
Experiments and Calculation on New N,N-bis-Tetrahydroacridines

 , , and
, , and
Abstract
1. Introduction
2. Results
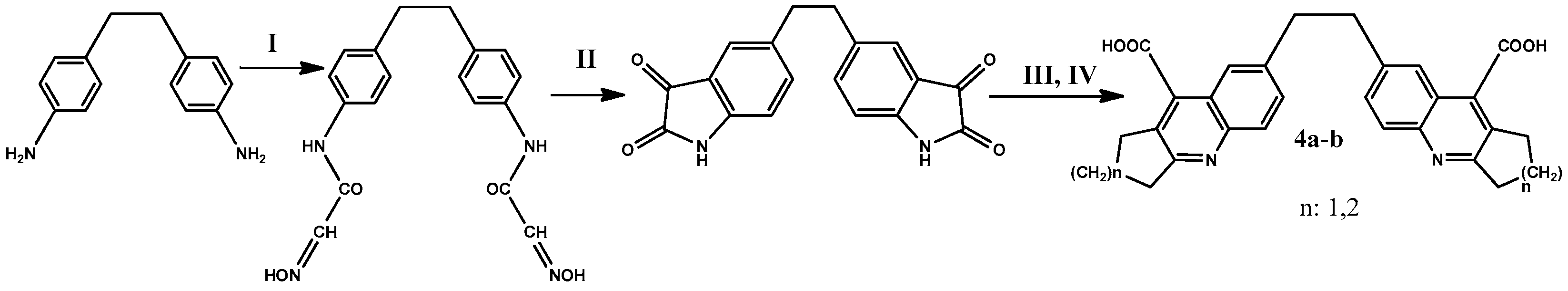
2.1. Synthesis and Physical-Chemical Characterization
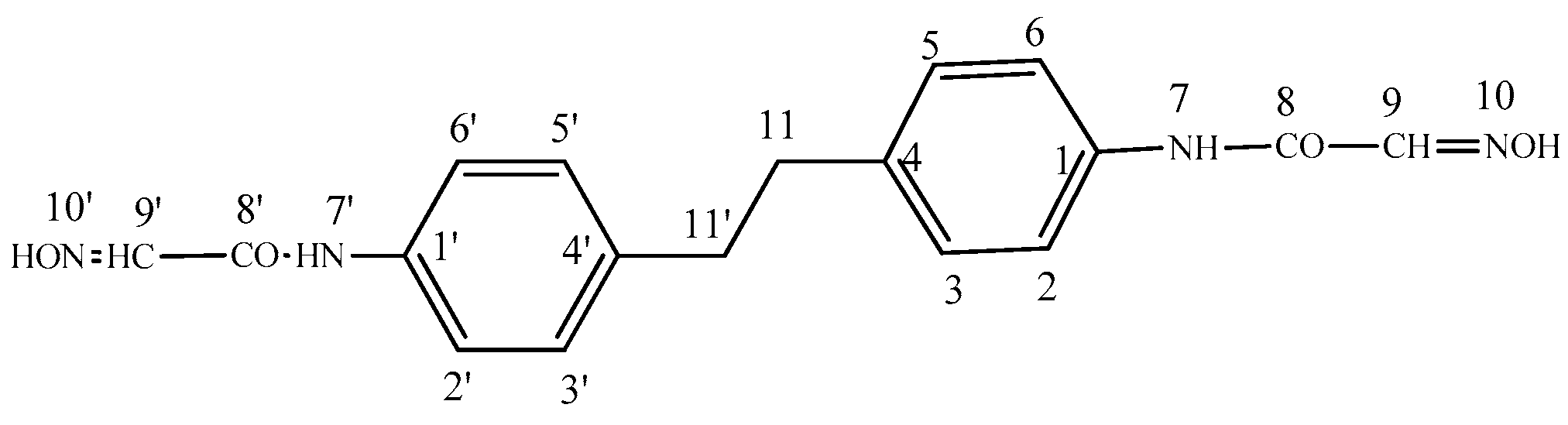
2.1.1. N,N′-(4,4′-(Ethane-1,2-diyl)bis(4,1-phenylene))bis(2-(hydroxyimino)acetamide) (2)
2.1.2. 5,5′-(Ethane-1,2-diyl)diindoline-2,3-dione (3)
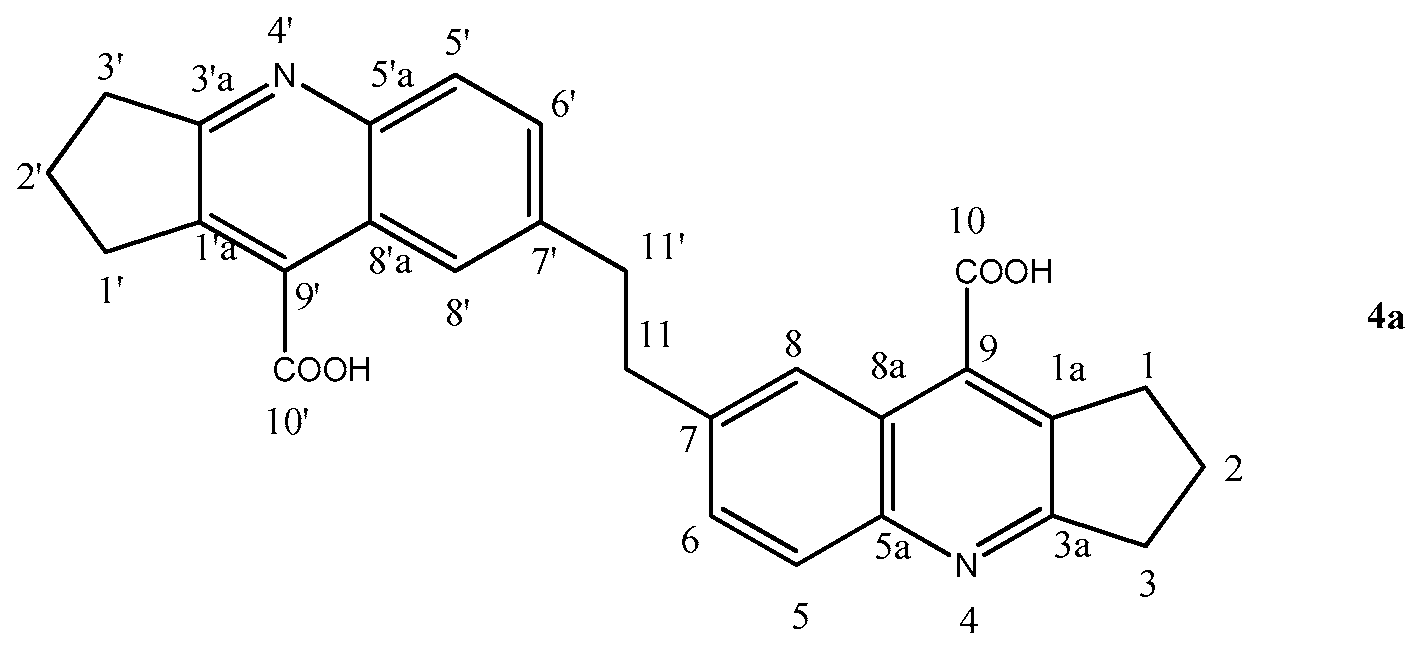
2.1.3. 7,7′-(Ethane-1,2-diyl)bis(2,3-dihydro-1H-cyclopenta[b]quinoline-9-carboxylic Acid) (4a)
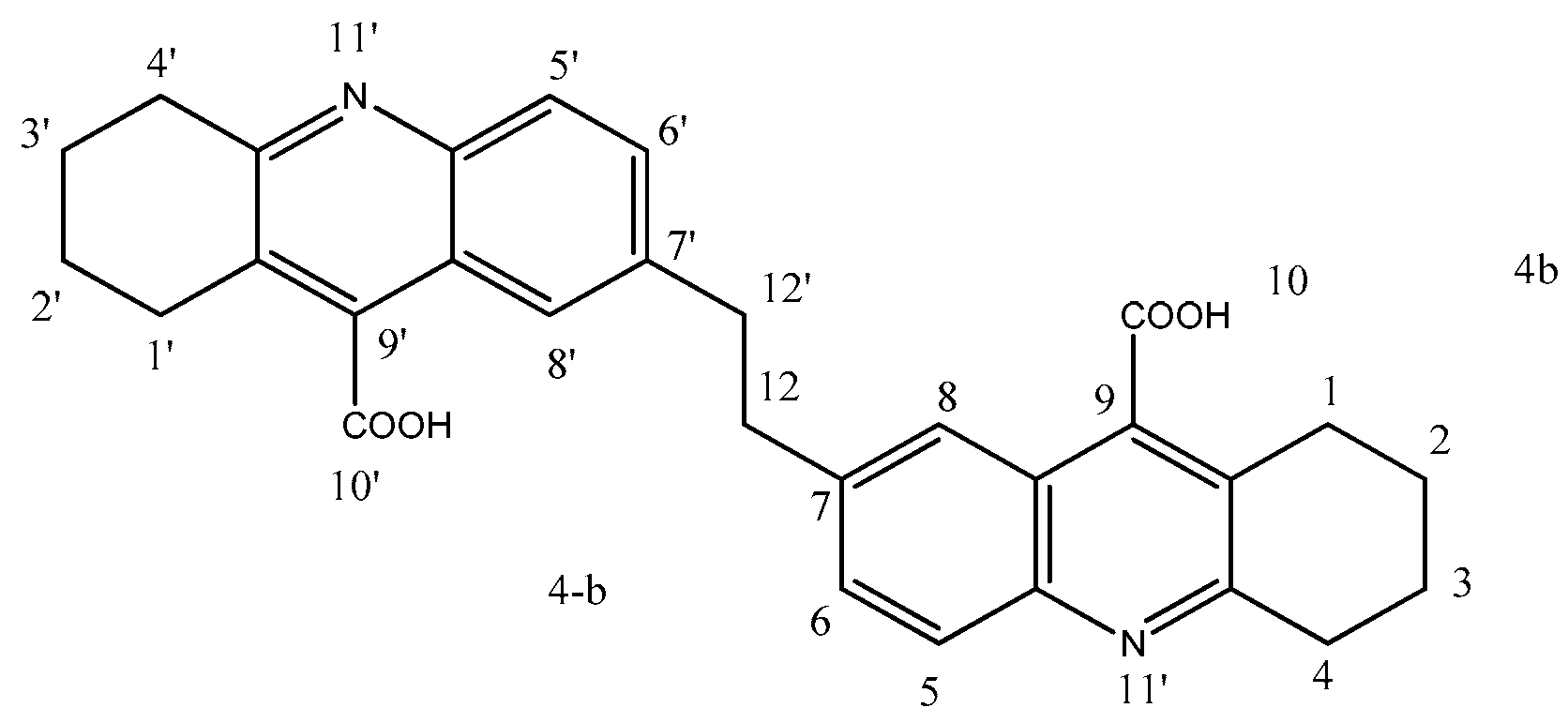
2.1.4. 7,7′-(Ethane-1,2-diyl)bis(1,2,3,4-tetrahydroacridine-9-carboxylic Acid) (4b)
2.2. Computations
2.3. Electrochemistry
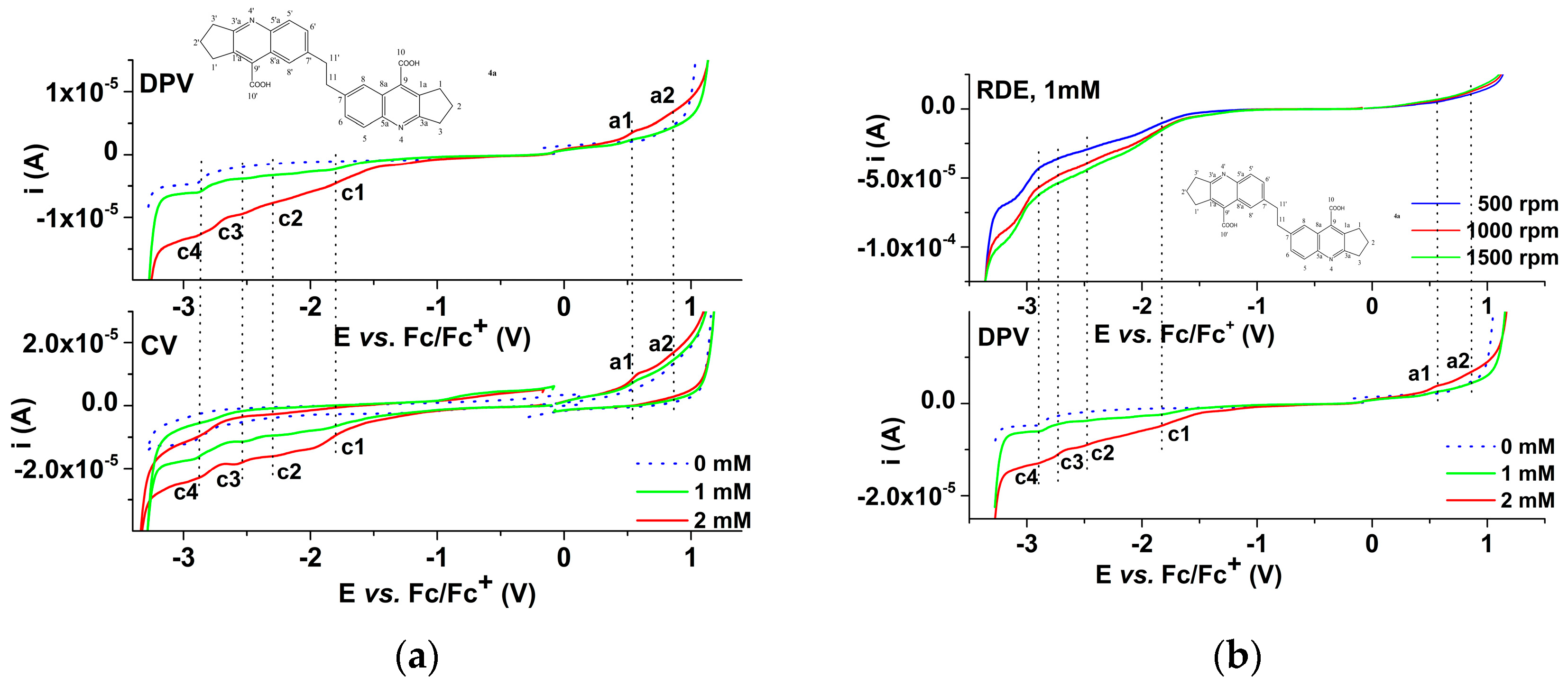
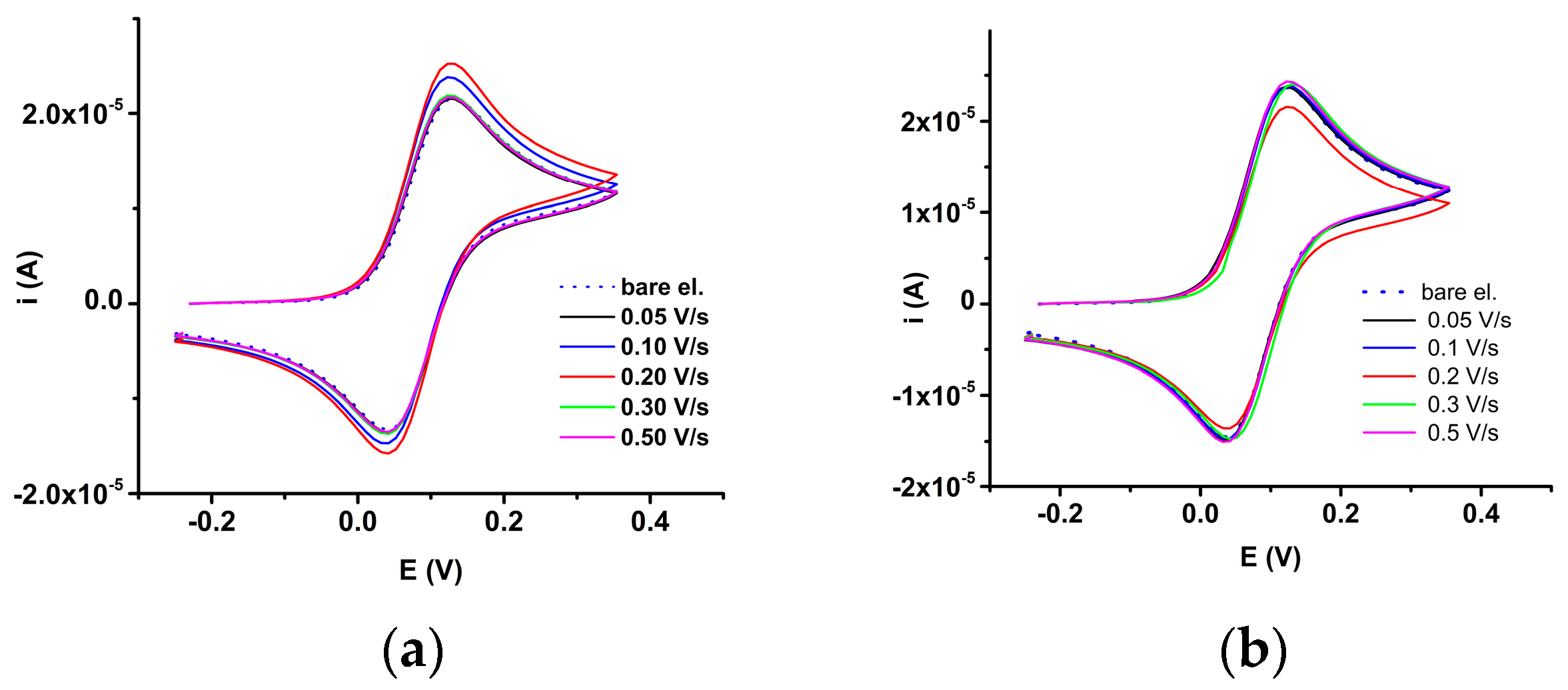
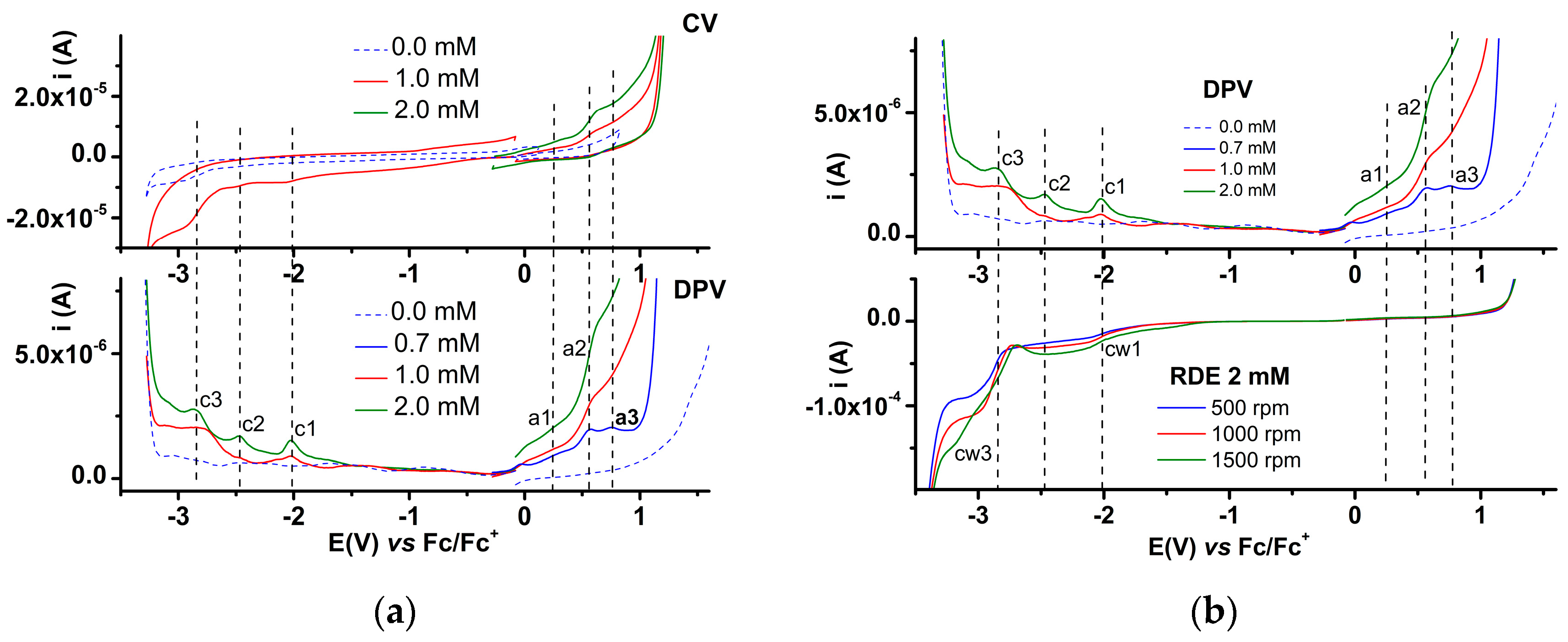
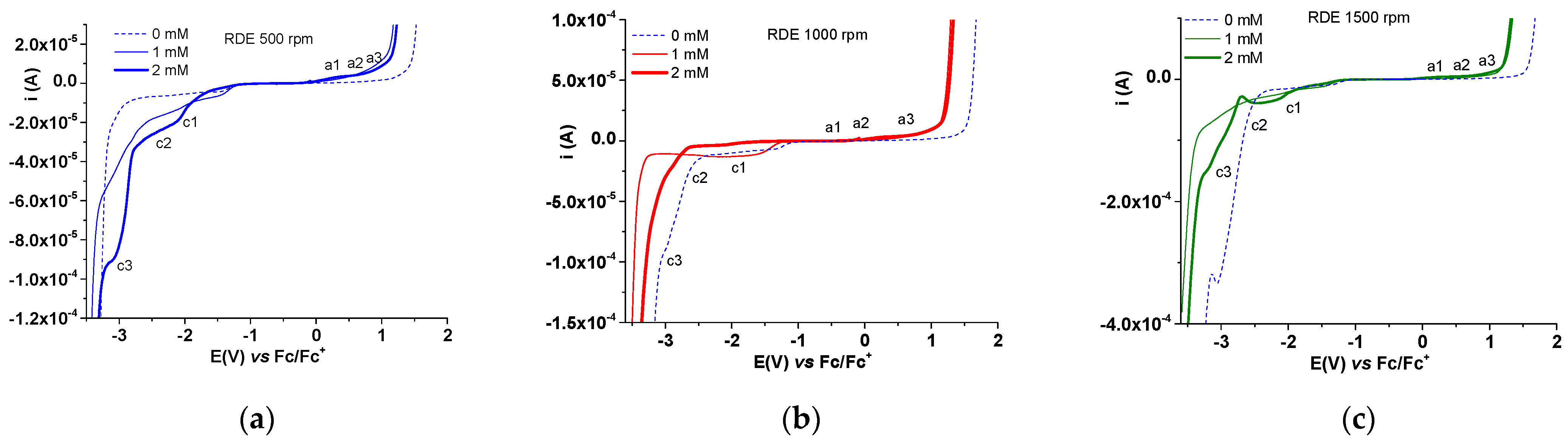
2.3.1. Electrochemical Study of 4a bis-Acid
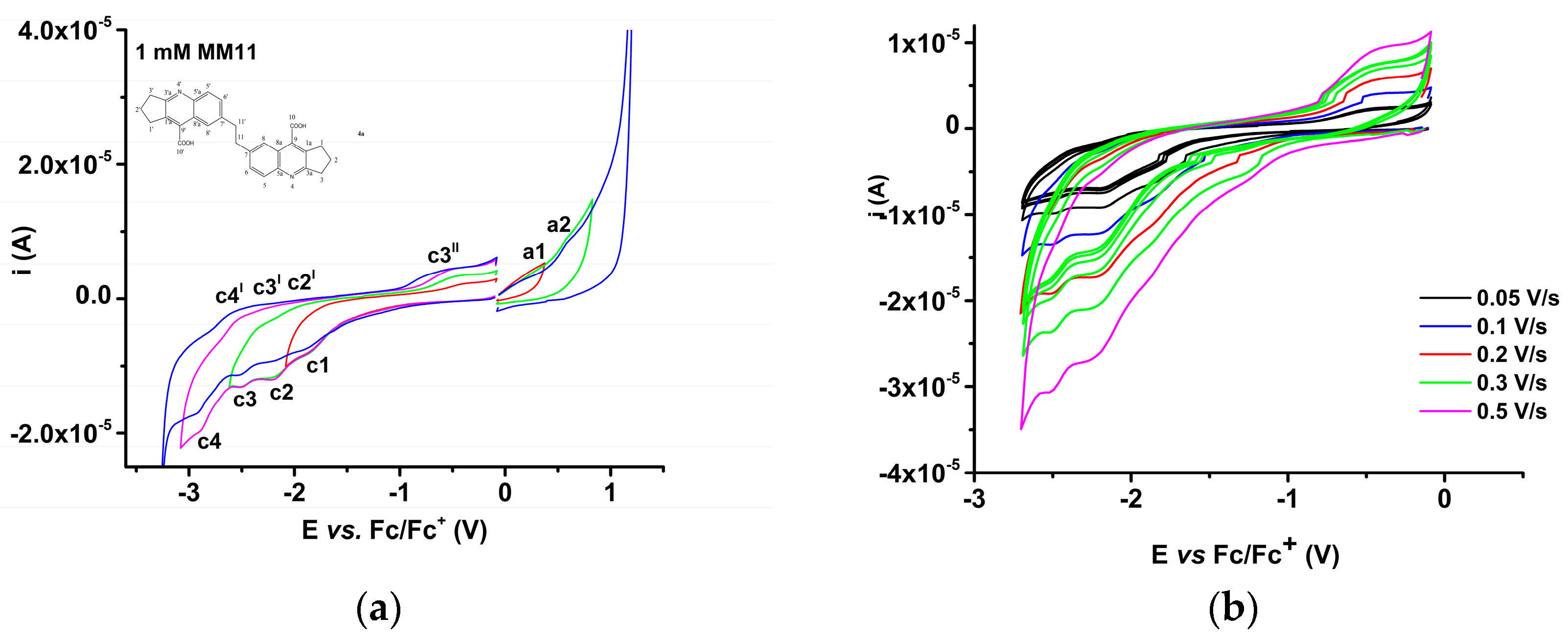
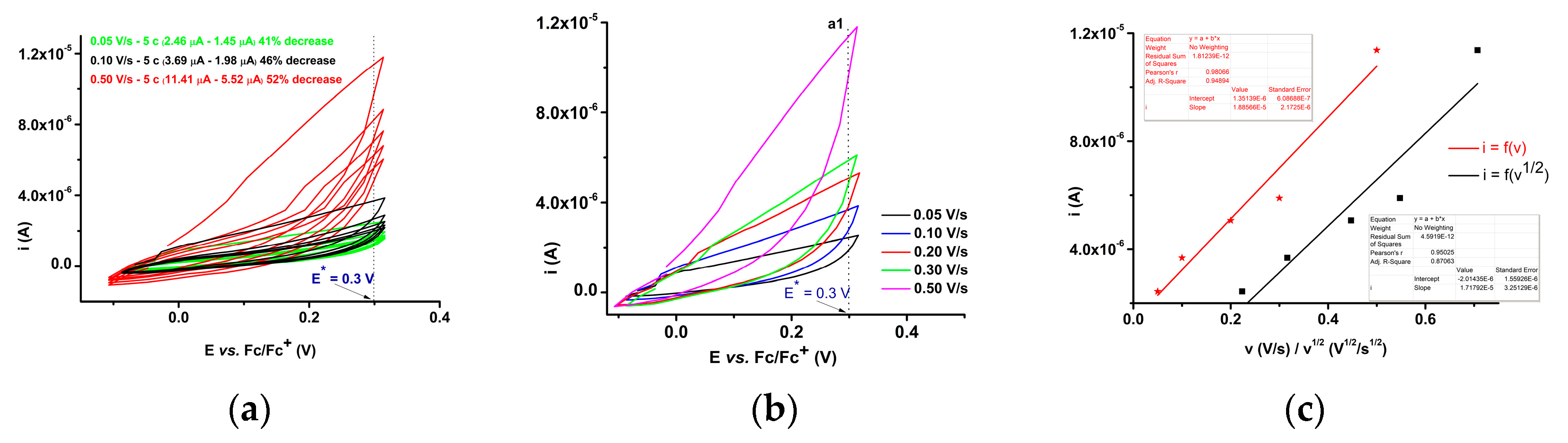
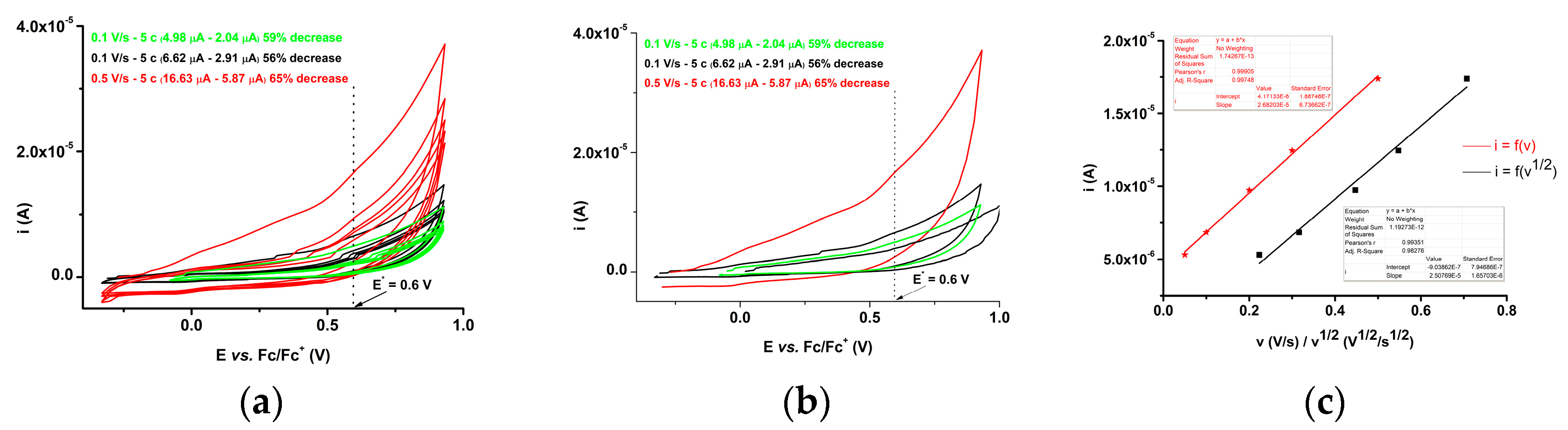
2.3.2. Electrochemical Study of 4b bis-Acid
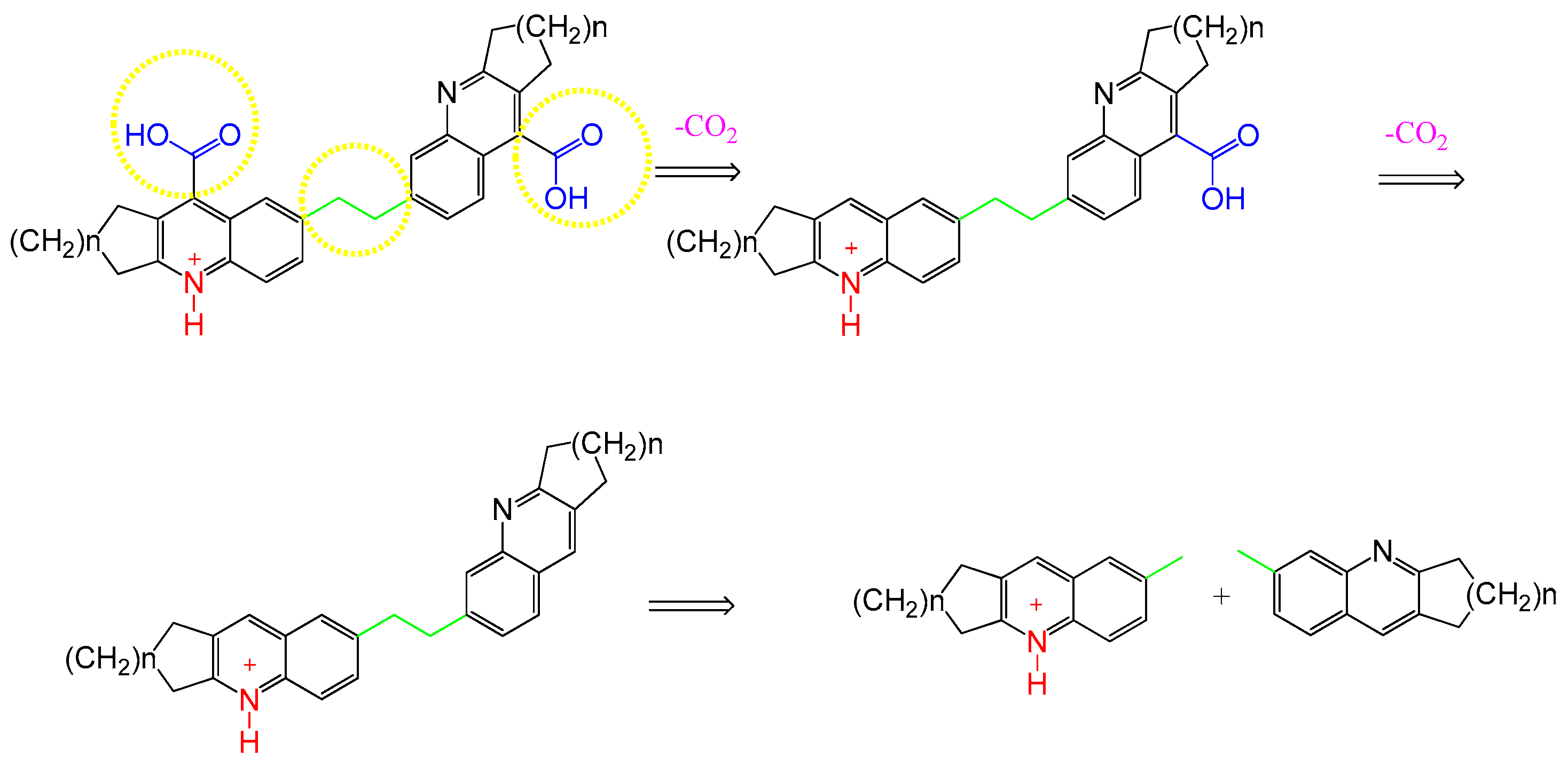
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Apparatus
4.3. Methods and Procedures
4.4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouffier, L.; Demeunynck, M.; Milet, A.; Dumy, P. Reactivity of Pyrido [4,3,2-kl]acridines: Regioselective Formation of 6-Substituted Derivatives. J. Org. Chem. 2004, 69, 8144–8147. [Google Scholar] [CrossRef] [PubMed]
- Chiron, J.; Galy, J.-P. Reactivity of the Acridine Ring: A Review. Synthesis 2004, 3, 313–325. [Google Scholar] [CrossRef]
- Belmont, P.; Andrez, J.-C.S.M. New methodology for acridine synthesis using a rhodium-catalyzed benzannulation. Tetrahedron Lett. 2004, 45, 2783–2786. [Google Scholar] [CrossRef]
- Wiseman, A.; Sims, L.A.; Snead, R.; Gronert, S.; Maclagan, R.G.A.R.; Meot-Ner, M. Protonation Energies of 1−5-Ring Polycyclic Aromatic Nitrogen Heterocyclics: Comparing Experiment and Theory. J. Phys. Chem. A 2015, 119, 118–126. [Google Scholar] [CrossRef]
- Chen, H. Application of fluorescence energy transfer system in biochemistry analysis-research on testing DNA with acridine orange-neutral red (NR) as fluorescence probe. HYYIFM 2000, 12, 164–168. [Google Scholar]
- Kręcisz, P.; Czarnecka, K.; Szymański, P. Physico-chemical evaluation of new tetrahydroacridine and iodobeznoic acid hybrids as the next step in the design of potential drugs in AD. Biomed. Chromatogr. 2020, 34, e4906. [Google Scholar] [CrossRef]
- Kostjukova, L.O.; Leontieva, S.V.; Kostjukov, V.V. The vibronic absorption spectra and electronic states of acridine yellow in aqueous solution. J. Mol. Liq. 2021, 326, 115312. [Google Scholar] [CrossRef]
- Sivan, S.; Tuchman, S.; Lotan, N. A biochemical logic gate using an enzyme and its inhibitor. Part II: The logic gate. Biosystems 2003, 70, 21–33. [Google Scholar] [CrossRef]
- Kumar, R.; Kaur, M.; Kumari, M. Acridine: A versatile heterocyclic nucleus. Acta Pol. Pharm. 2012, 69, 3–9. [Google Scholar]
- Ježek, J.; Hlaváček, J.; Šebestík, J. Syntheses. In Biomedical Applications of Acridines, Progress in Drug Research Series; Rainsford, R.D., Ed.; Springer: Cham, Switzerland, 2017; Volume 72, pp. 9–45. [Google Scholar]
- Song, B.; Li, M.; Imerhasan, M. Synthesis and Application of Acridine Derivatives. Chin. J. Org. Chem. 2018, 38, 594–611. [Google Scholar] [CrossRef]
- Flock, S.; Bailly, C.; Waring, M.-J.; Henichart, J.-P.; Colsom, P.; Houssier, C. Interaction of Two Peptide-Acridine Conjugates Containing the SPKK Peptide Motif with DNA and Chromatin. J. Biomol. Struct. Dyn. 1994, 11, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Sahiba, N.; Sethiya, A.; Soni, J.; Agarwal, S. Acridine-1,8-diones: Synthesis and Biological Applications. ChemistrySelect 2021, 6, 2210–2251. [Google Scholar] [CrossRef]
