Abstract
Its outstanding mechanical and thermodynamic characteristics make SrAl2O4 a highly desirable ceramic material for high-temperature applications. However, the effects of elevated pressure on the structural and other properties of SrAl2O4 are still poorly understood. This study encompassed structural, elastic, electronic, dynamic, and thermal characteristics. Band structure calculations indicate that the direct band gap of SrAl2O4 is 4.54 eV. In addition, the Cauchy pressures provide evidence of the brittle characteristics of SrAl2O4. The mechanical and dynamic stability of SrAl2O4 is evident from the accurate determination of its elastic constants and phonon dispersion relations. In addition, a comprehensive analysis was conducted of the relationship between specific heat and entropy concerning temperature variations.
1. Introduction
Many scientific studies have been conducted to analyze aluminate compounds possessing the structure AB2O4, owing to their exceptional properties, such as outstanding reflectivity and excellent resistance to chemical degradation at elevated temperatures. Furthermore, these compounds exhibit minimal electrical loss [1,2]. In addition, aluminates demonstrate environmentally friendly characteristics, display enhanced chemical durability, and can be easily manufactured at a cost that is deemed reasonable [3]. Consequently, more research investigations are being conducted to explore the synthesis and examination of phosphors based on aluminate, and there is a growing interest in conducting research studies to investigate the synthesis and analysis of aluminate-based phosphors [4,5,6]. In various fields of application, such as light-emitting diodes and imaging devices, there is a demand for inorganic phosphorus materials that exhibit exceptional color clarity and intensity [7,8,9]. Extensive research has been conducted on AB2O4 substances that display an orthorhombic configuration reminiscent of CaFe2O4 [10,11,12,13,14,15,16]. In particular, SrAl2O4 exhibits remarkable luminescent properties. These materials possess versatile applications in diverse sectors, including but not limited to display technologies, signage solutions, medical advancements, and storage innovations. Therefore, it is important to highlight the research conducted on SrAl2O4. Nazarov and colleagues [10] utilized density functional theory to examine the modifications in both structural and electronic characteristics of SrAl2O4 following the introduction of Eu3+ doping. They discovered that the band gap of SrAl2O4, measured at 4.52 eV, is lower than the experimental value of 6.5 eV. In a separate investigation, Rojas-Hernandez et al. examined the structural characteristics of SrAl2O4’s P21, P6322, and P63 phases utilizing both LDA and GGA techniques [14]. Recently, the hexagonal structure of XAl2O4 (X = Ca, Sr and Cd) has been investigated by Ref. [15]. In addition, research has been conducted on the structural, elastic, electronic, and vibrational properties of XAl2O4 (X = Ca, Sr and Cd) semiconductors with an orthorhombic structure [16].
However, there is currently a lack of comprehensive calculations available for orthorhombic SrAl2O4 under varying pressures. So, the primary objective of this study was to acquire a comprehensive understanding of the elastic, electronic, dynamic, and thermodynamic characteristics of SrAl2O4. Calculations based on density functional theory were utilized to achieve this objective. Pressure exerts an influence on both the lattice parameters and elastic constants of a substance, ultimately leading to alterations in its lattice parameters, elastic modulus, and melting point. Hence, it is imperative to investigate the impact of elevated pressure on the mechanical and thermodynamic characteristics of materials at high temperatures. The impact of elevated pressure on the structural, mechanical, and thermodynamic properties of SrAl2O4 has remained uncertain. To address that research gap, this study employed a first-principles approach to investigate how pressure influences the structural, elastic, electronic, dynamic, and thermal characteristics of SrAl2O4.
2. Results and Discussion
2.1. Structural Parameters
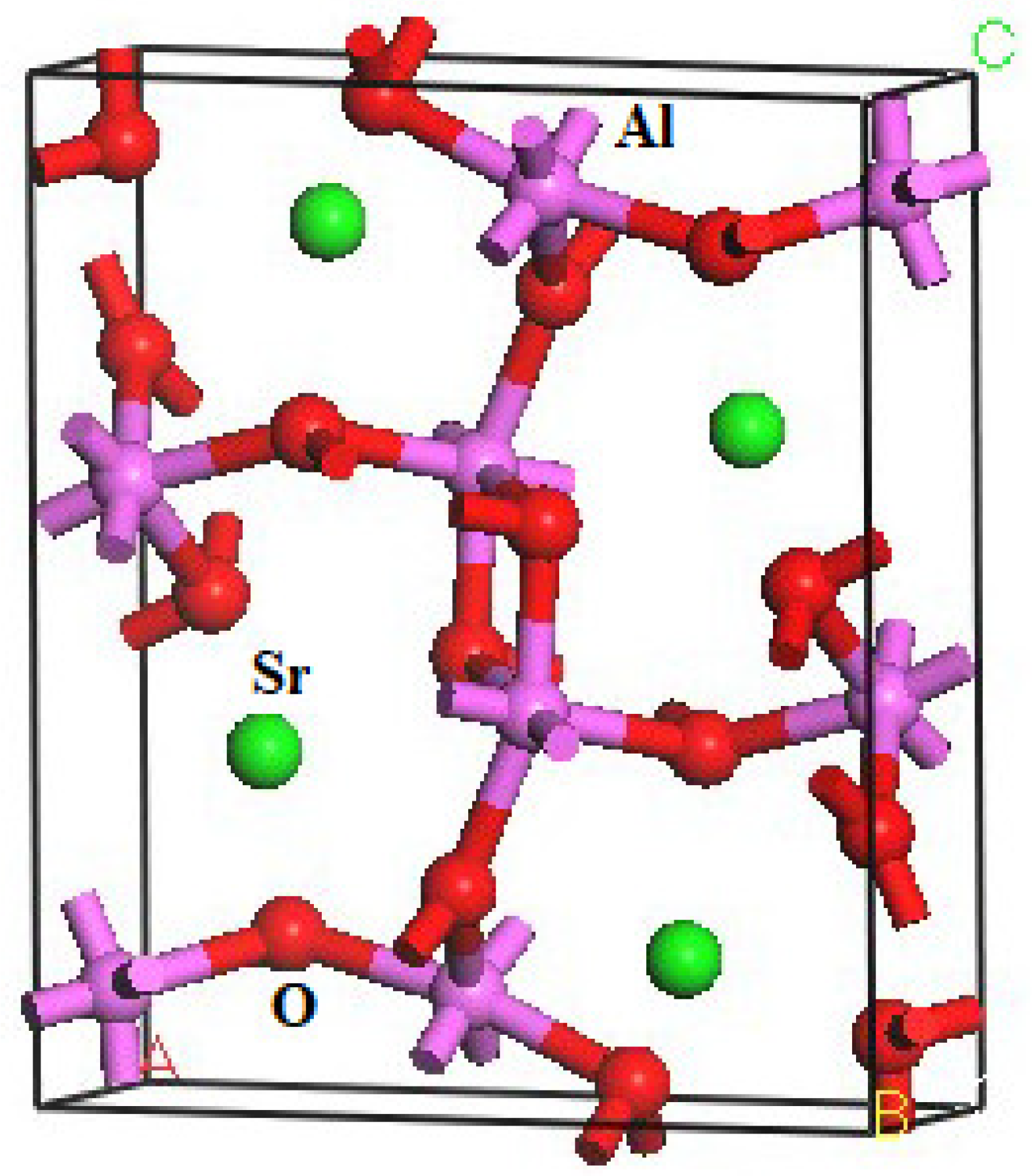
The crystal structure of SrAl2O4 is characterized by a CaFe2O4-type arrangement (Pnma). The DFT method was employed to compute the structural parameters of orthorhombic SrAl2O4. The structural parameters of SrAl2O4, and the corresponding theoretical data [16], are presented in Table 1. Furthermore, the atomic coordinates at equilibrium for SrAl2O4 in the Pnma phase can be found in Table S1. The crystal structure of SrAl2O4, as depicted in Figure 1, is derived from the computed positional arrangements. The results are consistent with the theoretical information presented in reference [16].

Table 1.
Calculated lattice parameters (a, b, c) (Å), density ρ (g/cm3), and volume V (Å3) of SrAl2O4.
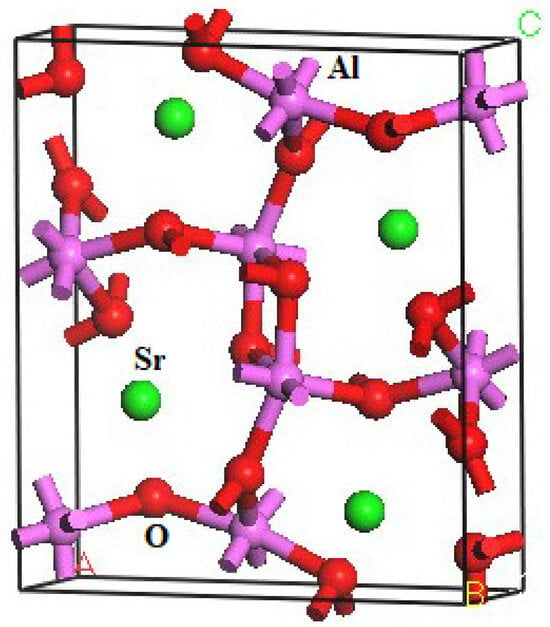
Figure 1.
Crystal structure of orthorhombic SrAl2O4 (The green, purple, and red balls are Sr, Al, and O atoms, respectively).
2.2. Mechanical Properties
In order to assess the stability of SrAl2O4 and gain deeper insights into its pressure-induced anisotropic structural characteristics, we conducted elasticity calculations for this compound. In particular, calculations were performed to determine the elastic constants Cij. For crystals with an orthorhombic structure, the elastic constants must meet certain criteria to ensure mechanical stability [17].
