
Homo- Versus Hetero- [2+2+2] Rhodium-Catalyzed Cycloaddition: Effect of a Self-Assembled Capsule on the Catalytic Outcome


Abstract
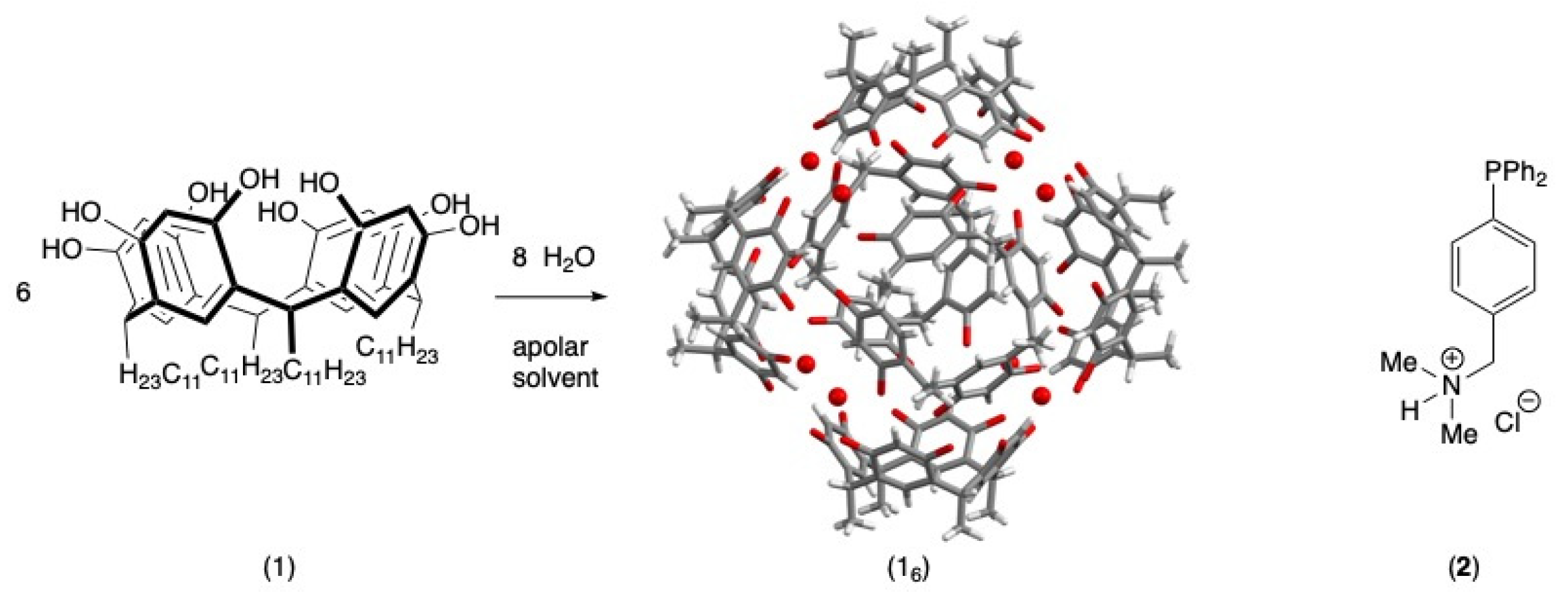
1. Introduction
2. Results and Discussion
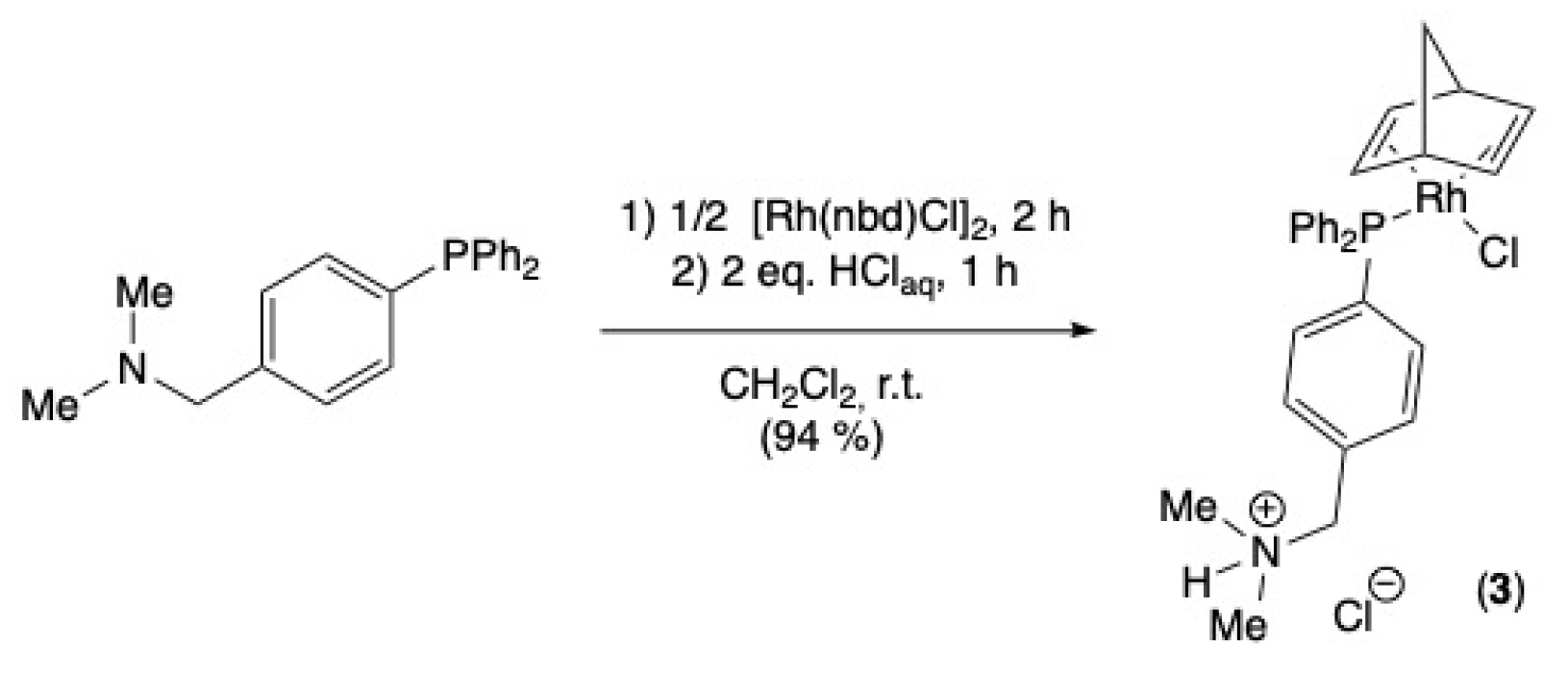
2.1. Synthesis of the Rhodium (I) Complex
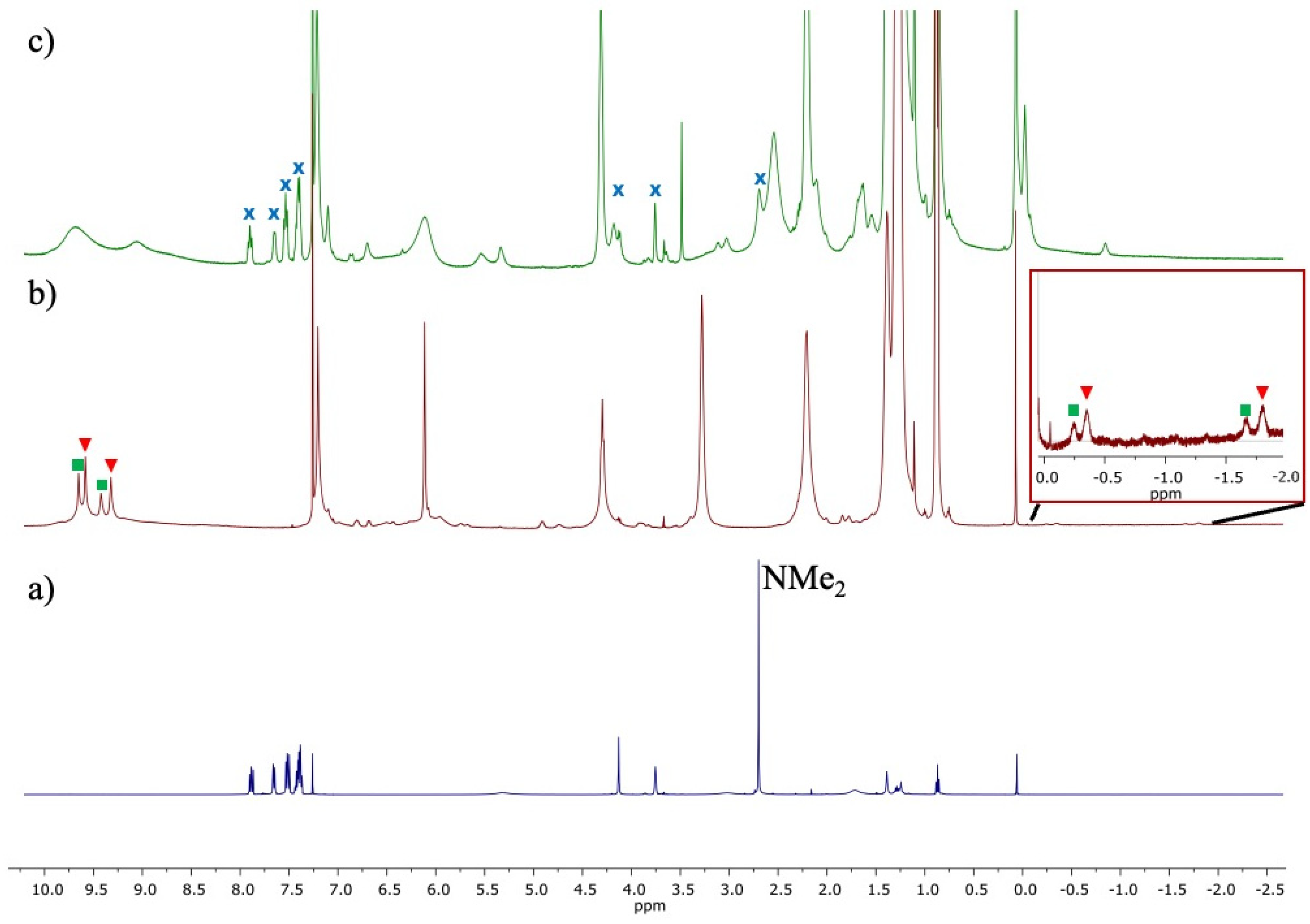
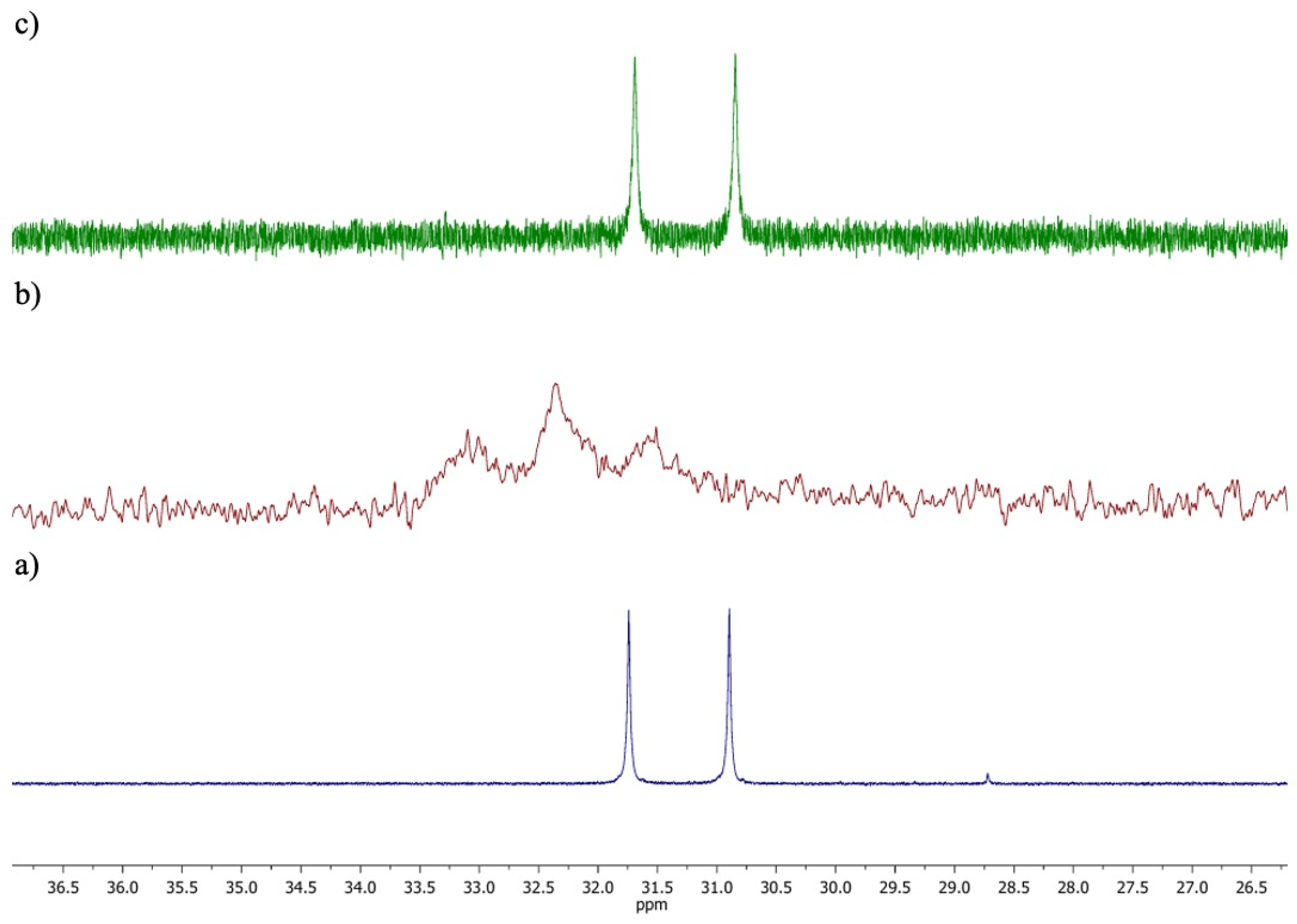
2.2. Formation of the Inclusion Complex
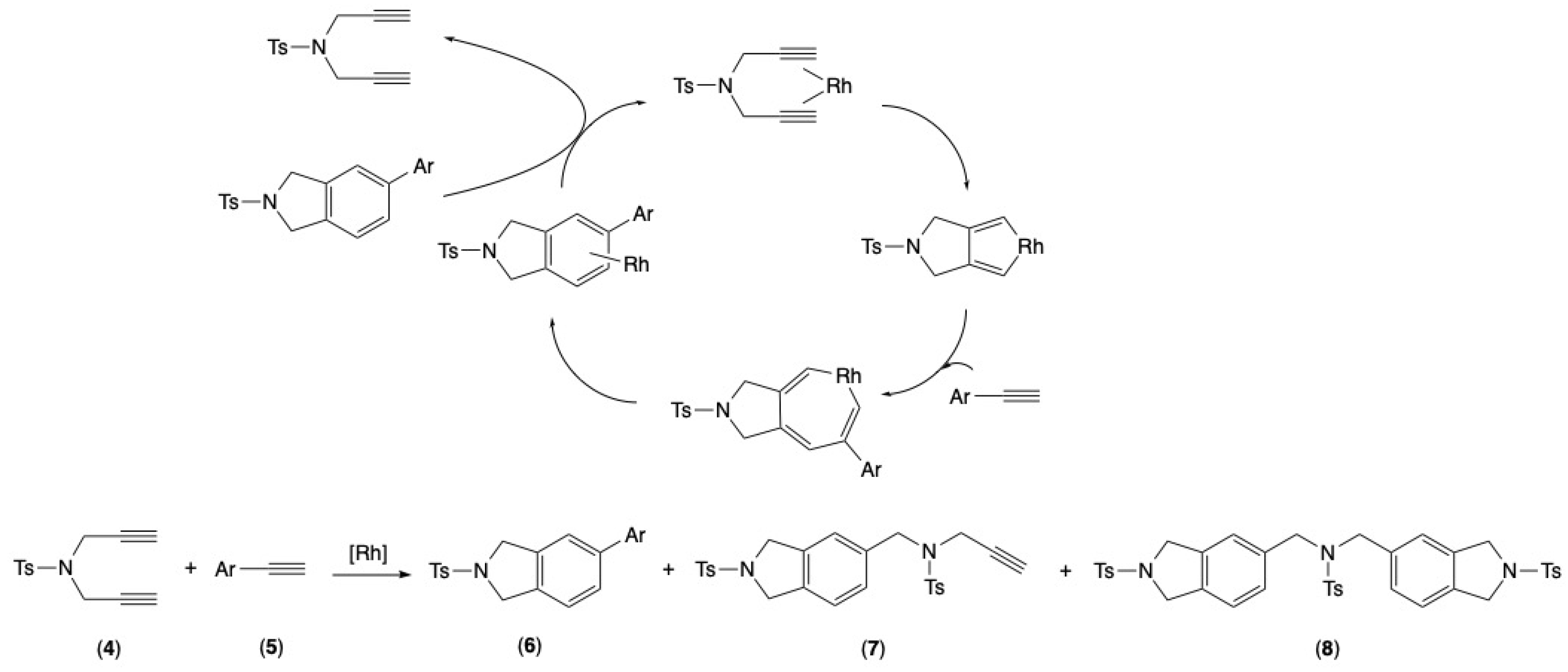
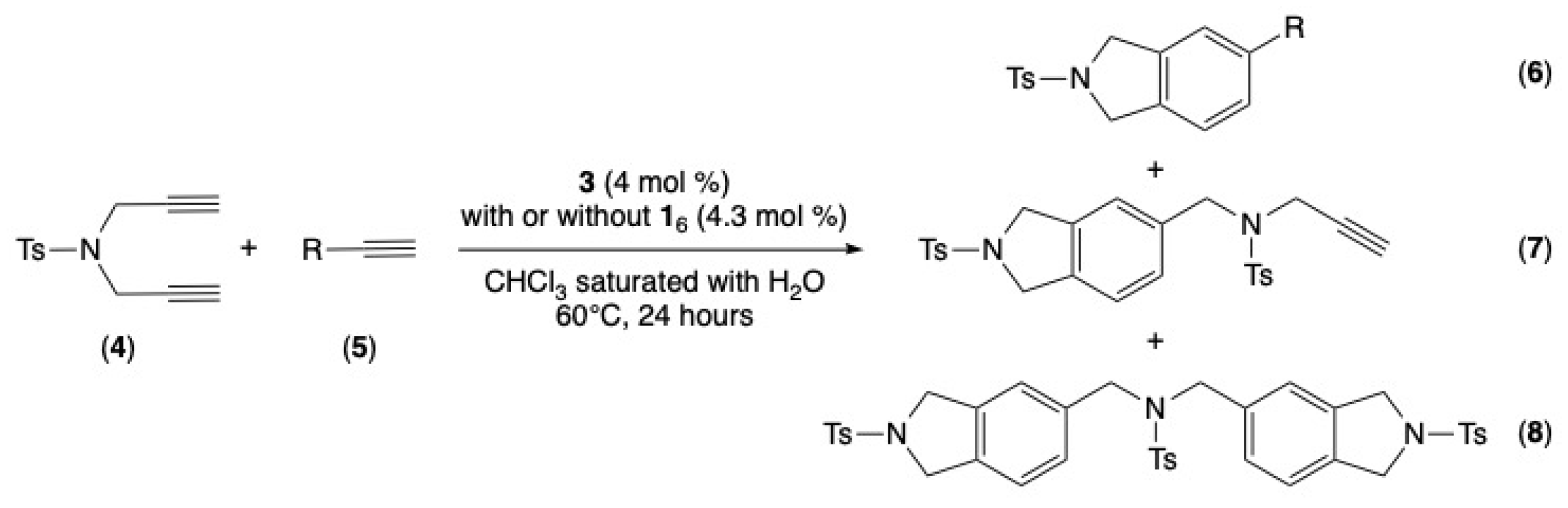
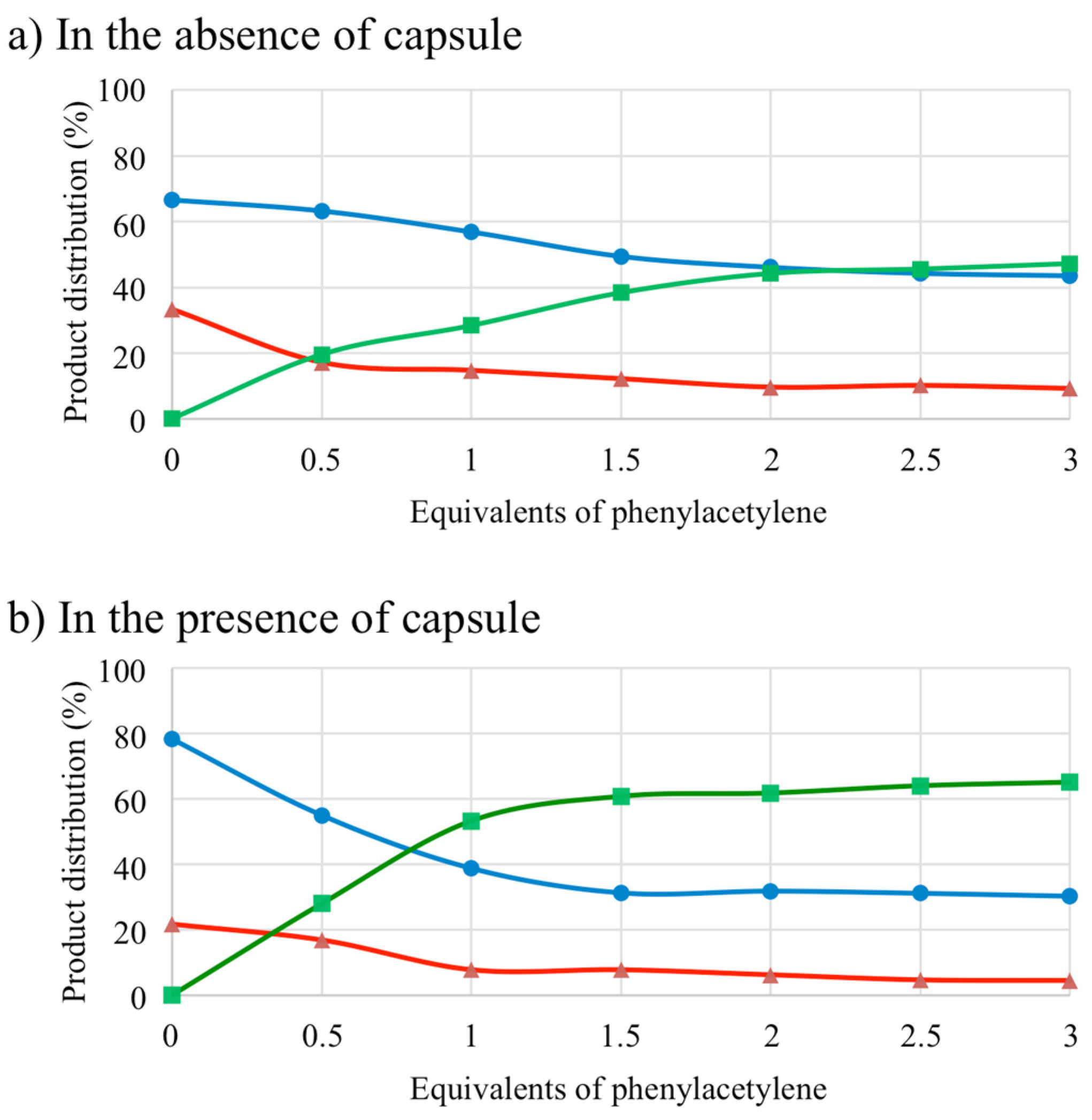
2.3. Rhodium-Catalyzed [2+2+2] Cycloaddition
3. Materials and Methods
3.1. General
3.2. Synthesis of Chloro-P-{[4-(Diphenylphosphanyl)phenyl]-N,N-dimethylmethanammonio} (Norbornadiene)rhodium(I) Chloride (3)
3.3. General Procedure for the Rhodium-Catalyzed Cycloaddition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diao, D.; Simaan, A.J.; Martinez, A.; Colomban, C. Bioinspired complexes confined in well-defined capsules: Getting closer to metalloenzyme functionalities. Chem. Commun. 2023, 59, 4288–4299. [Google Scholar] [CrossRef] [PubMed]
- Shteinman, A.A. Metallocavitins as advanced enzyme mimics and promising chemical catalysts. Catalysts 2023, 13, 415. [Google Scholar] [CrossRef]
- Li, W.-L.; Head-Gordon, T. Catalytic principles from natural enzymes and translational design strategies for synthetic catalysts. ACS Cent. Sci. 2021, 7, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, V.; Della Sala, P.; Talotta, C.; De Rosa, M.; Soriente, A.; Gaeta, C.; Neri, P. Supramolecular control on reactivity and selectivity inside the confined space of H-bonded hexameric capsules. Curr. Opin. Colloid Interface Sci. 2023, 65, 101692. [Google Scholar] [CrossRef]