- Prabakaran, K.; Manivannan, R.; Oh, H.; Parthiban, C.; Son, Y.-A. Synthesis and characterization of new acridine dye molecules combined UV absorber and exploring photophysical properties. Dye. Pigment. 2021, 192, 109391. [Google Scholar] [CrossRef]
- Mangueira, V.M.; de Sousa, T.K.G.; Batista, T.M.; de Abrantes, R.A.; Moura, A.P.G.; Ferreira, R.C.; Almeida, R.N.; Braga, R.M.; Leite, F.C.; Medeiros, K.C.P.; et al. 9-aminoacridine derivative induces growth inhibition of Ehrlich ascites carcinoma cells and antinociceptive effect in mice. Front. Pharmacol. 2022, 13, 963736. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Piplani, P. Acridine: A Scaffold for the Development of Drugs for Alzheimer’s Disease. Curr. Top. Med. Chem. 2023, 23, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Mojzych, I.; Zawadzka, A.; Kaczyn’ska, K.; Wojciechowski, P.; Zaja, D.; Chotkowski, M.; Wiktorska, K.; Maurinc, J.K.; Mazur, M. A tetrahydroacridine derivative and its conjugate with gold nanoparticles, promising agents for the treatment of Alzheimer’s disease. Phys. Chem. Chem. Phys. 2023, 25, 16796. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.J.; Lin, M.H.; Shiao, Y.J.; Yang, Y.C.; Chu, J.C.; Chen, C.Y.; Chen, Y.Y.; Lin, T.E.; Su, C.J.; Pan, S.L.; et al. Synthesis and biological evaluation of acridine-based histone deacetylase inhibitors as multitarget agents against Alzheimer’s disease. Eur. J. Med. Chem. 2020, 192, 112193. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.E.-S.; El-Kazak, A.M. Synthesis and Antimicrobial Activity of Some New 1,3-Thiazoles, 1,3,4-Thiadiazoles, 1,2,4-Triazoles and 1,3-Thiazines Incorporating Acridine and 1,2,3,4-Tetrahydroacridine Moieties. Eur. J. Chem. 2010, 1, 6–11. [Google Scholar] [CrossRef]
- Figgitt, D.; Denny, W.; Chavalitshewinkoon, P.; Wilairat, P.; Ralph, R. In vitro study of anticancer acridines as potential antitrypanosomal and antimalarial agents. Antimicrob. Agents Chemother. 1992, 36, 1644–1647. [Google Scholar] [CrossRef]
- Nelson, E.M.; Tewey, K.M.; Liu, L.F. Mechanism of antitumor drug action: Poisoning of mammalian DNA topoisomerase II on DNA by 4’-(9-acridinylamino)- methanesulfon-m-anisidide. Proc. Natl. Acad. Sci. USA 1984, 81, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Baguley, B.C. Dual topoisomerase I/II inhibitors in cancer therapy. Curr. Top. Med. Chem. 2003, 3, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.; Blake, A.J.; Benoit, A.; David, M.F.; Robert, A.K. Efficacy of substituted 9-aminoacridine derivatives in small cell lung cancer. Investig. New Drugs 2013, 31, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Jacobson, B.A.; Benoit, A.; Patel, M.R.; Jay-Dixon, J.; Hiasa, H.; Ferguson, D.M.; Kratzke, R.A. Novel acridine-based agents with topoisomerase II inhibitor activity suppress mesothelioma cell proliferation and induce apoptosis. Investig. New Drugs 2012, 30, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-L.; Wang, Z.-F.; Qin, Q.-P.; Tan, M.-X.; Luo, D.-M.; Zou, B.-Q.; Liu, Y.-C. A 9-chloro-5,6,7,8-tetrahydroacridine Pt(II) Complex Induces Apoptosis of Hep-G2 Cells via Inhibiting Telomerase Activity and Disrupting Mitochondrial Pathway. Inorg. Chem. Commun. 2019, 99, 77–81. [Google Scholar] [CrossRef]
- Belmont, P.; Bosson, J.; Godet, T.; Tiano, M. Acridine and acridone derivatives, anticancer properties and synthetic methods: Where are we now? Anticancer Agents. Med. Chem. 2007, 7, 139–169. [Google Scholar] [CrossRef]
- Girek, M.; Kłosiński, K.; Grobelski, B.; Pizzimenti, S.; Cucci, M.A.; Daga, M.; Barrera, G.; Pasieka, Z.; Czarnecka, K.; Szymański, P. Novel tetrahydroacridine derivatives with iodobenzoic moieties induce G0/G1 cell cycle arrest and apoptosis in A549 non-small lung cancer and HT-29 colorectal cancer cells. Mol. Cell. Biochem. 2019, 460, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, K.; Chufarova, N.; Halczuk, K.; Maciejewska, K.; Girek, M.; Skibiński, R.; Jończyk, J.; Bajda, M.; Kabziński, J.; Majsterek, I.; et al. Tetrahydroacridine derivatives with dichloronicotinic acid moiety as attractive, multipotent agents for Alzheimer’s disease treatment. Eur. J. Med. Chem. 2018, 145, 760–769. [Google Scholar] [CrossRef]