The obtained effective elastic constants are provided in Table 2, along with the theoretical data at a pressure of 0 GPa. The obtained data align with the findings reported in Ref. [16]. In the pressure range of 0 to 50 GPa, it can be observed that the mechanical stability of the orthorhombic phase of SrAl2O4 is maintained, as evidenced by meeting the criteria for stability. In the subsequent stage, we employed the Reuss–Voigt–Hill approximation techniques to determine the bulk modulus B, shear modulus G, and Young’s modulus E of SrAl2O4 [18,19,20]. In Table 3, we have included the pressure-dependent values of B, G, and E for SrAl2O4. The values of B, G, and E at 0 K and 0 GPa are slightly higher than the theoretical predictions of 163.1 GPa, 101.09 GPa, and 256.28 GPa, respectively [16], with measured values of approximately 180.52 GPa, 108.31 GPa, and 270.77 GPa. Poisson’s ratio v can be judged as a criterion of brittleness/ductility. Poisson’s ratio v of SrAl2O4 remains consistently below 0.26 throughout a pressure range of 0–50 GPa, indicating its inherent brittleness. The ratio B/G has also been used to judge the brittleness/ductility of a solid [21]. The B/G ratio of SrAl2O4 is less than 1.75, indicating that SrAl2O4 is a brittle structure. Table 3 displays the values of Poisson’s ratio (v) and B/G for SrAl2O4 under zero pressure conditions. These values, 0.25 and 1.67, respectively, exhibit a remarkable agreement with the results obtained from other simulation methods, as shown in Table 3, where the corresponding values were 0.2381 and 1.58. The pressure-dependent variations in v and B/G for SrAl2O4 are presented in Table 3, indicating an upward trend with increasing pressure. The findings indicate that SrAl2O4 demonstrates a tendency towards brittleness within the pressure intervals of 0–50 GPa.
Table 2.
The elastic constants Cij (GPa) of SrAl2O4 under different pressures.
Table 3.
Calculated bulk modulus B (GPa), shear modulus G (GPa), Young modulus E (GPa), B/G, Poisson’s ratio ν, wave velocities vl, vt, and vm (km/s), and Debye temperatures θ (K) of SrAl2O4 under pressures ranging from 0 to 50 GPa.
The anisotropic factors for shear on various crystallographic planes (A1, A2, and A3), the universal anisotropic index (AU), the percentage of anisotropy in shear and bulk moduli (AG and AB), as well as the directional bulk moduli (Ba, Bb, and Bc) were computed and are summarized in Table 4. It is possible to assess the mechanical anisotropy indexes through the following methods [22,23,24]
A1 = 4C44/(C11 + C33 − 2C13)
A2 = 4C55/(C22 + C33 − 2C23)
A3 = 4C66/(C11 + C22 − 2C12)
Table 4.
The shear anisotropy factors A1, A2, A3, the directional bulk modulus Ba, Bb, and Bc, the elastic anisotropy index AU, AB, and AG, hardness Hv (GPa), the melting temperature Tm (K), and thermal conductivity k (W·m−1·K−1) of SrAl2O4 under pressures ranging from 0 to 50 GPa.
The orthorhombic crystal’s elastic anisotropy can be characterized by the bulk moduli Ba, Bb, and Bc along the a, b, and c axes, respectively. These values are interrelated.
where
The anisotropy factor percentage for the orthorhombic crystal, derived from the bulk modulus (AB) and shear modulus (AG), can be determined using the equation proposed by Chung and Buessem [22,23,24].
