
Synthesis and Investigation of Biological Activity of New Betulonic Acid Derivatives Containing 1,2,3-Triazole Fragments

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
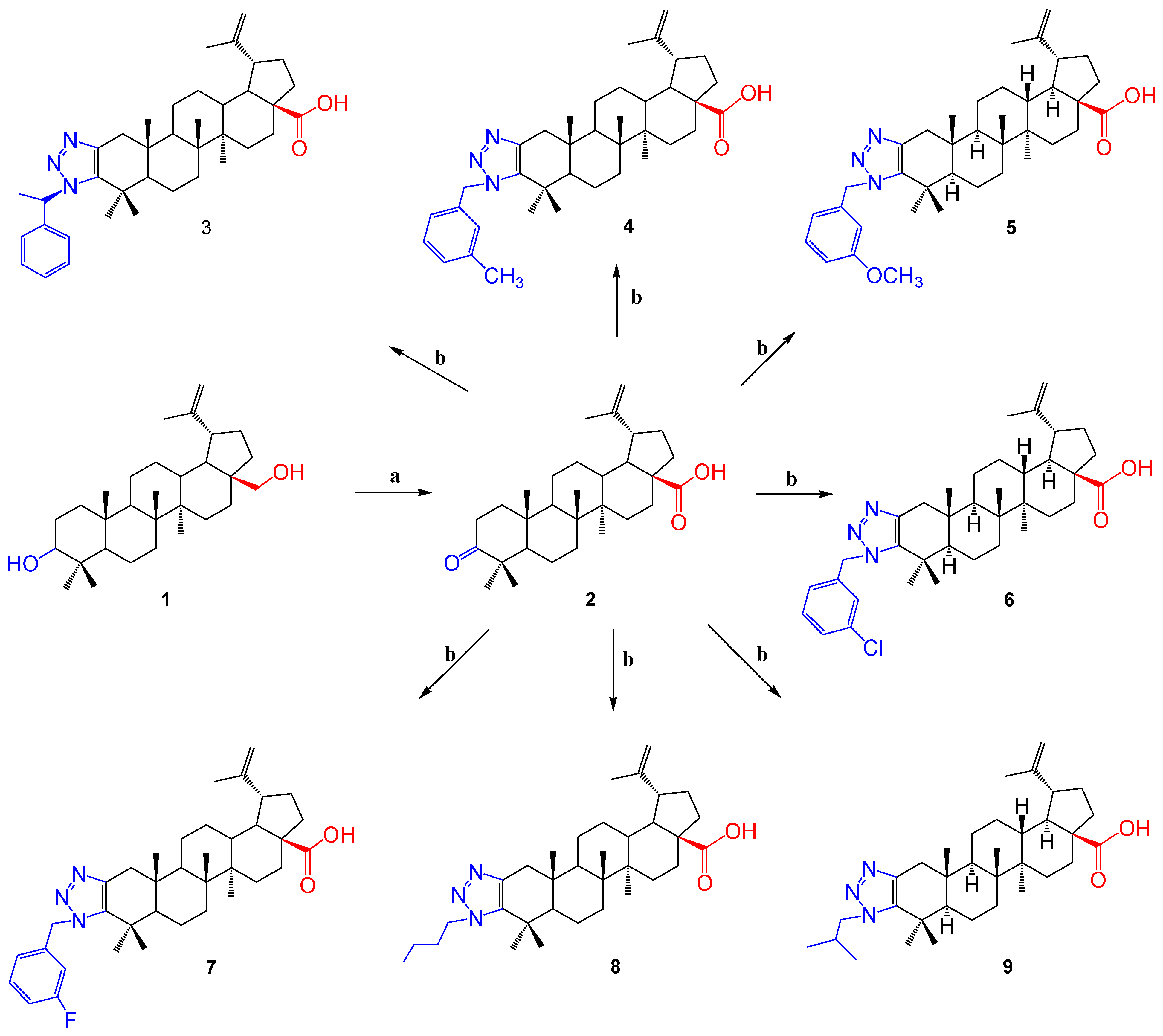
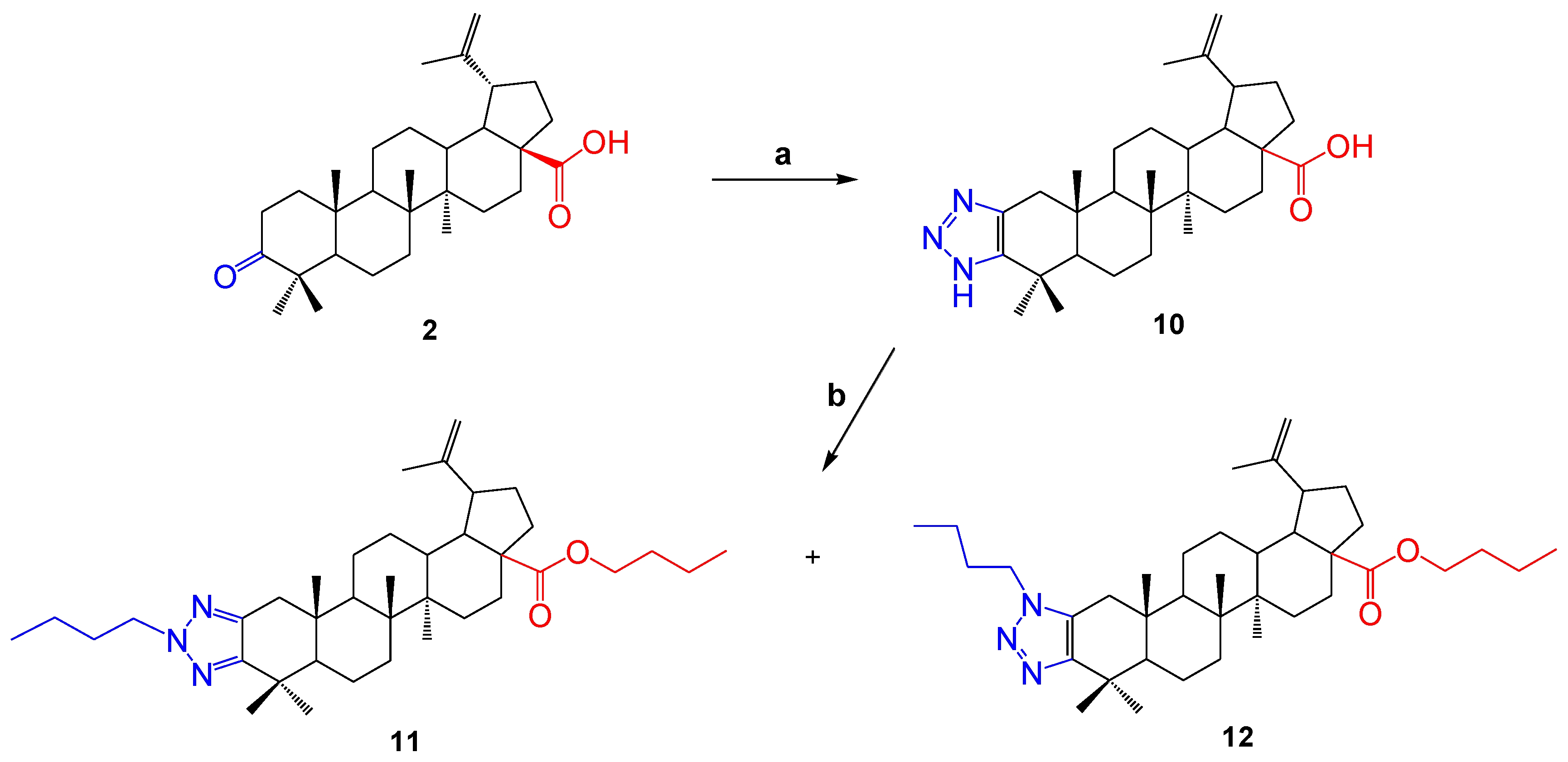
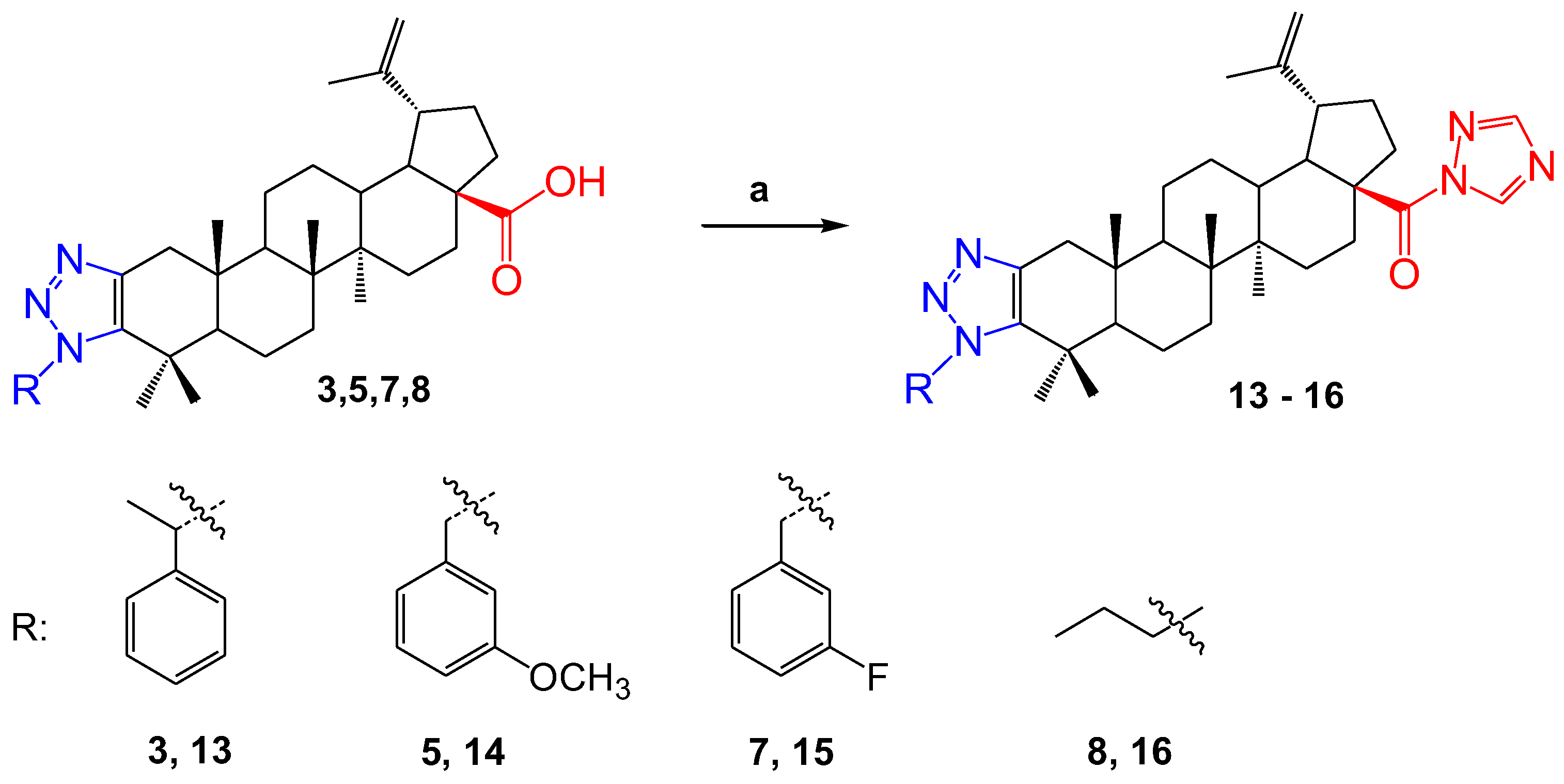
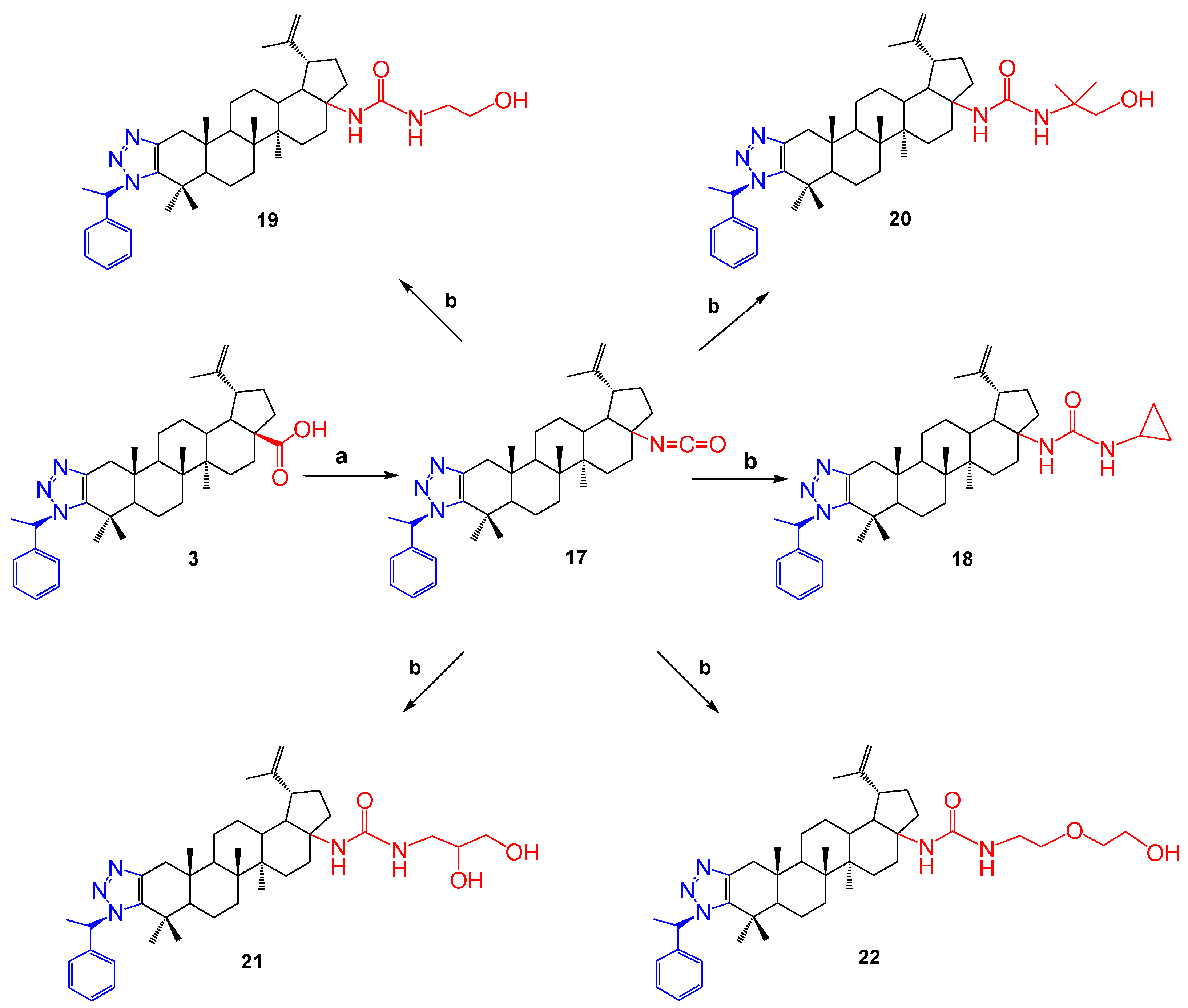
2.1. Chemistry
2.2. Evaluation of the Biological Activity
2.2.1. Antimicrobial Activity
2.2.2. Cytotoxic Activity
3. Materials and Methods
3.1. General Chemistry Section
3.2. Experimental Section
- 3-Oxo-lup-20(29)-en-28-oic Acid (Betulonic Acid, 2). Betulin (5.0 g, 11.3 mmol) was placed in a 0.5 l flask filled with acetone (150 mL) and dissolved in an ultrasonic water bath. Freshly prepared Jones reagent (6.65 g Na2Cr2O7 and 6 mL H2SO4 in 50 mL water) was added drop by drop to the cooled (on an ice bath) solution. The color of the solution began to change. The reaction mixture was allowed to heat up to room temperature and continued to be stirred for 5 h. The course of the reaction was checked by TLC. Then, MeOH (100 mL) was added to the reaction mixture, followed by water (50 mL). The resulting precipitate was filtered and washed with water (50 mL). The crude product was dried, then dissolved in Et2O (60 mL) and washed with water (30 mL), 7.5% hydrochloric acid (20 mL), water (20 mL), saturated aqueous solution NaHCO3 (20 mL) and water (20 mL). The ether layer was driven off on a rotary evaporator, and the remaining residue was purified by column chromatography (silica gel); a mixture of heptane and ethyl acetate (80:20) was used as an eluent. Compound 2 was a colorless powder. The product yield was 1.55 g (30%).