- Xu, A.; He, F.; Zhang, X.; Li, X.; Ran, Y.; Wei, C.; Chou, J.C.; Zhang, R.; Wu, J. Tacrine-hydroxamate derivatives as multitarget-directed ligands for the treatment of Alzheimer’s disease: Design, synthesis, and biological evaluation. Bioorg. Chem. 2020, 98, 103721. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Chau, J.H.-L.; Chen, J. Discovery of a novel tetrahydroacridine acetylcholinesterase inhibitor through an indexed combinatorial library. Chem. Biol. 1995, 2, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Bajda, M.; Jończyk, J.; Malawska, B.; Czarnecka, K.; Girek, M.; Olszewska, P.; Sikora, J.; Mikiciuk-Olasik, E.; Skibiński, R.; Gumieniczek, A.; et al. Synthesis, biological evaluation and molecular modeling of new tetrahydroacridine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 5610–5618. [Google Scholar] [CrossRef]
- Wang, X.-X.; Xie, F.; Jia, C.-C.; Yan, N.; Zeng, Y.-L.; Wu, J.-D.; Liu, Z.-P. Synthesis and biological evaluation of selective histone deacetylase 6 inhibitors as multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2021, 225, 113821. [Google Scholar] [CrossRef]
- Nishibori, M.; Oishi, R.; Itoh, Y.; Saeki, K. 9-Amino-1,2,3,4-Tetrahydroacridine Is a Potent Inhibitor of Histamine N-Methyltransferase. Jpn. J. Pharmacol. 1991, 55, 539–546. [Google Scholar] [CrossRef]
- Rydberg, E.H.; Brumshtein, B.; Greenblatt, H.M.; Wong, D.M.; Shaya, D.; Williams, L.D.; Carlier, P.R.; Pang, Y.-P.; Silman, I.; Sussman, J.L. Complexes of Alkylene-Linked Tacrine Dimers with Torpedo Californica Acetylcholinesterase: Binding of Bis(5)-Tacrine Produces a Dramatic Rearrangement in the Active-Site Gorge. J. Med. Chem. 2006, 49, 5491–5500. [Google Scholar] [CrossRef]
- Liu, Z.; Fang, L.; Zhang, H.; Gou, S.; Chen, L. Design, synthesis and biological evaluation of multifunctional tacrine-curcumin hybrids as new cholinesterase inhibitors with metal ions-chelating and neuroprotective property. Bioorg. Med. Chem. 2017, 25, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Cacabelos, R.; Oset-Gasque, M.J.; Samadi, A.; Marco-Contelles, J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013, 23, 1916–1922. [Google Scholar] [CrossRef]
- Czarnecka, K.; Girek, M.; Wójtowicz, P.; Kręcisz, P.; Skibiński, R.; Jończyk, J.; Łątka, K.; Bajda, M.; Walczak, A.; Galita, G.; et al. New Tetrahydroacridine Hybrids with Dichlorobenzoic Acid Moiety Demonstrating Multifunctional Potential for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3765. [Google Scholar] [CrossRef]
- Martins, C.; Carreiras, M.C.; León, R.; de los Ríos, C.; Bartolini, M.; Andrisano, V.; Iriepa, I.; Moraleda, I.; Gálvez, E.; García, M.; et al. Synthesis and biological assessment of diversely substituted furo [2,3-b]quinolin-4-amine and pyrrolo [2,3-b]quinolin-4-amine derivatives, as novel tacrine analogues. J. Eur. J. Med. Chem. 2011, 46, 6119–6130. [Google Scholar] [CrossRef]
- Olszewska, P.; Mikiciuk-Olasik, E.; Błaszczak-Świątkiewicz, K.; Szymański, J.; Szymański, P. Novel tetrahydroacridine derivatives inhibit human lung adenocarcinoma cell growth by inducing G1 phase cell cycle arrest and apoptosis. Biomed. Pharmacother. 2014, 68, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Kłosiński, K.; Girek, M.; Czarnecka, K.; Pasieka, Z.; Skibiński, R.; Szymański, P. Biological assessment of new tetrahydroacridine derivatives with fluorobenzoic moiety in vitro on A549 and HT-29 cell lines and in vivo on animal model. Hum. Cell. 2020, 33, 859–867. [Google Scholar] [CrossRef]
- Gensicka-Kowalewska, M.; Cholewiński, G.; Dzierzbicka, K. Recent Developments in the Synthesis and Biological Activity of Acridine/Acridone Analogues. RSC Adv. 2017, 7, 15776–15804. [Google Scholar] [CrossRef]
