
Microsolvation of a Proton by Ar Atoms: Structures and Energetics of ArnH+ Clusters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Details, Results and Discussion
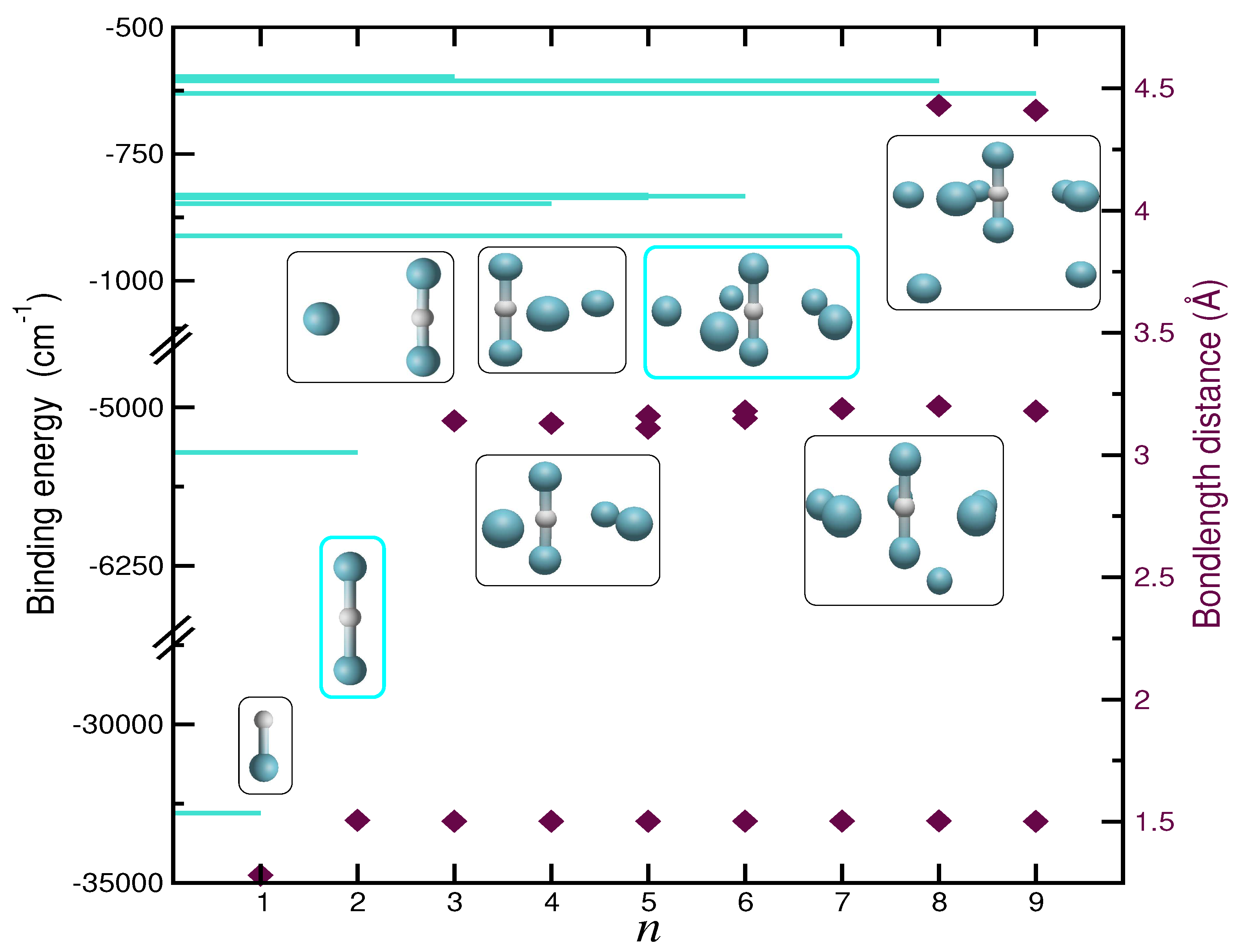
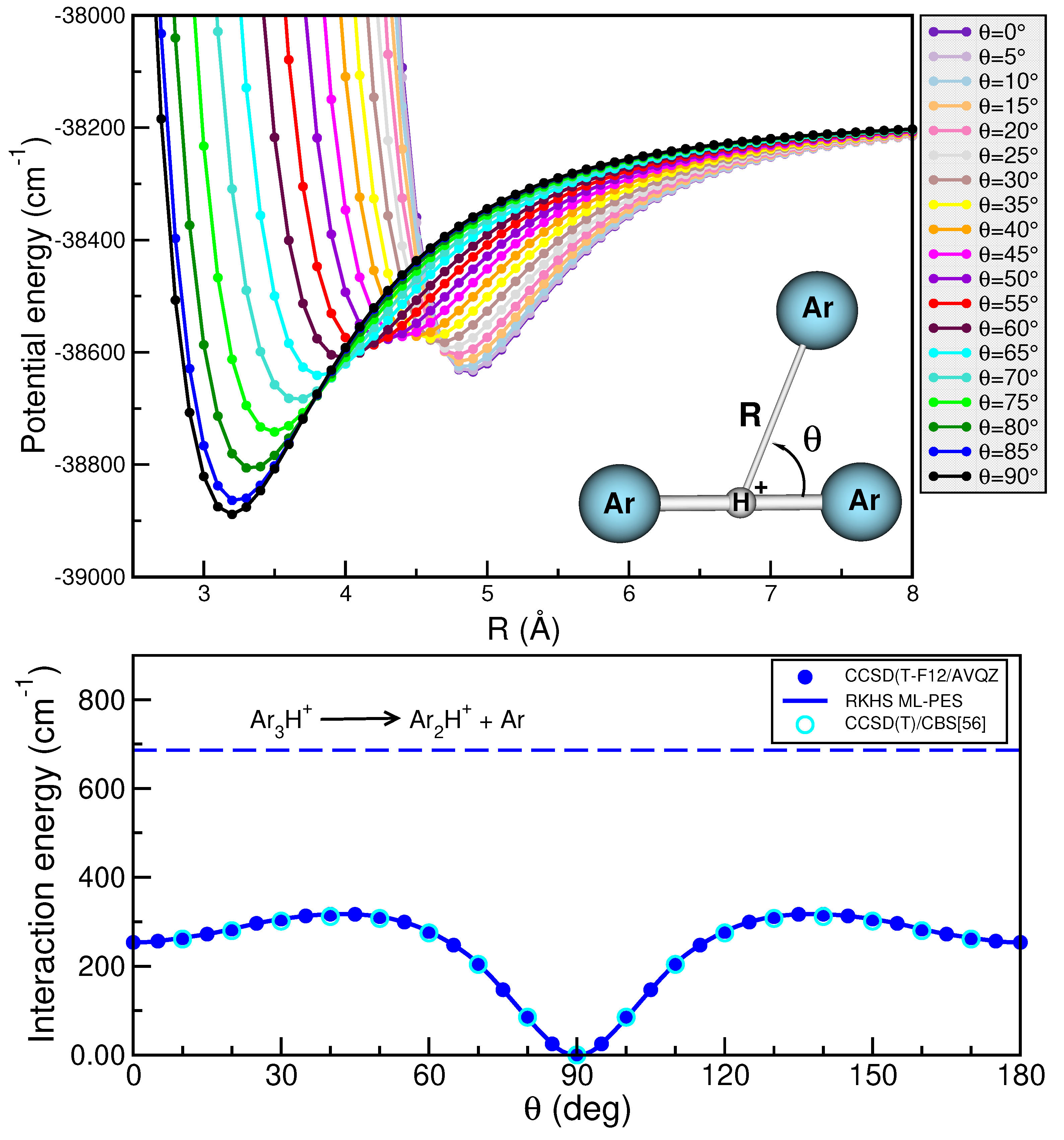
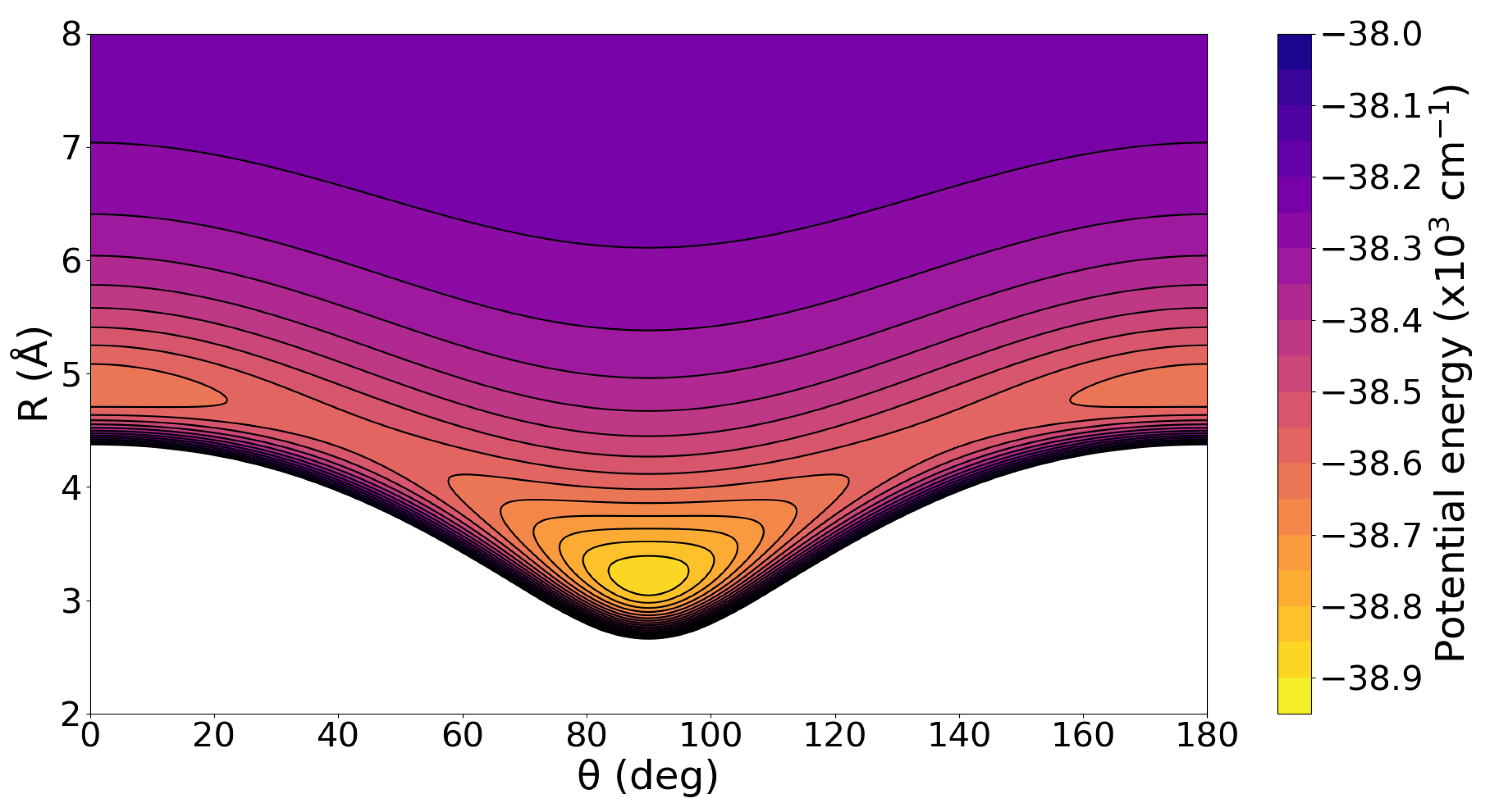
2.1. Binding and Structuring in Clusters from Electronic Structure Calculations
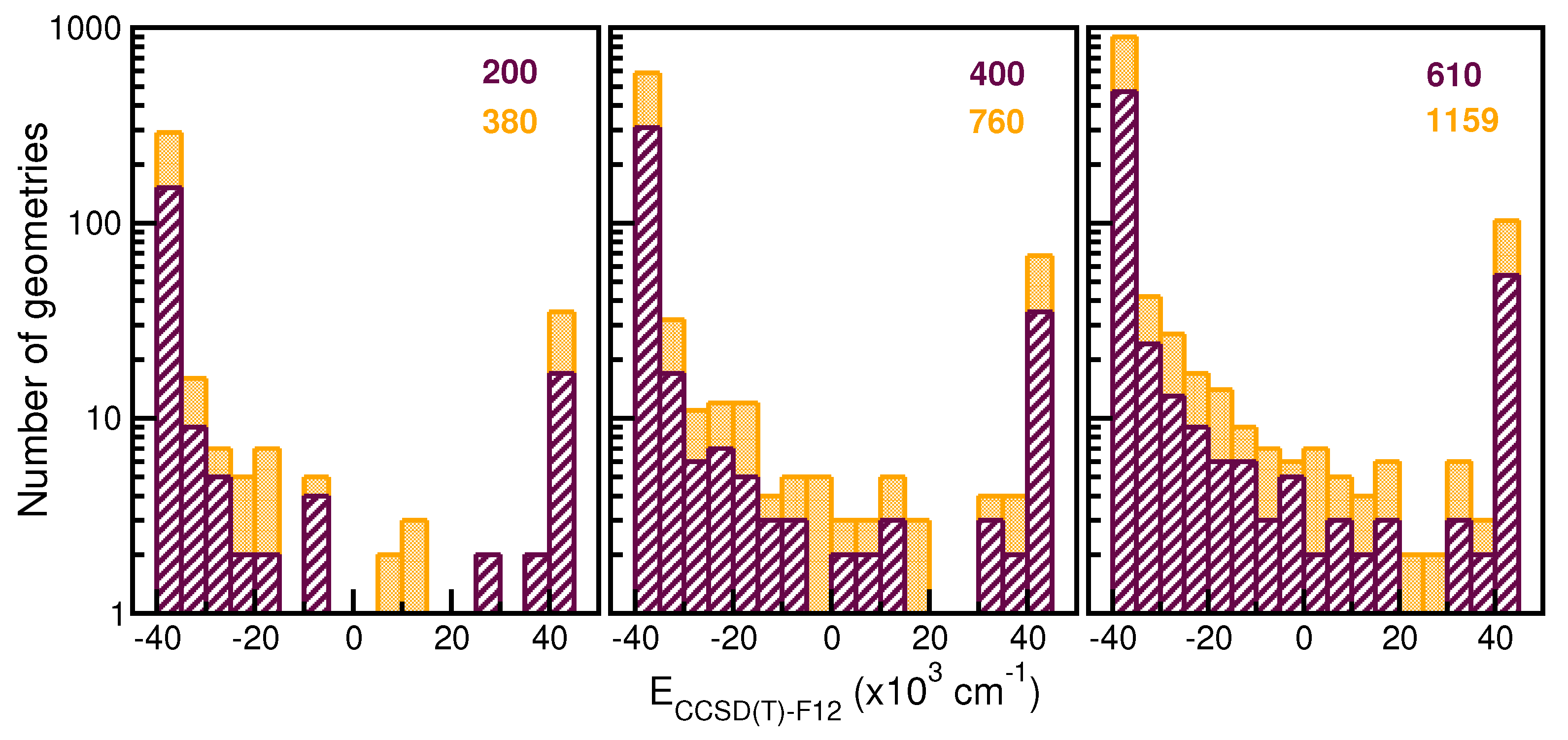
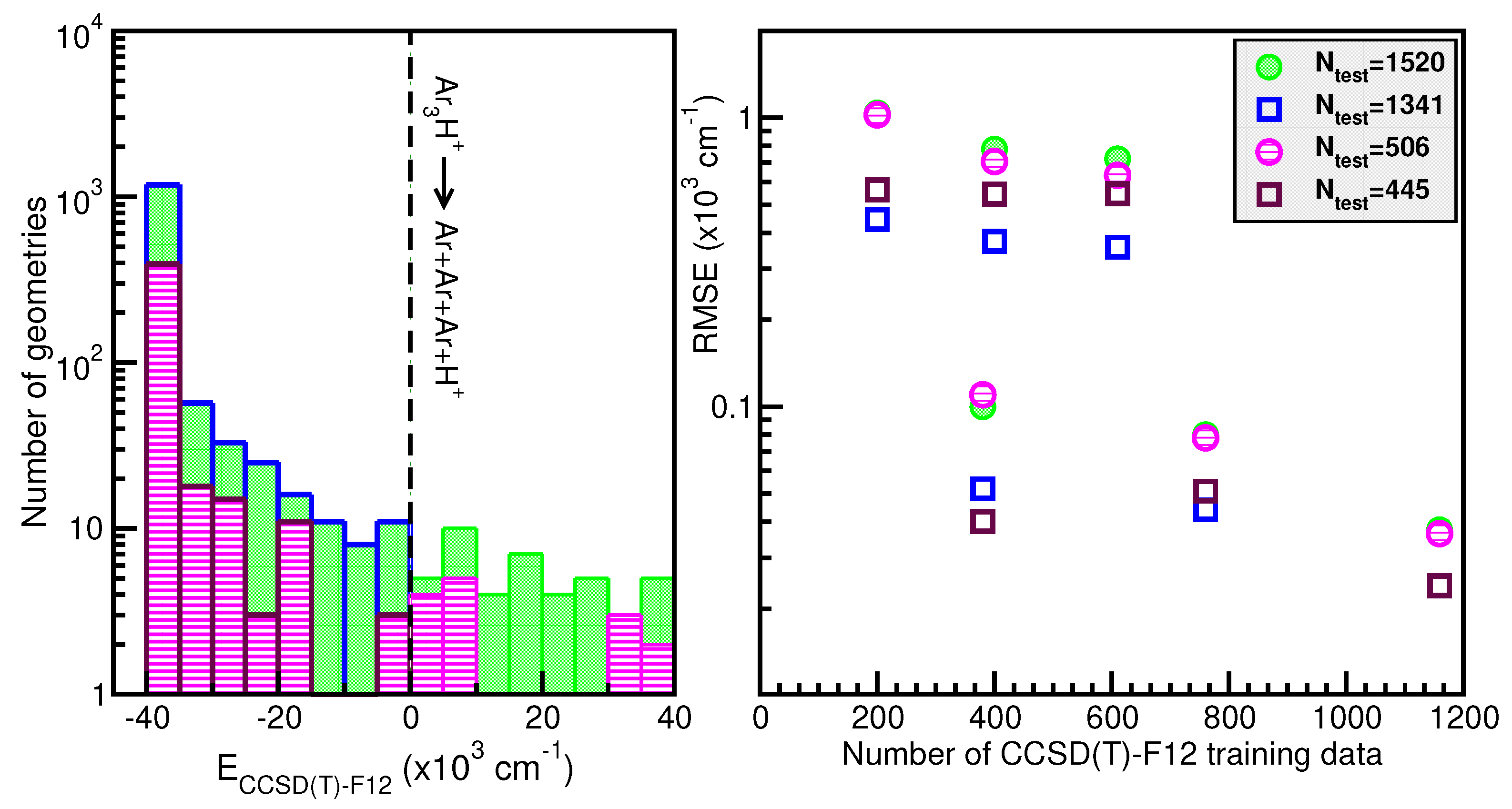
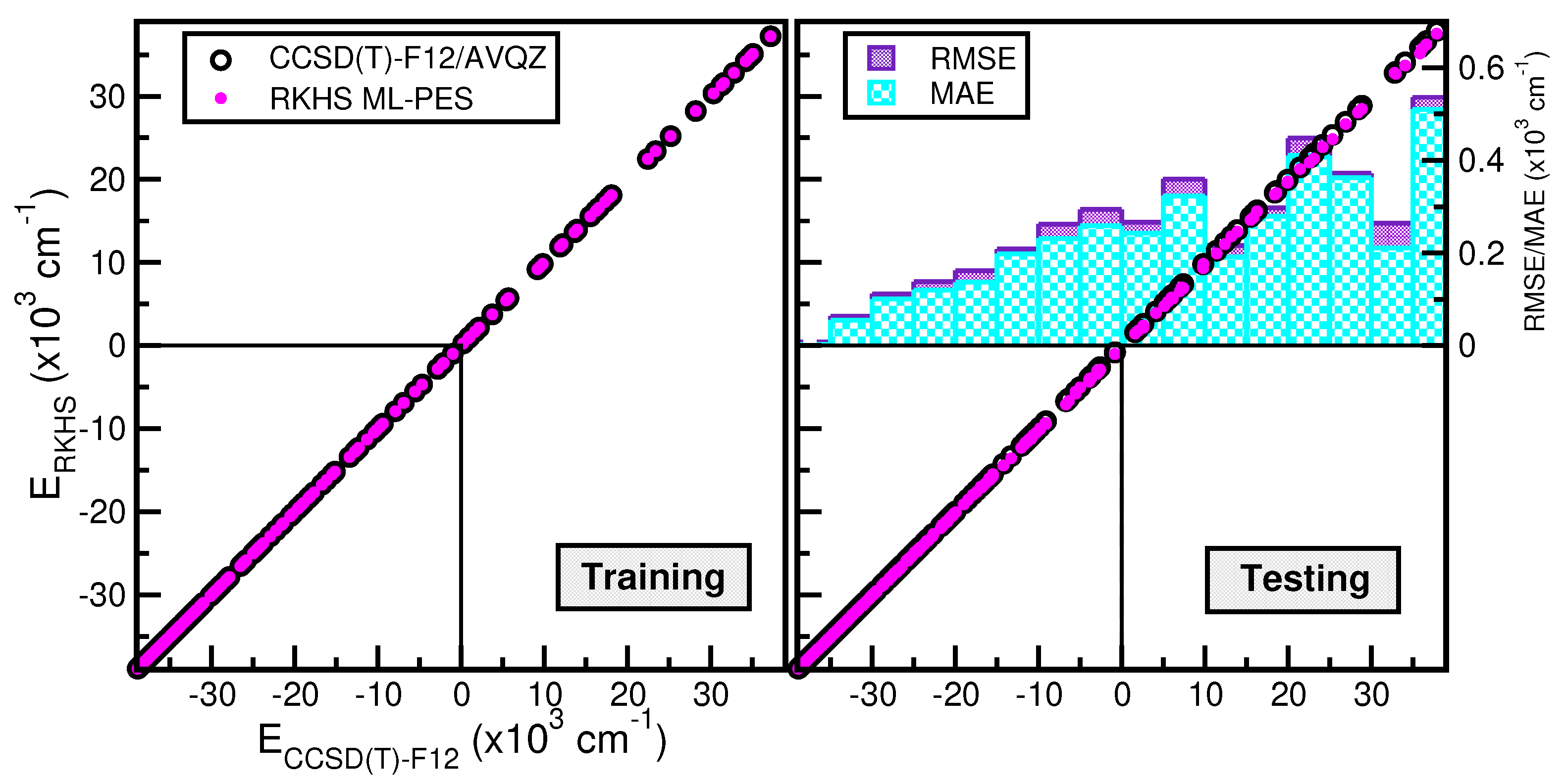
2.2. Building Up a ML PES for the
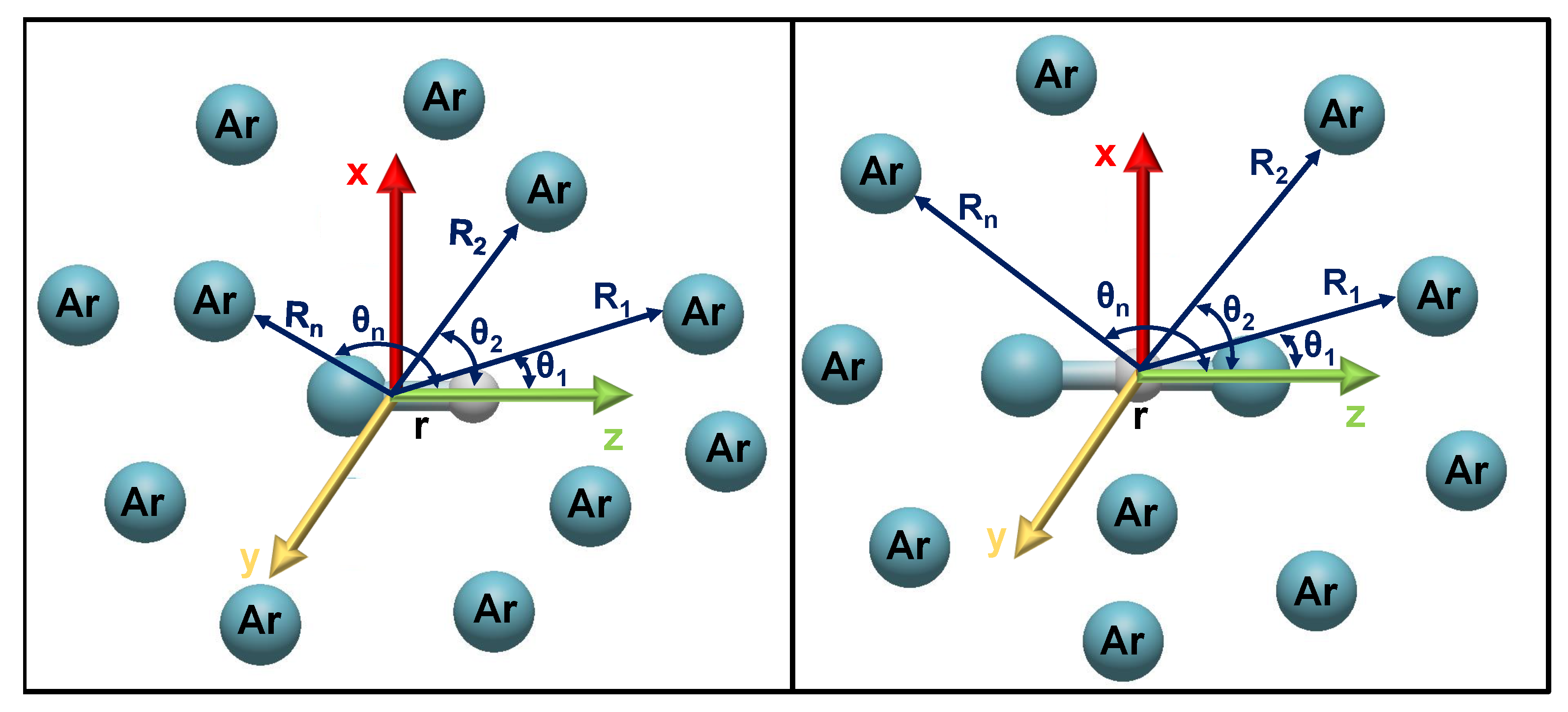
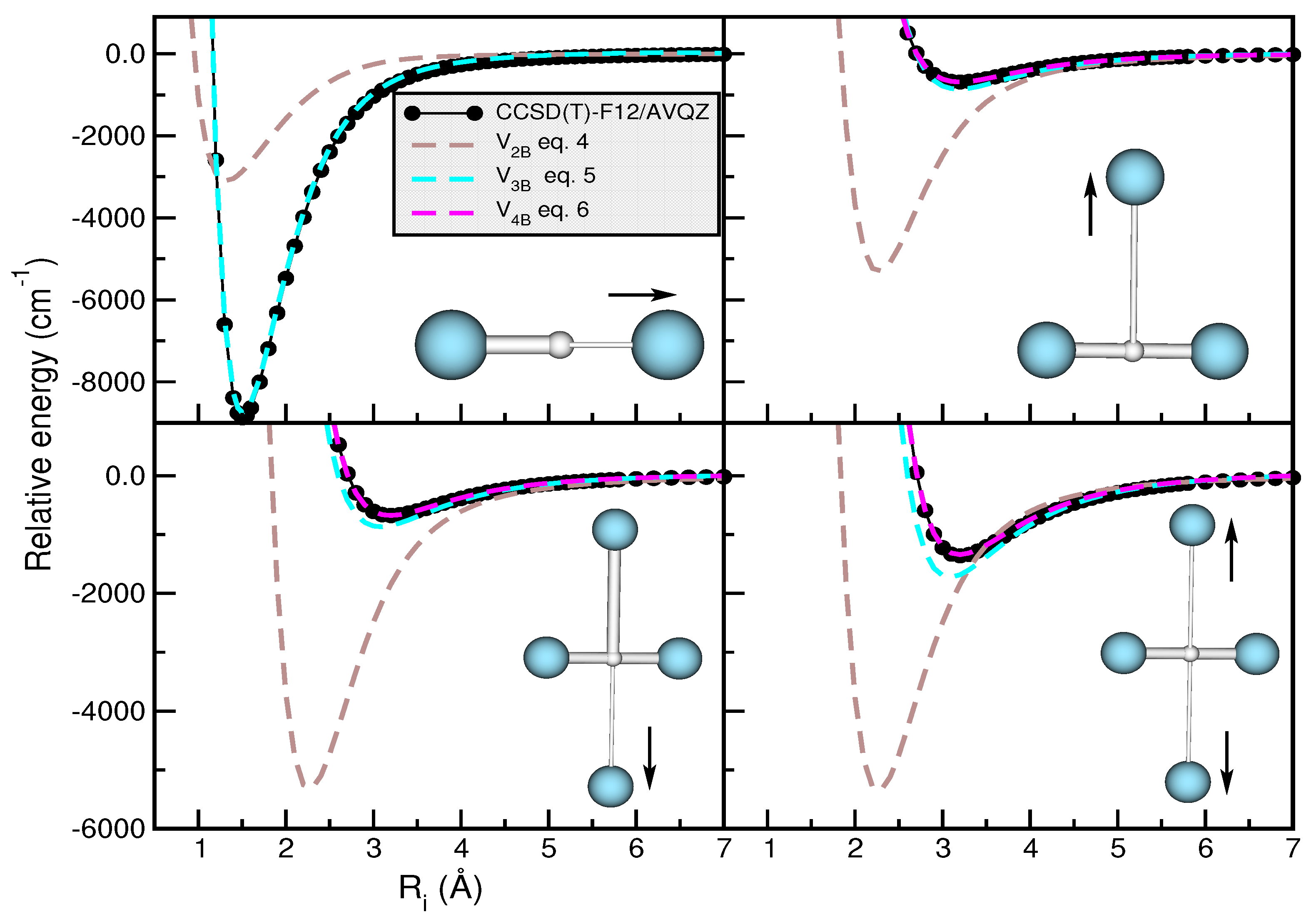
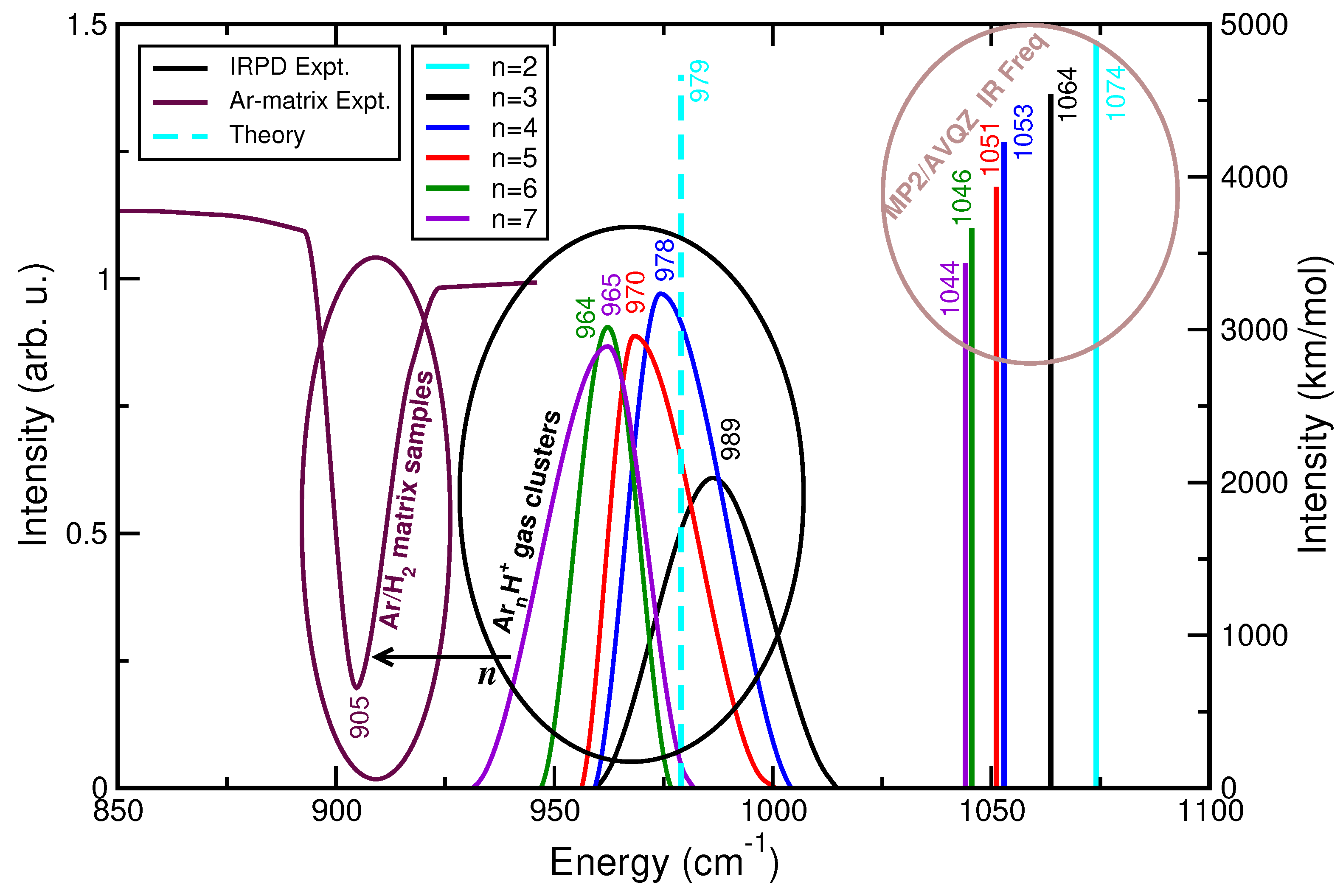
2.3. Modeling the Interaction in Higher-Order Clusters
2.4. Assessing the Sum-of-Potentials Models
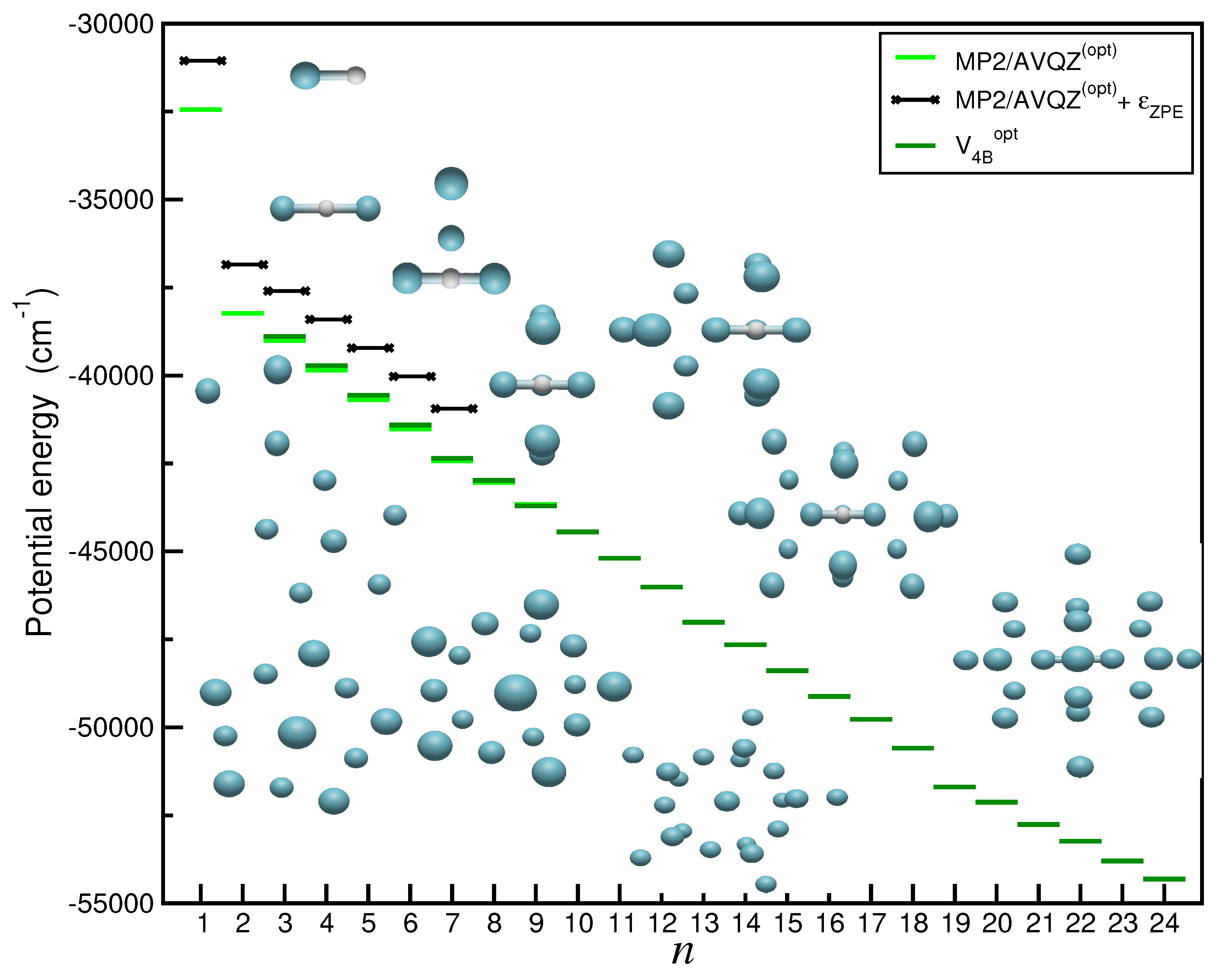
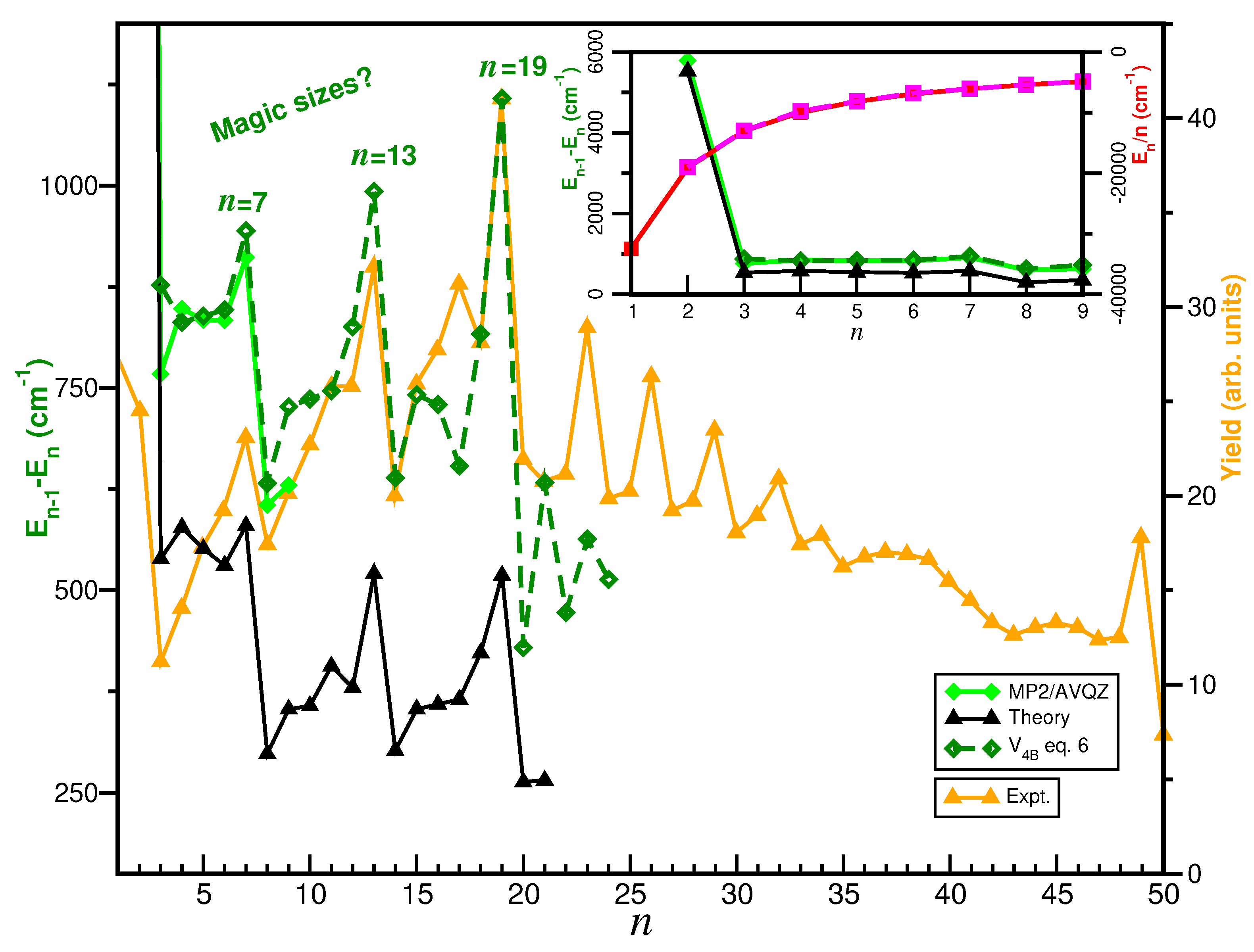
2.5. Searching for Potential Energy Minima and Microsolvated Structures
3. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grandinetti, F. Cationic Noble-Gas Hydrides: From Ion Sources to Outer Space. Front. Chem. 2020, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.A.; Kuo, J.L. Structure and Vibrational Spectra of ArnH+ (n = 2–3). J. Phys. Chem. A 2020, 124, 7726–7734. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, L.; Bartl, P.; Leidlmair, C.; Scheier, P.; Gatchell, M. Protonated and Cationic Helium Clusters. Molecules 2020, 25, 1066. [Google Scholar] [CrossRef]
