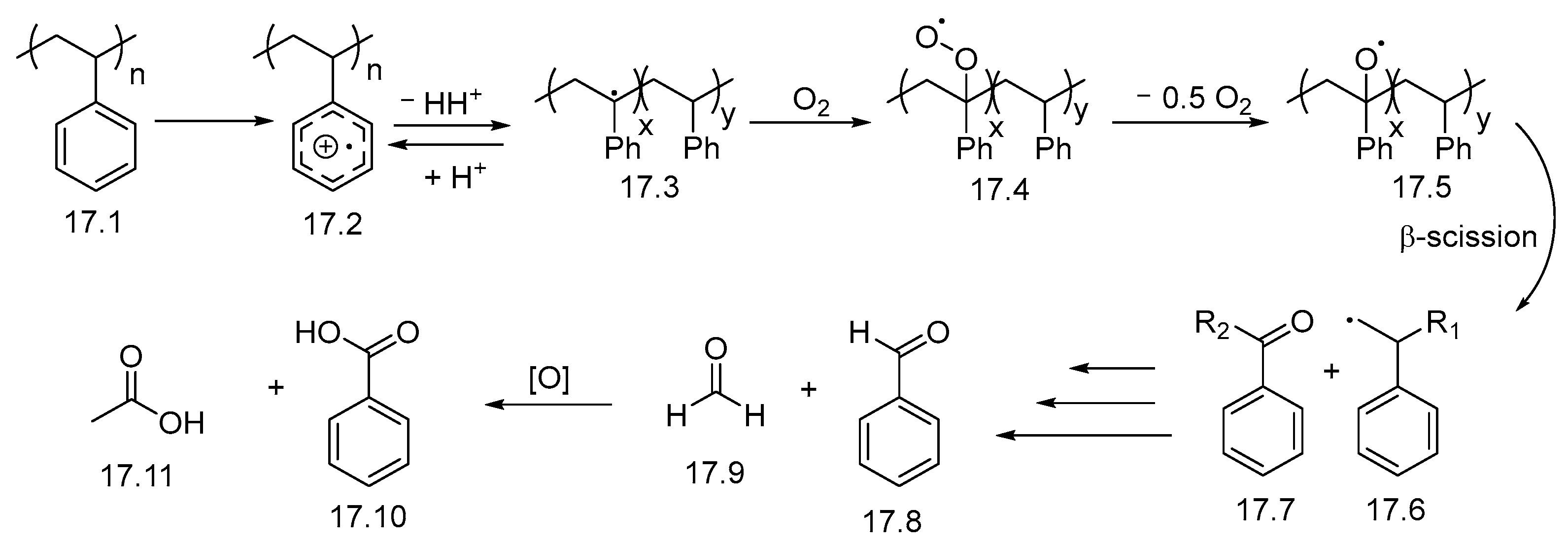
2.2. UV-Light-Induced Photocatalytic Upcycling
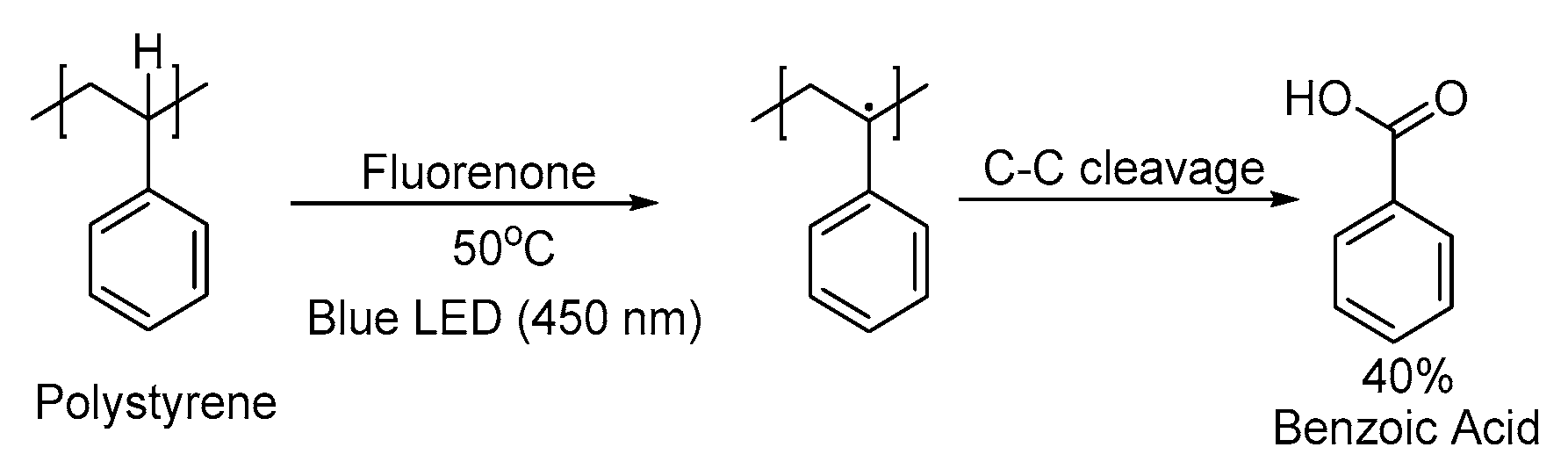
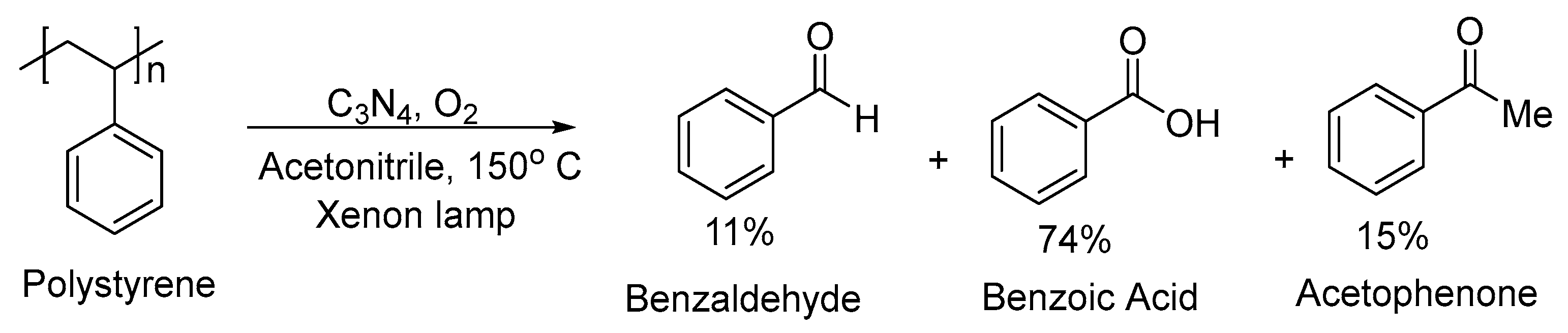
2.3. Broad-Spectrum Light Sources
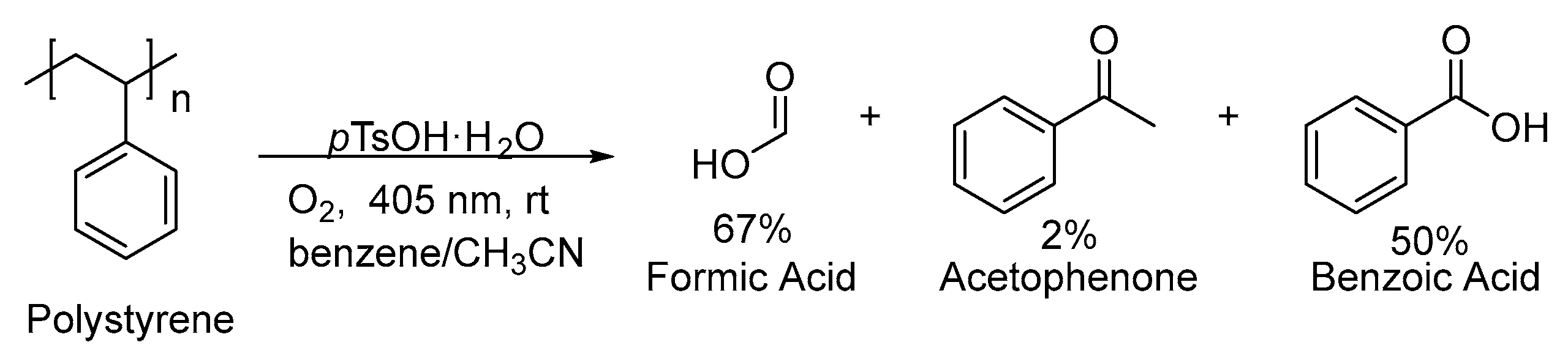