- Olivo, G.; Capocasa, G.; Del Giudice, D.; Lanzalunga, O.; Di Stefano, S. New horizons for catalysis disclosed by supramolecular chemistry. Chem. Soc. Rev. 2021, 50, 7681–7724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Catti, L.; Tiefenbacher, K. Catalysis inside the hexameric resorcinarene capsule. Acc. Chem. Res. 2018, 51, 2107–2114. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, C.; Talotta, C.; De Rosa, M.; La Manna, P.; Soriente, A.; Neri, P. The hexameric resorcinarene capsule at work: Supramolecular catalysis in confined spaces. Chem. Eur. J. 2019, 25, 4899–4913. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, A.; Puglisi, R.; Sfrazzetto, G.T. Catalysis inside supramolecular capsules: Recent developments. Catalysts 2019, 9, 630. [Google Scholar] [CrossRef]
- Catti, L.; Zhang, Q.; Tiefenbacher, K. Advantages of catalysis in self-assembled molecular capsules. Chem. Eur. J. 2016, 22, 9060–9066. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetto, T.; Bordignon, F.; Munarin, L.; Mancin, F.; Fabris, F.; Scarso, A. Substrate selectivity imparted by self-assembled molecular containers and catalysts. Chem. Eur. J. 2024, 30, e202301811. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, M.; Sémeril, D. Molecular modeling is key to understanding supramolecular resorcinarenyl capsules, inclusion complex formation and organic reactions in nanoconfined space. Molecules 2025, 30, 2549. [Google Scholar] [CrossRef] [PubMed]
- MacGillivray, L.R.; Atwood, J.L. A chiral spherical molecular assembly held together by 60 hydrogen bonds. Nature 1997, 389, 469–472. [Google Scholar] [CrossRef]
- Yamanaka, M.; Shivanyuk, A.; Rebek, J., Jr. Kinetics and thermodynamics of hexameric capsule formation. J. Am. Chem. Soc. 2004, 126, 2939–2943. [Google Scholar] [CrossRef] [PubMed]