- Császár, A.; Szidarovszky, T.; Asvany, O.; Schlemmer, S. Fingerprints of microscopic superfluidity in HHen+ clusters. Mol. Phys. 2019, 117, 1559–1583. [Google Scholar] [CrossRef]
- Gatchell, M.; Martini, P.; Kranabetter, L.; Rasul, B.; Scheier, P. Magic sizes of cationic and protonated argon clusters. Phys. Rev. A 2018, 98, 022519. [Google Scholar] [CrossRef]
- Stephan, C.J.; Fortenberry, R.C. The interstellar formation and spectra of the noble gas, proton-bound HeHHe+, HeHNe+ and HeHAr+ complexes. Mon. Not. R. Astron. Soc. 2017, 469, 339–346. [Google Scholar] [CrossRef]
- Fortenberry, R. Quantum astrochemical spectroscopy: Review. Int. J. Quant. Chem. 2017, 117, 81–91. [Google Scholar] [CrossRef]
- McDonald, D.C.; Mauney, D.T.; Leicht, D.; Marks, J.H.; Tan, J.A.; Kuo, J.L.; Duncan, M.A. Communication: Trapping a proton in Argon: Spectroscopy and theory of the proton-bound Argon dimer and its solvation. J. Chem. Phys. 2016, 145, 231101. [Google Scholar] [CrossRef]
- Güsten, R.; Wiesemeyer, H.; Neufeld, D.; Menten, K.; Graf, U.; Jacobs, K.; Klein, B.; Ricken, O.; Risacher, C.; Stutzki, J. Astrophysical detection of the helium hydride ion HeH+. Nature 2019, 568, 357–359. [Google Scholar] [CrossRef]