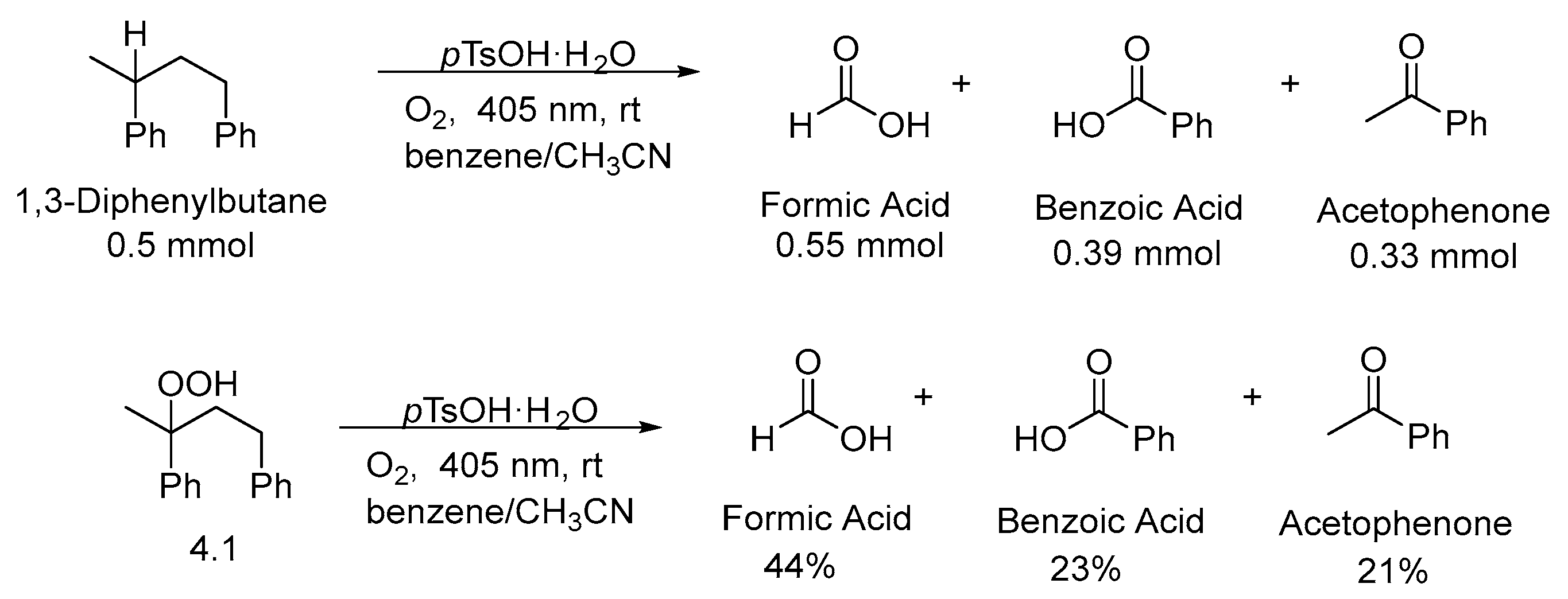
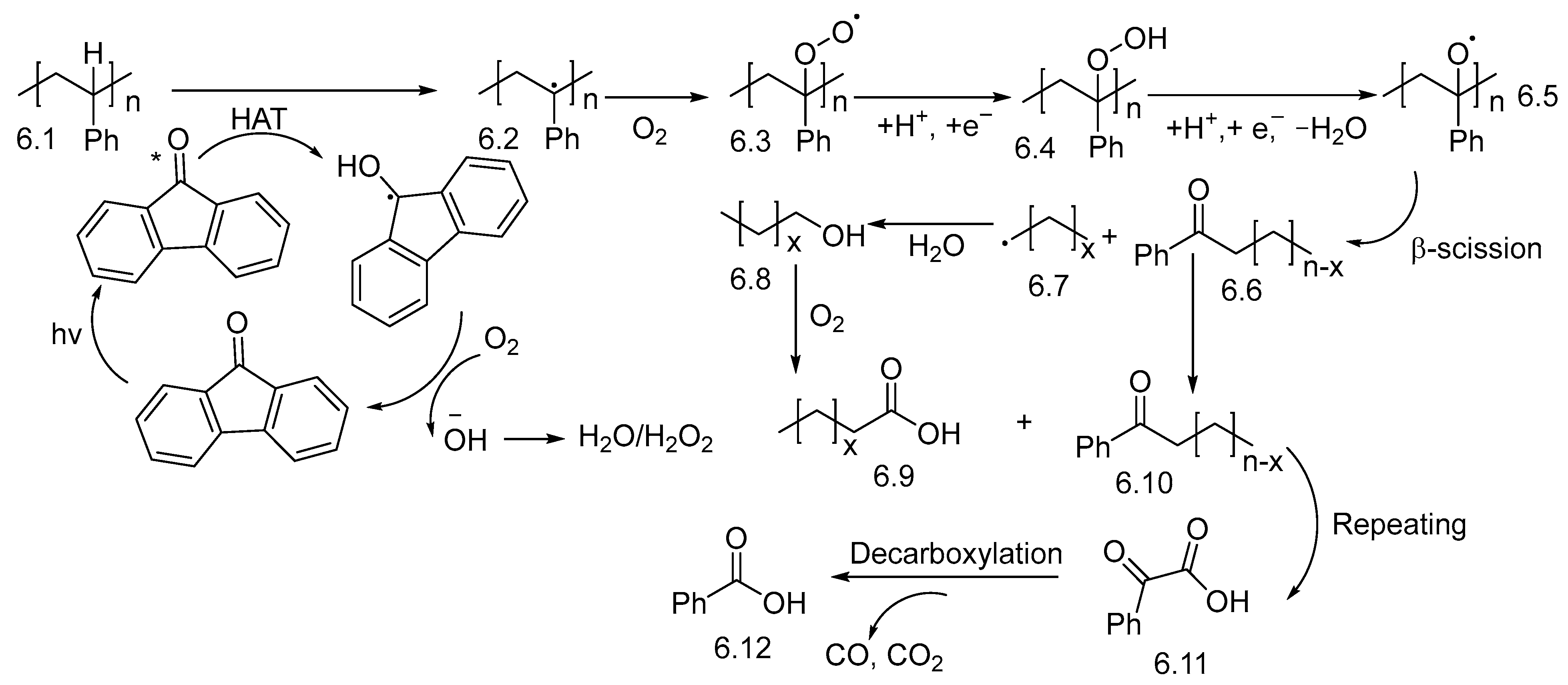
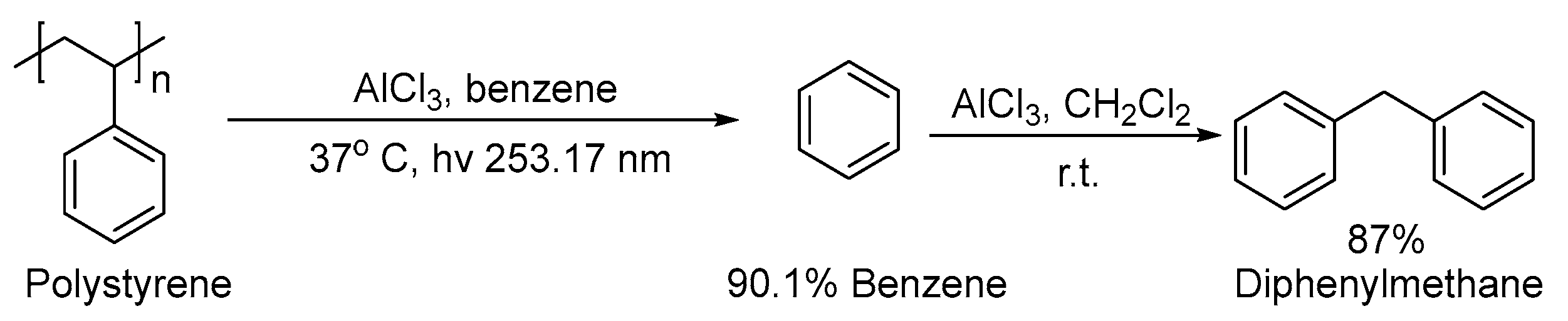
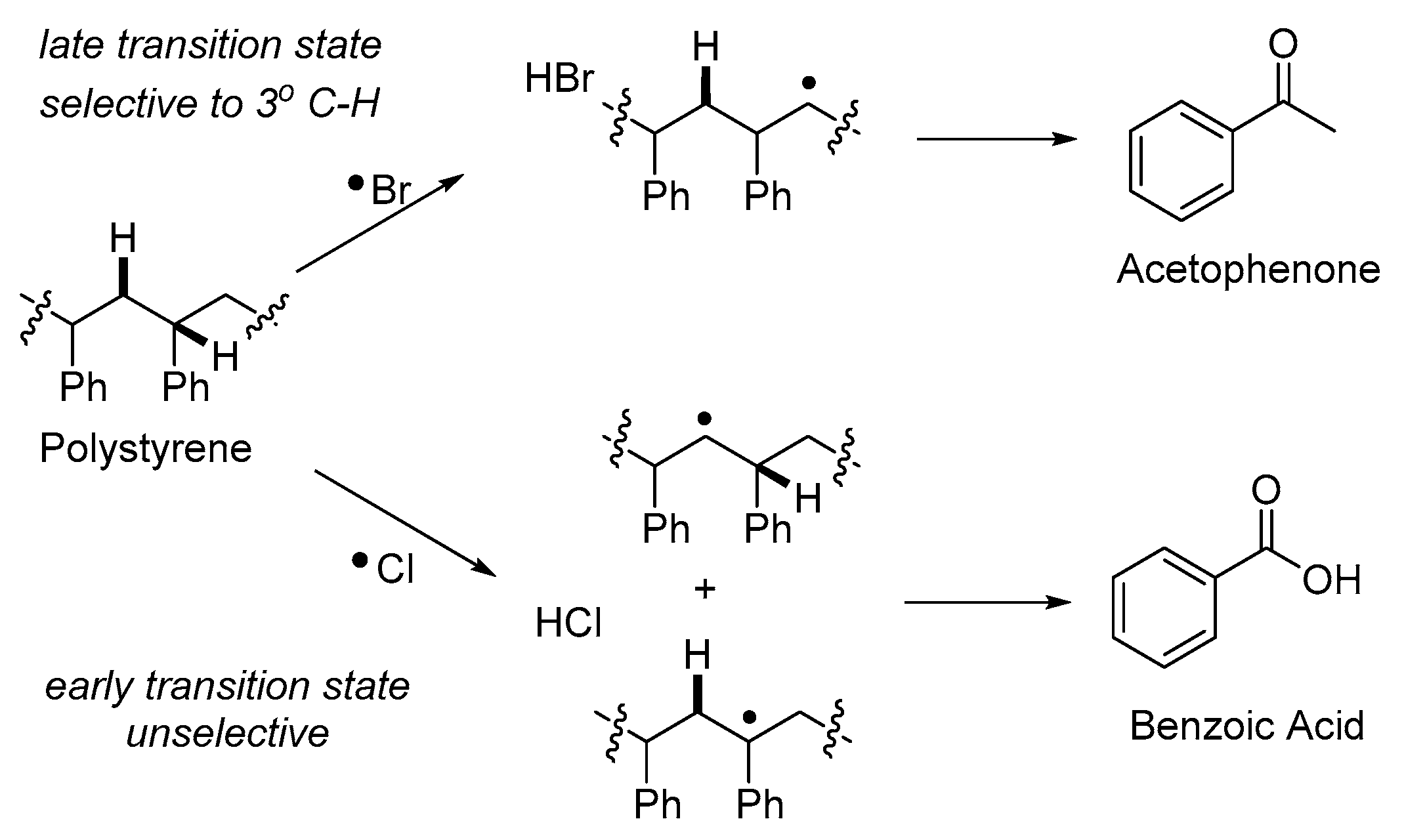
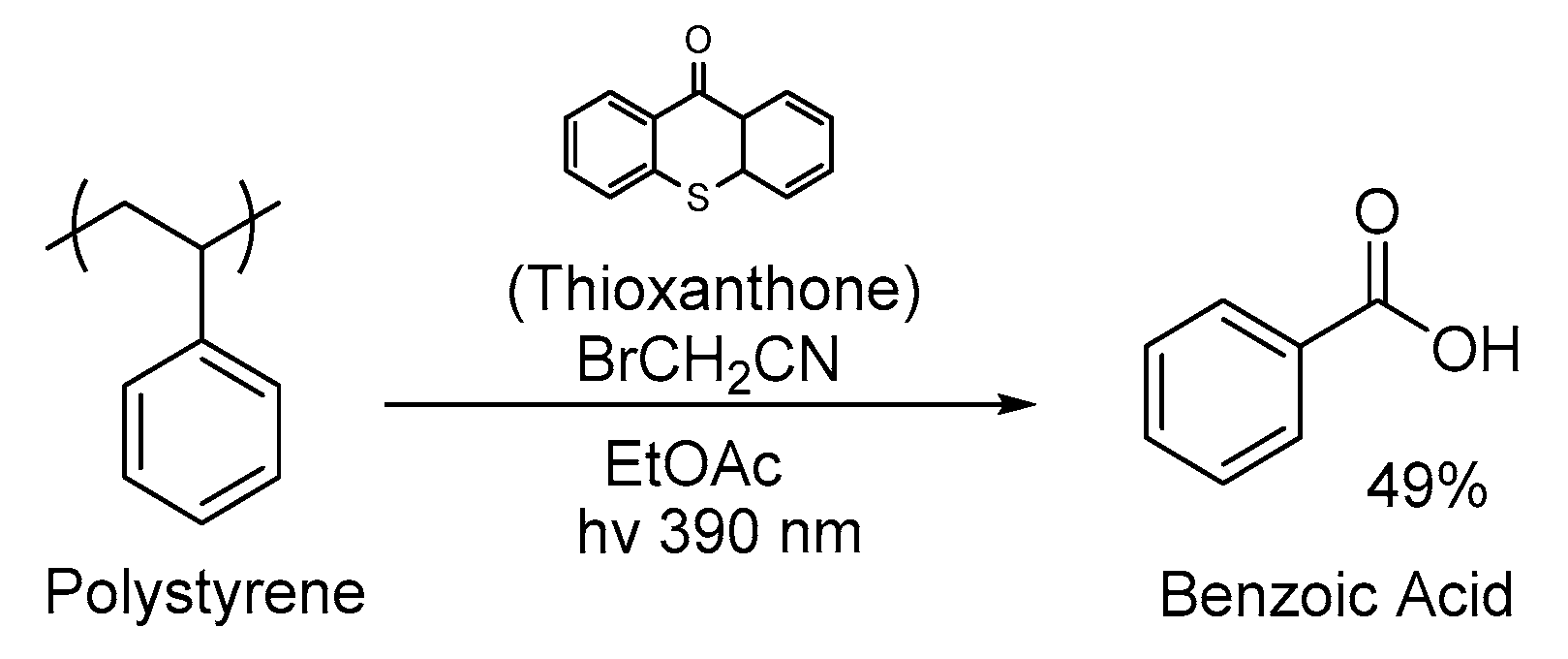
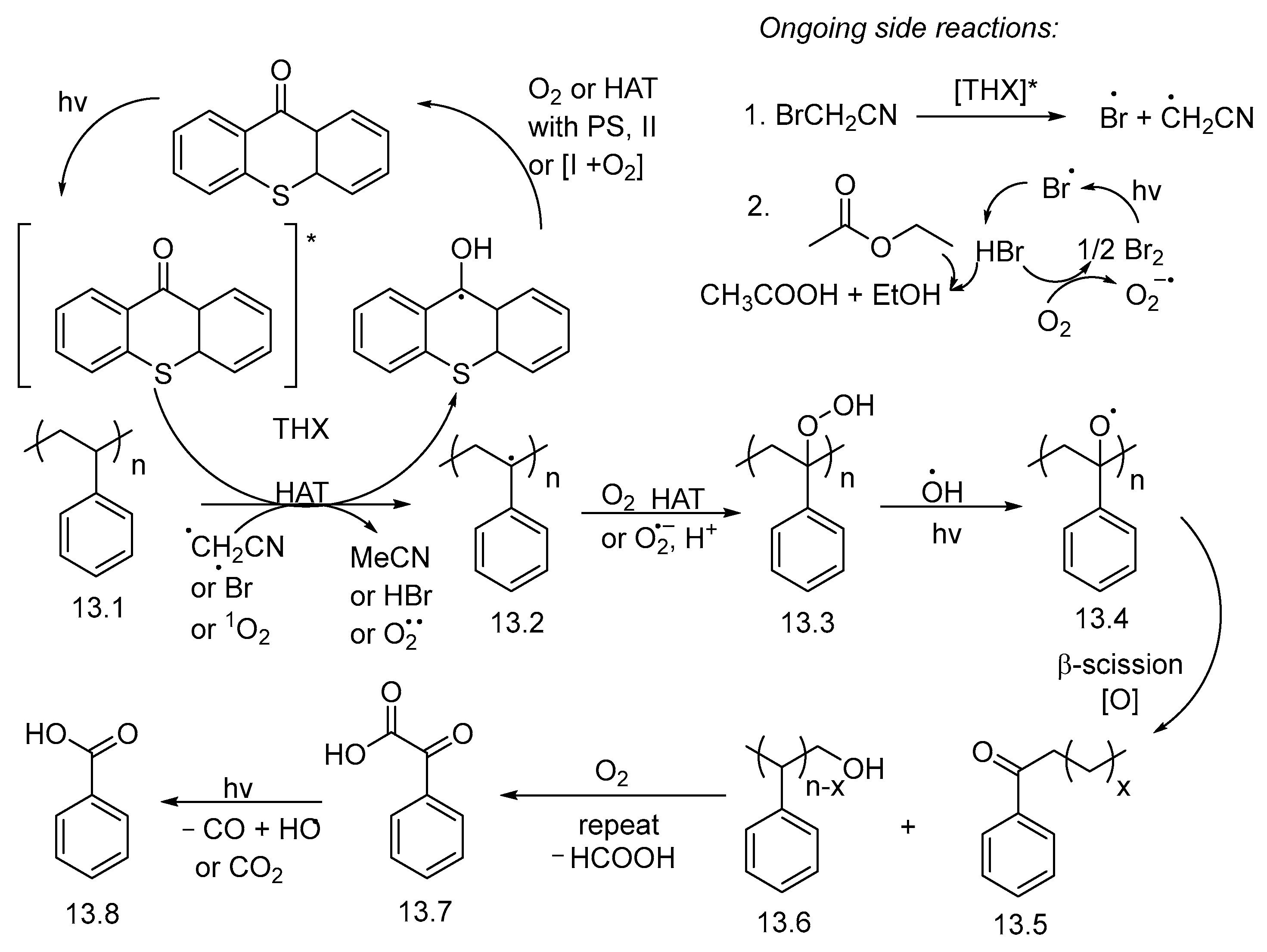
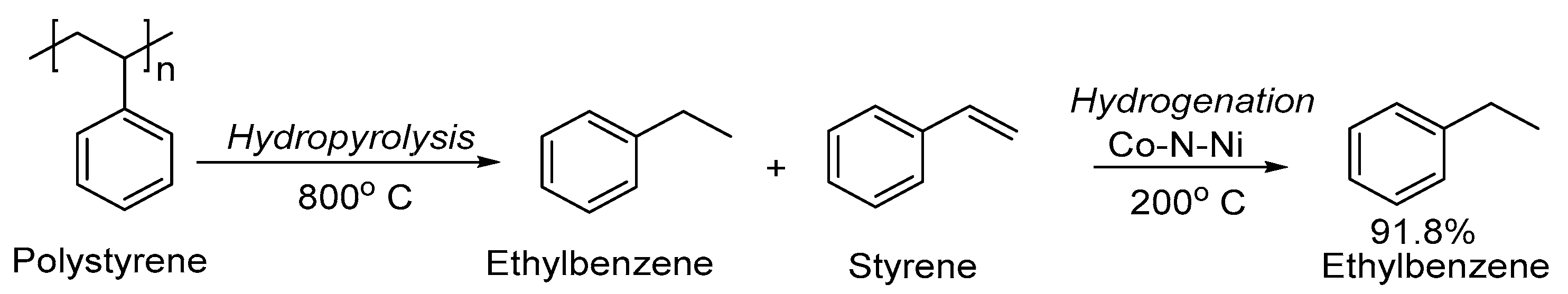