Table 4 presents the calculated values of A1, A2, and A3, indicating a deviation from unity. This suggests that SrAl2O4 demonstrates a significant elastic anisotropy, reaching up to 50 GPa. Meanwhile, the a-axis exhibits the highest directional bulk modulus for Ba compared to Bb and Bc. The observed trend indicates that the compressibility of SrAl2O4 is comparatively higher along the a and c axes, while it exhibits relatively lower compressibility along the b axis. Additionally, it can be noted that compression along the c axis is relatively more favorable. This observation is consistent with our previous findings in Figure S1, indicating that the lattice parameter b exhibits a more rapid decrease under pressure than parameters a and c. If the value of AU is equal to zero, it indicates that the material exhibits isotropic properties. Otherwise, a greater departure from zero results in increased anisotropic elasticity. The range of AB and AG values spans from zero to one, denoting the presence of elastic anisotropy in terms of compression and shear characteristics, respectively. In Table 4, we additionally present the pressure-dependent anisotropic indexes. The anisotropy of SrAl2O4 is more pronounced within the pressure range of 0–50 GPa, leading to higher AU values. The AU of SrAl2O4 exhibits a continuous increase until reaching 50 GPa.
The Debye temperature (θ) has a close correlation with both the specific heat and melting temperature. Acoustic vibrations solely contribute to the crystal vibrations in conditions of low temperatures. Hence, by utilizing the elastic constants as input parameters, it is possible to determine the Debye temperature at low temperatures through established relationships [25]
The given formula involves the utilization of various constants such as ħ, k, and NA, and variables including n, M, ρ, and vm. These elements are employed to calculate the average sound velocity in a manner that accounts for Planck’s constant divided by 2π, Boltzmann’s constant, Avogadro’s number, the number of atoms per formula unit, the molecular mass per formula unit, density, and the average sound velocity [26]
The calculation of longitudinal and transverse elastic wave velocities involves utilizing the following relationship, where vt represents the longitudinal velocity and vl denotes the transverse velocity [27,28]
The values for the longitudinal, transverse, and average sound velocities and Debye temperature of SrAl2O4 under different pressures are presented in Table 3, alongside the findings reported in ref. [16]. After conducting a thorough comparison, it can be concluded that our calculated outcomes align well with existing research findings, affirming the accuracy of our calculations. According to the data provided in Table 3, it can be deduced that an upward trend exists between pressure and the longitudinal, transverse, and average sound velocities, as well as the Debye temperature.
In addition, the hardness HV, melting point Tm, and thermal conductivity k of the material are calculated utilizing the subsequent formulas [29,30]
Tm = 354 + 4.5(2C11 + C33)/3
Here, kB is Boltzmann’s constant, ρ is the density, and Ma is the average mass per atom, respectively. At zero pressure, the material exhibits a hardness value of 14.2 GPa, a melting point temperature of 1646.10 K, and a thermal conductivity rate of 1.92 W·m−1·K−1. It is evident from the data presented in Table 4 that under increased pressure, Hv, Tm, and k exhibit noticeable increments.
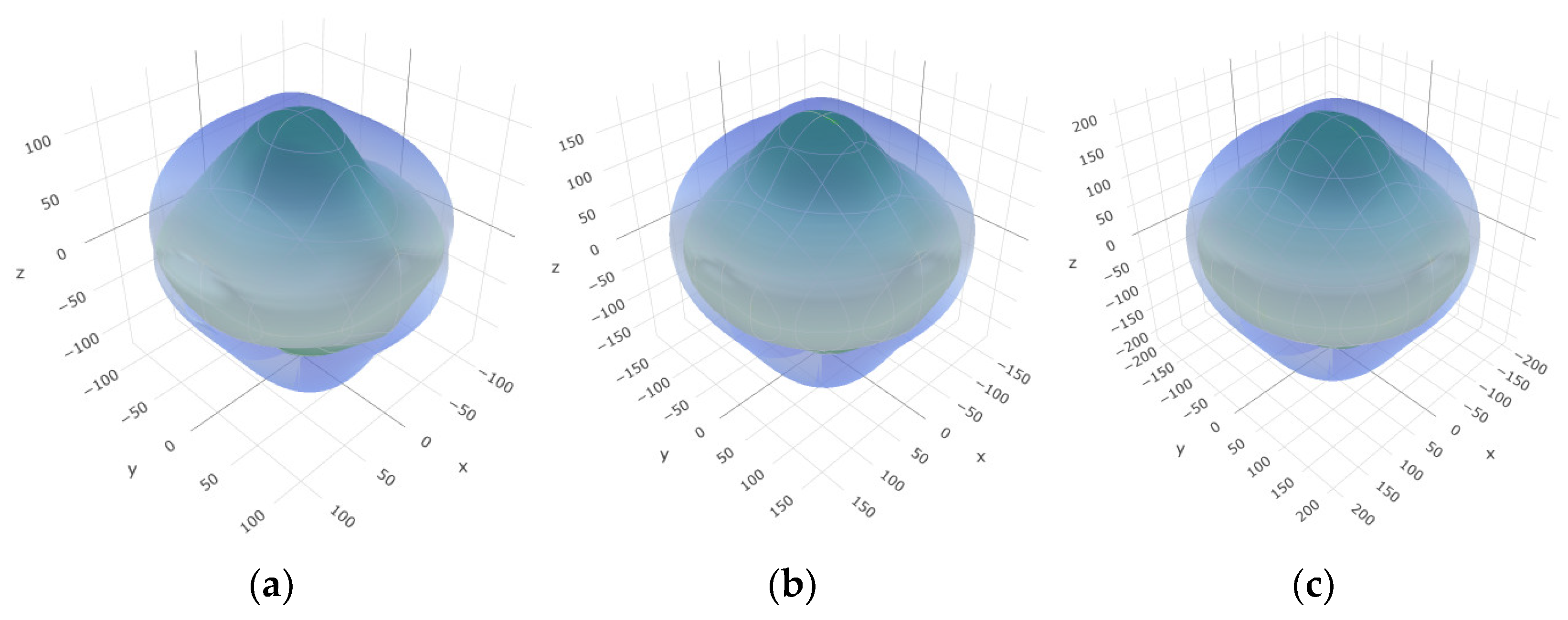
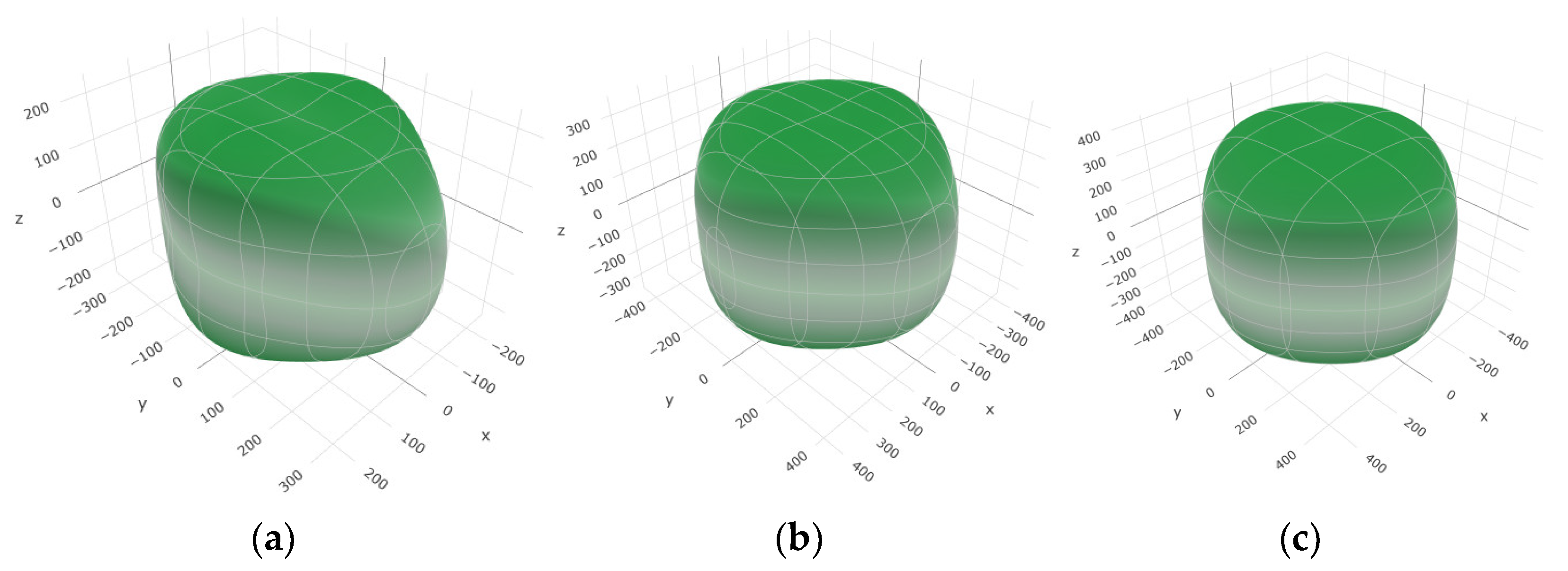
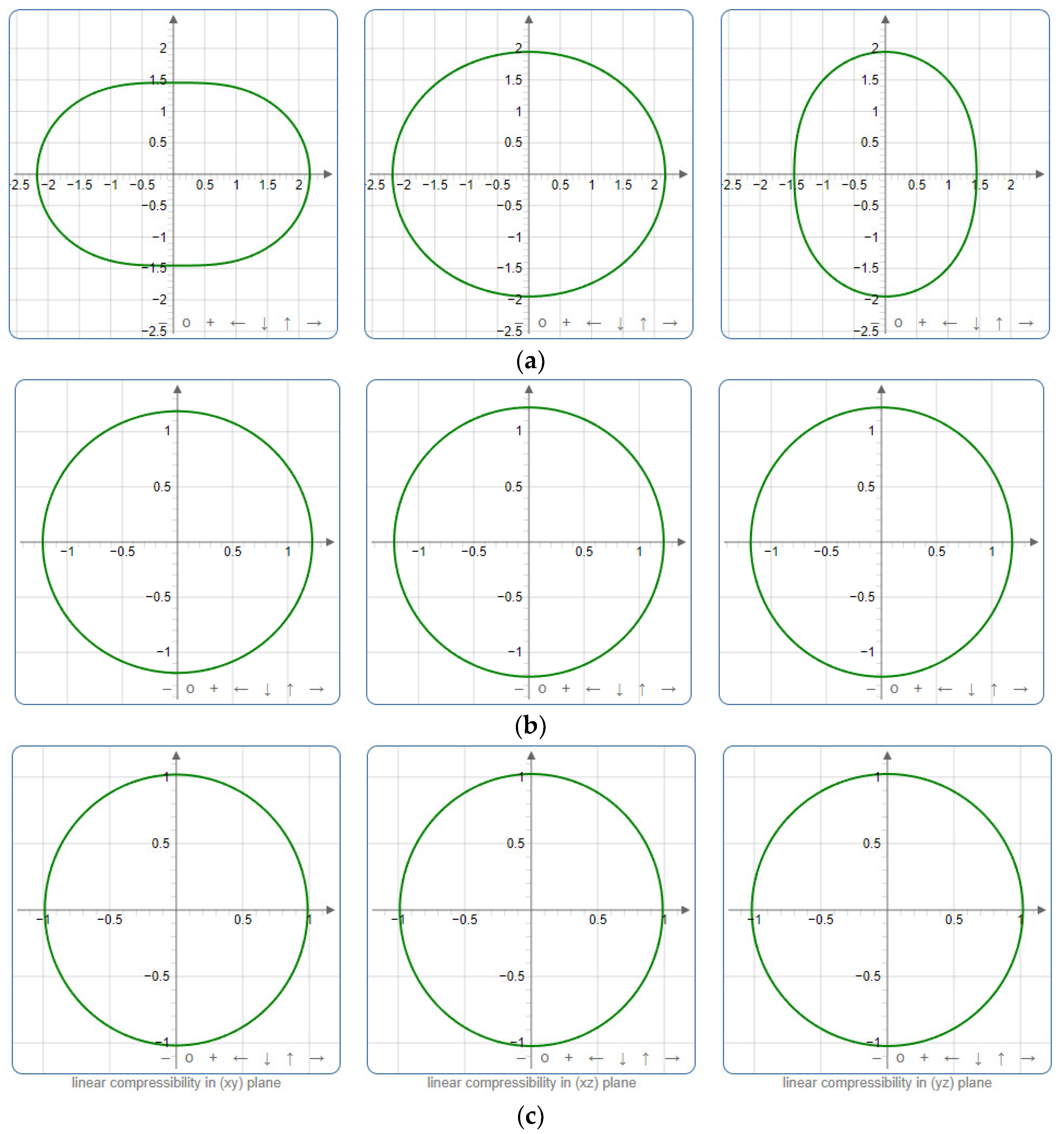
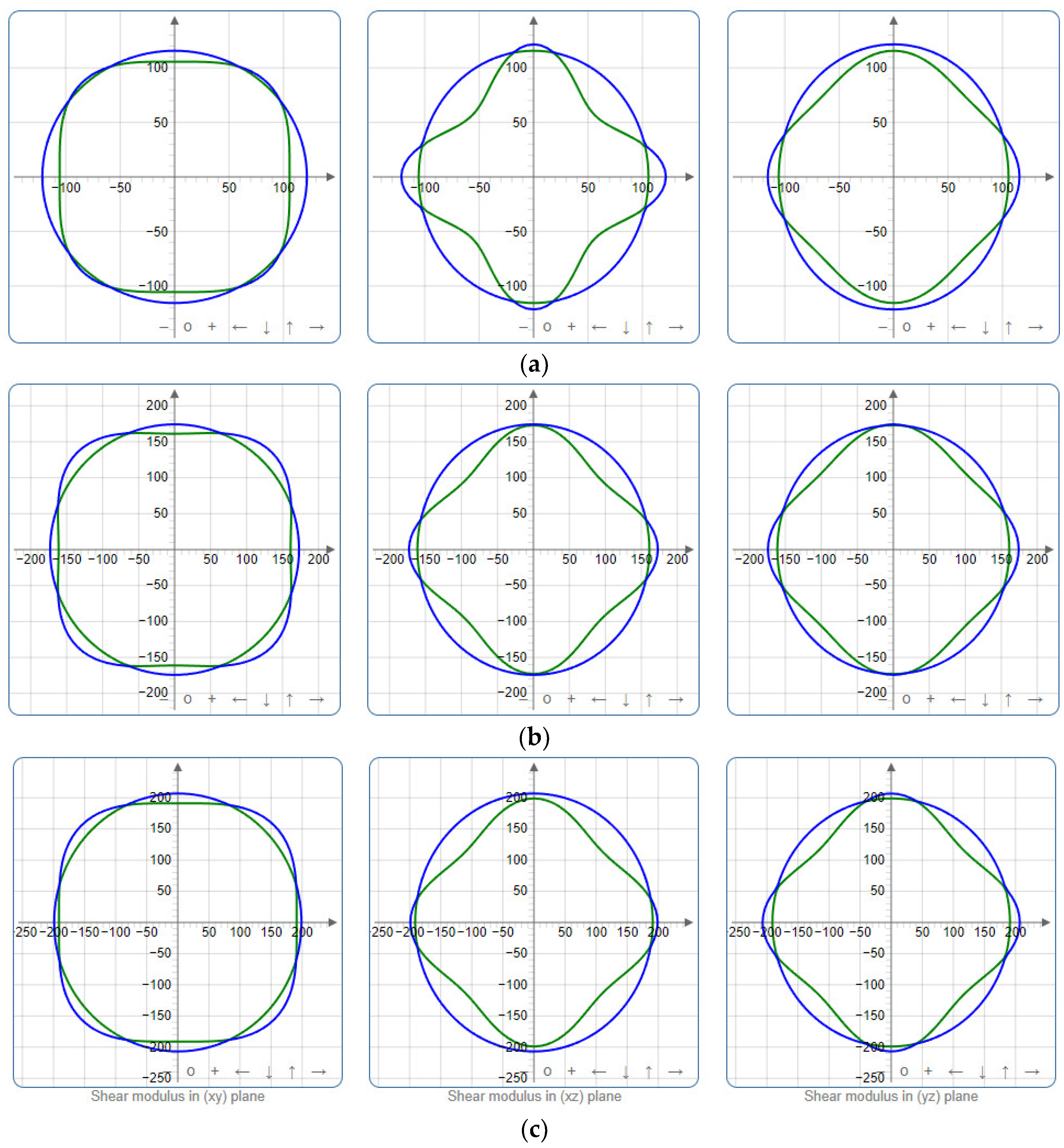
