
Old Acquaintances and Novel Complex Structures for the Ni(II) and Cu(II) Complexes of bis-Chelate Oxime–Amide Ligands


Abstract
1. Introduction
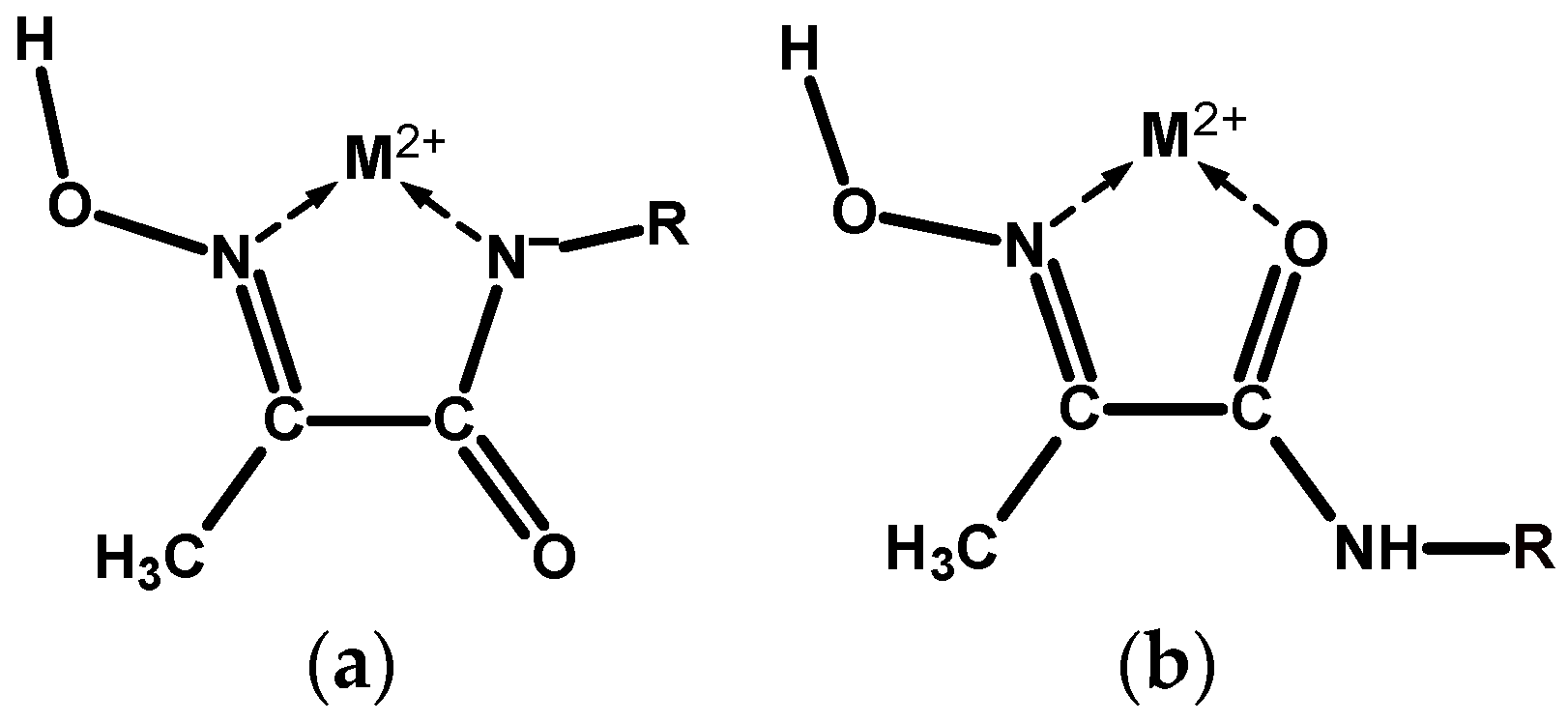
2. Results
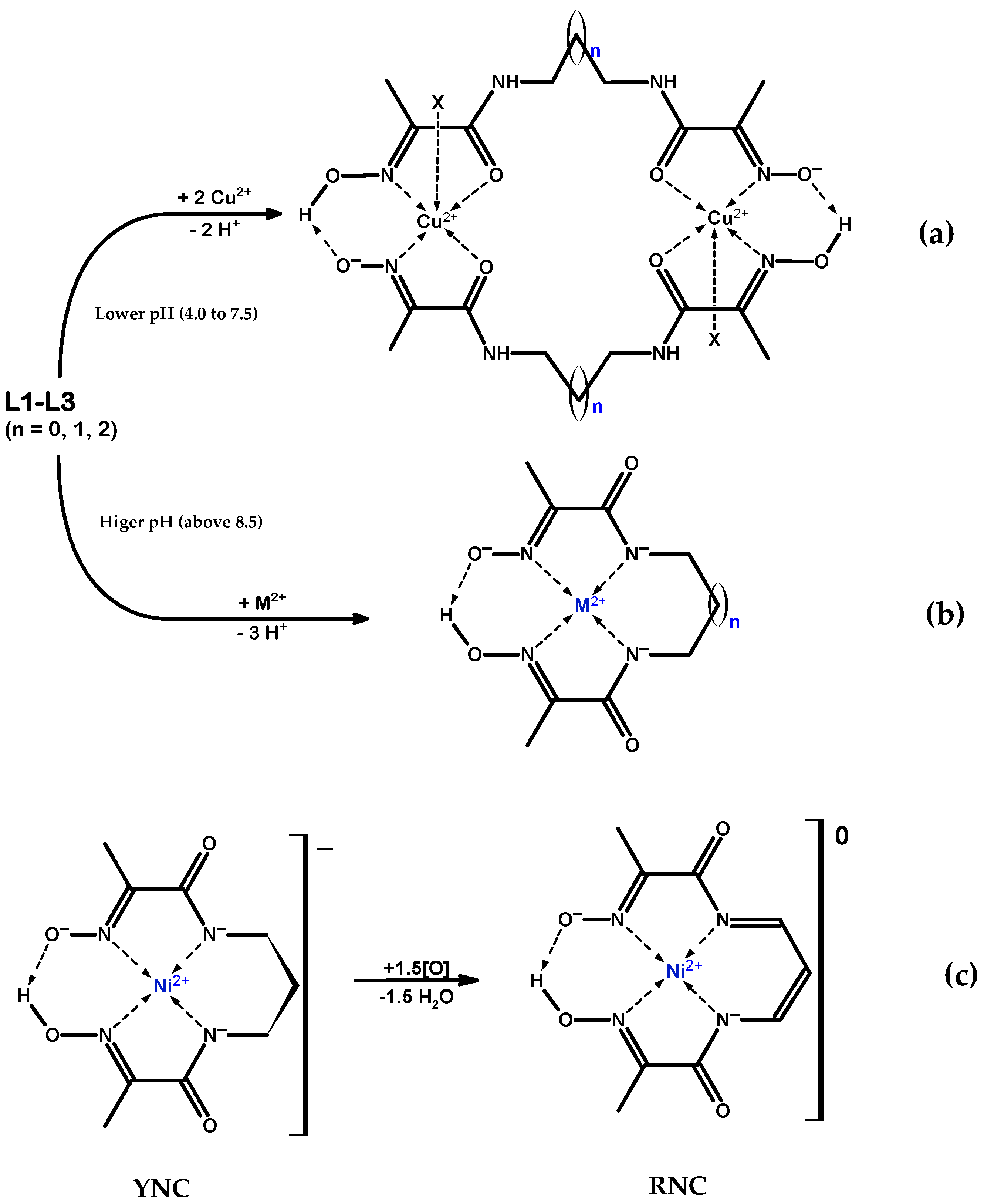
2.1. Synthesis and Characterisation
2.1.1. Cu(II) Complexes
2.1.2. Ni(II) Complexes
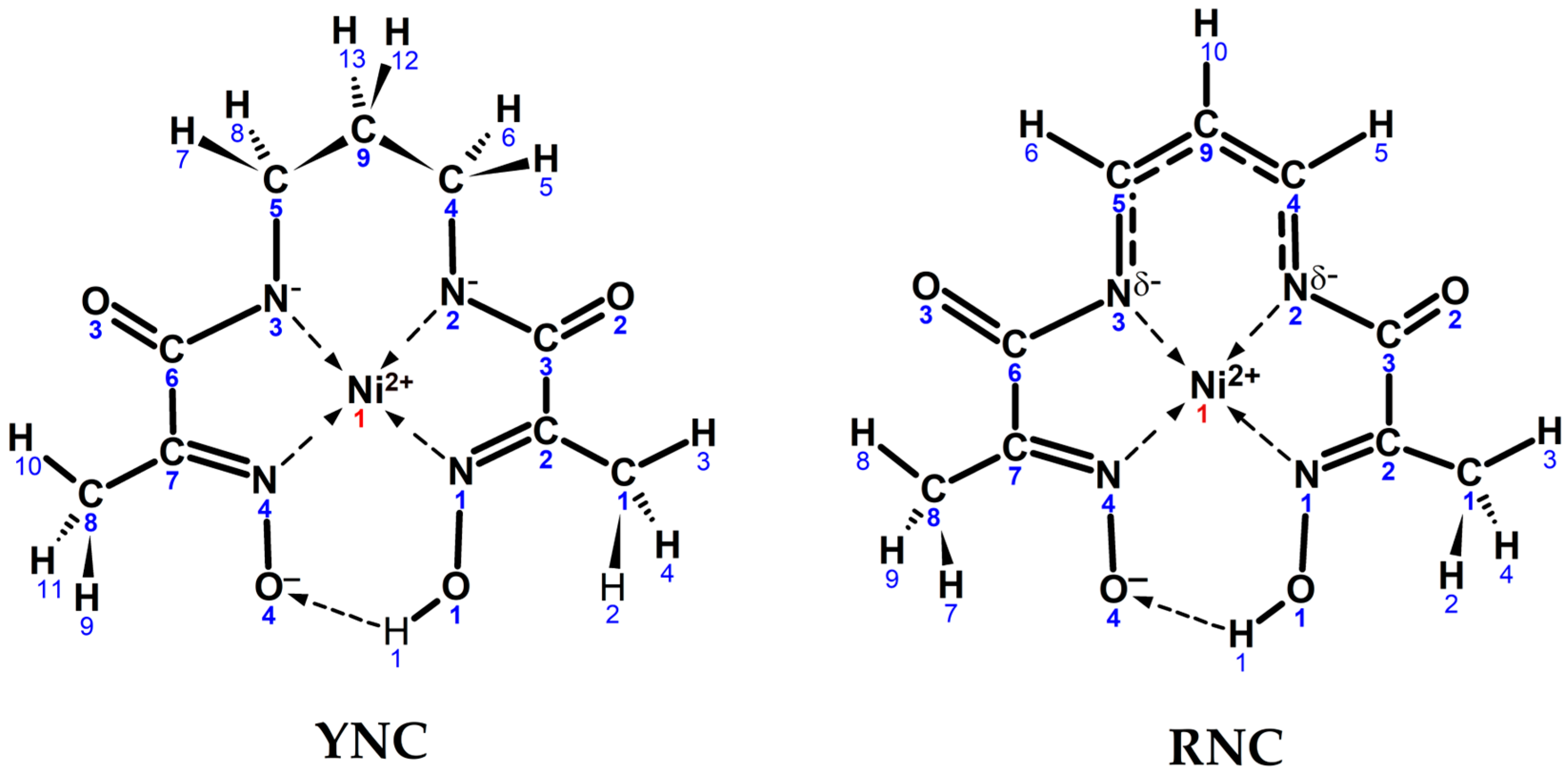
2.1.3. Novel Ni(II)-L2′ Red Nickel Complex (RNC)
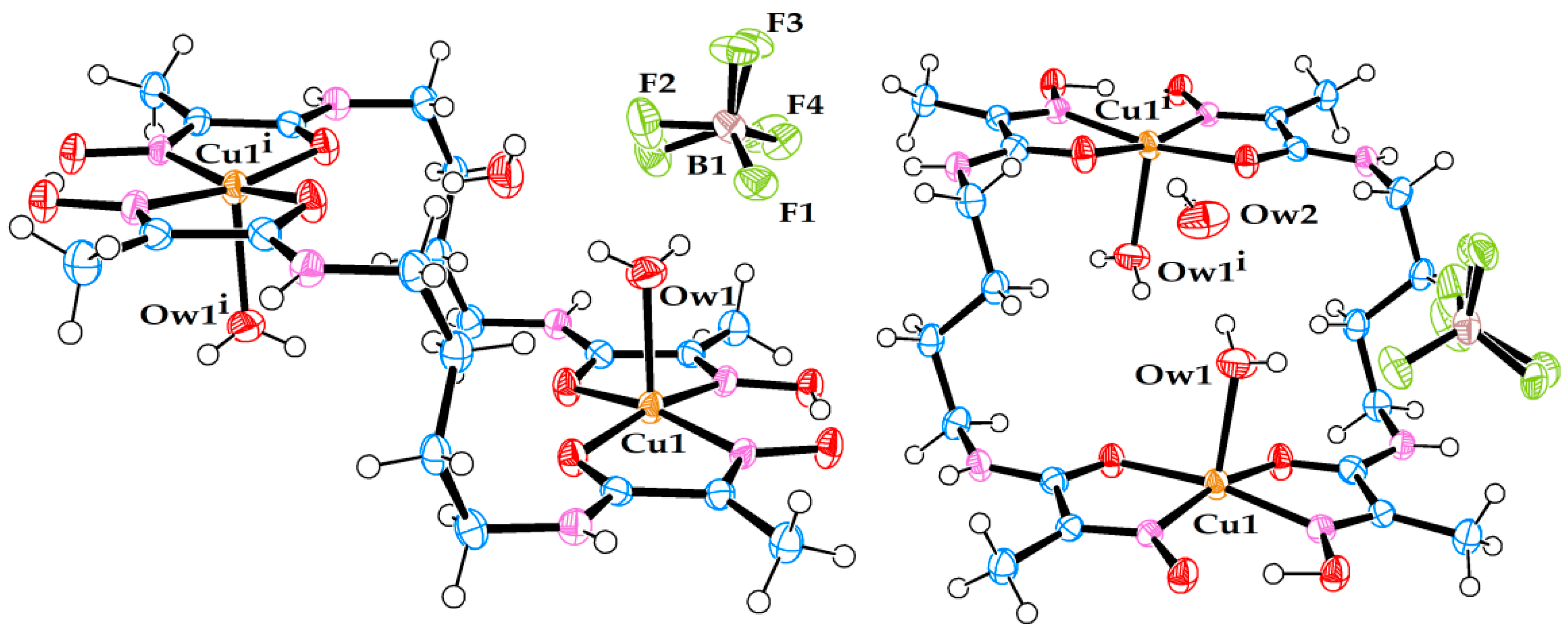
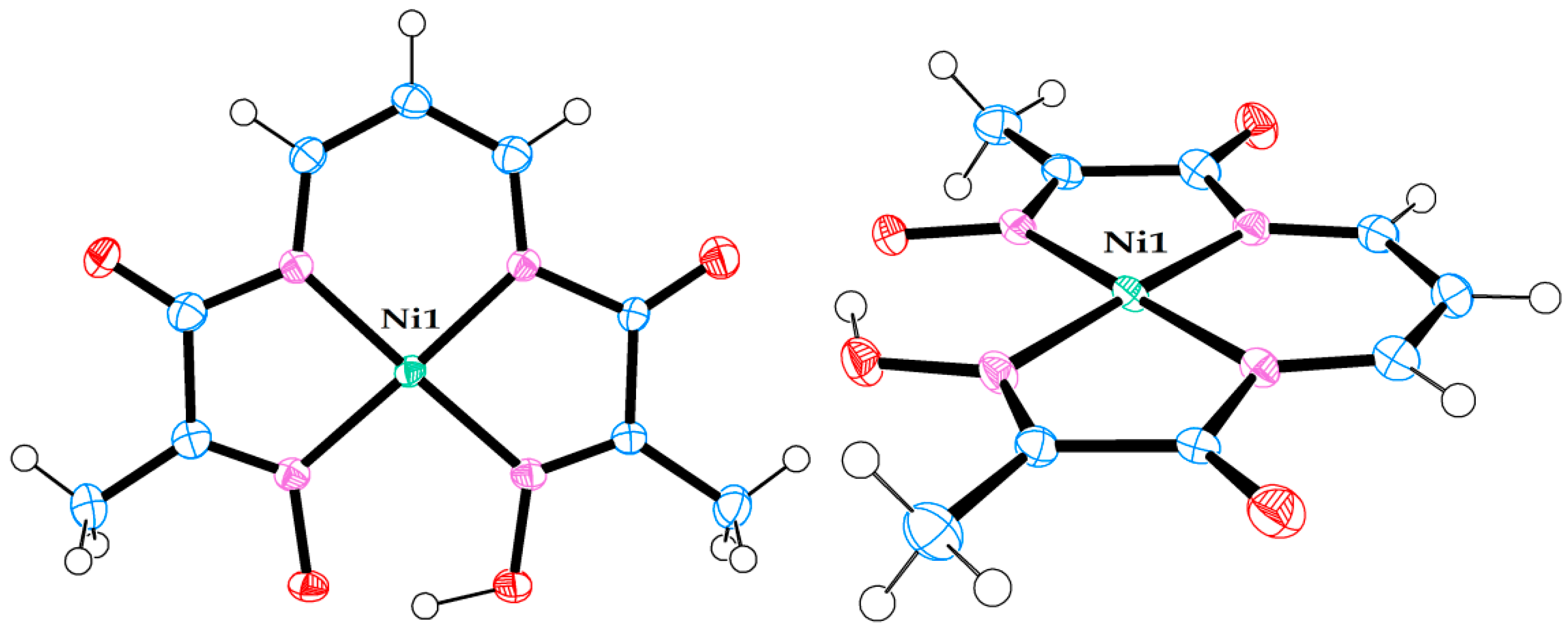
2.2. X-ray Diffraction
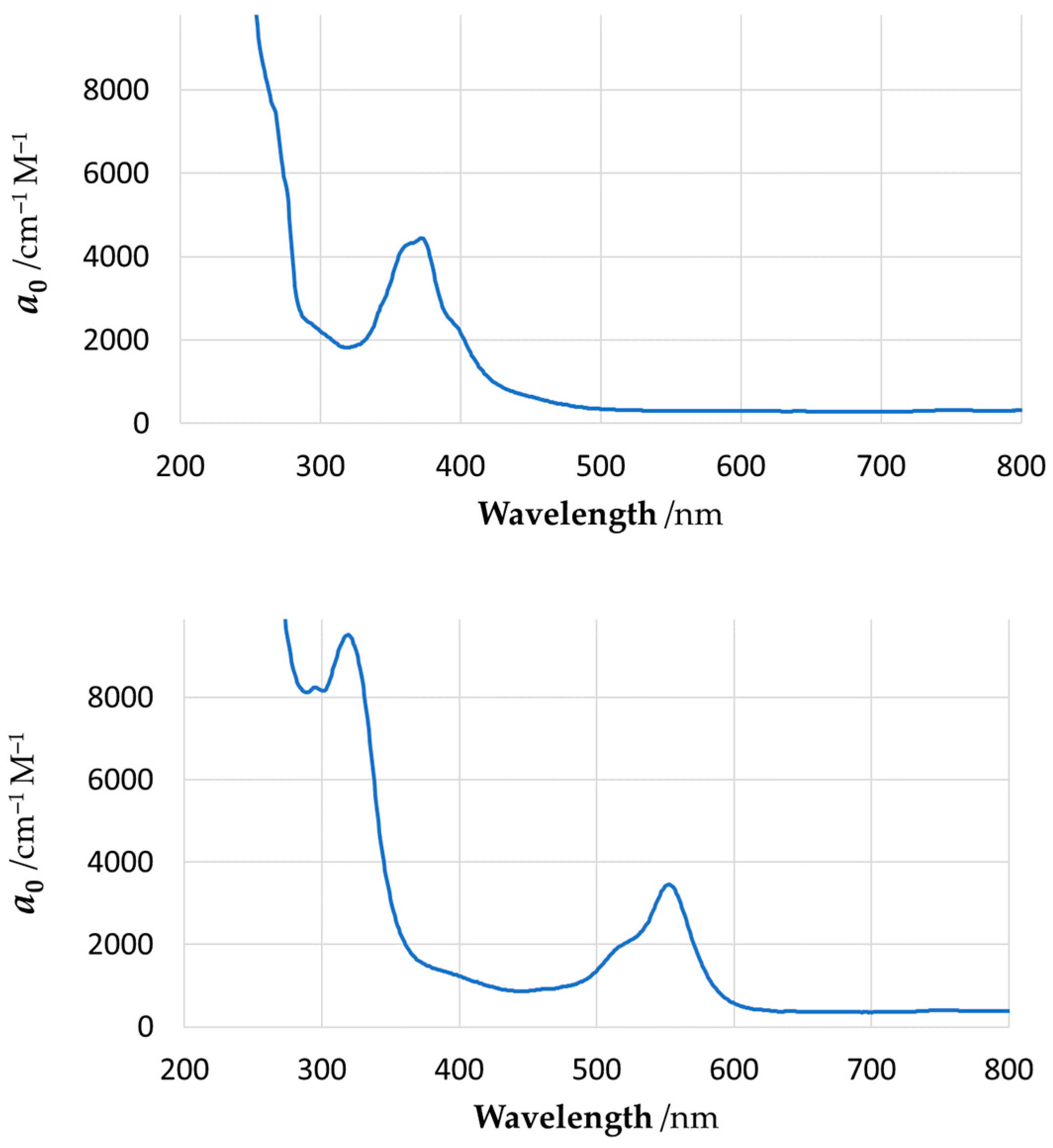
2.3. Quantum Chemical Modelling
3. Discussion
3.1. Cationic Complexes
3.2. Anionic Complexes
3.3. Neutral Complexes
3.4. Computational Results
4. Materials and Methods
4.1. Materials
4.2. Instrumental
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nikolayenko, I.V.; Barry, J.R.; Manival, A.; Theron, T.-J.; Grimmer, C. Peculiar sequence of the thermodynamic protonation parameters for bis-chelate ligands with methylhydroxyiminoethanamide moieties. Curr. Inorg. Chem. 2015, 5, 83–97. [Google Scholar] [CrossRef][Green Version]
- Moali, C.; Brollo, M.; Custot, J.; Sari, M.A.; Boucher, J.L.; Stuehr, D.J.; Mansuy, D. Recognition of α-amino acids bearing various C=NOH functions by nitric oxide synthase and arginase involves very different structural determinants. Biochemistry 2000, 39, 8208–8218. [Google Scholar] [CrossRef] [PubMed]
- Di Constanzo, L.; Moulin, M.; Haertlein, M.; Meilleur, F.; Christianson, D.W. Expression, purification, assay, and crystal structure of perdeuterated human arginase I. Arch. Biochem. Biophys. 2007, 465, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Pagonda, H.; Yogesh, P.P.; Katreddi, H.R.; Munirathinam, N. Novel tetranuclear distorted open-cubane copper complex containing oximate bridges: Synthesis, crystal structure, DNA binding and cleavage activity. Inorg. Chim. Acta 2012, 392, 478–482. [Google Scholar] [CrossRef]