- 1H NMR (400 MHz, CDCl3): δ 4.74 (d, J = 2.3 Hz, 1H), 4.66–4.58 (m, 1H), 3.01 (td, J = 10.7, 4.6 Hz, 1H), 2.56–2.35 (m, 2H), 2.33–2.17 (m, 2H), 2.07–1.83 (m, 2H), 1.70 (s, 4H), 1.64 (t, J = 11.4 Hz, 1H), 1.59–1.14 (m, 14H), 1.13–1.04 (m, 4H), 1.03–0.95 (m, 9H), 0.93 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 218.23, 181.72, 150.32, 109.79, 56.37, 54.94, 49.85, 49.19, 47.34, 46.89, 42.49, 40.64, 39.61, 38.51, 37.04, 36.92, 34.13, 33.60, 32.10, 30.55, 29.68, 26.64, 25.49, 21.37, 21.00, 19.63, 19.37, 15.96, 15.82, 14.63.
- 1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28 oic Acid (3). Betulonic acid (1.0 g, 1 equiv., 2.2 mmol), 4-nitrophenyl azide (360 mg, 1.3 equiv., 2.86 mmol), (S)-(–)-α-methylbenzylamine (350 mg, 1.3 equiv., 2.86 mmol) and 4 Å molecular sieves (200 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (5 mL), and the reaction mixture was stirred at 100 °C for 24 h. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 3 was powdery with a yellowish tinge, m.p. 325–327 °C. The yield was 790 mg (61%). The spectroscopic data for compound 3 were consistent with previously reported data for this compound [41].
- 1H NMR (400 MHz, CDCl3): δ 7.33–7.25 (m, 3H), 7.25–7.19 (m, 2H), 5.72 (q, J = 7.0 Hz, 1H), 4.77 (d, J = 2.3 Hz, 1H), 4.64 (m, 1H), 3.03 (td, J = 10.6, 4.5 Hz, 1H), 2.96 (d, J = 15.3 Hz, 1H), 2.32–2.20 (m, 2H), 2.15 (d, J = 15.3 Hz, 1H), 2.05–1.95 (m, 5H), 1.81–1.60 (m, 5H), 1.61–1.32 (m, 11H), 1.26 (s, 5H), 1.10 (s, 3H), 0.99 (s, 6H), 0.81 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 181.07, 150.22, 141.79, 141.05, 137.53, 128.64, 127.56, 126.24, 109.88, 59.28, 56.38, 54.78, 49.34, 49.21, 46.90, 42.45, 40.60, 38.91, 38.53, 38.32, 37.04, 33.80, 33.41, 32.08, 30.59, 29.80, 28.89, 25.50, 23.73, 21.36, 21.31, 19.42, 18.91, 16.25, 15.70, 14.65.
- 1′-(3-Methylbenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid (4). Betulonic acid 2 (100 mg, 1 equiv., 0.22 mmol), 4-nitrophenyl azide (72 mg, 2 equiv., 0.44 mmol), 3-methylbenzylamine (74.6 mg, 2.8 equiv., 0.616 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 mL), and the reaction mixture was stirred at 100 °C for 24 h. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate (10:1) was used as an eluent. Compound 4 was a pale-yellow powder, m.p. 261–264 °C. The yield was 72 mg (68%).
- 1H NMR (400 MHz, CDCl3): δ 7.17 (t, J = 7.6 Hz, 1H), 7.06 (d, J = 7.6 Hz, 1H), 6.85 (s, 1H), 6.81 (d, J = 7.7 Hz, 1H), 5.62 (m, 2H), 4.76 (d, J = 2.3 Hz, 1H), 4.64 (m, 1H), 4.12 (q, J = 7.2 Hz, OH), 3.04 (td, J = 10.6, 4.5 Hz, 1H), 2.96 (d, J = 15.3 Hz, 1H), 2.25 (m, 6H), 1.99 (m, 2H), 1.77 (d, J = 12.8 Hz, 1H), 1.71 (s, 3H), 1.45 (m, 13H), 1.17 (s, 3H), 1.04 (s, 3H), 0.99 (d, J = 9.2 Hz, 6H), 0.90 (m, 1H), 0.78 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 181.38, 150.25, 141.84, 138.49, 138.00, 136.38, 128.59, 128.52, 127.03, 123.46, 109.86, 56.40, 54.59, 52.81, 49.29, 49.19, 46.90, 42.44, 40.56, 38.97, 38.51, 38.36, 37.04, 33.71, 33.35, 32.08, 30.58, 29.79, 28.77, 25.50, 21.41, 21.37, 21.31, 19.42, 18.91, 16.08, 15.70, 14.65.
- HRMS (ESI+): m/z calculated for C38H54N3O2 [M + H]+: 584.42160, found: 584.4216.
- 1′-(3-Methoxybenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid (5). Betulonic acid (100 mg, 1 equiv., 0.22 mmol), 4-nitrophenyl azide (72 mg, 2 equiv., 0.44 mmol), 3-methoxybenzylamine (84.5 mg, 2.8 equiv., 0.616 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 mL), and the reaction mixture was stirred at 100 °C for 18 h. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 5 was powdery with a yellowish tinge, m.p. 205–208 °C. The yield was 78 mg (60%).
- 1H NMR (400 MHz, CDCl3): δ 7.21 (td, J = 7.9, 3.3 Hz, 1H), 6.83–6.75 (m, 1H), 6.65–6.54 (m, 3H), 5.62 (d, J = 3.8 Hz, 3H), 4.76 (d, J = 2.3 Hz, 1H), 4.67–4.63 (m, 1H), 3.74 (d, J = 1.6 Hz, 4H), 3.09–2.92 (m, 2H), 2.26 (ddd, J = 15.5, 9.7, 3.5 Hz, 2H), 2.21–2.14 (m, 1H), 2.06–1.82 (m, 3H), 1.81–1.75 (m, 2H), 1.71 (s, 3H), 1.68 (s, 0H), 1.61–1.36 (m, 10H), 1.35–1.18 (m, 4H), 1.17 (d, J = 2.6 Hz, 3H), 1.04 (s, 3H), 1.02–0.96 (m, 8H), 0.81 (s, 1H), 0.77 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 180.81, 159.97, 150.24, 141.89, 138.09, 138.04, 129.78, 118.65, 113.31, 111.96, 109.87, 56.36, 55.23, 54.79, 54.59, 52.74, 52.66, 49.30, 49.19, 46.88, 44.09, 42.45, 40.56, 40.36, 38.98, 38.48, 38.37, 37.03, 33.72, 33.35, 32.07, 30.57, 29.79, 28.73, 25.50, 21.38, 21.31, 19.42, 18.91, 16.08, 15.71, 15.19, 14.65.
- HRMS (ESI+): m/z calculated for C38H54N3O3 [M + H]+: 600.4160, found: 600.4194.
- 1′-(3-Chlorobenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid (6). Betulonic acid 2 (100 mg, 1 equiv., 0.22 mmol), 4-nitrophenyl azide (72 mg, 2 equiv., 0.44 mmol), 3-chlorobenzylamine (87 mg, 2.8 equiv., 0.616 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 mL), and the reaction mixture was stirred at 100 °C for 24 h. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate (1:1) was used as an eluent. Compound 6 was a pale-yellow powder, m.p. 288–290 °C. The yield was 65 mg (62%).