- Barlow, M.; Swinyard, B.; Owen, P.; Cernicharo, J.; Gomez, H.; Ivison, R.; Krause, O.; Lim, T.; Matsuura, M.; Miller, S.; et al. Detection of a Noble Gas Molecular Ion, 36ArH+, in the Crab Nebula. Science 2013, 342, 1343–1345. [Google Scholar] [CrossRef]
- Gianturco, F.; Filippone, F. Structure and dynamics of small protonated rare-gas clusters using quantum and classical methods. Comput. Phys. Commun. 2002, 145, 78–96. [Google Scholar] [CrossRef]
- Giju, K.T.; Roszak, S.; Leszczynski, J. A theoretical study of protonated Argon clusters: ArnH+ (n = 1–7). J. Chem. Phys. 2002, 117, 4803–4809. [Google Scholar] [CrossRef]
- Ritschel, T.; Zülicke, L.; Kuntz, P.J. Cationic van-der-Waals Complexes: Theoretical Study of Ar2H+ Structure and Stability. Z. Phys. Chem. 2004, 218, 377–389. [Google Scholar] [CrossRef]
- Ritschel, T.; Kuntz, P.J.; Zülicke, L. Structure and dynamics of cationic van-der-Waals clusters. Eur. Phys. J. D 2005, 33, 421–432. [Google Scholar] [CrossRef]
- Montes de Oca-Estévez, M.J.; Prosmiti, R. Quantum computations in heavy noble-gas hydride cations: Reference energies and new spectroscopic data. J. Mol. Graph. Model. 2023, 124, 108562. [Google Scholar] [CrossRef]
- Montes de Oca-Estévez, M.J.; Darna, B.; García-Ruíz, B.; Prosmiti, R.; González-Lezana, T.; Koner, D. Ar + ArH+ Reactive Collisions of Astrophysical Interest: The Case of 36Ar. ChemPhysChem 2023, 24, e202300450. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca-Estévez, M.J.; Prosmiti, R. Automated learning data-driven potential models for spectroscopic characterization of astrophysical interest noble gas-containing NgH2+ molecules. AIChem 2024, 2, 100059. [Google Scholar] [CrossRef]
- García-Vázquez, R.M.; Márquez-Mijares, M.; Rubayo-Soneira, J.; Denis-Alpizar, O. Relaxation of ArH+ by collision with He: Isotopic effects. A & A 2019, 631, A86. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals; Cornell University Press: Ithaca, NY, USA, 1954. [Google Scholar]
- Beyer, M.; Lammers, A.; Savchenko, E.V.; Niedner-Schatteburg, G.; Bondybey, V.E. Proton solvated by noble-gas atoms: Simplest case of a solvated ion. Phys. Chem. Chem. Phys. 1999, 1, 2213–2221. [Google Scholar] [CrossRef]
- Fortenberry, R.C. Rovibrational Characterization and Interstellar Implications of the Proton-Bound, Noble Gas Complexes: ArHAr+, NeHNe+, and ArHNe+. ACS Earth Space Chem. 2017, 1, 60–69. [Google Scholar] [CrossRef]
- Tan, J.A.; Kuo, J.L. A theoretical study on the infrared signatures of proton-bound rare gas dimers (Rg–H+–Rg), Rg = Ne, Ar, Kr, and Xe. J. Chem. Phys. 2019, 150, 124305. [Google Scholar] [CrossRef] [PubMed]