- Das, L.K.; Biswas, A.; Kinyon, J.S.; Dalal, N.S.; Zhou, H.; Ghosh, A. Di-, tri-, and tetranuclear nickel(II) complexes with oximato bridges: Magnetism and catecholase-like activity of two tetranuclear complexes possessing rhombic topology. Inorg. Chem. 2013, 52, 11744–11757. [Google Scholar] [CrossRef] [PubMed]
- Razali, M.R.; Urbatsch, A.; Langley, S.K.; MacLellan, J.G.; Deacon, G.B.; Moubaraki, B.; Murray, K.S.; Batten, S.R. Linear trinuclear copper(II) complexes derived from the nucleophilic addition products of dicyanonitrosomethanide [C(CN)2(NO)]–: Syntheses, structures, and magnetic properties. Aust. J. Chem. 2012, 65, 918–925. [Google Scholar] [CrossRef]
- Lau, H.-P.; Gutsche, C.D. Polyfunctional catalysis of acetyl phosphate decomposition. J. Am. Chem. Soc. 1978, 100, 1857–1865. [Google Scholar] [CrossRef]
- Fritsky, I.O.; Kozłowski, H.; Prisyazhnaya, E.V.; Rzączyńska, Z.; Karaczyn, A.; Sliva, T.Y.; Głowiak, T. Co-ordination ability of novel tetradentate amide-and-oxime ligands: Differential binding to CuII and NiII. J. Chem. Soc. Dalton. Trans. 1998, 21, 3629–3633. [Google Scholar] [CrossRef]
- Duda, A.M.; Karaczyn, A.; Kozłowski, H.; Fritsky, I.O.; Głowiak, T.; Prisyazhnaya, E.V.; Sliva, T.Y.; Świątek-Kozłowska, J. Co-ordination of copper(II) and nickel(II) ions by a novel open chain oxime ligand. Dalton. Trans. 1997, 20, 3853–3859. [Google Scholar] [CrossRef]
- Nomkoko, E.T.; Jackson, G.E.; Nakani, B.S. In vitro and in vivo stability investigations of Cu(II), Zn(II), Ca(II) and Gd(III) complexes with N,N’-bis(2-hydroxyiminopropionyl) propane-1,3-diamine. Dalton. Trans. 2004, 9, 1432–1440. [Google Scholar] [CrossRef]