- 1H NMR (400 MHz, CDCl3): δ 7.28–7.19 (m, 2H), 7.03 (d, J = 2.0 Hz, 1H), 6.89 (dt, J = 5.7, 2.1 Hz, 1H), 5.61 (d, J = 5.5 Hz, 2H), 4.76 (d, J = 2.3 Hz, 1H), 4.64 (t, J = 1.9 Hz, 1H), 3.04 (td, J = 10.6, 4.6 Hz, 1H), 2.96 (d, J = 15.4 Hz, 1H), 2.28 (ddd, J = 14.7, 9.0, 3.2 Hz, 2H), 2.23–2.15 (m, 1H), 2.00 (ddd, J = 17.8, 11.6, 5.5 Hz, 2H), 1.83–1.73 (m, 1H), 1.71 (s, 3H), 1.67–1.19 (m, 13H), 1.17 (s, 4H), 1.06–0.95 (m, 9H), 0.78 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 181.63, 150.23, 142.05, 130.06, 128.07, 126.53, 124.55, 109.87, 56.41, 54.51, 52.16, 49.27, 49.20, 46.91, 42.44, 40.55, 38.98, 38.53, 38.29, 37.03, 33.69, 33.31, 32.07, 30.58, 29.80, 28.83, 25.47, 21.41, 21.37, 19.42, 18.88, 16.09, 15.73, 14.64.
- HRMS (ESI+): m/z calculated for C37H51ClN3O2 [M + H]+: 604.36698, found: 604.3661.
- 1′-(3-Fluorobenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid (7). Betulonic acid (100 mg, 1 equiv., 0.22 mmol), 4-nitrophenyl azide (72 mg, 2 equiv., 0.44 mmol), 3-fluorobenzylamine (77 mg, 2.8 equiv., 0.616 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 mL), and the reaction mixture was stirred at 100 °C for 22 h. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 7 was powdery with a yellowish tinge, m.p. 302–305 °C. The yield was 82 mg (64%).
- 1H NMR (400 MHz, CDCl3): δ 7.27 (d, J = 5.5 Hz, 2H), 7.00–6.92 (m, 1H), 6.81 (ddd, JHF = 7.8 Hz, JHH =1.8, 0.9 Hz, 1H), 6.71 (dt, JHF = 9.6 Hz, JHH = 2.2 Hz, 1H), 5.68–5.58 (m, 2H), 4.76 (d, J = 2.3 Hz, 1H), 4.67–4.62 (m, 1H), 3.03 (td, J = 10.6, 4.6 Hz, 1H), 2.96 (d, J = 15.4 Hz, 1H), 2.27 (td, J = 12.7, 11.3, 3.3 Hz, 2H), 2.23–2.15 (m, 1H), 2.07–1.93 (m, 2H), 1.77 (dd, J = 11.4, 4.1 Hz, 1H), 1.71 (s, 3H), 1.66 (t, J = 11.4 Hz, 1H), 1.62–1.33 (m, 9H), 1.33–1.18 (m, 3H), 1.17 (s, 3H), 1.04 (s, 3H), 0.99 (d, J = 8.9 Hz, 6H), 0.78 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 181.18, 161.82 (d, 1JC-F = 245.5 Hz), 150.22, 142.05, 139.03 (d, 3JC-F = 7.2 Hz), 138.10, 130.36 (d, 3JC-F = 8.2 Hz), 121.99 (d, 4JC-F = 2.9 Hz), 114.80 (d, 2JC-F = 22.5 Hz), 113.50 (d, 2JC-F = 21.1 Hz), 109.88, 56.38, 54.53, 52.24, 49.28, 49.20, 46.91, 42.45, 40.56, 38.98, 38.53, 38.31, 37.02, 33.69, 33.32, 32.07, 30.58, 29.80, 28.78, 25.48, 21.37, 19.42, 18.89, 16.10, 15.73, 14.65.
- HRMS (ESI+): m/z calculated for C37H51FN3O2 [M + H]+: 588.3960, found: 588.3966.
- 1′-Butyl-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid (8). Betulonic acid (100 mg, 1 equiv., 0.22 mmol), 4-nitrophenyl azide (36 mg, 1 equiv., 0.22 mmol), n-butylamine (21 mg, 1.3 equiv., 0.286 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 mL), and the reaction mixture was stirred at 100 °C for 24 h. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate was used as an eluent. Compound 8 was powdery with a yellowish tinge, m.p. 288–290 °C. The yield was 74 mg (63%).
- 1H NMR (400 MHz, CDCl3): δ 4.76 (d, J = 2.3 Hz, 1H), 4.67–4.61 (m, 1H), 4.29 (td, J = 7.1, 1.6 Hz, 2H), 3.04 (td, J = 10.5, 4.5 Hz, 1H), 2.91 (d, J = 15.3 Hz, 1H), 2.34–2.20 (m, 2H), 2.13 (d, J = 15.4 Hz, 1H), 2.06–1.93 (m, 4H), 1.80–1.73 (m, 1H), 1.71 (s, 3H), 1.65 (d, J = 11.5 Hz, 1H), 1.62–1.33 (m, 11H), 1.30 (s, 3H), 1.26 (s, 4H), 1.18 (s, 3H), 1.04–0.93 (m, 10H), 0.93–0.80 (m, 1H), 0.77 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 181.21, 150.24, 141.03, 109.86, 56.39, 54.68, 49.37, 49.30, 49.21, 46.90, 42.46, 40.59, 38.98, 38.54, 38.27, 37.05, 33.66, 33.39, 32.85, 32.09, 30.60, 29.81, 29.71, 28.68, 25.50, 21.33, 20.18, 19.43, 18.98, 16.06, 15.72, 14.69, 13.70.
- HRMS (ESI+): m/z calculated for C34H54N3O2 [M + H]+: 536.4211, found: 536.4214.
- 1′-Isobutyl-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid (9). Betulonic acid 2 (100 mg, 1 equiv., 0.22 mmol), 4-nitrophenyl azide (36 mg, 1 equiv., 0.22 mmol), isobutylamine (21 mg, 1.3 equiv., 0.286 mmol) and 4 Å molecular sieves (50 mg) were added to a dried reaction tube with a screw cap equipped with a magnetic stirrer. The mixture was dissolved in dry toluene (1 mL), and the reaction mixture was stirred at 100 °C for 24 h. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate (10:1) was used as an eluent. Compound 9 was a pale-yellow powder, m.p. 299–301 °C. The yield was 68 mg (65%).
- 1H NMR (400 MHz, CDCl3): δ 4.76 (d, J = 2.3 Hz, 1H), 4.64 (t, J = 1.9 Hz, 1H), 4.16–4.03 (m, 2H), 3.04 (td, J = 10.6, 4.6 Hz, 1H), 2.92 (d, J = 15.3 Hz, 1H), 2.54 (dq, J = 13.8, 6.9 Hz, 1H), 2.35–2.19 (m, 2H), 2.14 (d, J = 15.3 Hz, 1H), 2.08–1.94 (m, 2H), 1.81–1.73 (m, 1H), 1.71 (s, 3H), 1.69–1.32 (m, 10H), 1.30 (s, 3H), 1.24 (d, J = 16.4 Hz, 3H), 1.17 (s, 3H), 1.15–1.03 (m, 1H), 1.03–0.93 (m, 13H), 0.93–0.83 (m, 1H), 0.77 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 181.59, 150.26, 140.94, 137.79, 109.83, 56.56, 56.42, 54.74, 49.30, 49.22, 46.91, 42.45, 40.58, 38.90, 38.55, 38.28, 37.05, 33.73, 33.38, 32.10, 30.61, 29.82, 29.38, 28.90, 25.50, 21.57, 21.36, 20.25, 20.21, 19.43, 19.00, 16.04, 15.74, 14.68.
- HRMS (ESI+): m/z calculated for C34H54N3O2 [M + H]+: 536.42160, found: 536.4209.
- 1H′-Lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid (10). Betulonic acid 2 (100 mg, 1 equiv., 0.22 mmol), ammonium acetate (84.8 mg, 5 equiv., 1.1 mmol) and 4-nitrophenyl azide (45.8 mg, 1.3 equiv., 0.28 mmol) were dissolved in dry DMF (1 mL). The reaction mixture was stirred at 80 °C for 24 h. A mixture of H2SO4 and ethanol (10:90) was used to visualize the TLC plates. The crude reaction mixture was purified directly using column chromatography (silica gel), first using dichloromethane to remove all 4-nitroaniline formed during the reaction, and then a mixture of petroleum ether and ethyl acetate (10:1) was used as an eluent. Compound 10 was a pale-yellow powder, m.p. 158 °C. The yield was 48 mg (46%). The spectroscopic data for compound 10 were consistent with previously reported data for this compound [41].