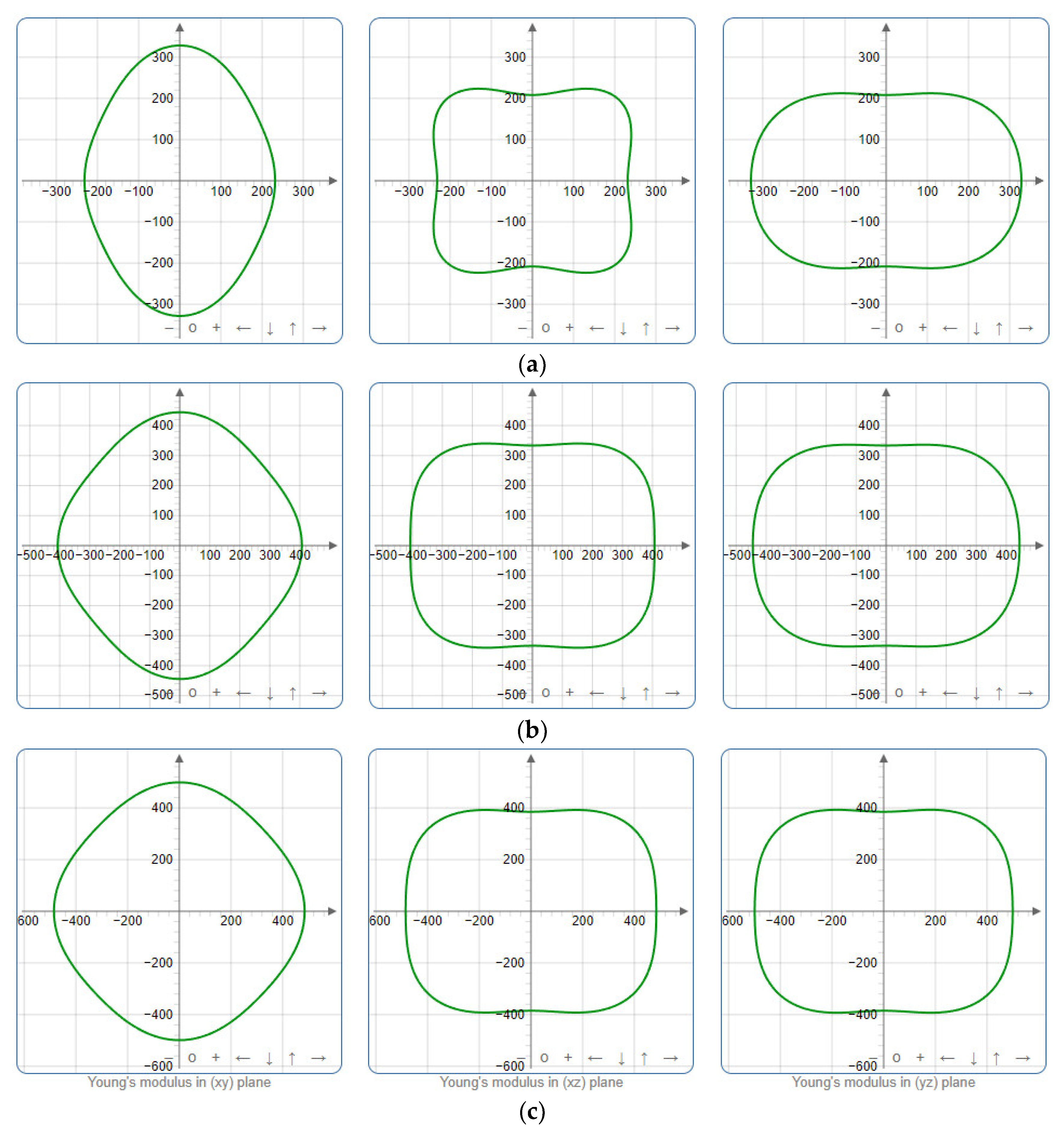
To provide a comprehensive analysis of the mechanical anisotropy, a study was conducted on the spatial variation in linear compressibility (β), shear modulus (G), and Young’s modulus (E) in SrAl2O4 at varying pressure levels [31]. Figure 2, Figure 3 and Figure 4 illustrate the spatial correlation between linear compressibility (β), shear modulus (G), and Young’s modulus (E) in SrAl2O4. In the event of a structure exhibiting isotropy, the spatial correlation will be evident in a spherical configuration, whereby any deviation from this particular shape can be utilized as an indicator for identifying anisotropy. The non-spherical spatial distribution of linear compressibility (β), shear modulus (G), and Young’s modulus (E) at varying pressures in SrAl2O4 suggests a significant level of material anisotropy. For a more extensive view of the directional responsiveness of SrAl2O4’s linear compressibility (β), shear modulus (G), and Young’s modulus (E) under varying pressure conditions, Figure 5, Figure 6 and Figure 7 present the planar projections of linear compressibility (β), shear modulus (G), and Young’s modulus (E) in the xy, yz, and xz planes. For materials with properties that are the same in all directions, the curve of projection displays a circular form. For the xy and xz planes, there is a noticeable increase in deviation for the linear compressibility (β), shear modulus (G), and Young’s modulus (E), as shown in Figure 5, Figure 6 and Figure 7. This indicates a significant variation in elastic properties between the xy and xz planes for these solids. It suggests a notable disparity in the elastic characteristics between the xy and xz orientations within these materials. Simultaneously, the elastic anisotropy of SrAl2O4 was thoroughly examined under different pressure conditions by employing βmax/βmin, Gmax/Gmin, and Emax/Emin ratios. Higher linear compression anisotropy can be inferred from a greater βmax/βmin and the corresponding Gmax/Gmin and Emax/Emin ratios. The βmax/βmin values are 1.50, 1.18, 1.08, 1.03, 1.02, and 1.04 at 0 GPa, 10 GPa, 20 GPa, 30 GPa, 40 GPa, and 50 GPa, respectively; the Gmax/Gmin values are 1.59, 1.48, 1.44, 1.43, 1.41, and 1.41 at 0 GPa, 10 GPa, 20 GPa, 30 GPa, 40 GPa, and 50 GPa, respectively; and the Emax/Emin values are 1.58, 1.43, 1.37, 1.35, 1.35, and 1.35 at 0 GPa, 10 GPa, 20 GPa, 30 GPa, 40 GPa, and 50 GPa, respectively. According to the data provided, anisotropy decreases with increasing pressure within the range of 0–50 GPa.
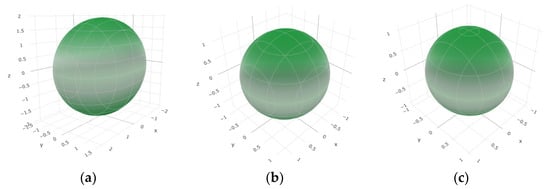
Figure 2.
The surface contours of linear compressibility β (TPa−1) of SrAl2O4. (a) 0 GPa, (b) 30 GPa, (c) 50 GPa.
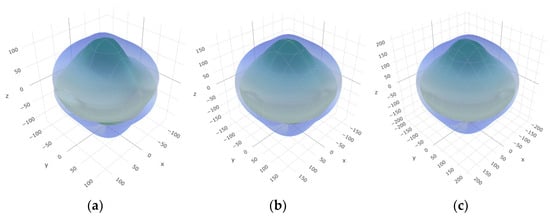
Figure 3.
The surface contours of shear modulus G (GPa) of SrAl2O4. (a) 0 GPa, (b) 30 GPa, (c) 50 GPa.
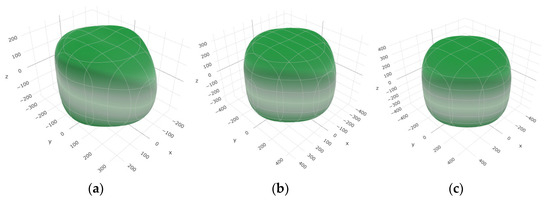
Figure 4.
The surface contours of Young’s modulus E (GPa) of SAl2O4. (a) 0 GPa, (b) 30 GPa, (c) 50 GPa.
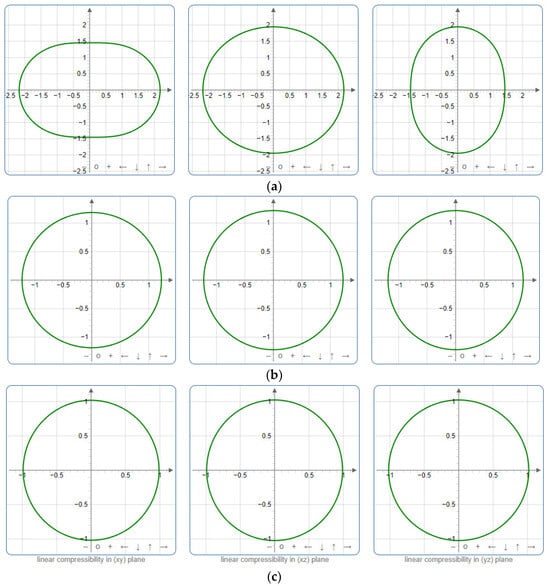
Figure 5.
Projections of linear compressibility β (TPa−1) in xy, xz, and yz planes of SrAl2O4. (a) 0 GPa, (b) 30 GPa, (c) 50 GPa.
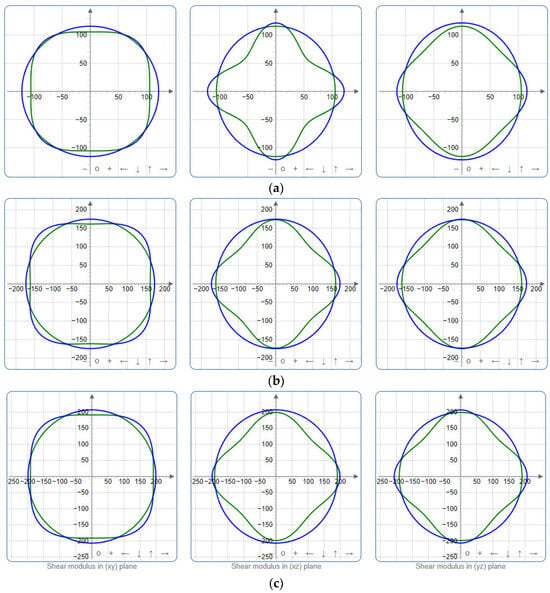
Figure 6.
Projections of shear modulus G (GPa) in xy, xz, and yz planes of SrAl2O4. (a) 0 GPa, (b) 30 GPa, (c) 50 GPa.
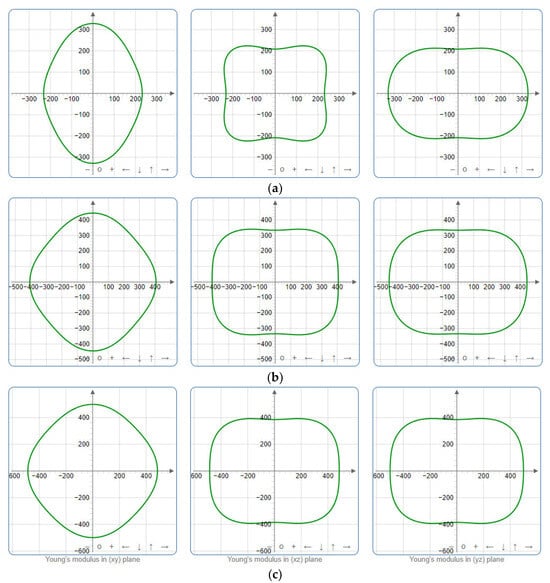
Figure 7.
Projections of Young’s modulus G (GPa) in xy, xz, and yz planes of SrAl2O4. (a) 0 GPa, (b) 30 GPa, (c) 50 GPa.