- Fritsky, I.O.; Karaczyn, A.; Kozłowski, H.; Głowiak, T.; Prisyazhnaya, E.V. Crystal and molecular structure of two tetradentate “oxime-and-amide” ligands. Z. Naturforsch. 1999, 54b, 456–460. [Google Scholar] [CrossRef]
- Fritsky, I.O.; Kozłowski, H.; Prisyazhnaya, E.V.; Karaczyn, A.; Kalibabchuk, V.A.; Głowiak, T. A short intramolecular hydrogen bond is a key factor in the self-assembly of a dimeric complex with a 22-membered metallamacrocyclic cavity. J. Chem. Soc. Dalton Trans. 1998, 10, 1535–1536. [Google Scholar] [CrossRef]
- Fritsky, I.O.; Swiatek-Kozlowska, J.; Dobosz, A.; Sliva, T.Y.; Dudarenko, N.M. Hydrogen bonded supramolecular structures of cationic and anionic module assemblies containing square-planar oximate complex anions. Inorg. Chim. Acta 2004, 357, 3746–3752. [Google Scholar] [CrossRef]
- Kanderal, O.M.; Kozlowski, H.; Dobosz, A.; Swiatek-Kozlowska, J.; Meyer, F.; Fritsky, I.O. Effect of metal ionic radius and chelate ring alternation motif on stabilization of trivalent nickel and copper in binuclear complexes with double cis-oximato bridges. Dalton. Trans. 2005, 8, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Fritsky, I.O.; Swiatek-Kozlowska, J.; Kapshuk, A.A.; Kozlowski, H.; Sliva, T.Y.; Gumienna-Kontecka, E.; Prisyazhnaya, E.V.; Iskenderov, T.S. Preparation and crystal structure of a mixed metal assembly [Ni(phen)3][Cu(H-1pap)]2(NO3) · 8 H2O featuring octahedral cationic and square-planar anionic modules. Z. Naturforsch 2000, 55b, 966–970. [Google Scholar] [CrossRef]
- Fritsky, I.O.; Kozlowski, H.; Kanderal, O.M.; Haukka, M.; Swiatek-Kozlowska, J.; Gumienna-Kontecka, E.; Meyer, F. Efficient stabilization of copper(III) in tetraaza pseudo-macrocyclic oxime-and-hydrazide ligands with adjustable cavity size. Chem. Commun. 2006, 39, 4125–4127. [Google Scholar] [CrossRef] [PubMed]