- 1H NMR (400 MHz, CDCl3): δ 4.77 (d, J = 2.5 Hz, 1H), 4.64 (d, J = 1.9 Hz, 1H), 3.05 (td, J = 10.7, 4.6 Hz, 1H), 2.90 (d, J = 15.5 Hz, 1H), 2.36–2.22 (m, 2H), 2.19–2.09 (m, 1H), 2.08–1.93 (m, 2H), 1.83–1.74 (m, 1H), 1.72 (s, 3H), 1.68–1.36 (m, 15H), 1.32 (s, 4H), 1.30–1.18 (m, 4H), 1.18–1.04 (m, 1H), 1.01 (d, J = 10.2 Hz, 6H), 0.97–0.80 (m, 6H), 0.77 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 181.38, 150.35, 149.92, 140.40, 109.80, 56.40, 53.42, 49.22, 49.09, 46.95, 42.51, 41.35, 40.75, 39.04, 38.51, 37.32, 37.07, 33.72, 33.38, 33.30, 32.16, 31.01, 30.62, 29.80, 29.06, 27.67, 25.51, 23.76, 22.63, 21.41, 20.45, 19.42, 19.16, 16.27, 15.69, 14.68.
- Compound (11) and (12). Compound 10 (181 mg) was dissolved in dry methanol (1.5 mL), then potassium tret-butoxide (tButOK) (100.08 mg, 0.91 mmol) was added. The reaction mixture was stirred at room temperature for 3 h, after which 1-bromobutane was slowly added and stirred at 60 °C for 12 h. The solvent was distilled in a rotary evaporator. The crude reaction mixture was purified directly using column chromatography (silica gel), using a mixture of petroleum ether and ethyl acetate as an eluent. Compound 11 was a colorless substance. The yield was 103 mg (46%).
- 1H NMR (400 MHz, CDCl3): δ 4.76 (d, J = 2.3 Hz, 1H), 4.63 (t, J = 1.8 Hz, 1H), 4.32 (t, J = 7.3 Hz, 2H), 4.09 (qt, J = 10.8, 6.6 Hz, 2H), 3.04 (td, J = 10.8, 4.5 Hz, 1H), 2.83 (d, J = 15.4 Hz, 1H), 2.29 (td, J = 12.4, 11.7, 3.6 Hz, 2H), 2.07 (d, J = 15.4 Hz, 1H), 1.96–1.85 (m, 4H), 1.80–1.72 (m, 1H), 1.71 (s, 3H), 1.68–1.30 (m, 15H), 1.29 (d, J = 4.0 Hz, 4H), 1.21 (s, 2H), 1.17 (d, J = 5.7 Hz, 4H), 1.15–1.02 (m, 1H), 1.00 (s, 3H), 0.98 (d, J = 4.3 Hz, 4H), 0.96–0.81 (m, 11H), 0.79 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 176.22, 150.89, 150.54, 141.49, 109.63, 63.72, 56.58, 54.41, 53.50, 49.35, 49.17, 47.01, 42.46, 40.76, 38.90, 38.35, 37.62, 37.05, 33.50, 33.39, 32.14, 32.05, 31.08, 30.81, 30.68, 29.72, 25.61, 23.82, 21.44, 19.84, 19.44, 19.32, 19.21, 16.26, 15.63, 14.67, 13.72, 13.59.
- HRMS (ESI+): m/z calculated for C38H62N3O2 [M + H]+: 592.48420, found: 592.4850.
- Compound 12 was a colorless substance. The yield was 45 mg (20%).
- 1H NMR (400 MHz, CDCl3) δ 4.75 (d, J = 2.4 Hz, 1H), 4.61 (m, 1H), 4.17 (m, 1H), 4.10 (m, 3H), 3.05 (td, J = 10.8, 4.4 Hz, 1H), 2.61 (d, J = 15.3 Hz, 1H), 2.37–2.24 (m, 2H), 2.05–1.98 (m, 1H), 1.97–1.85 (m, 2H), 1.84–1.73 (m, 3H), 1.70 (s, 3H), 1.68–1.37 (m, 13H), 1.33 (d, J = 10.5 Hz, 4H), 1.27–1.20 (m, 6H), 1.18–1.03 (m, 2H), 1.01 (s, 3H), 0.98 (d, J = 5.2 Hz, 3H), 0.95 (d, J = 6.8 Hz, 4H), 0.93–0.82 (m, 5H), 0.79 (s, 3H).
- 13C NMR (101 MHz, CDCl3) δ 176.17, 150.68, 150.15, 129.16, 109.61, 63.76, 56.52, 53.37, 49.34, 49.27, 47.36, 47.00, 42.49, 40.87, 39.24, 38.31, 37.03, 36.06, 33.65, 33.36, 32.16, 32.09, 30.81, 30.70, 30.59, 29.70, 25.58, 23.08, 21.59, 19.86, 19.38, 19.31, 19.00, 16.63, 15.63, 14.65, 13.72, 13.60.
- HRMS (ESI+): m/z calculated for C38H62N3O2 [M + H]+: 592.48420, found: 592.4844.
- 1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-(1H-triazol-1-yl) (13). Compound 3 (100 mg, 1 equiv., 0.171 mmol) was dissolved in 2 mL of THF, and 1,1′-carbonyldi-(1,2,4-triazole) (112.1 mg, 4 equiv., 0.684 mmol) was added. The reaction mixture was stirred at 70 °C. The reaction was carried out for 5 h. The solvent was distilled in a rotary evaporator. The remainder was chromatographed with silica gel in a column. When the column was eluted with a mixture of petroleum ether and ethyl acetate (5:3), compound 13 was isolated. Compound 13 was a colorless powder, m.p. 232–235 °C. The yield was 116 mg (107%).
- 1H NMR (400 MHz, CDCl3): δ 8.93 (s, 1H), 7.99 (s, 1H), 7.34–7.26 (m, 3H), 7.24–7.18 (m, 3H), 5.72 (q, J = 7.0 Hz, 1H), 4.79 (d, J = 2.1 Hz, 1H), 4.68 (t, J = 1.8 Hz, 1H), 3.04–2.90 (m, 3H), 2.74–2.55 (m, 2H), 2.20–2.09 (m, 2H), 2.02 (d, J = 7.0 Hz, 3H), 1.90–1.75 (m, 3H), 1.73 (s, 3H), 1.72–1.57 (m, 2H), 1.57–1.30 (m, 6H), 1.30–1.13 (m, 9H), 1.09 (s, 3H), 0.99 (d, J = 10.7 Hz, 6H), 0.95–0.86 (m, 1H), 0.85 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 152.19, 145.19, 141.02, 128.64, 127.55, 126.23, 110.20, 59.28, 58.45, 54.84, 50.92, 49.54, 45.65, 42.33, 40.62, 38.94, 38.41, 37.19, 36.26, 33.81, 33.38, 31.47, 30.54, 29.97, 28.89, 25.53, 23.74, 21.52, 21.32, 19.40, 18.90, 16.29, 15.67, 14.65.
- HRMS (ESI+): m/z calculated for C40H55N6O [M + H]+: 635.4432, found: 635.4393.
- 1′-(3-Methoxybenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-(1H-triazol-1-yl) (14). Compound 5 (93 mg, 1 equiv., 0.155 mmol) was dissolved in 2 mL of THF, and 1,1′-carbonyldi-(1,2,4-triazole) (152.6 mg, 6 equiv., 0.93 mmol) was added. The reaction mixture was stirred at 70 °C. The reaction was carried out for 24 h. The solvent was distilled in a rotary evaporator. The remainder was chromatographed with silica gel in a column. When the column was eluted with a mixture of petroleum ether and ethyl acetate (4:1), compound 14 was isolated. Compound 14 was a colorless powder, m.p. 279–282 °C. The yield was 52 mg (52%).