2.3. Electronic Properties
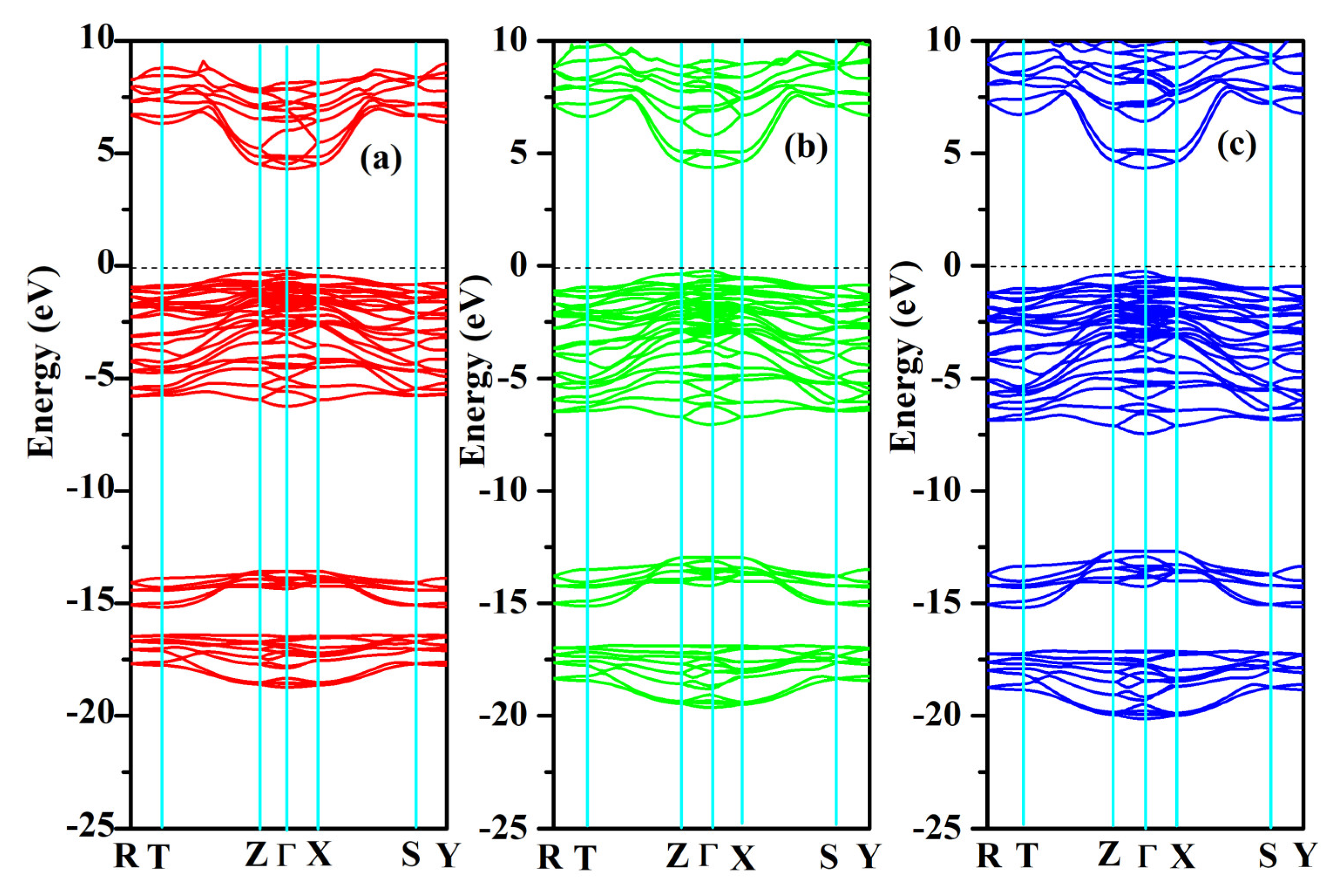
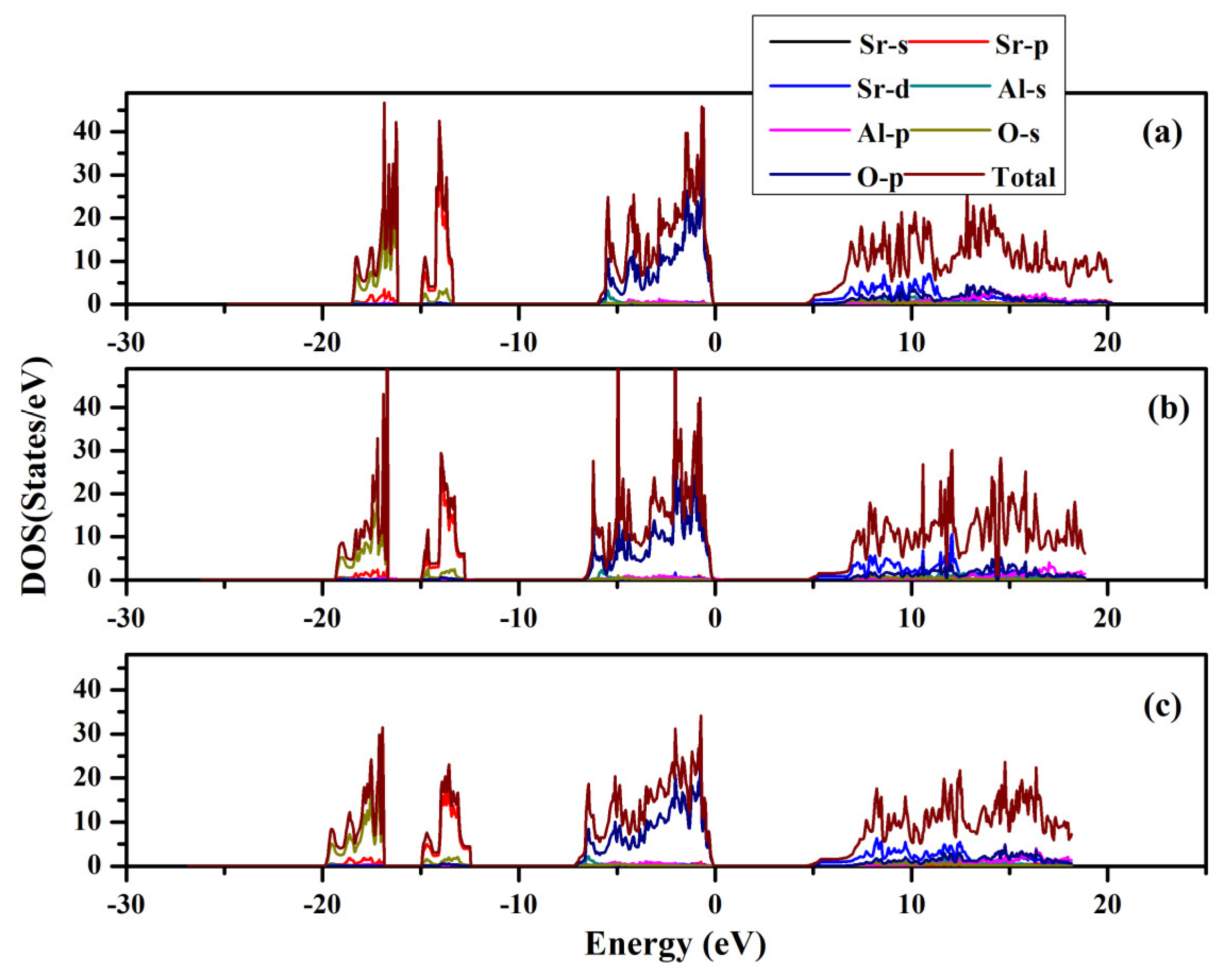
The band structures and partial and total density of states were computed for SrAl2O4. Graphs illustrating these properties along high symmetry directions can be observed in Figure 8 and Figure 9. From these figures, it can be seen that there is a direct band gap of 4.54 eV for SrAl2O4. Our calculated band gap value for SrAl2O4 aligns well with the band gap (4.54 eV) reported in ref. [16]. Based on the analysis of the total and partial density of states for SrAl2O4 (Figure 9), it can be inferred that the primary contribution to the valence band of SrAl2O4 comes from the p orbitals of O atoms, with minor contributions from both the d and p orbitals of strontium atoms, as well as the p orbitals of Al atoms. Moreover, the remaining orbitals of the constituent atoms make negligible contributions. Based on the observations in Figure 9, it can be noted that there is an increase in the involvement of the O atom’s p orbitals as we progress from the lower region of the valence band toward the Fermi surface. The primary composition of the conduction band in SrAl2O4 is attributed to the d orbitals of Sr atoms, with minor involvement from the p orbitals of O atoms and negligible contributions from other constituent atom orbitals.
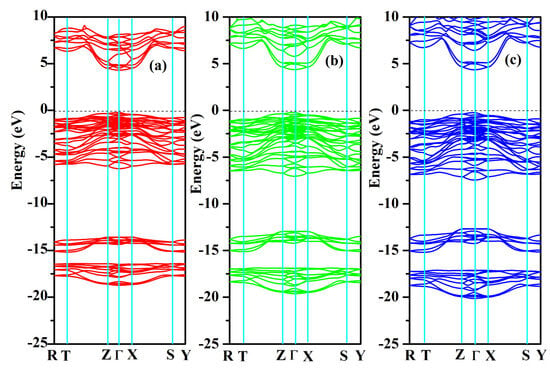
Figure 8.
Electronic band structures for SrAl2O4. (a) 0 GPa, (b) 30 GPa, and (c) 50 GPa.
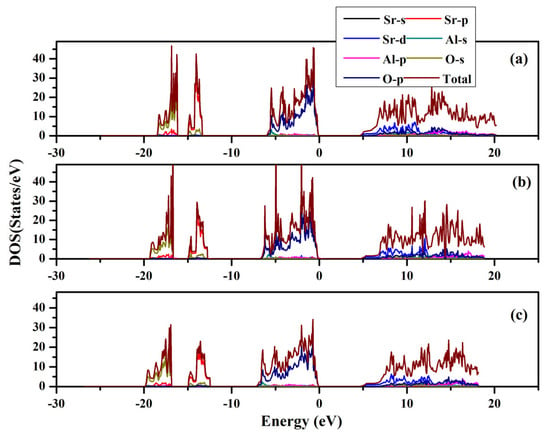
Figure 9.