- Buvailo, A.I.; Gumienna-Kontecka, E.; Pavlova, S.V.; Fritsky, I.O.; Haukka, M. Dimeric versus polymeric coordination in copper(II) cationic complexes with bis(chelating) oxime and amide ligands. Dalton Trans. 2010, 39, 6266–6275. [Google Scholar] [CrossRef]
- Kalibabchuk, V.A.; Usenko, N.I.; Golenya, I.A.; Iskenderov, T.S.; Haukka, M. Poly[bis [μ4-N-(2-hydroxyiminopropionyl)-N’-(2-oxidoimino propionyl)propane-1,3-diaminato]dimethano lcalciumdicopper(II)]. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, m1139. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomie distances in halides and chaleogenides. Acta Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Banks, C.V.; Barnum, D.W. Intermolecular Metal-Metal Bonds and Absorption Spectra of Some Nickel(II) and Palladium(II) Complexes of vic-Dioximes1. J. Am. Chem. Soc. 1958, 80, 4767–4772. [Google Scholar] [CrossRef]
- Williams, D.E.; Wohlauer, G.; Rundle, R.E. Crystal structures of nickel and palladium dimethylglyoximes. J. Am. Chem. Soc. 1959, 81, 755–756. [Google Scholar] [CrossRef]
- Egneus, B. The solution chemistry of ethylmethylglyoxime. Anal. Chim. Acta 1969, 48, 291–307. [Google Scholar] [CrossRef]
- Li, D.-X.; Xu, D.-J.; Xu, Y.-Z. Redetermination of bis(dimethylglyoximato-κ2N,N′)nickel(II). Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, m1094–m1095. [Google Scholar] [CrossRef]
- Czapik, A.; Gdaniec, M. CSD Communication; University of Cambridge: Cambridge, UK, 2009. [Google Scholar]
- Bruce-Smith, I.F.; Zakharov, B.A.; Stare, J.; Boldyreva, E.V.; Pulham, C.R. Structural properties of nickel dimethylglyoxime at high pressure: Single-crystal X-ray diffraction and dft studies. J. Phys. Chem. C 2014, 118, 24705–24713. [Google Scholar] [CrossRef]