- Cavarzan, A.; Scarso, A.; Sgarbossa, P.; Strukul, G.; Reek, J.N.H. Supramolecular control on chemo- and regioselectivity via encapsulation of (NHC)-Au catalyst within a hexameric self-assembled host. J. Am. Chem. Soc. 2011, 133, 2848–2851. [Google Scholar] [CrossRef] [PubMed]
- Cavarzan, A.; Reek, J.N.H.; Trentin, F.; Scarso, A.; Strukul, G. Substrate selectivity in the alkyne hydration mediated by NHC-Au(I) controlled by encapsulation of the catalyst within a hydrogen bonded hexameric host. Catal. Sci. Technol. 2013, 3, 2898–2901. [Google Scholar] [CrossRef]
- Jans, A.C.H.; Gómez-Suárez, A.; Nolan, S.P.; Reek, J.N.H. A switchable gold catalyst by encapsulation in a self-assembled cage. Chem. Eur. J. 2016, 22, 14836–14839. [Google Scholar] [CrossRef] [PubMed]
- Jongkind, L.J.; Rahimi, M.; Poole III, D.; Ton, S.J.; Fogg, D.E.; Reek, J.N.H. Protection of ruthenium olefin metathesis catalysts by encapsulation in a self-assembled resorcinarene capsule. ChemCatChem 2020, 12, 4019–4023. [Google Scholar] [CrossRef]
- Adriaenssens, L.; Escribano-Cuesta, A.; Homs, A.; Echavarren, A.M.; Ballester, P. Encapsulation studies of cationic gold complexes within a self-assembled hexameric resorcin[4] arene capsule. Eur. J. Org. Chem. 2013, 2013, 1494–1500. [Google Scholar] [CrossRef]
- Philip, I.E.; Kaifer, A.E. Electrochemically driven formation of a molecular capsule around the ferrocenium ion. J. Am. Chem. Soc. 2002, 124, 12678–12679. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Yamaguchi, T.; Tessarolo, J.; Tanaka, H.; Sakuda, E.; Arikawa, Y.; Meggers, E.; Clever, G.H.; Umakoshi, K. Symmetry-breaking host-guest assembly in a hydrogen-bonded supramolecular system. Nat. Commun. 2023, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Philip, I.; Kaifer, A.E. Noncovalent encapsulation of cobaltocenium inside resorcinarene molecular capsules. J. Org. Chem. 2005, 70, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Scarso, A.; Sorella, G.L.; Strukul, G. Switching the activity of a photoredox catalyst through reversible encapsulation and release. Chem. Commun. 