Total and partial density of states for SrAl2O4. (a) 0 GPa, (b) 30 GPa, and (c) 50 GPa.
2.4. Phonon Properties
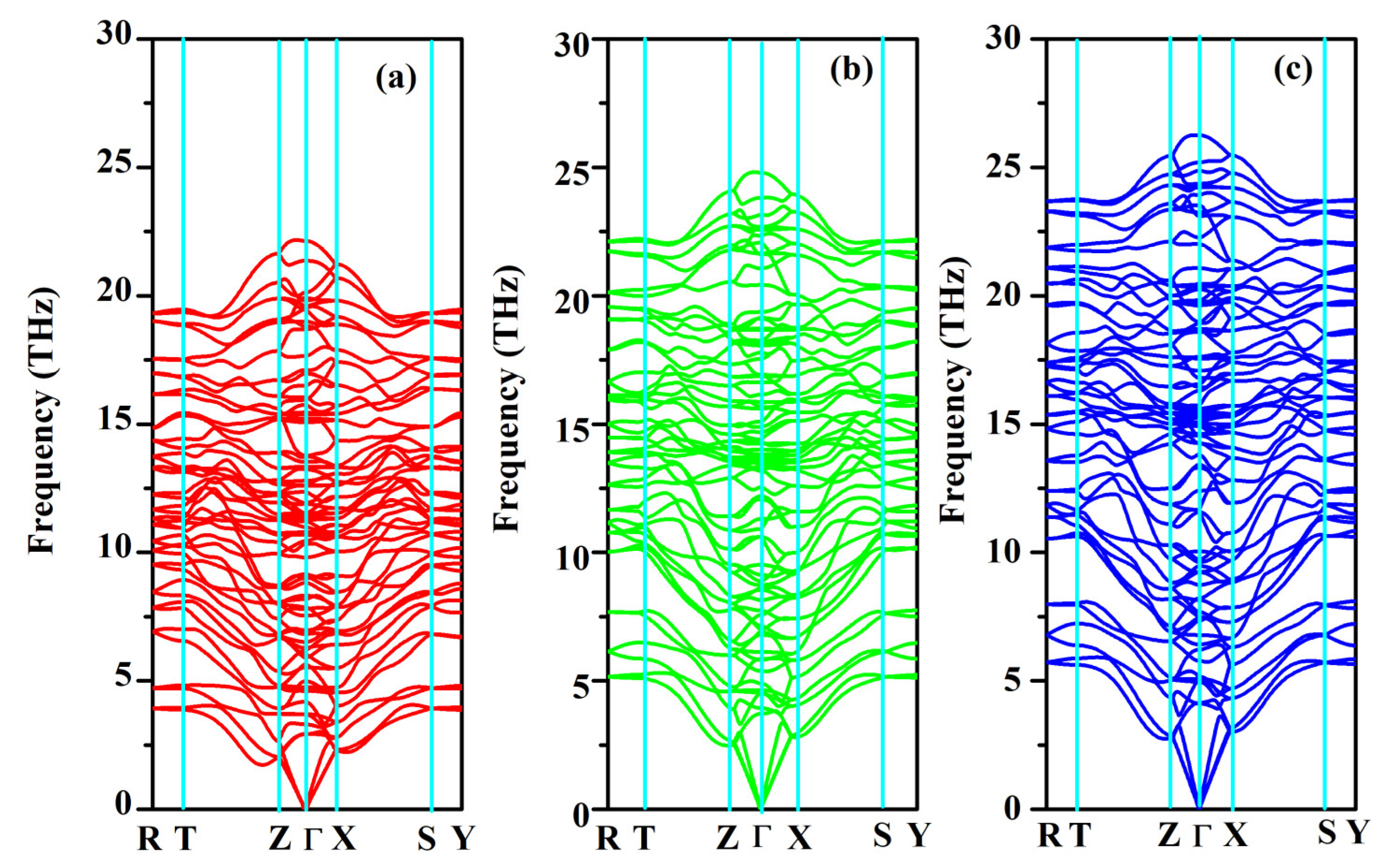
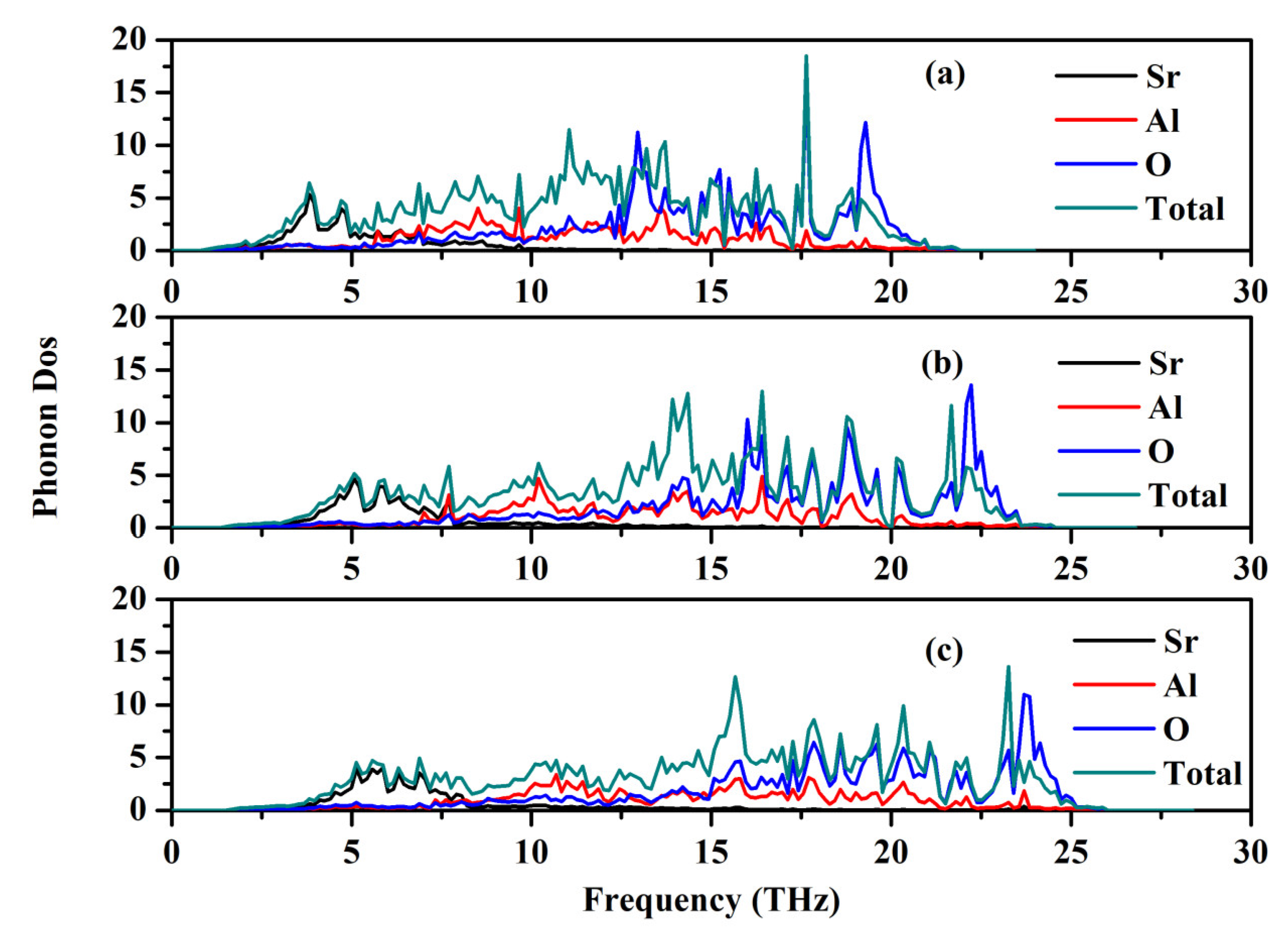
The phonon dispersion relation and the total and partial phonon density of states for SrAl2O4 in its orthorhombic structure are depicted in Figure 10 and Figure 11, respectively. The dispersion relation curves indicate the absence of any gap between the overall spectra, and no negative phonon frequencies can be observed. Hence, our findings suggest that the investigated SrAl2O4 exhibits favorable dynamic stability. According to the results depicted in Figure 10, the phonon dispersion curves of SrAl2O4 exhibit similarities to those observed at 0 GPa, even under increased pressures of 30 GPa and 50 GPa. As the pressure rises, there is a shift in the phonon frequency towards higher energy levels. It is evident that the low energy region depicted in Figure 11a primarily exhibits vibrations within the frequency range of 0–7.5 THz, predominantly originating from Sr atoms with a minor contribution from Al and O atoms. As the frequency rises, the influence of Sr atom vibrations diminishes while the impact of O and Al atom vibrations amplifies. In the mid-frequency range of 7.5–15.0 THz, the dominant vibrations are attributed to O and Al atoms, while the involvement of Sr atom vibrations diminishes. In the higher frequency range of 15.0–22.5 THz, the dominant vibrations are primarily attributed to the vibrational contributions from O and Al atoms, while the involvement of Sr atom vibrations is minimal in this high energy region. When examining Figure 11b,c, it becomes apparent that the vibration frequencies of Sr atoms, O atoms, and Al atoms progressively shift towards higher values as the applied pressure increases. In the low frequency range, there is a reduction in the vibrational contribution of Sr atoms as pressure levels increase.
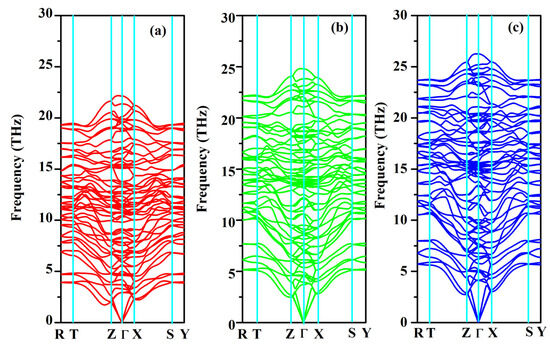
Figure 10.
Phonon band structure for SrAl2O4. (a) 0 GPa, (b) 30 GPa, and (c) 50 GPa.