- Foust, A.S.; Soderberg, R.H. Complexes of bromine and iodine with bis-(diphenylglyoximato)nickel(II) and bis(diphenylglyoximato)palladium(II). J. Am. Chem. Soc. 1967, 89, 5507–5508. [Google Scholar] [CrossRef]
- Gleizes, A.; Marks, T.J.; Ibers, J.A. Solid state structure and oxidation states in bis(diphenylglyoximato)nickel and -palladium iodides. J. Am. Chem. Soc. 1975, 97, 3545–3546. [Google Scholar] [CrossRef]
- Cowie, M.; Gleizes, A.; Grynkewich, G.W.; Kalina, D.W.; McClure, M.S.; Scaringe, R.P.; Teitelbaum, R.C.; Ruby, S.L.; Ibers, J.A.; Kannewurf, C.R.; et al. Rational synthesis of unidimensional mixed valence solids. Structural, spectral, and electrical studies of charge distribution and transport in partially oxidized nickel and palladium bisdiphenylglyoximates. J. Am. Chem. Soc. 1979, 101, 2921–2936. [Google Scholar] [CrossRef]
- Schlemper, E.O.; Murmann, R.K. Bis[3-(hydroxyamino)-3-methyl-2-butanone oximato(2-)-N,N’]nickel. Structure and properties of a [Ni(DMG)2]0-related complex. Inorg. Chem. 1983, 22, 1077–1081. [Google Scholar] [CrossRef]
- Opalade, A.A.; Labadi, I.; Gomez-Garcia, C.; Hietsoi, O.; Gerasimchuk, N. Nickel(II) aqua complexes with chelating ligands: What happens when water is gone? Cryst. Growth Des. 2022, 22, 6168–6182. [Google Scholar] [CrossRef]
- CrysAlis RED and Scakle3 Abspack, Version 1.171.32.29; Oxford Diffraction Ltd.: Abingdon, UK.
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Lübben, J.; Wandtke, C.M.; Hübschle, C.B.; Ruf, M.; Sheldrick, G.M.; Dittrich, B. Aspherical scattering factors for SHELXL—Model, implementation and application. Acta Crystallogr. A Found. Adv. 2019, A75, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Mercury 2023.1.0 (Build 376230). Copyright © CCDC, 2001–2023. Available online: https://www.ccdc.cam.ac.uk/solutions/software/mercury/ (accessed on 1 March 2023).