- Tripathi, R.P.; Verma, S.S.; Pandey, J.; Agarwal, K.C.; Chaturvedi, V.; Manju, Y.K.; Srivastva, A.K.; Gaikwad, A.; Sinha, S. Search of antitubercular activities in tetrahydroacridines: Synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2006, 16, 5144–5147. [Google Scholar] [CrossRef] [PubMed]
- Girault, S.; Grellier, P.; Berecibar, A.; Maes, L.; Mouray, E.; Lemière, P.; Debreu, M.A.; Davioud-Charvet, E.; Sergheraert, C. Antimalarial, antitrypanosomal, and activities and cytotoxicity of bis(9-amino-6-chloro-2-methoxyacridines): Influence of the linker. J. Med. Chem. 2000, 43, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Ojha, P.K.; Roy, K. First report on exploring structural requirements of 1,2,3,4- tetrahydroacridin-9(10h)-one analogs as antimalarials using multiple QSAR approaches: Descriptor-based QSAR, CoMFA-CoMSIA 3DQSAR, HQSAR and G-QSAR approaches. CCHTS 2012, 16, 7–21. [Google Scholar] [CrossRef]
- Silva, C.F.M.; Leão, T.; Dias, F.; Tomás, A.M.; Pinto, D.C.G.A.; Oliveira, E.F.T.; Oliveira, A.; Fernandes, P.A.; Silva, A.M.S. Structure-Activity Relationship Studies of 9- Alkylamino 1,2,3,4-tetrahydroacridines against Leishmania (Leishmania) infantum Promastigotes. Pharmaceutics 2023, 15, 669. [Google Scholar] [CrossRef]
- Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Acridine and Its Derivatives: A Patent Review (2009–2013). Expert. Opin. Ther. Pat. 2014, 24, 647–664. [Google Scholar] [CrossRef]
- Silva, C.F.M.; Pinto, D.C.G.A.; Fernandes, P.A.; Silva, A.M.S. Evolution of Acridines and Xanthenes as a Core Structure for the Development of Antileishmanial Agents. Pharmaceuticals 2022, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, I. ‘Acridines’ as New Horizons in Antifungal Treatment. Molecules 2020, 25, 1480. [Google Scholar] [CrossRef]
- de Oliveira Viana, J.; Silva e Souza, E.; Sbaraini, N.; Vainstein, M.H.; Gomes, J.N.S.; de Moura, R.O.; Barbosa, E.G. Scafold repositioning of spiro-acridine derivatives as fungi chitinase inhibitor by target fishing and in vitro studies. Sci. Rep. 2023, 13, 7320. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, C.R.; Steverding, D.; Swenerton, R.K.; Kelly, B.; Walshe, D.; Debnath, A.; Zhou, Y.-M.; Doyle, P.S.; Fafarman, A.T.; Zorn, J.A.; et al. Bis-Acridines as Lead Antiparasitic Agents: Structure-Activity Analysis of a Discrete Compound Library In Vitro. AAC 2007, 51, 2164–2172. [Google Scholar] [CrossRef]
- Chen, Y.L.; Lu, C.M.; Chen, I.L.; Tsao, L.T.; Wang, J.P. Synthesis and antiinflammatory evaluation of 9-anilinoacridine and 9-phenoxyacridine derivatives. J. Med. Chem. 2002, 45, 4689–4694. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Chen, I.L.; Lu, C.M.; Treng, C.C.; Tsao, L.T.; Wang, J.P. Synthesis and Anti-Inflammatory Evaluation of 9-Phenoxyacridine and 4-Phenoxyfuro [2,3-b] quinoline Derivatives. Part 2. Bioorg. Med. Chem. 2003, 11, 3921–3927. [Google Scholar] [CrossRef] [PubMed]
- Gaydukevich, A.N.; Kazakov, G.P.; Kravchenko, A.A.; Porokhnyak, L.A.; Dinchuk, V.V. Synthesis and biological activity of acridinyl-9-thioacetic acids and their derivatives. Pharm. Chem. J. 1987, 21, 633–636. [Google Scholar] [CrossRef]
- Zhao, Z.; Song, J.; Shang, Y.; Shan, M.; Tang, L.; Pang, T.; Ye, Q.; Cheng, G.; Jiang, G. Design, Synthesis, Biological Evaluation and Docking Study of A New Series of 9-aminoacridine Derivatives as Anti-inflammatory Agents. ChemistrySelect 2024, 9, e202302387. [Google Scholar] [CrossRef]
- Keenan, R.M.; Miller, W.H.; Lago, M.A.; Ali, F.E.; Bondinell, W.E.; Callahan, J.F.; Calvo, R.R.; Cousins, R.D.; Hwang, S.-M.; Jakas, D.R.; et al. Benzimidazole Derivatives as Arginine Mimetics in 1,4-Benzodiazepine Nonpeptide Vitronectin Receptor (Avβ3) Antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 3165–3170. [Google Scholar] [CrossRef]
- Schmidt, A.; Liu, M. Recent Advances in the Chemistry of Acridines. Adv. Heterocycl. Chem. 2015, 115, 287–353. [Google Scholar] [CrossRef]