- Koner, D. Quantum and quasiclassical dynamical simulations for the Ar2H+ on a new global analytical potential energy surface. J. Chem. Phys. 2021, 154, 054303. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca-Estévez, M.J.; Valdés, A.; Prosmiti, R. A kernel-based machine learning potential and quantum vibrational state analysis of the cationic Ar hydride (Ar2H+). Phys. Chem. Chem. Phys. 2024, 26, 7060–7071. [Google Scholar] [CrossRef]
- Montes de Oca-Estévez, M.J.; Valdés, A.; Koner, D.; González-Lezana, T.; Prosmiti, R. Quantum computations on a new neural network potential for the proton-bound noble-gas Ar2H+ complex: Isotopic effects. Chem. Phys. Lett. 2024. [Google Scholar]
- Botschwina, P.; Dutoi, T.; Mladenović, M.; Oswald, R.; Schmatz, S.; Stoll, H. Theoretical investigations of proton-bound cluster ions. Faraday Discuss. 2001, 118, 433–453. [Google Scholar] [CrossRef]
- Fortenberry, R.C.; Yu, Q.; Mancini, J.S.; Bowman, J.M.; Lee, T.J.; Crawford, T.D.; Klemperer, W.F.; Francisco, J.S. Communication: Spectroscopic consequences of proton delocalization in OCHCO+. J. Chem. Phys. 2015, 143, 071102. [Google Scholar] [CrossRef]
- Yu, Q.; Bowman, J.M.; Fortenberry, R.C.; Mancini, J.S.; Lee, T.J.; Crawford, T.D.; Klemperer, W.; Francisco, J.S. Structure, Anharmonic Vibrational Frequencies, and Intensities of NNHNN+. J. Phys. Chem. A 2015, 119, 11623–11631. [Google Scholar] [CrossRef] [PubMed]
- Bondybey, V.E.; Pimentel, G.C. Infrared Absorptions of Interstitial Hydrogen Atoms in Solid Argon and Krypton. J. Chem. Phys. 1972, 56, 3832–3836. [Google Scholar] [CrossRef]