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- EnCIFer 2023.1.0 (Build 376230). Copyright © CCDC. 2001–2023. Available online: https://www.ccdc.cam.ac.uk/solutions/software/encifer/ (accessed on 1 March 2023).
- Olex2 v1.5 Copyright © OlexSys Ltd. 2004–2023. Available online: https://www.olexsys.org/olex2/ (accessed on 1 March 2023).
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision 9.0; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1; 2001, Pittsburg. Available online: http://www.ccl.net/cca/software/MS-WIN95-NT/mopac6/nbo.htm (accessed on 1 March 2023).
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Coordination Mode | Complex |
|---|---|---|
| L1 | 2(NoxOad) | = (1) sp-tpp 1 |
| L3 | 2(NoxOad) | = (2) sp-tpp |
| L3 | 2(NoxOad) | = (3) sp-tpp |
| L3 | 2(NoxOad) | = (4) sp-tpp |
| L2 | 2(NoxNad) | = (5) 2 sppm 3 |
| Ligand | Coordination Mode | Complex |
|---|---|---|
| L2 | 2(NoxNad) | = (6) 1 sppm |
| L2′ | (NoxNad)(NoxNki) | = (7) 2 dohp 3 |
| L2′ | (NoxNad)(NoxNki) | = (8) 2 dohdp 4 |
| Complex | (1) | (2) | (3) | (4) | (5) | (7) | (8) |
|---|---|---|---|---|---|---|---|
| Empirical formula | C16H32N10O17Cu2 | C20H42N8O12B2F8Cu2 | C20H42N8O12Cl2Cu2 | C30H56N12O14Cl2Cu2 | C9H21N4O8CuLi | C9H10N4O4Ni | C9H10N4O4Ni |
| M | 763.57 | 887.30 | 784.59 | 1006.84 | 383.77 | 296.89 | 296.89 |
| T/K | 293 (2) | 293(2) | 293(2) | 293(2) | 293 (2) | 293(2) | 293(2) |
| Crystal system | monoclinic | triclinic | triclinic | triclinic | triclinic | monoclinic | triclinic |
| Space group | I 2/a | P | P | P | P | C c | P |
| a/Å | 8.027(1) | 8.664(5) | 8.141(1) | 8.8321(1) | 7.450(2) | 9.5938(13) | 6.5672(6) |
| b/Å | 23.663(3) | 9.516(5) | 8.887(2) | 10.8660(1) | 8.862(1) | 19.587(2) | 10.5171(10) |
| c/Å | 15.194(5) | 11.745(5) | 11.414(2) | 11.8382(2) | 11.922(2) | 6.5058(8) | 16.1337(14) |
| α/° | 90 | 102.016(5) | 95.47(1) | 105.766(1) | 103.19(1) | 90 | 85.084(7) |
| β/° | 96.135(14) | 91.879(5) | 94.41(1) | 96.551(2) | 97.31(1) | 117.346(9) | 82.041(7) |
| γ/° | 90 | 108.828(5) | 102.823(5) | 100.627(2) | 90.39(1) | 90 | 80.262(7) |
| V/Å−3 | 2869.5(10) | 891.2(8) | 797.4(2) | 1058.30(3) | 759.6(3) | 1085.9(2) | 1085.47(17) |
| Z | 4 | 1 | 1 | 1 | 2 | 4 | 4 |
| dc/g cm−3 | 1.768 | 1.653 | 1.634 | 1.580 | 1.678 | 1.816 | 1.817 |
| μ/mm−1 | 1.577 | 1.300 | 1.570 | 1.208 | 2.481 | 2.757 | 1.801 |
| F(0 0 0) | 1568 | 454 | 406 | 524 | 398 | 608 | 608 |
| Crystal size /mm | 0.1 × 0.05 × 0.05 | 0.2 × 0.15 × 0.15 | 0.15 × 0.15 × 0.1 | 0.23 × 0.18 × 0.1 | 0.25 × 0.1 × 0.1 | 0.22 × 0.15 × 0.1 | 0.17 × 0.13 × 0.1 |
| Colour | dark green | dark green | dark green | dark green | red | raspberry red | wine red |
| λ/Å | 0.71069 | 0.71069 | 0.71069 | 0.71069 | 1.5418 | 1.5418 | 0.71069 |
| θ range/° | 4.137 to 22.541 | 4.130 to 28.431 | 3.884 to 28.370 | 1.999 to 25.871 | 5.130 to 58.947 | 4.515 to 72.105 | 3.815 to 27.503 |
| Index range | −6 ≤ h ≤ 8, | −11 ≤ h ≤ 11, | −10 ≤ h ≤ 10, | −10 ≤ h ≤ 10, | −8 ≤ h ≤ 8, | −11 ≤ h ≤ 9, | −8 ≤ h ≤ 8, |
| −21 ≤ k ≤ 25, | −12 ≤ k ≤ 12, | −10 ≤ k ≤ 11, | −12 ≤ k ≤ 13, | −9 ≤ k ≤ 8, | −23 ≤ k ≤ 22, | −13 ≤ k ≤ 12, | |
| −16 ≤ l ≤ 16 | −14 ≤ l ≤ 15 | −15 ≤ l ≤ 14 | −14 ≤ l ≤ 14 | −13 ≤ l ≤ 13 | −6 ≤ l ≤ 7 | −17 ≤ l ≤ 20 | |
| Reflections collected | 5734 | 9350 | 8239 | 19106 | 5677 | 2091 | 9259 |
| Independent Reflections/Rint, Rsigma | 1886/ 0.0662, 0.117 | 3807/ 0.0281, 0.0421 | 3408/ 0.0371, 0.075 | 3972/ 0.0263, 0.0183 | 2109/ 0.0350, 0.0318 | 1291/ 0.0443, 0.0376 | 4162/ 0.0371, 0.0897 |
| Data/ restraints/ parameters | 1886/0/211 | 3807/0/292 | 3408/0/207 | 3972/1/283 | 2109/0/218 | 1291/149/166 | 4162/0/343 |