2012, 48, 12082. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Tanaka, H.; Sakuda, E.; Arikawa, Y.; Umakoshi, K. Encapsulation and enhanced luminescence properties of IrIII complexes within a hexameric self-assembled capsule. Chem. Eur. J. 2016, 22, 17533–17537. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Matsuo, C.; Sakuda, E.; Arikawa, Y.; Clever, G.H.; Umakoshi, K. Anion-mediated encapsulation-induced emission enhancement of an IrIII complex within a resorcin[4]arene hexameric capsule. Dalton Trans. 2020, 49, 8472–8477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Le Corre, L.; Reinaud, O.; Colasson, B. A promising approach for controlling the second coordination sphere of biomimetic metal complexes: Encapsulation in a dynamic hydrogen-bonded capsule. Chem. Eur. J. 2021, 27, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Chopade, P.R.; Louie, J. [2+2+2] Cycloaddition reactions catalyzed by transition metal complexes. Adv. Synth. Catal. 2006, 348, 2307–2327. [Google Scholar] [CrossRef]
- Pla-Quintana, A.; Roglans, A. [2+2+2] Cycloaddition reactions of macrocyclic systems catalyzed by transition metals. A review. Molecules 2010, 15, 9230–9251. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Tanaka, K. Rhodium-catalyzed [2+2+2] cycloaddition of alkynes for the synthesis of substituted benzenes: Catalysts, reaction scope, and synthetic applications. Synthesis 2012, 44, 323–350. [Google Scholar] [CrossRef]
- Transition-Metal-Mediated Aromatic Ring Construction; Tanaka, K., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Roglans, A.; Pla-Quintana, A.; Solà, M. Mechanistic Ssudies of transition-metal-catalyzed [2+2+2] cycloaddition reactions. Chem. Rev. 2021, 121, 1894–1979. [Google Scholar] [CrossRef] [PubMed]