- Milligan, D.E.; Jacox, M.E. Infrared spectroscopic evidence for the stabilization of HArn+ in solid Argon at 14 K. J. Mol. Spectrosc. 1973, 46, 460–469. [Google Scholar] [CrossRef]
- Fridgen, T.D.; Parnis, J.M. Electron bombardment matrix isolation of Rg/Rg/methanol mixtures (Rg = Ar, Kr, Xe): Fourier-transform infrared characterization of the proton-bound dimers Kr2H+, Xe2H+, (ArHKr)+ and (ArHXe)+ in Ar matrices and (KrHXe)+ and Xe2H+ in Kr matrices. J. Chem. Phys. 1998, 109, 2155–2161. [Google Scholar] [CrossRef]
- Ho, T.; Rabitz, H. A general method for constructing multidimensional molecular potential energy surfaces from ab initio calculations. J. Chem. Phys. 1996, 104, 2584–2597. [Google Scholar] [CrossRef]
- Hollebeek, T.; Ho, T.S.; Rabitz, H. A fast algorithm for evaluating multidimensional potential energy surfaces. J. Chem. Phys. 1997, 106, 7223–7227. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- van Mourik, T.; Wilson, A.K.; Dunning, T.H. Benchmark calculations with correlated molecular wavefunctions. XIII. Potential energy curves for He2, Ne2 and Ar2 using correlation consistent basis sets through augmented sextuple zeta. Mol. Phys. 1999, 96, 529–547. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibsom, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Iwamatsu, M. Applying evolutionary programming to structural optimization of atomic clusters. Comput. Phys. Commun. 2001, 142, 214–218. [Google Scholar] [CrossRef]