| Goodness of fit on F2 | 0.817 | 1.052 | 0.927 | 0.978 | 0.973 | 1.086 | 0.872 |
| R indices | |||||||
| [I > 2σ(I)] | |||||||
| R1 | 0.0501 | 0.0384 | 0.0334 | 0.0370 | 0.0424 | 0.0419 | 0.0350 |
| wR2 | 0.1020 | 0.0935 | 0.0800 | 0.1006 | 0.1147 | 0.1121 | 0.0754 |
| R indices | |||||||
| (all data) | |||||||
| R1 | 0.0927 | 0.0530 | 0.0548 | 0.0399 | 0.0499 | 0.0467 | 0.0696 |
| wR2 | 0.1150 | 0.1016 | 0.0767 | 0.1043 | 0.1203 | 0.1197 | 0.0804 |
| Max/min electron density/e Å−3 | 0.73/−0.44 | 0.98/−0.37 | 0.66/−0.48 | 0.59/−0.93 | 0.65/−0.76 | 0.39/−0.81 | 0.67/−0.39 |
| (1) | (2) | (3) | (4) | ||||
|---|---|---|---|---|---|---|---|
| Cu N1 | 1.960(5) | Cu N1 | 1.957(2) | Cu N1 | 1.960(2) | Cu N1 | 1.969(3) |
| Cu N4 | 1.946(5) | Cu N4 | 1.939(2) | Cu N4 | 1.958(2) | Cu N4 | 1.964(2) |
| Cu O2 | 1.962(4) | Cu O2 | 1.984(2) | Cu O2 | 1.974(2) | Cu O2 | 1.988(2) |
| Cu O3 | 1.978(4) | Cu O3 | 1.963(2) | Cu O3 | 1.972(2) | Cu O3 | 1.983(2) |
| Cu OW1 | 2.194(5) | Cu OW1 | 2.175(3) | Cu Cl | 2.4624(8) | Cu Cl | 2.5678(8) |
| C3 O2 | 1.267(7) | C3 O2 | 1.258(3) | C3 O2 | 1.266(3) | C3 O2 | 1.264(3) |
| C6 O3 | 1.261(8) | C6 O3 | 1.265(3) | C6 O3 | 1.267(3) | C6 O3 | 1.266(3) |
| N1 C2 | 1.29(1) | N1 C2 | 1.283(3) | N1 C2 | 1.272(3) | N1 C2 | 1.285(3) |
| N4 C7 | 1.283(8) | N4 C7 | 1.290(3) | N4 C7 | 1.291(3) | N4 C7 | 1.292(3) |
| O1 N1 | 1.331(8) | O1 N1 | 1.354(3) | O1 N1 | 1.358(3) | O1 N1 | 1.352(3) |
| O4 N4 | 1.371(6) | O4 N4 | 1.332(3) | O4 N4 | 1.331(3) | O4 N4 | 1.331(3) |
| C3 N2 | 1.330(9) | C3 N2 | 1.318(4) | C3 N2 | 1.303(3) | C3 N2 | 1.320(3) |
| C6 N3 | 1.332(8) | C6 N3 | 1.317(3) | C6 N3 | 1.303(3) | C6 N3 | 1.316(4) |
| O1 O4 | 2.500 | O1 O4 | 2.495 | O1 O4 | 2.500 | O1 O4 | 2.470 |
| O1 H1 | 1.226 | O1 H1 | 1.040 | O1 H1 | 1.032 | O1 H1 | 0.885 |
| O4 H1 | 1.282 | O4 H1 | 1.459 | O4 H1 | 1.476 | O4 H1 | 1.588 |
| BP 1—Cu | 0.2458(8) | BP—Cu | 0.2010(4) | BP—Cu | 0.3200(4) | BP—Cu | 0.2247(4) |
| N1 Cu O2 | 81.8(2) | N1 Cu O2 | 80.50(8) | N1 Cu O2 | 80.38(8) | N1 Cu O2 | 81. 03(10) |
| O2 Cu O3 | 99.1(2) | O2 Cu O3 | 99.07(8) | O2 Cu O3 | 96.99(7) | O2 Cu O3 | 99.98(8) |
| O3 Cu N4 | 80.6(2) | O3 Cu N4 | 81.96(8) | O3 Cu N4 | 81.27(8) | O3 Cu N4 | 81.64(9) |
| N4 Cu N1 | 94.9(2) | N4 Cu N1 | 96.06(9) | N4 Cu N1 | 95.25(9) | N4 Cu N1 | 94.4(1) |
| N1 Cu O3 | 165.0(2) | N1 Cu O3 | 169.26(9) | N1 Cu O3 | 162.30(8) | N1 Cu O3 | 166.73(9) |
| O2 Cu N4 | 165.9(2) | O2 Cu N4 | 166.98(8) | O2 Cu N4 | 160.17(8) | O2 Cu N4 | 166.54(9) |
| N1 Cu OW1 | 101.6(2) | N1 Cu OW1 | 94.40(10) | N1 Cu Cl | 98.87(7) | N1 Cu Cl | 96.65(7) |
| O2 Cu OW1 | 101.1(2) | O2 Cu OW1 | 93.85(9) | O2 Cu Cl | 99.65(5) | O2 Cu Cl | 95.23(6) |
| O3 Cu OW1 | 93.0(2) | O3 Cu OW1 | 96.33(9) | O3 Cu Cl | 98.83(5) | O3 Cu Cl | 96.43(6) |
| N4 Cu OW1 | 93.1(2) | N4 Cu OW1 | 98.96(9) | N4 Cu Cl | 100.13(7) | N4 Cu Cl | 97.88(7) |
| BP Cu-OW1 | 84.5(1) | BP Cu-OW1 | 87.51(8) | BP Cu-Cl | 89.86(4) | BP Cu-Cl | 88.69(4) |
| (5) | |||||||
|---|---|---|---|---|---|---|---|
| Cu N1 | 1.963(2) | N1 C2 | 1.273(4) | O1 O4 | 2.580 | N1 Cu N2 | 81.6(1) |
| Cu N2 | 1.930(3) | N4 C7 | 1.281(4) | O1 H1 | 0.839 | N2 Cu N3 | 99.2(1) |
| Cu N3 | 1.920(2) | O1 N1 | 1.384(4) | O4 H1 | 1.751 | N3 Cu N4 | 82.5(1) |
| Cu N4 | 1.953(3) | O4 N4 | 1.374(3) | O2 Li | 1.953(6) | N4 Cu N1 | 96.49(11) |
| C3 O2 | 1.251(4) | C3 N2 | 1.325(4) | BP—Cu | 0.0658(5) | N1 Cu N3 | 174.5(1) |
| C6 O3 | 1.271(3) | C6 N3 | 1.296(4) | N2 Cu N4 | 177.0(1) | ||
| (7) | (8) | ||||
|---|---|---|---|---|---|
| Ni N1 | 1.877(8) | Ni1 N1 | 1.858(3) | Ni2 N5 | 1.867(2) |
| Ni N2 | 1.861(7) | Ni1 N2 | 1.841(3) | Ni2 N6 | 1.848(2) |