- 1H NMR (400 MHz, CDCl3): δ 8.92 (s, 1H), 7.99 (s, 1H), 7.21 (t, J = 7.9 Hz, 1H), 6.82–6.77 (m, 1H), 6.63–6.58 (m, 1H), 6.56 (t, J = 2.1 Hz, 1H), 5.62 (s, 2H), 4.80 (d, J = 2.2 Hz, 1H), 4.71–4.64 (m, 1H), 3.74 (s, 3H), 3.05–2.90 (m, 3H), 2.68 (td, J = 12.4, 3.4 Hz, 1H), 2.60 (dd, J = 12.9, 7.3 Hz, 1H), 2.17 (s, 5H), 1.92–1.75 (m, 3H), 1.74 (s, 3H), 1.72–1.64 (m, 1H), 1.63 (d, J = 1.9 Hz, 2H), 1.60–1.34 (m, 5H), 1.35–1.19 (m, 2H), 1.18 (s, 3H), 1.06 (s, 3H), 1.03 (s, 3H), 0.97 (s, 4H), 0.80 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 173.39, 159.97, 152.21, 149.83, 145.18, 141.87, 138.10, 138.00, 129.78, 118.65, 113.27, 112.00, 110.20, 58.44, 55.23, 54.66, 52.74, 50.91, 49.49, 45.65, 42.33, 40.60, 39.01, 38.48, 37.18, 36.27, 33.74, 33.32, 31.47, 30.54, 29.96, 28.73, 25.53, 21.54, 21.35, 19.41, 18.91, 16.13, 15.65, 14.66.
- HRMS (ESI+): m/z calculated for C40H55N6O2 [M + H]+: 651.4381, found: 651.4378.
- 1′-(3-Fluorobenzyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-(1H-triazol-1-yl) (15). Compound 7 (50 mg, 1 equiv., 0.085 mmol) was dissolved in 2 mL of THF, and 1,1′-carbonyldi-(1,2,4-triazole) (55.7 mg, 4 equiv., 0.34 mmol) was added. The reaction mixture was stirred at 70 °C. The reaction was carried out for 2.5 h. The solvent was distilled in a rotary evaporator. The formed compound was washed three times with cold acetone. Compound 15 was a colorless powder, m.p. 307–310 °C. The yield was 61 mg (112%).
- 1H NMR (400 MHz, CDCl3): δ 8.92 (s, 1H), 7.99 (s, 1H), 7.31–7.23 (m, 2H), 7.00–6.91 (m, 1H), 6.81 (dt, JHF = 7.8 Hz, JHH = 1.3 Hz, 1H), 6.72 (dt, JHF = 9.6 Hz, JHH =2.1 Hz, 1H), 5.64 (d, J = 3.0 Hz, 2H), 4.80 (d, J = 2.2 Hz, 1H), 4.68 (t, J = 1.8 Hz, 1H), 3.75 (tdd, J = 5.8, 2.5, 1.6 Hz, 1H), 3.04–2.90 (m, 3H), 2.68 (td, J = 12.4, 3.4 Hz, 1H), 2.64–2.55 (m, 1H), 2.20 (d, J = 15.4 Hz, 1H), 1.91–1.75 (m, 4H), 1.74 (s, 3H), 1.70–1.34 (m, 8H), 1.25 (ddd, J = 25.0, 8.5, 3.2 Hz, 3H), 1.17 (s, 4H), 1.07 (s, 3H), 1.03 (s, 3H), 0.97 (s, 3H), 0.81 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 13C NMR (101 MHz, CDCl3): δ 173.38, 161.83 (d, 1JC-F = 245.5 Hz), 152.21, 149.82, 145.18, 142.04, 139.01 (d, 3JC-F = 7.2 Hz), 138.07, 130.35 (d, 3JC-F = 8.3 Hz), 121.94 (d, 4JC-F = 2.9 Hz), 114.83 (d, 2JC-F = 20.9 Hz), 113.52 (d, 2JC-F = 22.6 Hz), 110.21, 67.98, 58.44, 54.61, 52.25, 50.91, 49.49, 45.65, 42.34, 40.60, 39.01, 38.42, 37.18, 36.27, 33.72, 33.30, 31.46, 30.53, 29.96, 28.79, 25.52, 21.54, 21.41, 19.40, 18.90, 16.16, 15.65, 14.66.
- HRMS (ESI+): m/z calculated for C39H52FN6O [M + H]+: 639.4181, found: 639.4180.
- 1′-Butyl-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-(1H-triazol-1-yl) (16). Compound 8 (50 mg, 1 equiv., 0.0934 mmol) was dissolved in 2 mL of THF, and 1,1′-carbonyldi-(1,2,4-triazole) (61.2 mg, 4 equiv., 0.3736 mmol) was added. The reaction mixture was stirred at 70 °C. The reaction was carried out for 2 h. The solvent was distilled in a rotary evaporator. The formed compound was washed three times with cold acetone. Compound 16 was a colorless powder, m.p. 281–283° C. The yield was 59 mg (108%).
- 1H NMR (400 MHz, CDCl3): δ 8.93 (s, 1H), 8.00 (s, 1H), 7.27 (s, 1H), 4.79 (d, J = 2.2 Hz, 1H), 4.71–4.63 (m, 1H), 4.29 (td, J = 7.1, 1.4 Hz, 2H), 3.05–2.89 (m, 3H), 2.68 (td, J = 12.4, 3.4 Hz, 1H), 2.63–2.56 (m, 1H), 2.19–2.10 (m, 2H), 1.97 (pd, J = 7.1, 1.4 Hz, 2H), 1.89–1.75 (m, 3H), 1.71 (d, J = 17.7 Hz, 5H), 1.66–1.34 (m, 7H), 1.30 (s, 3H), 1.27–1.18 (m, 4H), 1.12 (td, J = 13.0, 4.3 Hz, 1H), 1.03 (s, 3H), 1.01–0.94 (m, 6H), 0.80 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 173.39, 152.21, 149.82, 145.19, 141.02, 137.26, 110.20, 58.45, 54.75, 50.92, 49.50, 49.36, 45.65, 42.34, 40.62, 39.01, 38.39, 37.19, 36.27, 33.68, 33.36, 32.86, 31.48, 30.54, 29.98, 28.68, 25.53, 21.52, 21.36, 20.18, 19.40, 18.97, 16.11, 15.67, 14.70, 13.70.
- HRMS (ESI+): m/z calculated for C36H55N6O [M + H]+: 587.4432, found: 587.4438.
- 1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-isocyanate (17). Compound 3 (200 mg, 1 equiv., 0.34 mmol) was dissolved in toluene (6 mL) in an ultrasonic bath, then triethylamine (0.0464 mL, 1 equiv., 0.34 mmol) and diphenylphosphoryl azide (0.0772 mL, 1 equiv., 0.34 mmol) were added. The reaction was carried out at room temperature (21 °C) for 6 h. At the end of the reaction, the solvent was distilled in a rotary evaporator. The remainder was chromatographed with silica gel in a column. Compound 17 was a colorless powder, m.p. 197–200 °C. The yield was 120 mg (61%).
- 1H NMR (400 MHz, CDCl3): δ 7.29 (m, 3H), 7.22 (m, 2H), 5.73 (q, J = 7.0 Hz, 1H), 4.77 (d, J = 2.1 Hz, 1H), 4.67 (m, 1H), 2.97 (d, J = 15.3 Hz, 1H), 2.56 (td, J = 10.9, 5.8 Hz, 1H), 2.13 (m, 2H), 2.02 (d, J = 7.0 Hz, 3H), 1.84 (m, 5H), 1.70 (d, J = 1.2 Hz, 3H), 1.52 (m, 8H), 1.29 (s, 3H), 1.20 (m, 2H), 1.11 (d, J = 1.8 Hz, 6H), 0.94 (s, 3H), 0.84 (m, 3H).
- 13C NMR (101 MHz, CDCl3): δ 148.71, 141.83, 140.99, 137.51, 128.64, 127.55, 126.24, 121.63, 110.63, 71.59, 59.27, 54.80, 49.32, 49.18, 48.10, 42.06, 40.66, 39.27, 39.17, 38.91, 38.42, 33.81, 33.57, 33.46, 29.29, 28.91, 27.81, 24.91, 23.74, 21.36, 21.34, 19.52, 18.89, 16.29, 15.69, 14.44.
- HRMS (ESI+): m/z calculated for C38H53N4O [M + H]+: 581.4214, found: 581.4218.
- 1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid derivative-1. (18). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 mL) in an ultrasonic bath, then triethylamine (0.024 mL, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 mL, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (20 °C) for 7 h, after which cyclopropylamine (0.177 mL, 10 equiv., 1.7 mmol) in 0.5 mL of toluene was added to the reaction mixture. Then, it was heated at 110 °C for 4 h. At the end of the reaction, the solvent was distilled in a rotary evaporator. The remainder was chromatographed with silica gel in a column. During elution of the column with ethyl acetate, a colorless substance (compound 18) with a melting point of 200–203 °C was isolated. The yield was 66 mg (61%).