- Fogel, D.B. Evolutionary Computation: Toward a New Philosophy of Machine Intelligence; John Wiley and Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Werner, H.J.; Knowles, P.; Knizia, G.; Manby, F.; Schütz, M. MOLPRO, Version 2022.2. a Package of Ab Initio Programs. 2022. Available online: http://www.molpro.net (accessed on 15 February 2024).
- DENEB 1.30 Beta: The Nanotechnology Software by Atelgraphics. Available online: http://www.atelgraphics.com (accessed on 30 January 2024).
- Grabowski, S.J.; Ugalde, J.M.; Andrada, D.M.; Frenking, G. Comparison of Hydrogen and Gold Bonding in [XHX]−, [XAuX]−, and Isoelectronic [NgHNg]+, [NgAuNg]+ (X = Halogen, Ng = Noble Gas). Eur. J. Chem. 2016, 22, 11317–11328. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Unke, O.T.; Meuwly, M. Toolkit for the construction of reproducing kernel-based representations of data: Application to multidimensional potential energy surfaces. J. Chem. Inf. Model. 2017, 57, 1923–1931. [Google Scholar] [CrossRef]
- Murrell, J.N. Molecular Potential Energy Functions; Wiley: Hoboken, NJ, USA, 1984. [Google Scholar]
- Montes de Oca-Estévez, M.J.; Prosmiti, R. Computational Characterization of Astrophysical Species: The Case of Noble Gas Hydride Cations. Front. Chem. 2021, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.-F.; Yang, C.-L.; Xiao, J.; Wang, M.-S.; Ma, X.-G. Theoretical study on the complexes of He, Ne and Ar. Chin. Phys. B 2010, 19, 123102. [Google Scholar] [CrossRef]
- Perez de Tudela, R.; Barragan, P.; Valdes, A.; Prosmiti, R. Energetics and solvation structure of a dihalogen dopant (I2) in 4He clusters. J. Phys. Chem. A 2014, 118, 6492–6500. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montes de Oca-Estévez, M.J.; Prosmiti, R. Microsolvation of a Proton by Ar Atoms: Structures and Energetics of ArnH+ Clusters. Molecules 2024, 29, 4084. https://doi.org/10.3390/molecules29174084
Montes de Oca-Estévez MJ, Prosmiti R. Microsolvation of a Proton by Ar Atoms: Structures and Energetics of ArnH+ Clusters. Molecules. 2024; 29(17):4084. https://doi.org/10.3390/molecules29174084
Chicago/Turabian StyleMontes de Oca-Estévez, María Judit, and Rita Prosmiti. 2024. "Microsolvation of a Proton by Ar Atoms: Structures and Energetics of ArnH+ Clusters" Molecules 29, no. 17: 4084. https://doi.org/10.3390/molecules29174084
APA StyleMontes de Oca-Estévez, M. J., & Prosmiti, R. (2024). Microsolvation of a Proton by Ar Atoms: Structures and Energetics of ArnH+ Clusters. Molecules, 29(17), 4084. https://doi.org/10.3390/molecules29174084