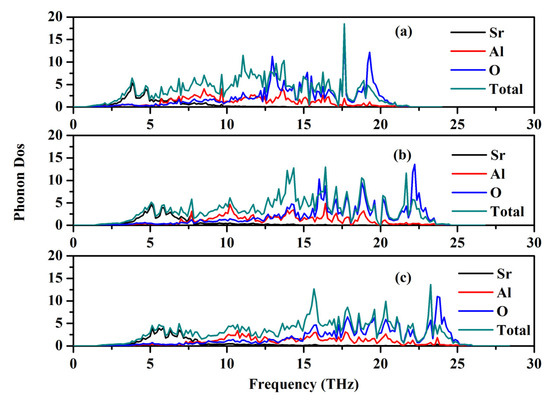
Figure 11.
Total and partial phonon density of states (PDOS) for SrAl2O4. (a) 0 GPa, (b) 30 GPa, and (c) 50 GPa.
The Pnma space group is associated with the orthorhombic crystal system of SrAl2O4. The irreducible representation of crystals with the Pnma structure in the center of the Brillouin zone (k = 0) is as follows:
Γ = 13Ag + 13B2g + 13B1u + 13B3u + 8Au + 8B1g + 8B3g + 8B2u
The Raman activity is observed in the 13Ag, 8B1g, 13B2g, and 8B3g modes, while the infrared activity is detected in the 12B1u, 7B2u, and 12B3u modes. One B1u, one B2u, and one B3u mode exhibit acoustic characteristics, whereas the 8Au mode does not display Raman or infrared activity. Consequently, the Au mode can be classified as an acoustic mode. Table 5, Table 6 and Table 7 present the phonon frequencies at the Γ point and other computed values for SrAl2O4 under varying pressure conditions. In contrast, the computed values obtained under zero pressure concur with alternative theoretical findings [16].
Table 5.
Phonon frequencies (THz) at the Γ point for orthorhombic SrAl2O4 at 0 GPa.
Table 6.
Phonon frequencies (THz) at the Γ point for orthorhombic SrAl2O4 at 30 GPa.
Table 7.
Phonon frequencies (THz) at the Γ point for orthorhombic SrAl2O4 at 50 GPa.
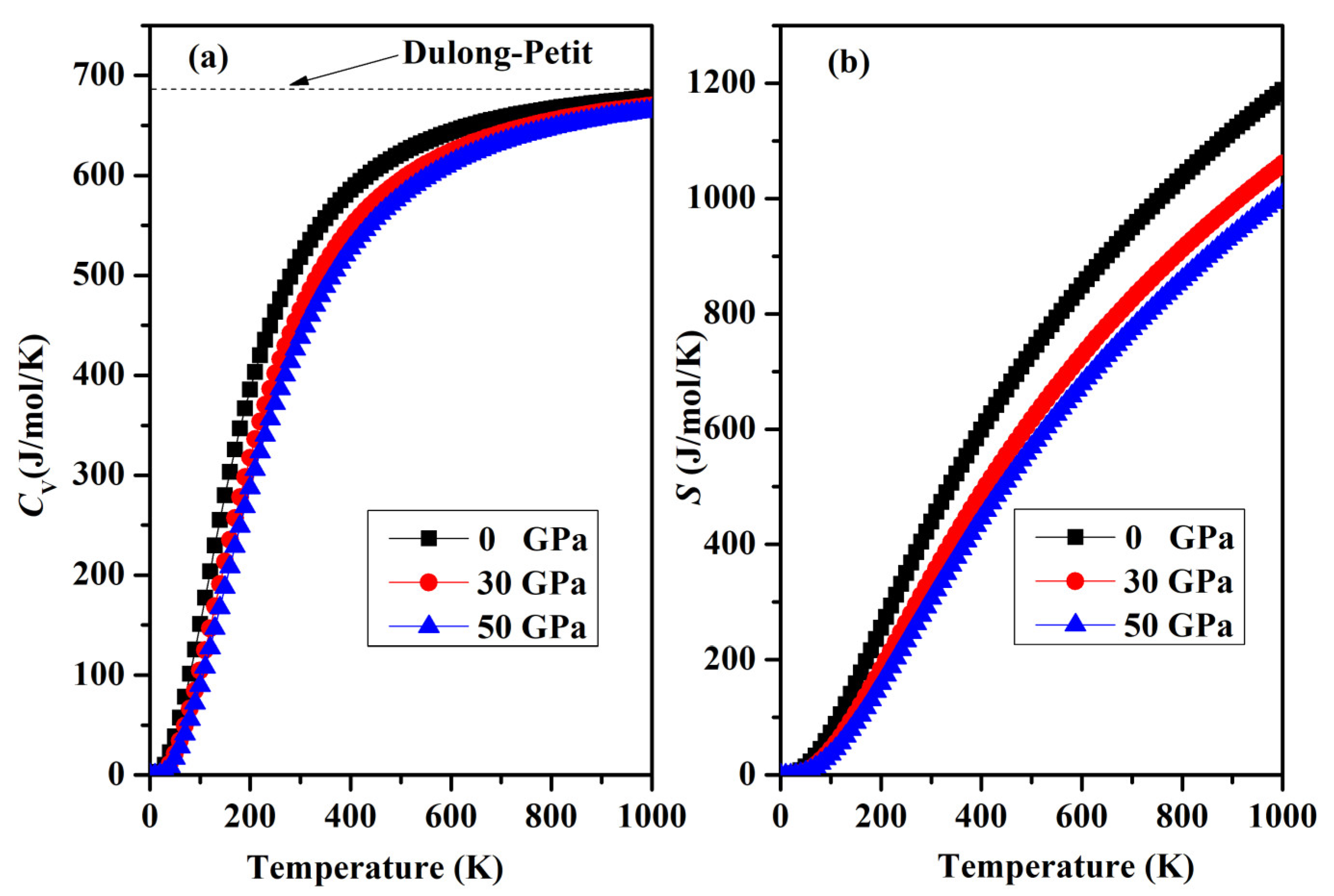
Finally, a graphical representation of the temperature-dependent thermodynamic properties, including specific heat CV and entropy S, for SrAl2O4 can be observed in Figure 12a,b. As the temperature increases, there is a rapid increase in heat capacity until it reaches approximately 400 K. Beyond this temperature, the rate of increase becomes smaller. The heat capacity reaches a stable value called the Dulong–Petit limit when temperatures are elevated. In addition, the increase in temperature shown in Figure 12b is accompanied by an increase in entropy. Regrettably, there is a lack of empirical and theoretical information available for the comparison of thermodynamic properties.
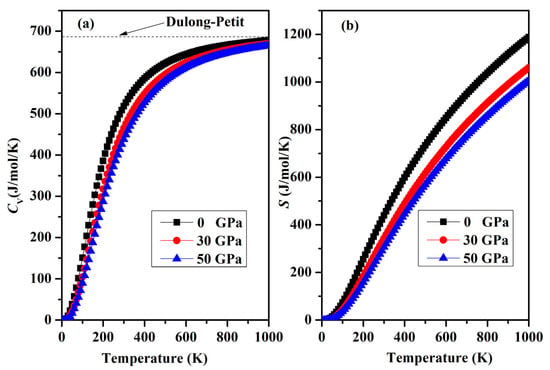
Figure 12.
(a) Variation in the specific heat capacity CV and (b) entropy S for orthorhombic SrAl2O4.
3. Computational Methods
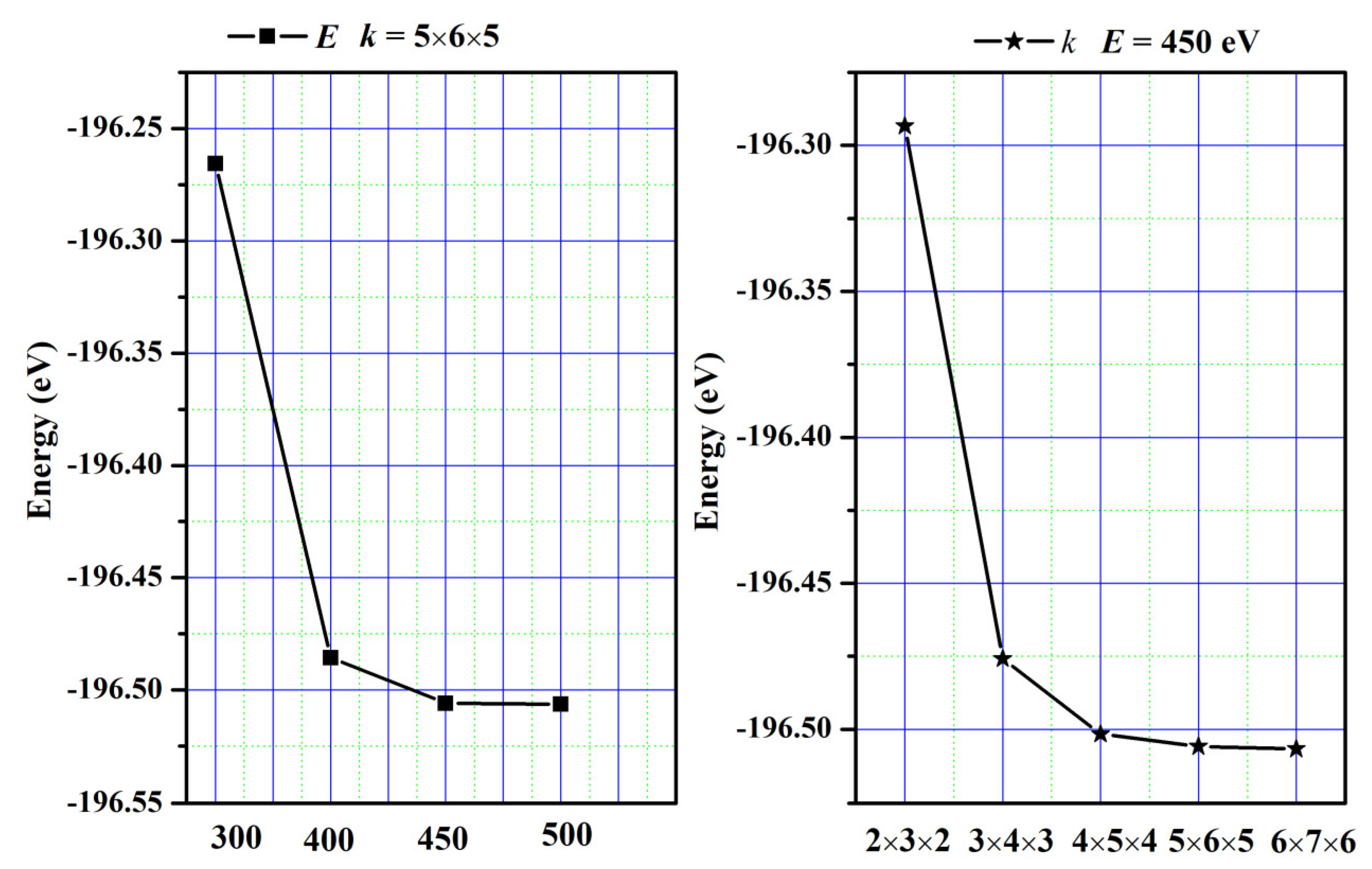
Relevant calculations were conducted utilizing the VASP code for the first principles of density functional theory [32,33]. To perform computations, we utilize Projected Augmented Wave (PAW) pseudo-potentials [34] in conjunction with the Generalized Gradient Approximation developed by Perdew et al. (GGA-PBE) [35]. The integration of the Brillouin zone was achieved by employing Monkhorst–Pack-generated sets of k-points. For SrAl2O4, a grid of 5 × 6 × 5 k-points and a kinetic energy cutoff value of 450 eV were used to achieve the desired level of convergence. The convergence curves are plotted in Figure 13. To solve the Kohn–Sham equation, an energy tolerance of 10−4 eV/cell was selected, and a force tolerance of 0.001 eV/Å was chosen to minimize the Hellman–Feynman force. The valence electron configurations were as follows: Sr atom is 4s2p65s2, Al atom is 3s2p1, and O atom is 2s2p4. Moreover, phonon properties were investigated using the phonopy code [36].