| Ni N3 | 1.851(8) | Ni1 N3 | 1.842(3) | Ni2 N7 | 1.837(2) |
| Ni N4 | 1.873(8) | Ni1 N4 | 1.862(3) | Ni2 N8 | 1.875(3) |
| C3 O2 | 1.228(11) | C3 O2 | 1.193(4) | C12 O6 | 1.200(4) |
| C6 O3 | 1.210(13) | C6 O3 | 1.207(4) | C15 O7 | 1.205(3) |
| N1 C2 | 1.282(11) | N1 C2 | 1.294(4) | N5 C11 | 1.293(4) |
| N4 C7 | 1.274(13) | N4 C7 | 1.293(4) | N8 C16 | 1.297(4) |
| O1 N1 | 1.339(10) | O1 N1 | 1.348(3) | O5 N5 | 1.343(3) |
| O4 N4 | 1.342(10) | O4 N4 | 1.350(3) | O8 N8 | 1.335(3) |
| C3 N2 | 1.441(11) | C3 N2 | 1.421(4) | C12 N6 | 1.413(4) |
| C6 N3 | 1.412(11) | C6 N3 | 1.418(4) | C15 N7 | 1.427(4) |
| N2 C4 | 1.315(14) | N2 C4 | 1.339(4) | N6 C13 | 1.331(4) |
| N3 C5 | 1.325(13) | N3 C5 | 1.331(4) | N7 C14 | 1.326(4) |
| C4 C9 | 1.369(14) | C4 C9 | 1.383(4) | C13 C18 | 1.391(4) |
| C5 C9 | 1.406(12) | C5 C9 | 1.377(5) | C14 C18 | 1.380(5) |
| O1 O4 | 2.439 | O1 O4 | 2.449 | O5 O8 | 2.446 |
| O1 H1 | 1.024 | O1 H1 | 0.992 | O5 H2 | 1.125 |
| O4 H1 | 1.415 | O4 H1 | 1.472 | O8 H2 | 1.340 |
| BP—Ni | 0.010(2) | BP—Ni1 | 0.0047(4) | BP—Ni2 | 0.0017(4) |
| N1 Ni N2 | 84.0(3) | N1 Ni1 N2 | 83.51(11) | N5 Ni2 N6 | 83.15(11) |
| N2 Ni N3 | 94.4(4) | N2 Ni1 N3 | 95.22(11) | N6 Ni2 N7 | 94.83(11) |
| N3 Ni N4 | 83.7(3) | N3 Ni1 N4 | 83.40(11) | N7 Ni2 N8 | 84.20(11) |
| N4 Ni N1 | 97.8(3) | N4 Ni1 N1 | 97.88(12) | N8 Ni2 N5 | 97.82(12) |
| N1 Ni N3 | 178.2(4) | N1 Ni1 N3 | 178.54(11) | N5 Ni2 N7 | 177.95(12) |
| N2 Ni N4 | 178.0(5) | N2 Ni1 N4 | 178.61(11) | N6 Ni2 N8 | 179.02(11) |
| YNC | RNC | ||||||
|---|---|---|---|---|---|---|---|
| Ni−N1 | 1.903 | C7−C6 | 1.502 | Ni−N1 | 1.905 | C7−C6 | 1.473 |
| Ni−N4 | 1.900 | N2−C4 | 1.453 | Ni−N4 | 1.895 | N2−C4 | 1.322 |
| Ni−N2 | 1.898 | N3−C5 | 1.455 | Ni−N2 | 1.880 | N3−C5 | 1.321 |
| Ni−N3 | 1.883 | C4−C9 | 1.527 | Ni−N3 | 1.864 | C4−C9 | 1.387 |
| O1−N1 | 1.354 | C5−C9 | 1.528 | O1−N1 | 1.340 | C5−C9 | 1.398 |
| O4−N4 | 1.324 | O1−O4 | 2.461 | O4−N4 | 1.257 | O1−O4 | 2.486 |
| C3−O2 | 1.246 | O1−H1 | 1.067 | C3−O2 | 1.216 | O1−H1 | 1.049 |
| C6−O3 | 1.248 | O4⋯H1 | 1.402 | C6−O3 | 1.217 | O4⋯H1 | 1.449 |
| N1−C2 | 1.283 | N1−Ni−N2 | 82.60 | N1−C2 | 1.287 | N1−Ni−N2 | 83.00 |
| N4−C7 | 1.292 | N4−Ni−N3 | 83.57 | N4−C7 | 1.304 | N4−Ni−N3 | 83.93 |
| C3−N2 | 1.334 | N1−Ni−N4 | 96.59 | C3−N2 | 1.399 | N1−Ni−N4 | 97.85 |
| C6−N3 | 1.339 | N2−Ni−N3 | 97.23 | C6−N3 | 1.417 | N2−Ni−N3 | 95.22 |
| C1−C2 | 1.488 | N1−Ni−N3 | 179.28 | C1−C2 | 1.486 | N1−Ni−N3 | 178.33 |
| C8−C7 | 1.489 | N4−Ni−N2 | 179.02 | C8−C7 | 1.488 | N4−Ni−N2 | 179.15 |
| C2−C3 | 1.513 | O1−H1⋯O4 | 170.04 | C2−C3 | 1.498 | O1−H1⋯O4 | 168.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzicalupi, C.; Grimmer, C.; Nikolayenko, I.V. Old Acquaintances and Novel Complex Structures for the Ni(II) and Cu(II) Complexes of bis-Chelate Oxime–Amide Ligands. Molecules 2024, 29, 522. https://doi.org/10.3390/molecules29020522
Bazzicalupi C, Grimmer C, Nikolayenko IV. Old Acquaintances and Novel Complex Structures for the Ni(II) and Cu(II) Complexes of bis-Chelate Oxime–Amide Ligands. Molecules. 2024; 29(2):522. https://doi.org/10.3390/molecules29020522
Chicago/Turabian StyleBazzicalupi, Carla, Craig Grimmer, and Igor Vasyl Nikolayenko. 2024. "Old Acquaintances and Novel Complex Structures for the Ni(II) and Cu(II) Complexes of bis-Chelate Oxime–Amide Ligands" Molecules 29, no. 2: 522. https://doi.org/10.3390/molecules29020522
APA StyleBazzicalupi, C., Grimmer, C., & Nikolayenko, I. V. (2024). Old Acquaintances and Novel Complex Structures for the Ni(II) and Cu(II) Complexes of bis-Chelate Oxime–Amide Ligands. Molecules, 29(2), 522. https://doi.org/10.3390/molecules29020522