- Sondhi, S.M.; Sharma, V.K.; Singhal, N.; Verma, R.P.; Shukla, R.; Raghubir, R.; Dubey, M.P. Synthesis and anti-inflammatory activity evaluation of some acridinyl amino antipyrine, acridinyl amino anthraquinone, acridinothiourea and thiazolinothiourea derivatives. Phosphorus Sulphur Silicon 2000, 156, 21–33. [Google Scholar] [CrossRef]
- Sondhi, S.M.; Rajvanshi, S.; Singh, N.; Jain, S.; Lahoti, A.M. Heterocyclic Inflammation Inhibitors. Cent. Eur. J. Chem. 2004, 2, 141–187. [Google Scholar] [CrossRef]
- Sondhi, S.M.; Singh, J.; Rani, R.; Gupta, P.P.; Agrawal, S.K.; Saxena, A.K. Synthesis, anti-inflammatory and anticancer activity evaluation of some novel acridine derivatives. EJMECH 2010, 45, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Taraporewala, I.B.; Kauffman, J.M. Synthesis and structure-activity relationships of anti-inflammatory 9,10-dihydro-9-oxo-2-acridine-alkanoic acids and 4-(2-carboxyphenyl) aminobenzenealkanoic acids. J Pharm Sci. 1990, 79, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, S.M.; Singh, N.; Lahoti, A.M.; Bajaj, K.; Kumar, A.; Lozachc, O.; Meijer, L. Synthesis of acridinyl-thiazolino derivatives and their evaluation for anti-inflammatory, analgesic and kinase inhibition activities. Bioorg. Med. Chem. 2005, 13, 4291–4299. [Google Scholar] [CrossRef] [PubMed]
- Putz, C.K.; Strassburger, W.W.A.; Zimmer, O.; Englberger, W.G. Acridine derivatives. CA Patent Application No. 02,276,764, 02 July 1999. [Google Scholar]
- Kothamunireddy, R.G. Synthesis, Characterization and Biological Evaluation of Novel Acridine Derivatives for Anti-Inflammatory and Analgesic Activities. Indian J. Pharm. Sci. 2021, 83, 1016. [Google Scholar] [CrossRef]
- Demeunynck, M. Antitumour acridines. Expert. Opin. Ther. Patents 2004, 14, 55–70. [Google Scholar] [CrossRef]
- Moghal, Z.K.B.; Manivannan, R.; Son, Y.-A. Acridine-based fluorophores with improved lightfastness properties. Dye. Pigment. 2022, 197, 109924. [Google Scholar] [CrossRef]
- Gong, X.; Lu, C.-H.; Lee, W.-K.; Li, P.; Huang, Y.-H.; Chen, Z.; Zhan, L.; Wu, C.-C.; Gong, S.; Yang, C. High-efficiency red thermally activated delayed fluorescence emitters based on benzothiophene-fused spiro-acridine donor. J. Chem. Eng. 2021, 405, 126663. [Google Scholar] [CrossRef]
- Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117, 3479–3716. [Google Scholar] [CrossRef] [PubMed]
- Goni, L.K.M.O.; Mazumder, M.A.J.; Tripathy, D.B.; Quraishi, M.A. Acridine and Its Derivatives: Synthesis, Biological, and Anticorrosion Properties. Materials 2022, 15, 7560. [Google Scholar] [CrossRef]
- Dai, Q.; Gao, C.; Liu, Y.; Liu, H.; Xiao, B.; Chen, C.; Chen, J.; Yuan, Z.; Jiang, Y. Highly sensitive and selective “naked eye” sensing of Cu(II) by a novel acridine-based sensor both in aqueous solution and on the test kit. Tetrahedron 2018, 74, 6459–6464. [Google Scholar] [CrossRef]
- Wang, C.; Fu, J.; Yao, K.; Xue, K.; Xu, K.; Pang, X. Acridine-based fluorescence chemosensors for selective sensing of Fe3+ and Ni2+ ions. Spectrochim. Acta Part A 2018, 199, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Bazzicalupi, C.; Bencini, A.; Matera, I.; Puccioni, S.; Valtancoli, B. Selective binding and fluorescence sensing of ZnII with acridine-based macrocycles. Inorg. Chim. Acta 2012, 381, 162–169. [Google Scholar] [CrossRef]
- Chen, J.-X.; Tao, W.-W.; Wang, K.; Zheng, C.-J.; Liu, W.; Li, X.; Ou, X.-M.; Zhang, X.-H. Highly efficient thermally activated delayed fluorescence emitters based on novel Indolo [2,3-b] acridine electron-donor. Org. Electron. 2018, 57, 327–334. [Google Scholar] [CrossRef]
- Rasool, A.; Basha, B.; Elmushyakhi, A.; Hossain, I.; ur Rehman, A.; Ans, M. Tuning the optoelectronic properties of acridine-triphenylamine (ACR-TPA) based novel hole transporting material for high efficiency perovskite and organic solar cell. J. Mol. Graphics Modell. 2023, 123, 108526. [Google Scholar] [CrossRef]