- 1H NMR (400 MHz, CDCl3): δ 7.34–7.25 (m, 3H), 7.23–7.18 (m, 2H), 5.73 (q, J = 7.0 Hz, 1H), 4.95 (s, 1H), 4.75 (d, J = 2.1 Hz, 1H), 4.69–4.60 (m, 2H), 2.96 (d, J = 15.2 Hz, 1H), 2.67 (dt, J = 13.2, 3.3 Hz, 1H), 2.54 (dd, J = 12.5, 8.1 Hz, 1H), 2.44 (tq, J = 7.9, 4.6, 4.1 Hz, 2H), 2.16 (d, J = 15.2 Hz, 1H), 2.07–1.91 (m, 4H), 1.82–1.64 (m, 7H), 1.62–1.16 (m, 11H), 1.10 (d, J = 9.6 Hz, 6H), 0.99 (s, 3H), 0.95–0.75 (m, 6H), 0.69–0.56 (m, 2H).
- 13C NMR (101 MHz, CDCl3): δ 158.04, 149.40, 141.76, 140.94, 137.53, 128.66, 127.59, 126.23, 110.27, 63.72, 59.28, 54.74, 49.31, 49.23, 48.33, 42.07, 40.64, 38.89, 38.42, 38.37, 35.19, 33.83, 33.15, 29.86, 29.33, 28.89, 27.34, 25.11, 23.72, 22.70, 21.48, 21.35, 19.28, 18.90, 16.29, 15.58, 14.48, 7.82, 7.35.
- HRMS (ESI+): m/z calculated for C41H60N5O [M + H]+: 638.4792, found: 638.4750.
- 1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid derivative-2. (19). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 mL) in an ultrasonic bath, then triethylamine (0.024 mL, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 mL, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (21 °C) for 2 h, after which ethanolamine (0.1026 mL, 10 equiv., 1.7 mmol) in 0.5 mL of toluene was added to the reaction mixture. Then, it was heated at 110 °C for 7 h. At the end of the reaction, the solvent was distilled in a rotary evaporator. The remainder was chromatographed with silica gel in a column. During elution of the column with ethyl acetate, a colorless substance (compound 19) with a melting point of 202–204 °C was isolated. The yield was 126 mg (116%).
- 1H NMR (400 MHz, CDCl3): δ 7.35–7.23 (m, 2H), 7.27–7.14 (m, 2H), 5.79 (s, 1H), 5.75 (q, J = 6.9 Hz, 1H), 5.18 (s, 1H), 4.69 (d, J = 2.3 Hz, 1H), 4.62 (t, J = 1.9 Hz, 1H), 4.12 (q, J = 7.2 Hz, 1H), 3.77–3.60 (m, 3H), 3.60–3.45 (m, 1H), 3.24 (s, 2H), 2.91 (d, J = 15.3 Hz, 1H), 2.68–2.60 (m, 1H), 2.55 (td, J = 11.0, 5.0 Hz, 1H), 2.46 (dd, J = 12.2, 8.1 Hz, 1H), 2.21–2.15 (m, 1H), 2.14 (d, J = 14.3 Hz, 1H), 2.08 (s, 1H), 2.03 (d, J = 12.8 Hz, 2H), 2.02–1.92 (m, 2H), 1.86–1.75 (m, 1H), 1.74 (s, 1H), 1.70 (s, 3H), 1.65 (d, J = 11.6 Hz, 1H), 1.62–1.54 (m, 1H), 1.57–1.47 (m, 4H), 1.47–1.35 (m, 1H), 1.38–1.28 (m, 1H), 1.28 (s, 4H), 1.26 (s, 9H), 1.30–1.19 (m, 1H), 1.13 (s, 3H), 1.19–1.03 (m, 2H), 1.02 (s, 3H), 0.96 (s, 3H), 0.96–0.85 (m, 2H), 0.89–0.81 (m, 2H), 0.79 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 174.49, 159.57, 149.75, 141.44, 140.91, 137.89, 128.73, 127.74, 126.14, 110.03, 64.03, 63.73, 59.28, 54.73, 49.53, 49.31, 47.47, 43.51, 42.07, 40.65, 38.86, 38.31, 37.51, 35.61, 33.85, 33.23, 31.93, 29.79, 29.71, 29.66, 29.37, 28.88, 27.43, 25.02, 23.58, 22.70, 21.49, 21.35, 19.23, 18.90, 16.35, 15.70, 14.41, 14.13.
- HRMS (ESI+): m/z calculated for C40H60N5O2 [M + H]+: 642.4742, found: 642.470.
- 1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid derivative-3. (20). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 mL) in an ultrasonic bath, then triethylamine (0.024 mL, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 mL, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (21 °C) for 3 h, after which 2-amino-2-methyl-1-propanol (0.162 mL, 10 equiv., 1.7 mmol) in 0.5 mL of toluene was added to the reaction mixture. Then, it was heated at 110 °C for 3.5 h. At the end of the reaction, the solvent was distilled in a rotary evaporator. The remainder was chromatographed with silica gel in a column. During elution of the column with ethyl acetate, a colorless substance (compound 20) with a melting point of 176–178 °C was isolated. The yield was 96 mg (84%).
- 1H NMR (400 MHz, CDCl3): δ 7.30 (dd, J = 8.2, 6.3 Hz, 3H), 7.23–7.16 (m, 2H), 6.45 (s, 1H), 5.75 (q, J = 7.0 Hz, 1H), 5.25 (s, 1H), 5.05 (s, 1H), 4.65 (d, J = 2.3 Hz, 1H), 4.60 (t, J = 1.9 Hz, 1H), 3.55 (s, 2H), 2.92 (d, J = 15.2 Hz, 1H), 2.67 (dt, J = 13.4, 3.4 Hz, 1H), 2.44 (ddd, J = 20.1, 14.0, 9.6 Hz, 2H), 2.17 (d, J = 15.2 Hz, 1H), 2.08 (s, 0H), 2.02 (d, J = 7.0 Hz, 3H), 1.99–1.90 (m, 1H), 1.75 (td, J = 14.7, 14.1, 8.5 Hz, 2H), 1.68 (s, 3H), 1.62 (d, J = 11.7 Hz, 1H), 1.62–1.40 (m, 5H), 1.39–1.24 (m, 7H), 1.23 (d, J = 1.9 Hz, 6H), 1.09 (d, J = 22.1 Hz, 8H), 0.96 (s, 3H), 0.93–0.78 (m, 7H).
- 13C NMR (101 MHz, CDCl3): δ 158.88, 149.68, 141.54, 140.91, 128.70, 127.69, 126.18, 109.99, 72.29, 63.89, 59.30, 54.78, 54.76, 49.51, 49.35, 47.37, 42.04, 40.62, 38.89, 38.36, 37.52, 35.61, 33.85, 33.24, 29.84, 29.55, 28.89, 27.40, 25.26, 25.22, 25.04, 23.60, 21.50, 21.35, 19.29, 18.89, 16.33, 15.74, 14.45.
- HRMS (ESI+): m/z calculated for C42H64N5O2 [M + H]+: 670.5055, found: 670.5093.
- 1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28- oic Acid derivative-4. (21). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 mL) in an ultrasonic bath, then triethylamine (0.024 mL, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 mL, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (22 °C) for 3 h, after which 3-amino-1,2-propanediol (0.1317 mL, 10 equiv., 1.7 mmol) was added to the reaction mixture. Then, it was heated at 110 °C for 2 h. At the end of the reaction, the solvent was distilled in a rotary evaporator. The remainder was chromatographed with silica gel in a column. Elution of the column with a mixture of ethyl acetate and methanol (10:1) isolated a colorless substance (compound 21) with a melting point of 182–185 °C. The yield was 89 mg (78%).