- Pla-Quintana, A.; Roglans, A. The choice of rhodium catalysts in [2+2+2] cycloaddition reaction: A personal account. Molecules 2022, 27, 1332. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Li, W.; Chen, Y.; Wu, R.; Zhu, S. Dirhodium-catalyzed [2+2+2] cycloaddition of 1,6-diynes and alkynes. J. Org. Chem. 2024, 89, 17248–17259. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, X.Y.; Kang, S.X. Rhodium-catalyzed selective [2+2+2] cyclizations of 1,6-diynes with monoynes leading to isoindolines and isobenzofurans. Chin. Chem. Lett. 2010, 21, 18–22. [Google Scholar] [CrossRef]
- García-Lacuna, J.; Domínguez, G.; Blanco-Urgoiti, J.; Pérez-astells, J. Cobalt octacarbonyl-catalyzed scalable alkyne cyclotrimerization and crossed [2+2+2]-cycloaddition reaction in a plug flow reactor. Org. Lett. 2018, 20, 5219–5223. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Si, X.J.; Wang, X.N.; Kou, H.D.; Chen, D.M.; Liu, C.S.; Du, M. A high-activity cobalt-based MOF catalyst for [2+2+2] cycloaddition of diynes and alkynes: Insights into alkyne affinity and selectivity control. RSC Adv. 2018, 8, 4895–4899. [Google Scholar] [CrossRef] [PubMed]
- Féo, M.; Bakas, N.J.; Radović, A.; Parisot, W.; Clisson, A.; Chamoreau, L.M.; Haddad, M.; Ratovelomanana-Vidal, V.; Neidig, M.L.; Lefèvre, G. Thermally stable redox noninnocent bathocuproine-iron complex for cycloaddition reactions. ACS Catal. 2023, 13, 4882–4893. [Google Scholar] [CrossRef]