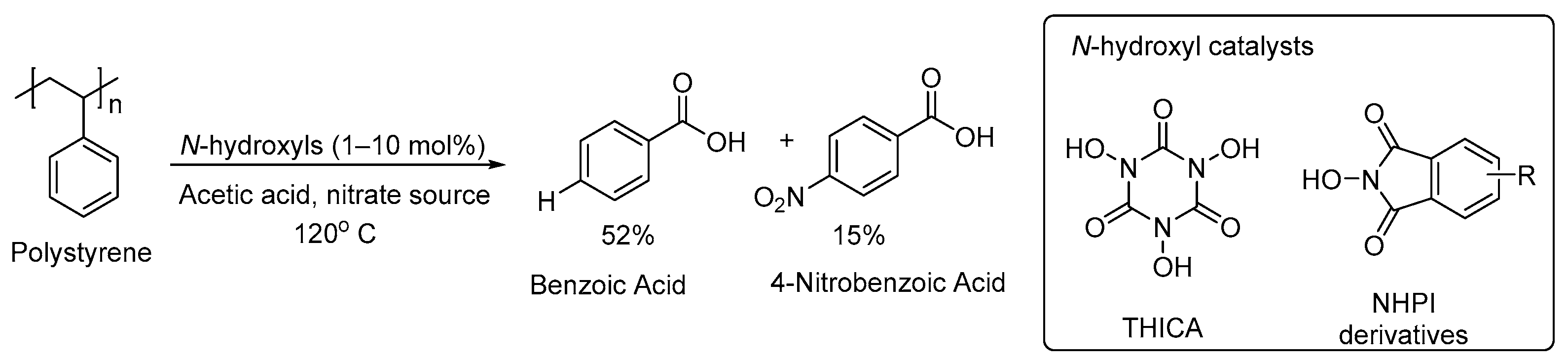
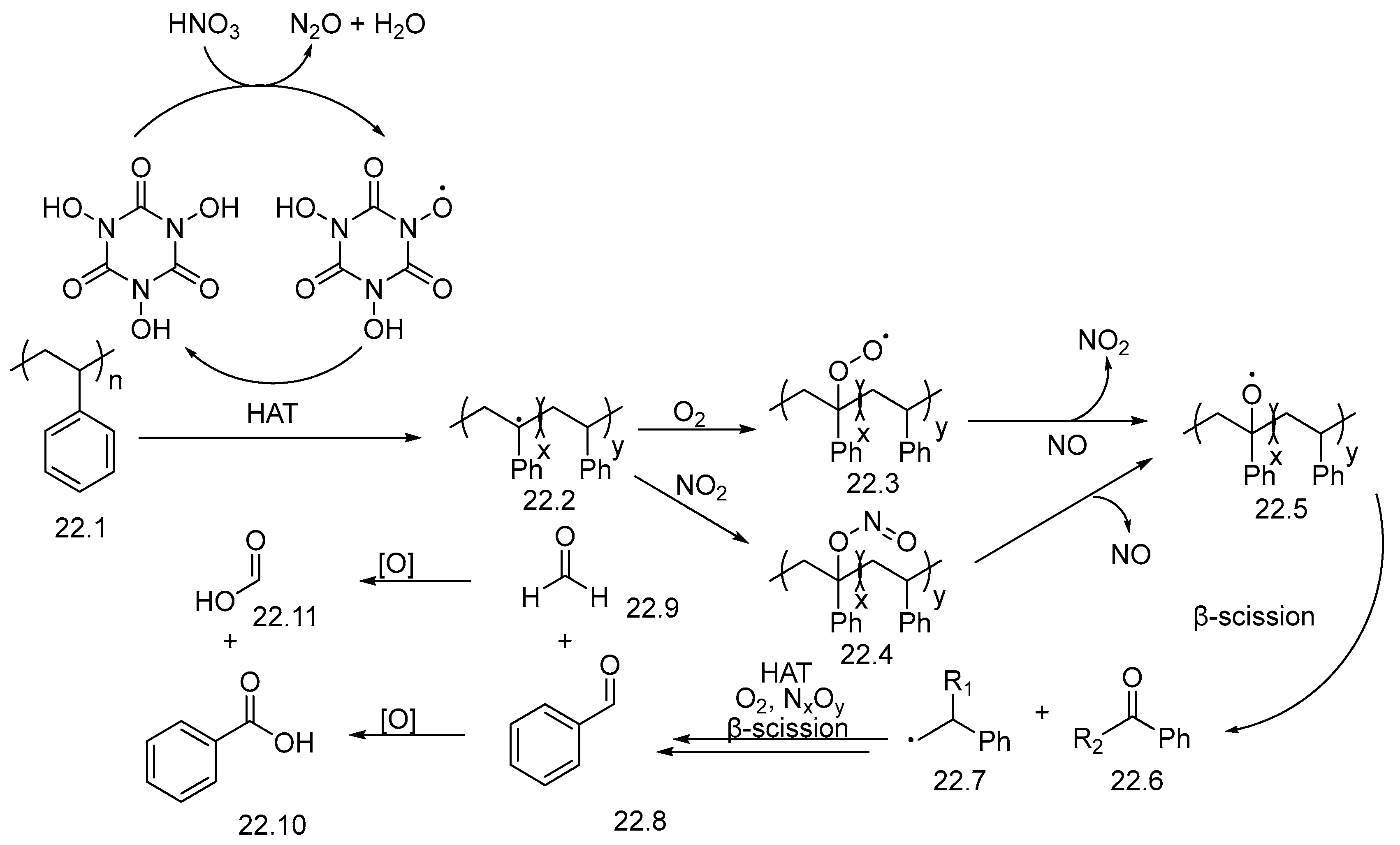
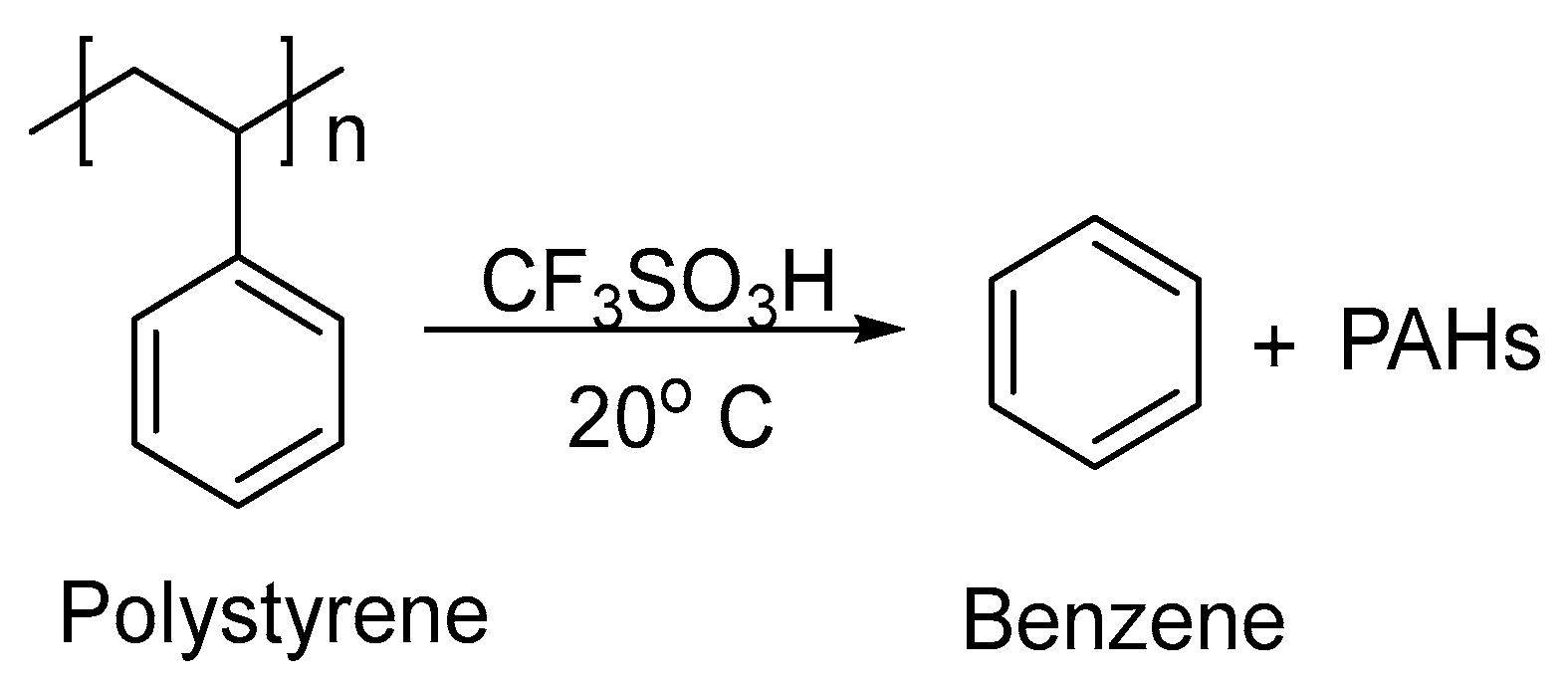
3. Non-Photocatalytic Upcycling
4. Conclusions
Funding
Conflicts of Interest
References
- Scheirs, J.; Priddy, D.B. Modern Styrenic Polymers: Polystyrenes and Styrenic Copolymers; Wiley: West Sussex, UK, 2003. [Google Scholar]
- Priddy, D.B. Styrene polymers. In Encyclopedia of Polymer Science and Technology, 4th ed.; Mark, H.F., Ed.; Wiley: Hoboken, NJ, USA, 2002; pp. 247–336. [Google Scholar]
- Assaad, J.J.; El Mir, A. Durability of polymer-modified lightweight flowable concrete made using expanded polystyrene. Constr. Build. Mater. 2020, 249, 118764–118779. [Google Scholar] [CrossRef]
- Yeung, C.W.S.; Teo, J.Y.Q.; Loh, X.J.; Lim, J.Y.C. Polyolefins and Polystyrene as Chemical Resources for a Sustainable Future: Challenges, Advances, and Prospects. ACS Mater. Lett. 2021, 3, 1660–1676. [Google Scholar] [CrossRef]
- Global Thermoplastic Production by Type 2050; Statista: New York, NY, USA, 2024.