- Adad, A.; Hmammouchi, R.; Lakhlifi, T.; Bouachrine, M. Theoretical investigation on the optoelectronic properties of low-band-gap acridine and carbazole derivatives for photovoltaic devices. Chem. Pharm. Res. 2013, 5, 26–33. [Google Scholar]
- Moroni, G.; Calabria, D.; Quintavalla, A.; Lombardo, M.; Mirasoli, M.; Roda, A.; Gioiello, A. Thermochemiluminescence-Based Sensitive Probes: Synthesis and Photophysical Characterization of Acridine Containing 1,2-Dioxetanes Focusing on Fluorophore PushPull Effects. ChemPhotoChem 2022, 6, e202100152. [Google Scholar] [CrossRef]
- Sadki, H.; Chtita, S.; Bennani, M.N.; Lakhlifi, T.; Bouachrine, M. New Materials Based on Acridine: Correlation Structure—Properties and Optoelectronic Applications. JCMR 2014, 1, 112–122. [Google Scholar]
- Tka, N.; Ayed, M.A.H.; Braiek, M.B.; Jabli, M.; Chaaben, N.; Alimi, K.; Jopp, S.; Lange, P. 2,4-Bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridines: Synthesis and photophysical properties. Beilstein J. Org. Chem. 2021, 17, 1629–1640. [Google Scholar] [CrossRef]
- Goel, A.; Kumar, V.; Singh, S.P.; Sharma, A.; Prakash, S.; Singh, C.; Anand, R.S. Non-aggregating solvatochromic bipolar benzo[f]quinolines and benzo[a] acridines for organic electronics. J. Mater. Chem. 2012, 22, 14880. [Google Scholar] [CrossRef]
- Boudreault, P.-L.T.; Alleyne, B.; Xia, C. Organic Electroluminescent Materials and Devices. US Patent Application No. 10,199,581 B2, 05 February 2019. [Google Scholar]
- Kim, M.; Lee, J.Y. Improved power efficiency in deep blue phosphorescent organic light-emitting diodes using an acridine core based hole transport material. Org. Electron. 2012, 13, 1245–1249. [Google Scholar] [CrossRef]
- Seo, J.-A.; Jeon, S.K.; Lee, J.Y. Acridine derived stable host material for long lifetime blue phosphorescent organic light-emitting diodes. Org. Electron. 2016, 34, 33–37. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Wang, Z.; Zhao, B.; Chen, L.; Li, J.; Wang, H.; Hao, Y.; Li, W. Efficient management of excitons in red and white organic light-emitting diodes by employing blue thermally activated delayed fluorescent emitter based acridine/sulfone derivative as the host. Org. Electron. 2018, 57, 311–316. [Google Scholar] [CrossRef]
- Zeng, W.; Lai, H.-Y.; Lee, W.-K.; Jiao, M.; Shiu, Y.-J.; Zhong, C.; Gong, S.; Zhou, T.; Xie, G.; Sarma, M.; et al. Achieving Nearly 30% External Quantum Efficiency for Orange–Red Organic Light Emitting Diodes by Employing Thermally Activated Delayed Fluorescence Emitters Composed of 1,8-Naphthalimide-Acridine Hybrids. Adv. Mater. 2017, 30, 1704961. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Chen, Y.; Xie, Y.; Zhang, F.; Li, X.; Huang, A.; Fan, Y.; Fan, Y.; Gong, Y.; Peng, Q.; et al. 1.42-Fold Enhancement of Blue OLED Device Performance by Simply Changing Alkyl Groups on the Acridine Ring. Cell. Rep. Phys. Sci. 2020, 1, 100252. [Google Scholar] [CrossRef]
- Sun, W.; Zhou, N.; Xiao, Y.; Wang, S.; Li, X. Novel carbazolyl-substituted spiro[acridine-9,9′-fluorene] derivatives as deep-blue emitting materials for OLED applications. Dye. Pigment. 2018, 154, 30–37. [Google Scholar] [CrossRef]
- Hrubaru, M.-M.; Kamga, F.A.N.; Murat, N.; Draghici, C.; Bujduveanu, M.-R.; Ungureanu, E.-M.; Diacu, E. Electrochemical investigation on 1,2,3,4-tetrahydroacridine-9-carboxamide in different organic solvents. Sci. Bull. UPB Ser. B 2024, 86, 31–46. [Google Scholar]
- Hrubaru, M.-M.; Kamga, F.-A.-N.; Murat, N.; Stefaniu, A.; Ungureanu, E.-M.; Diacu, E. Electrochemical and optical investigation and DFT calculation on two tetrahydroacridines. Sci. Bull. UPB Ser. B 2024, 86, 3–18. [Google Scholar]
- Bielawsky, J. Analogues of 9-amino-1,2,3,4-tetrahydroacridine. Coll. Czech. Chim. Commun. 1977, 42, 2802–2808. [Google Scholar] [CrossRef]
- Carje, M. Űber 1,2,3 4-Tetrahydroakridin-9-Carboxyl-Säure-derivate. Rev. Roum. Chem. 1973, 18, 1013–1016. [Google Scholar]