- Parisot, W.; Haddad, M.; Phansavath, P.; Lefèvre, G.; Ratovelomanana-Vidal, V. A versatile, functional group-tolerant, and bench-stable Iron precatalyst for building arene and triazine rings by [2+2+2] cycloadditions. Chem. Eur. J. 2024, 30, e202400096. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mao, Y.; Sun, Y.; Ma, B.; Wang, Y.; Zhou, G.; Zhang, Y.; Zeng, F.; Wang, Z.; Li, B. Bench-stable nickel(II)-aryl complex-catalyzed cyclotrimerisation of diynes and alkynes to synthesis multisubstituted benzene derivatives. Results Chem. 2024, 7, 101523. [Google Scholar] [CrossRef]
- Hkiri, S.; Steinmetz, M.; Schurhammer, R.; Sémeril, D. Encapsulated neutral ruthenium catalyst for substrate-selective oxidation of alcohols. Chem. Eur. J. 2022, 28, e202201887. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, M.; Sémeril, D. Confined catalysis involving a palladium complex and a self-assembled capsule for the dimerization of vinyl arenes and the formation of indane and tribenzo-pentaphene derivatives. Catalysts 2025, 15, 585. [Google Scholar] [CrossRef]
- Kawagoe, Y.; Moriyama, K.; Togo, H. Facile preparation of amides from carboxylic acids and amines with ion-supported Ph3P. Tetrahedron 2013, 69, 3971–3977. [Google Scholar] [CrossRef]
- Takenaka, Y.; Osakada, K. Preparation and structure of [RhCl(nbd)(dmap)] (nbd = bicyclo[2.2.1]hepta-2,5-diene; dmap = 4-(N,N-dimethylamino)pyridine). Bull. Chem. Soc. Jpn. 2000, 73, 129–130. [Google Scholar] [CrossRef]
- Poole III, D.A.; Mathew, S.; Reek, J.N.H. Just add water: Modulating the structure-derived acidity of catalytic hexameric resorcinarene capsules. J. Am. Chem. Soc. 2021, 143, 16419–16427. [Google Scholar] [CrossRef] [PubMed]
- Mecozzi, S.; Rebek, J., Jr. The 55 % solution: A formula for molecular recognition in the liquid state. Chem. Eur. J. 1998, 4, 1016–1022. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. Self-recognition, structure, stability, and guest affinity of pyrogallol[4]arene and resorcin[4]arene capsules in solution. J. Am. Chem. Soc. 2004, 126, 11556–11563. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, C.; Manna, P.L.; Rosa, M.D.; Talotta, C.; Rescifina, A.; Floresta, G.; Soriente, A.; Neri, P. Synergic interplay between halogen bonding and hydrogen bonding in the activation of a neutral substrate in a nanoconfined space. Angew. Chem. Int. Ed. 2019, 59, 811–818. [Google Scholar]
- Merget, S.; Catti, L.; Piccini, G.; Tiefenbacher, K. Requirements for terpene cyclizations inside the supramolecular resorcinarene capsule: Bound water and its protonation determine the catalytic activity. J. Am. Chem. Soc. 2020, 142, 4400–4410. [Google Scholar] [CrossRef] [PubMed]
- Vila, J.; Vinardell, R.; Solà, M.; Pla-Quintana, A.; Roglans, A. A Rh(I)-catalyzed cascade cyclization of 1,5-bisallenes and alkynes for the formation of cis-3,4-arylvinyl pyrrolidines and cyclopentanes. Adv. Synth. Catal. 2022, 364, 206–217. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 3 | Capsule (16) | Additive | Conversion (%) 2 | Product Distribution 2 | ||
|---|---|---|---|---|---|---|---|
| 6a (%) | 7 (%) | 8 (%) | |||||
| 1 | Yes | No | / | 100 | 44 | 46 | 10 |
| 2 | Yes | Yes | / | 100 | 62 | 32 | 6 |
| 3 | No | No | / | 0 | 0 | 0 | 0 |
| 4 | No | Yes | / | 0 | 0 | 0 | 0 |
| 5 | Yes | Yes | NEt4Cl 3 | 100 | 53 | 40 | 7 |
| 6 | Yes | Yes | D2O 4 | 100 | 65 | 30 | 5 |
| 7 | Yes | No | MeOH 5 | 100 | 44 | 50 | 6 |
| 8 | Yes | Yes | MeOH 5 | 100 | 43 | 50 | 7 |
| Entry | Alkyne | Capsule (16) | Product Distribution 2 | |||
|---|---|---|---|---|---|---|
| 6 (%) | 7 (%) | 8 (%) | ||||
| 1 | 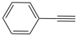 | 5a | No | 44 | 46 | 10 |
| 2 | Yes | 62 | 32 | 6 | ||
| 3 |  | 5b | No | 39 | 50 | 11 |
| 4 | Yes | 57 | 37 | 6 | ||
| 5 | 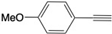 | 5c | No | 48 | 41 | 11 |