- Jakab, E.; Uddin, M.A.; Bhaskar, T.; Sakata, Y. Thermal decomposition of flame-retarded high-impact polystyrene. J. Anal. Appl. Pyrolysis. 2003, 68–69, 83–99. [Google Scholar] [CrossRef]
- Schyns, Z.O.G.; Shaver, M.P. Mechanical Recycling of Packaging Plastics: A Review. Macromol. Rapid Commun. 2020, 42, 2000415. [Google Scholar] [CrossRef] [PubMed]
- Maafa, L. Pyrolysis of polystyrene waste: A review. Polymers 2021, 13, 225. [Google Scholar] [CrossRef]
- Inayat, A.; Fasolini, A.; Basile, F.; Fridrichova, D.; Lestinsky, P. Chemical recycling of waste polystyrene by thermo-catalytic pyrolysis: A description for different feedstocks, catalysts and operation modes. Polym. Degrad. Stab. 2022, 201, 109981. [Google Scholar] [CrossRef]
- Zhuo, C.; Levendis, Y.A. Upcycling Waste Plastics into Carbon Nanomaterials: A Review. J. Appl. Polym. Sci. 2013, 131, 39931. [Google Scholar] [CrossRef]
- Gautam, B.; Tsai, T.-H.; Chen, J.-T. Towards Sustainable Solutions: A Review of Polystyrene Upcycling and Degradation Techniques. Polym. Degrad. Stab. 2024, 225, 110779. [Google Scholar] [CrossRef]
- Ran, J.; Talebian-Kiakalaieh, A.; Zhang, S.; Hashem, E.M.; Guo, M.; Qiao, S.-Z. Recent Advancement on Photocatalytic Plastic Upcycling. Chem. Sci. 2024, 15, 1611–1637. [Google Scholar] [CrossRef]
- Mountanea, O.G.; Skolia, E.; Kokotos, C.G. Photochemical Upcycling and Recycling of Plastics: Achievements and Future Opportunities. Green Chem. 2024, 26, 8528–8549. [Google Scholar] [CrossRef]
- Wimberger, L.; Ng, G.; Boyer, C. Light-driven polymer recycling to monomers and small molecules. Nat. Commun. 2024, 15, 2510. [Google Scholar] [CrossRef]
- Capricho, J.C.; Prasad, K.; Hameed, N.; Nikzad, M.; Slim, N. Upcycling Polystyrene. Polymers 2022, 14, 5010. [Google Scholar] [CrossRef] [PubMed]
- Grassie, N.; Weir, N.A. The Photooxidation of Polymers. II. Photolysis of Polystyrene. J. Appl. Polym. Sci. 1965, 9, 975–986. [Google Scholar] [CrossRef]
- Grassie, N.; Weir, N.A. The Photooxidation of Polymers. III. Photooxidation of Polystyrene. J. Appl. Polym. Sci. 1965, 9, 987–998. [Google Scholar] [CrossRef]
- Grassie, N.; Weir, N.A. The Photooxidation of Polymers. IV. A Note on the Coloration of Polystyrene. J. Appl. Polym. Sci. 1965, 9, 999–1003. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Basak, G.C. Studies on photocatalytic degradation of polystyrene. Mater. Sci. Technol. 2007, 23, 307–314. [Google Scholar] [CrossRef]
- Kaczmarek, H.; Kaminska, A.; Swiatek, M.; Sanyal, S. Photoinitiated degradation of polystyrene in the presence of lowmolecular organic compounds. Eur. Polym. J. 2000, 36, 1167–1173. [Google Scholar] [CrossRef]
- Yousif, E.; Haddad, R. Photodegradation and photostabilization of polymers, especially polystyrene: Review. SpringerPlus 2013, 2, 398. [Google Scholar] [CrossRef]
- Wang, M.; Wen, J.; Huang, Y.; Hu, P. Selective Degradation of Styrene-Related Plastics Catalyzed by Iron under Visible Light. ChemSusChem 2021, 14, 5049–5056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Z.; Zeng, R. Photoinduced FeCl3-Catalyzed Alkyl Aromatics Oxidation toward Degradation of Polystyrene at Room Temperature. Chin. J. Chem. 2021, 39, 3225–3230. [Google Scholar] [CrossRef]
- Oh, S.; Stache, E.E. Chemical Upcycling of Commercial Polystyrene via Catalyst-Controlled Photooxidation. J. Am. Chem. Soc. 2022, 144, 5745–5749. [Google Scholar] [CrossRef]
- Shang, J.; Chai, M.; Zhu, Y. Photocatalytic Degradation of Polystyrene Plastic under Fluorescent Light. Environ. Sci. Technol. 2003, 37, 4494–4499. [Google Scholar] [CrossRef] [PubMed]
- Rohe, S.; Morris, A.O.; McCallum, T.; Barriault, L. Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation. Angew. Chem. Int. Ed. 2018, 57, 15664–15669. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, Z.; Liang, X.; Shen, J.; Wang, J.; Sun, W.; Wang, D.; Jiang, J.; Li, Y. Polystyrene Waste Thermochemical Hydrogenation to Ethylbenzene by a N-Bridged Co, Ni Dual-Atom Catalyst. J. Am. Chem. Soc. 2023, 145, 16218–16227. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shanmugam, M.; Liu, Z.; Brookfield, A.; Bennett, E.L.; Guan, R.; Vega Herrera, D.E.; Lopez-Sanchez, J.A.; Slater, A.G.; McInnes, E.J.L.; et al. Chemical Recycling of Polystyrene to Valuable Chemicals via Selective Acid-Catalyzed Aerobic Oxidation under Visible Light. J. Am. Chem. Soc. 2022, 144, 6532–6542. [Google Scholar] [CrossRef]
- Sagadevan, A.; Hwang, K.C.; Su, M.-D. Singlet Oxygen-Mediated Selective C–H Bond Hydroperoxidation of Ethereal Hydrocarbons. Nat. Commun. 2017, 8, 1812. [Google Scholar] [CrossRef]