- Hrubaru, M.M.; Maganu, M.; Vulpes, D.; Alexandru, M.G.; Plaveti, M. Synthesis and spectral characterization of new methylene-bis-polymethylenquinoline-bis-carboxamides. Rev. Chim. 2009, 60, 859–862. [Google Scholar]
- Hrubaru, M.M.; Badiceanu, C.D.; Ekennia, A.C.; Okafor, S.N.; Enache, C.; Tarko, L.; Tecuceanu, V.; Maganu, M. Synthesis, Spectral Characterization and Molecular Docking Studies of Novel Benzidin-bis-tetrahydroacridine Analogues. Rev. Chim. 2020, 71, 163–181. [Google Scholar] [CrossRef]
- Janani, S.; Rajagopal, H.; Muthu, S.; Aayisha, S.; Raja, M. Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR), HOMO-LUMO, chemical reactivity, AIM, ELF, LOL and Molecular docking studies on 1-Benzyl-4-(N-Boc-amino) piperidine. J. Mol. Struct. 2021, 1230, 129657. [Google Scholar] [CrossRef]
- Vasanthakumari, R.; Nirmala, W.; Sagadevan, S.; Mugeshini, S.; Rajeswari, N.; Balu, R.; Santhakumari, R. Synthesis, growth, crystal structure, vibrational, DFT and HOMO, LUMO analysis on protonated molecule-4-aminopyridinium nicotinate. J. Mol. Struct. 2021, 1239, 130449. [Google Scholar] [CrossRef]
- Basha, F.; Khan, F.L.A.; Muthu, S.; Raja, M. Computational evaluation on molecular structure (Monomer, Dimer), RDG, ELF, electronic (HOMO-LUMO, MEP) properties, and spectroscopic profiling of 8-Quinolinesulfonamide with molecular docking studies. Comput. Theor. Chem. 2021, 1198, 113169. [Google Scholar]
- Agwupuye, J.A.; Neji, P.A.; Louis, H.; Odey, J.O.; Unimuke, T.O.; Bisiong, E.A.; Ntui, T.N. Investigation on electronic structure, vibrational spectra, NBO analysis, and molecular docking studies of aflatoxins and selected emerging mycotoxins against wild-type androgen receptor. Heliyon 2021, 7, e07544. [Google Scholar] [CrossRef] [PubMed]
- Bisong, E.A.; Louis, H.; Unimuke, T.O.; Odey, J.O.; Ubana, E.I.; Edim, M.M.; Utsu, P.M. Vibrational, electronic, spectroscopic properties, and NBO analysis of p-xylene, 3, 6-difluoro-p-xylene, 3, 6-dichloro-p-xylene and 3, 6-dibromo-pxylene: DFT study. Heliyon 2020, 6, e05783. [Google Scholar] [CrossRef] [PubMed]
- Nemykin, V.N.; Nevonen, D.E.; Ferch, L.S.; Shepit, M.; Herbert, D.E.; van Lierop, J. Accurate Prediction of Mossbauer Hyperfine Parameters in Bis-Axially Coordinated Iron (II) Phthalocyanines Using Density Functional Theory Calculations: A Story of a Single Orbital Revealed by Natural Bond Orbital Analysis. Inorg. Chem. 2021, 60, 3690–3706. [Google Scholar] [CrossRef] [PubMed]
- Akman, F. A comparative study based on molecular structure, spectroscopic, electronic, thermodynamic and NBO analysis of some nitrogen-containing monomers. Polym. Bull. 2021, 78, 663–693. [Google Scholar] [CrossRef]
- Zamora, P.P.; Bieger, K.; Cuchillo, A.; Tello, A.; Muena, J.P. Theoretical determination of a reaction intermediate: Fukui function analysis, dual reactivity descriptor and activation energy. J. Mol. Struct. 2021, 1227, 129369. [Google Scholar] [CrossRef]
- Isravel, A.D.; Jeyaraj, J.K.; Thangasamy, S.; John, W.J. DFT, NBO, HOMO-LUMO, NCI, stability, Fukui function and hole–Electron analyses of tolcapone. Comput. Theor. Chem. 2021, 1202, 113296. [Google Scholar] [CrossRef]
- Mu, L.; Fan, W.; Yuan, X.A.; Huang, C.; Li, D.; Bi, S. Mechanistic insights into the C (sp3)-H heteroarylation of amides and Fukui function analysis of regioselectivity. Mol. Catal. 2021, 502, 111394. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. II. The effect of the Perdew–Wang generalized-gradient correlation correction. J. Chem. Phys. 1992, 97, 9173. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.P.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0. 16; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. [Google Scholar] [CrossRef]
- Ma, L.; Gao, X.; Liu, X.; Gu, X.; Li, B.; Mao, B.; Sun, Z.; Gao, W.; Jia, X.; Chen, J. Recent advances in organic electrosynthesis using heterogeneous catalysts modified electrodes. Chin. Chem. Lett. 2023, 34, 107735. [Google Scholar] [CrossRef]