| 6 | Yes | 69 | 28 | 3 | ||
| 7 |  | 5d | No | 38 | 50 | 12 |
| 8 | Yes | 58 | 36 | 6 | ||
| 9 |  | 5e | No | 47 | 45 | 8 |
| 10 | Yes | 60 | 34 | 6 | ||
| 11 |  | 5f | No | 40 | 49 | 11 |
| 12 | Yes | 60 | 34 | 6 | ||
| 13 |  | 5g | No | 32 | 54 | 14 |
| 14 | Yes | 54 | 37 | 9 | ||
| 15 | 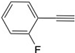 | 5h | No | 37 | 51 | 12 |
| 16 | Yes | 59 | 32 | 9 | ||
| 17 |  | 5i | No | 52 | 40 | 8 |
| 18 | Yes | 63 | 31 | 6 | ||
| 19 |  | 5j | No | 48 | 43 | 9 |
| 20 | Yes | 63 | 32 | 6 | ||
| 21 | 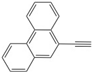 | 5k | No | 43 | 46 | 11 |
| 22 | Yes | 53 | 38 | 9 | ||
| 23 | 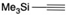 | 5l | No | 40 | 50 | 10 |
| 24 | Yes | 41 | 47 | 12 | ||
| 25 | 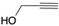 | 5m | No | 85 | 15 | 0 |
| 26 | Yes | 85 | 15 | 0 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinmetz, M.; Sémeril, D. Homo- Versus Hetero- [2+2+2] Rhodium-Catalyzed Cycloaddition: Effect of a Self-Assembled Capsule on the Catalytic Outcome. Molecules 2025, 30, 3052. https://doi.org/10.3390/molecules30143052
Steinmetz M, Sémeril D. Homo- Versus Hetero- [2+2+2] Rhodium-Catalyzed Cycloaddition: Effect of a Self-Assembled Capsule on the Catalytic Outcome. Molecules. 2025; 30(14):3052. https://doi.org/10.3390/molecules30143052
Chicago/Turabian StyleSteinmetz, Maxime, and David Sémeril. 2025. "Homo- Versus Hetero- [2+2+2] Rhodium-Catalyzed Cycloaddition: Effect of a Self-Assembled Capsule on the Catalytic Outcome" Molecules 30, no. 14: 3052. https://doi.org/10.3390/molecules30143052
APA StyleSteinmetz, M., & Sémeril, D. (2025). Homo- Versus Hetero- [2+2+2] Rhodium-Catalyzed Cycloaddition: Effect of a Self-Assembled Capsule on the Catalytic Outcome. Molecules, 30(14), 3052. https://doi.org/10.3390/molecules30143052