- Li, T.; Vijeta, A.; Casadevall, C.; Gentleman, A.S.; Euser, T.; Reisner, E. Bridging Plastic Recycling and Organic Catalysis: Photocatalytic Deconstruction of Polystyrene via a C–H Oxidation Pathway. ACS Catal. 2022, 12, 8155–8163. [Google Scholar] [CrossRef]
- Xia, J.-B.; Zhu, C.; Chen, C. Visible Light-Promoted Metal-Free C–H Activation: Diarylketone-Catalyzed Selective Benzylic Mono- and Difluorination. J. Am. Chem. Soc. 2013, 135, 17494–17500. [Google Scholar] [CrossRef]
- Xu, Z.; Pan, F.; Sun, M.; Xu, J.; Munyaneza, N.E.; Croft, Z.L.; Cai, G.; Liu, G. Cascade Degradation and Upcycling of Polystyrene Waste to High-Value Chemicals. Proc. Natl. Acad. Sci. USA 2022, 119, e2203346119. [Google Scholar] [CrossRef]
- Oh, S.; Stache, E.E. Mechanistic Insights Enable Divergent Product Selectivity in Catalyst-Controlled Photooxidative Degradation of Polystyrene. ACS Catal. 2023, 13, 10968–10975. [Google Scholar] [CrossRef]
- Gonzalez, M.I.; Gygi, D.; Qin, Y.; Zhu, Q.; Johnson, E.J.; Chen, Y.-S.; Nocera, D.G. Taming the Chlorine Radical: Enforcing Steric Control over Chlorine-Radical-Mediated C–H Activation. J. Am. Chem. Soc. 2022, 144, 1464–1472. [Google Scholar] [CrossRef]
- Zhao, J.; Nanjo, T.; de Lucca, E.C.; White, M.C. Chemoselective Methylene Oxidation in Aromatic Molecules. Nat. Chem. 2018, 11, 213–221. [Google Scholar] [CrossRef]
- Jia, P.; Li, Q.; Poh, W.C.; Jiang, H.; Liu, H.; Deng, H.; Wu, J. Light-Promoted Bromine-Radical-Mediated Selective Alkylation and Amination of Unactivated C(Sp3)–H Bonds. Chem 2020, 6, 1766–1776. [Google Scholar] [CrossRef]
- Peng, Z.; Chen, R.; Li, H. Heterogeneous Photocatalytic Oxidative Cleavage of Polystyrene to Aromatics at Room Temperature. ACS Sustain. Chem. Eng. 2023, 11, 10688–10697. [Google Scholar] [CrossRef]
- Xu, S.; Liu, S.; Song, W.; Zheng, N. Metal-Free Upcycling of Plastic Waste: Photo-Induced Oxidative Degradation of Polystyrene in Air. Green Chem. 2024, 26, 1363–1369. [Google Scholar] [CrossRef]
- Ong, A.; Wong, Z.C.; Osmund, L.; Loh, W.W.; Chua, M.H.; Ang, S.J.; Lim, J.Y.C. Enhancing the Photocatalytic Upcycling of Polystyrene to Benzoic Acid: A Combined Computational-Experimental Approach for Acridinium Catalyst Design. Chem. Sci. 2023, 15, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhang, M.-Q.; Hu, C.; Xiao, D.; Wang, M.; Ma, D. Catalytic Oxidation of Polystyrene to Aromatic Oxygenates over a Graphitic Carbon Nitride Catalyst. Nat. Commun. 2022, 13, 4809. [Google Scholar] [CrossRef]
- Li, R.; Wang, D. Understanding the Structure-Performance Relationship of Active Sites at Atomic Scale. Nano Res. 2022, 15, 6888–6923. [Google Scholar] [CrossRef]
- Ong, A.; Teo, J.Y.Q.; Feng, Z.; Tan, T.T.Y.; Lim, J.Y.C. Organocatalytic Aerobic Oxidative Degradation of Polystyrene to Aromatic Acids. ACS Sustain. Chem. Eng. 2023, 11, 12514–12522. [Google Scholar] [CrossRef]
- Melone, L.; Punta, C. Metal-Free Aerobic Oxidations Mediated by N-Hydroxyphthalimide. A Concise Review. Beilstein J. Org. Chem. 2013, 9, 1296–1310. [Google Scholar] [CrossRef] [PubMed]
- Recupero, F.; Punta, C. Free Radical Functionalization of Organic Compounds Catalyzed by N-Hydroxyphthalimide. Chem. Rev. 2007, 107, 3800–3842. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, S.; Nishiwaki, Y.; Sakaguchi, S.; Ishii, Y. Nitration of Alkanes with Nitric Acid Catalyzed by N-Hydroxyphthalimide. Chem. Commun. 2001, 1352–1353. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Shibamoto, A.; Ishii, Y. Remarkable Effect of Nitrogen Dioxide for N-Hydroxyphthalimide-Catalyzed Aerobic Oxidation of Methylquinolines. Chem. Commun. 2002, 2, 180–181. [Google Scholar] [CrossRef] [PubMed]
- Thigale, A.; Trant, C.; Ahn, J.; Nelson, M.; Ryu, C.Y.; Bae, C.; Lee, S. Superacid-Assisted Degradation of Polystyrene. Macromolecules 2023, 56, 5117–5126. [Google Scholar] [CrossRef]

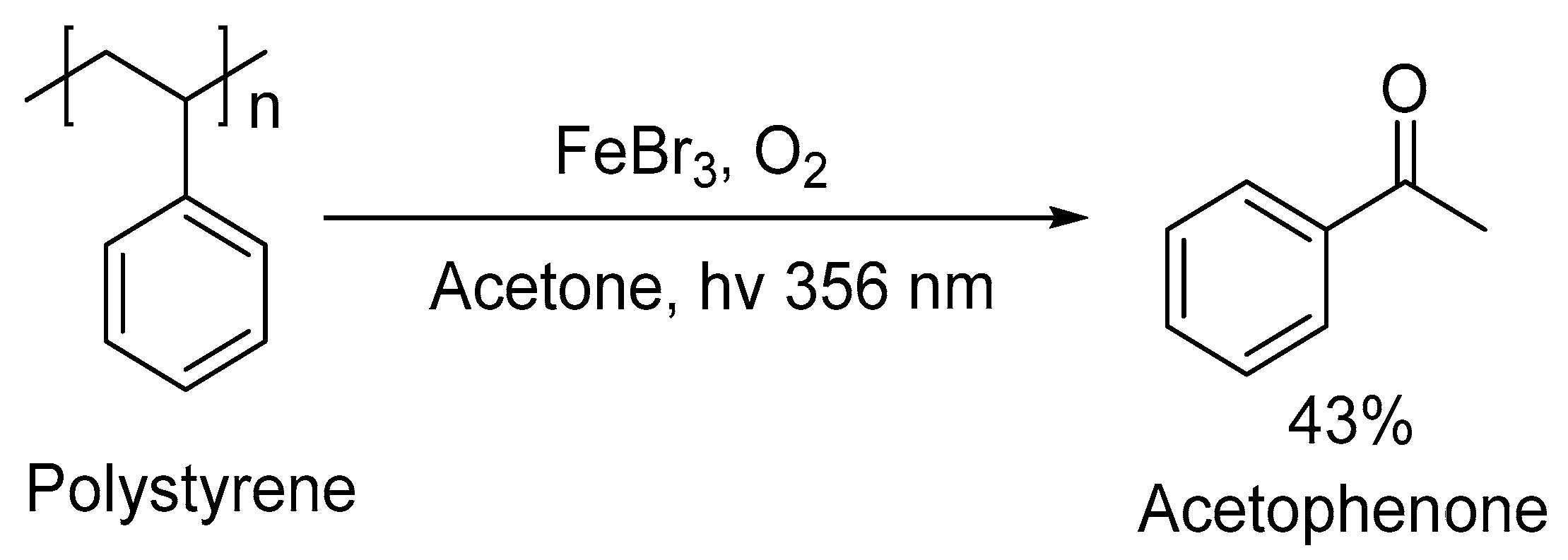


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.; Xing, Y. Polystyrene Upcycling via Photocatalytic and Non-Photocatalytic Degradation. Molecules 2025, 30, 3165. https://doi.org/10.3390/molecules30153165
Yang T, Xing Y. Polystyrene Upcycling via Photocatalytic and Non-Photocatalytic Degradation. Molecules. 2025; 30(15):3165. https://doi.org/10.3390/molecules30153165
Chicago/Turabian StyleYang, Terry, and Yalan Xing. 2025. "Polystyrene Upcycling via Photocatalytic and Non-Photocatalytic Degradation" Molecules 30, no. 15: 3165. https://doi.org/10.3390/molecules30153165
APA StyleYang, T., & Xing, Y. (2025). Polystyrene Upcycling via Photocatalytic and Non-Photocatalytic Degradation. Molecules, 30(15), 3165. https://doi.org/10.3390/molecules30153165