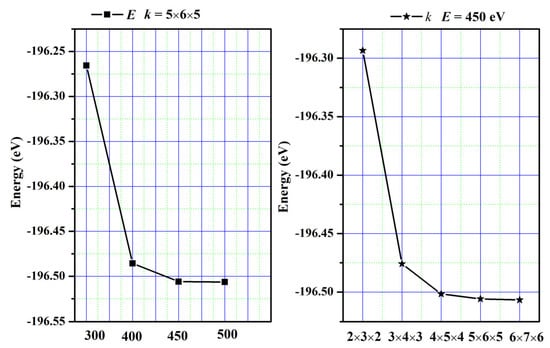
Figure 13.
The convergence curves.
4. Conclusions
In this study, we employed the first-principles method to investigate the effects of pressure on the structural, elastic, electronic, and dynamic properties of orthorhombic SrAl2O4. The elastic constants acquired indicate that SrAl2O4 possesses mechanical stability as they adhere to the criteria established by Born–Huang. Moreover, the Cauchy pressures indicate that the SrAl2O4 tends towards brittleness. The compressibility of SrAl2O4 is higher along the a and c axes, while it exhibits lower compressibility along the b axis. The elastic properties of SrAl2O4 are anisotropic. Finally, this study examined the dynamic characteristics and confirmed that the phonon dispersion curves of SrAl2O4 demonstrate dynamic stability. An investigation of the thermodynamic characteristics of SrAl2O4 was conducted as well. We noted that as the temperature increases, the specific heat values tend to approach the Dulong–Petit limit.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29215192/s1, Table S1. The calculated atomic positions of SrAl2O4 in fractional coordinates; Figure S1. The variations in lattice parameters (a, b, c) and their ratios (a/a0, b/b0, c/c0) under different pressure conditions..
Author Contributions
Methodology, H.G., H.Y., S.D. and H.H.; Software, S.Z.; Formal analysis, H.H.; Investigation, H.G., H.Y. and S.Z.; Writing—original draft, H.G.; Writing—review & editing, H.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Science and Technology Project of Chongqing Municipal Commission of Education (No. KJQN202301429).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jones, W.I.; Miles, L.J. Production of β-Al2O3 electrolyte. Proc. Br. Ceram. Soc. 1971, 19, 161. [Google Scholar]
- Leger, J.M.; Haines, J.; Schmidt, M.; Petitet, J.P.; Pereira, A.S.; Da Jordana, J.A.H. Discovery of hardest known oxide. Nature 1996, 383, 401. [Google Scholar] [CrossRef]
- Mothudi, B.M.; Ntwaeaborwa, O.M.; Pitale, S.S.; Swart, H.C. Luminescent properties of Ca0.97Al2O4:Eu0.012+, Dy0.023+ phosphors prepared by combustion method at different initiating temperatures. J. Alloys Compd. 2010, 508, 262–265. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Li, Y.; Gong, Y. Energy transfer between Eu2+ and Mn2+ in long-afterglow phosphor CaAl2O4:Eu2+, Nd3+, and Mn2+. J. Appl. Phys. 2009, 105, 083502. [Google Scholar] [CrossRef]
- Singh, V.; Zhu, J.J.; Bhide, M.K.; Natarajan, V. Synthesis, characterisation and luminescence investigations of Eu activated CaAl2O4 phosphor. Opt. Mater. 2007, 30, 446–450. [Google Scholar] [CrossRef]
- Niittykoski, J.; Aitasalo, T.; Hölsä, J.; Jungner, H.; Lastusaari, M.; Parkkinen, M.; Tukia, M. Effect of boron substitution on the preparation and luminescence of Eu2+ doped strontium aluminates. J. Alloys Compd. 2004, 374, 108. [Google Scholar] [CrossRef]
- Fan, L.; Zhao, X.; Zhang, S.; Ding, Y.; Li, Z.; Zou, Z. Enhanced luminescence intensity of Sr3B2O6:Eu2+ phosphor prepared by sol-gel method. J. Alloys Compd. 2013, 579, 432–437. [Google Scholar] [CrossRef]
- Rambabu, U.; Han, S.D. Enhanced luminescence intensity and color purity of the red emitting LnVO4:Eu3+@ SiO2 (Ln = Gd, Y and Gd/Y) powder phosphors. Mater. Res. Bull. 2013, 48, 512–520. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, J.; Xu, X.; Wang, Y.; Zhou, L.; Li, B. White LED with high color rendering index based on Ca8Mg(SiO4)4Cl2:Eu2+and ZnCdTe/CdSe quantum dot hybrid phosphor. Mater. Lett. 2012, 84, 24–26. [Google Scholar] [CrossRef]
- Nazarov, M.; Brik, M.G.; Spassky, D.; Tsukerblat, B.; Nazida, A.N.; Ahmad Fauzi, M.N. Structural and electronic properties of SrAl2O4:Eu2+ from density functional theory calculations. J. Alloys Compd. 2013, 573, 6–10. [Google Scholar] [CrossRef]
- Shi, R.; Qi, M.; Ning, L.; Pan, F.; Zhou, L.; Zhou, W.; Huang, Y.; Liang, H. Combined Experimental and ab Initio Study of Site Preference of Ce3+ in SrAl2O4. J. Phys. Chem. C 2015, 119, 19326–19332. [Google Scholar] [CrossRef]
- Fu, Z.; Zhou, S.; Yu, Y.; Zhang, S. Combustion synthesis and luminescence properties of nanocrystalline monoclinic SrAl2O4:Eu2+. Chem. Phys. Lett. 2004, 395, 285–289. [Google Scholar] [CrossRef]
- Zhai, B.G.; Huang, Y.M. Green photoluminescence and afterglow of Tb-doped SrAl2O4. J. Mater. Sci. 2017, 52, 1813–1822. [Google Scholar] [CrossRef]
- Rojas-Hernandez, R.E.; More, W.; Rubio-Marcos, F.; Fernandez, J.F. Investigating Raman spectra and density functional theory calculations on SrAl2O4 polymorphs. J. Raman Spectrosc. 2019, 50, 91–101. [Google Scholar] [CrossRef]
- Kushwaha, A.K.; Akbudak, S.; Candan, A.; Yadav, A.C.; Uğur, G.; Uğur, Ş. First Principles Investigations of the Structural, Elastic, Electronic, Vibrational and Thermodynamic Properties of Hexagonal XAl2O4 (X = Cd, Ca and Sr). Mater. Res. Express 2019, 6, 085518. [Google Scholar] [CrossRef]
- Akbudak, S.; Candan, A.; Kushwaha, A.K.; Yadav, A.C.; Uğur, G.; Uğur, Ş. Structural, elastic, electronic and vibrational properties of XAl2O4 (X =Ca, Sr and Cd) semiconductors with orthorhombic structure. J. Alloys Compd. 2019, 809, 151773. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Voigt, W. Handbook of Crystal Physics; Taubner: Leipzig, Germany, 1928. [Google Scholar]
- Reuss, A.; Angew, Z. Calculation of the flow limits of mixed crystals on the basis of the plasticity of monocrystals. Math. Mech. 1929, 9, 49. [Google Scholar]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal Elastic Anisotropy Index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.H.; Buessem, W.R.; Vahldiek, F.W.; Mersol, S.A. (Eds.) Anisotropy in Single Crystal Refractory Compound; Plenum; Springer: New York, NY, USA, 1968. [Google Scholar]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B. Density functional theory for calculation of elastic properties of orthorhomcic crystals: Applications to TiSi. J. Appl. Phys. 1998, 84, 4891. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Chen, J.; Du, Y.; Yu, J.; Zhou, R. Stability, thermal and mechanical properties of PtxAly compounds. Mater. Des. 2011, 32, 3231–3239. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the Debye temperature from elastic constants. J. Phys. Chem. Solid. 1963, 2490, 9–17. [Google Scholar] [CrossRef]
- Arab, F.; Sahraoui, F.A.; Haddadi, K.; Bouhemadou, A.; Louail, L. Phase stability, mechanical and thermodynamic properties of orthorhombic and trigonal MgSiN2: An ab initio study. Phase Transit. 2016, 89, 480–513. [Google Scholar] [CrossRef]
- Chen, S.; Sun, Y.; Duan, Y.H.; Huang, B.; Peng, M.J. Phase stability, structural and elastic properties of C15-type Laves transition-metal compounds MCo2 from first principles calculations. J. Alloys Compd. 2015, 630, 202–208. [Google Scholar] [CrossRef]
- Kim, W. Strategies for engineering phonon transport in thermoelectrics. J. Mater. Chem. C 2015, 3, 10336. [Google Scholar] [CrossRef]
- Yang, J.; Shahid, M.; Wan, C.L.; Feng, J.; Pan, W. Anisotropy in elasticity, sound velocities and minimum thermal conductivity of zirconia from first-principles calculations. J. Eur. Ceram. Soc. 2017, 37, 689. [Google Scholar] [CrossRef]
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio Molecular Dynamics for Liquid Metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and -type at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).