- Lv, S.; Han, X.; Wang, J.-Y.; Zhou, M.; Wu, Y.; Ma, L.; Niu, L.; Gao, W.; Zhou, J.; Hu, W.; et al. Tunable Electrochemical C-N versus N-N Bond Formation of Nitrogen-Centered Radicals Enabled by Dehydrogenative Dearomatization: Biological Applications. Angew. Chem. Int. Ed. 2020, 59, 11583–11590. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Formula | 4a | 4b |
|---|---|---|---|
| Energy of the HOMO orbital (EHOMO), eV | −6.4722 | −6.4322 | |
| Energy of the LUMO orbital (ELUMO), eV | −2.2888 | −2.0490 | |
| Energy between HOMO and LUMO (Eg), eV | EHOMO − ELUMO | 4.1834 | 4.3832 |
| Ionization potential (I), eV | I = −EHOMO | 6.4722 | 6.4322 |
| Electron affinity (A), eV | A = −ELUMO | 2.2888 | 2.0490 |
| Electronegativity (χ), eV | χ = (I + A)/2 | 4.3805 | 4.2406 |
| Hardness (η), eV | η = (I − A)/2 | 2.0917 | 2.1916 |
| Softness (σ), (eV)−1 | σ = 1/η | 0.4780 | 0.4562 |
| Chemical potential (µ), (eV)−1 | μ= −((I + A)/2) | −4.3805 | −4.2406 |
| Donor (ἰ) | Acceptor (j) | E(2) (Kcal/mol) | E(j) − E(ἰ) (a.u) | F(ἰ,j) (a.u) |
|---|---|---|---|---|
| σC42–H50 | σ*C37–H43 | 75,640.17 | 6.26 | 19.549 |
| σO55–H56 | σ*C29–C34 | 14,509.10 | 0.05 | 0.785 |
| σC42–H50 | σ*C40–C45 | 13,898.42 | 16.14 | 13.464 |
| πC29–C31 | σ*C40–H45 | 13,437.61 | 17.48 | 9.427 |
| σC1–H7 | σ*C39–H54 | 11,842.02 | 0.11 | 1.011 |
| σC29–C31 | σ*C34–O55 | 11,526.30 | 7.97 | 8.798 |
| σC42–H50 | σ*C34–O55 | 10,631.21 | 16.64 | 12.063 |
| σC42–H50 | σ*O57–H58 | 8913.69 | 13.06 | 9.702 |
| πC29–C31 | σ*O57–H58 | 7806.22 | 4.40 | 5.512 |
| σC42–H49 | σ*C29–C34 | 5097.24 | 0.28 | 1.073 |
| Donor (ἰ) | Acceptor (j) | E(2) (Kcal/mol) | E(j) − E(ἰ) (a.u) | F(ἰ,j) (a.u) |
|---|---|---|---|---|
| σC32–O33 | σ*C42–C43 | 8691.80 | 0.08 | 0.760 |
| σC34–O63 | σ*C39–H59 | 8113.54 | 0.03 | 0.439 |
| σC34–O35 | σ*C37–H46 | 3609.32 | 0.01 | 0.188 |
| σC32–O33 | σC34–O35 | 2771.92 | 0.23 | 0.714 |
| σC32–O33 | σ*C63–H64 | 2544.73 | 0.37 | 0.0869 |
| σC32–O33 | σ*C37–H46 | 1734.11 | 0.32 | 0.663 |
| σC34–O35 | σ*C39–H59 | 499.24 | 0.24 | 0.3112 |
| σC34–O35 | σ*C63–H64 | 401.63 | 0.07 | 0.147 |
| σC29–C34 | σ*C44–H45 | 390.80 | 0.57 | 0.423 |
| σC42–H49 | σ*C44–H45 | 313.94 | 0.43 | 0.327 |
| Parameter\Peak | a1 | a2 | c1 | c2 | c3 | c4 |
|---|---|---|---|---|---|---|
| Ep (V) | 0.6 | 0.9 | −1.8 | −2.3 | −2.5 | −2.9 |
| Parameter\Peak | a1 | a2 | a3 | c1 | c2 | c3 |
|---|---|---|---|---|---|---|
| Ep (V) | 0.303 | 0.565 | 0.767 | −2.025 | −2.466 | −2.851 |
| Parameter | a1 | a2 | a3 | c1 |
|---|---|---|---|---|
| Slope for i = a + b·v | 1.303·10₋5 | 2.984·10₋5 | 4.969·10₋5 | −3.235·10₋5 |
| Pearson’s r for i = a + b·v | 0.989 | 0.987 | 0.994 | 0.989 |
| Slope for i = a + b·v 1/2 | 1.238·10₋5 | 2.841·10₋5 | 4.692·10₋5 | −3.058·10₋5 |
| Pearson’s r for i = a + b·v 1/2 | 0.999 | 0.9997 | 0.999 | 0.995 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrubaru, M.-M.; Draghici, C.; Ngounoue Kamga, F.A.; Diacu, E.; Egemonye, T.C.; Ekennia, A.C.; Ungureanu, E.-M. Experiments and Calculation on New N,N-bis-Tetrahydroacridines. Molecules 2024, 29, 4082. https://doi.org/10.3390/molecules29174082
Hrubaru M-M, Draghici C, Ngounoue Kamga FA, Diacu E, Egemonye TC, Ekennia AC, Ungureanu E-M. Experiments and Calculation on New N,N-bis-Tetrahydroacridines. Molecules. 2024; 29(17):4082. https://doi.org/10.3390/molecules29174082
Chicago/Turabian StyleHrubaru, Madalina-Marina, Constantin Draghici, Francis Aurelien Ngounoue Kamga, Elena Diacu, ThankGod C. Egemonye, Anthony C. Ekennia, and Eleonora-Mihaela Ungureanu. 2024. "Experiments and Calculation on New N,N-bis-Tetrahydroacridines" Molecules 29, no. 17: 4082. https://doi.org/10.3390/molecules29174082
APA StyleHrubaru, M.-M., Draghici, C., Ngounoue Kamga, F. A., Diacu, E., Egemonye, T. C., Ekennia, A. C., & Ungureanu, E.-M. (2024). Experiments and Calculation on New N,N-bis-Tetrahydroacridines. Molecules, 29(17), 4082. https://doi.org/10.3390/molecules29174082