- 1H NMR (400 MHz, CDCl3): δ 7.35–7.23 (m, 3H), 7.18 (td, J = 7.3, 1.9 Hz, 2H), 5.95 (s, 1H), 5.76 (q, J = 7.3 Hz, 1H), 5.35–5.18 (m, 1H), 4.68 (s, 1H), 4.64–4.57 (m, 1H), 4.12 (q, J = 7.2 Hz, 1H), 3.82–3.70 (m, 1H), 3.70–3.58 (m, 1H), 3.59–3.39 (m, 2H), 3.26 (dt, J = 16.1, 5.3 Hz, 2H), 2.92 (dd, J = 20.5, 15.2 Hz, 1H), 2.69–2.59 (m, 1H), 2.54 (ddq, J = 16.3, 11.8, 6.9, 5.2 Hz, 1H), 2.42 (dd, J = 12.0, 8.1 Hz, 1H), 2.24–2.10 (m, 1H), 2.06 (d, J = 10.6 Hz, 1H), 2.01 (d, J = 6.9 Hz, 3H), 1.97–1.71 (m, 1H), 1.69 (s, 3H), 1.67–1.30 (m, 5H), 1.30–1.19 (m, 7H), 1.12 (d, J = 8.0 Hz, 4H), 1.03–0.89 (m, 6H), 0.89–0.80 (m, 1H), 0.78 (s, 2H).
- 13C NMR (101 MHz, CDCl3): δ 159.68, 149.68, 141.37, 140.94, 138.02, 128.76, 128.68, 127.79, 126.18, 126.08, 110.09, 71.98, 63.85, 63.83, 63.30, 59.27, 54.71, 49.45, 49.31, 47.39, 42.50, 42.06, 40.63, 38.86, 38.29, 37.50, 35.72, 33.86, 33.22, 30.84, 29.71, 28.85, 27.41, 25.62, 25.01, 23.63, 23.52, 21.52, 21.34, 19.22, 18.89, 16.40, 15.68, 14.43, 14.20.
- HRMS (ESI+): m/z calculated for C41H62N5O3 [M + H]+: 672.4847, found: 672.4858.
- 1′-((S)-1-Phenylethyl)-1H′-lup-2-eno-[2,3-d]-[1,2,3]-triazole-28-oic Acid derivative-5. (22). Compound 3 (100 mg, 1 equiv., 0.17 mmol) was dissolved in toluene (3 mL) in an ultrasonic bath, then triethylamine (0.024 mL, 1 equiv., 0.17 mmol) and diphenylphosphoryl azide (0.039 mL, 1 equiv., 0.17 mmol) were added. The reaction was carried out at room temperature (22 °C) for 3 h, after which 2-(2-aminoethoxy)ethanol (0.1705 mL, 10 equiv., 1.7 mmol) in 0.5 mL of toluene was added to the reaction mixture. Then, it was heated at 110 °C for 2 h. At the end of the reaction, the solvent was distilled in a rotary evaporator. The remainder was chromatographed with silica gel in a column. Elution of the column with a mixture of ethyl acetate and methanol (20:1) isolated a colorless substance (compound 22) with a melting point of 165–167 °C. The yield was 76 mg (65%).
- 1H NMR (400 MHz, CDCl3): δ 7.37–7.23 (m, 3H), 7.22–7.16 (m, 2H), 5.74 (q, J = 7.0 Hz, 1H), 5.49 (s, 1H), 4.76–4.67 (m, 2H), 4.62 (t, J = 1.9 Hz, 1H), 3.83–3.68 (m, 3H), 3.68–3.60 (m, 3H), 3.60–3.51 (m, 4H), 3.36 (d, J = 5.1 Hz, 2H), 2.93 (d, J = 15.2 Hz, 1H), 2.71–2.61 (m, 1H), 2.49 (ddt, J = 20.7, 12.4, 6.5 Hz, 2H), 2.16 (d, J = 15.2 Hz, 1H), 2.06 (d, J = 8.7 Hz, 0H), 2.01 (d, J = 7.0 Hz, 3H), 1.76 (d, J = 11.2 Hz, 2H), 1.70 (s, 3H), 1.68–1.28 (m, 7H), 1.27 (s, 7H), 1.19–1.02 (m, 7H), 0.96 (s, 3H), 0.90–0.82 (m, 5H), 0.80 (s, 3H).
- 13C NMR (101 MHz, CDCl3): δ 158.08, 149.69, 141.63, 140.96, 137.71, 128.69, 127.65, 126.19, 110.06, 72.36, 70.81, 63.60, 61.66, 61.14, 59.30, 54.74, 49.50, 49.27, 47.52, 42.07, 41.35, 40.63, 40.18, 38.87, 38.32, 37.63, 35.71, 33.83, 33.25, 29.81, 29.77, 28.88, 27.39, 25.03, 23.67, 22.63, 21.46, 21.35, 19.26, 18.91, 16.32, 15.71, 14.42.
- HRMS (ESI+): m/z calculated for C42H64N5O3 [M + H]+: 686.5004, found: 686.4954.
3.3. In Vitro Biological Assays
3.3.1. Antimicrobial Activity
3.3.2. Cytotoxic Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Myagchilov, A.V. Triterpenoids of Inflorescences of Synurus deltoides (Asteraceae) from Primorskii Krai. Dokl. Biol. Sci. 2023, 512, 333–335. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, E.B.; Luo, J.H.; Gu, W.J.; Tao, M.; Geng, H. Terpenoids from the Petroleum Ether Extract of Artemisia argyi. Chem. Nat. Compd. 2024, 60, 68–71. [Google Scholar] [CrossRef]
- Babekov, A.U.; Yuldasheva, N.M.; Ismailova, A.M.; Komilov, B.D.; Turgunov, K.K.; Tashkhodzhaev, B.; Eshbakova, K.A. Terpenoids from the Plant Ferula ferganensis. Chem. Nat. Compd. 2022, 58, 967–969. [Google Scholar] [CrossRef]
- Fan, Y.Z.; Tian, C.; Tong, S.Y.; Liu, Q.; Xu, F.; Shi, B.-B.; Ai, H.-L.; Liu, J.-K. The antifungal properties of terpenoids from the endophytic fungus Bipolaris eleusines. Nat. Prod. Bioprospect. 2023, 13, 43. [Google Scholar] [CrossRef]
- Tholl, D. Biosynthesis and Biological Functions of Terpenoids in Plants. Advances in Biochemical Engineering. In Biotechnology of Isoprenoids. Advances in Biochemical Engineering/Biotechnology; Springer International: Cham, Switzerland, 2015; pp. 63–106. [Google Scholar] [CrossRef]
- Castañeta, G.; Cifuentes, N.; Sepulveda, B.; Bárcenas-Pérez, D.; Cheel, J.; Areche, C. Untargeted Metabolomics by Using UHPLC–ESI–MS/MS of an Extract Obtained with Ethyl Lactate Green Solvent from Salvia rosmarinus. Separations 2022, 9, 327. [Google Scholar] [CrossRef]
- Ivakhnov, A.D.; Selivanova, N.V.; Krasikova, A.A.; Stavrianidi, A.N.; Gusakova, M.A.; Bogolitsyn, K.G. Extraction of Terpenes of Common Juniper Greenery Under Sub- and Supercritical Conditions. Russ. J. Phys. Chem. B 2022, 16, 1354–1360. [Google Scholar] [CrossRef]
- Venkatesh, K.S.; Krishnamoorthi, S.R.; Palani, N.S.; Thirumal, V.; Jose, S.P.; Wang, F.-M.; Ilangovan, R. Facile one step synthesis of novel TiO2 nanocoral by sol–gel method using Aloe vera plant extract. Indian J. Phys. 2015, 89, 445–452. [Google Scholar] [CrossRef]
- Pirsaheb, M.; Gholami, T.; Seifi, H.; Dawi, E.A.; Said, E.A.; Hamoody, A.-H.M.; Altimari, U.S.; Salavati-Niasari, M. Green synthesis of nanomaterials by using plant extracts as reducing and capping agents. Environ. Sci. Pollut. Res. 2024, 31, 24768–24787. [Google Scholar] [CrossRef]
- Cao, Y.; Xian, M. Recent Advances in Microbial Production of Terpenoids from Biomass-derived Feedstocks. Chem. Res. Chin. Univ. 2024, 40, 20–28. [Google Scholar] [CrossRef]
- Liu, J.K. Natural products in cosmetics. Nat. Prod. Bioprospect. 2022, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Gammatantrawet, N.; Nguyễn, C.T.; Susawaengsup, C.; Ramli, A.N.M.; Tongkoom, K.; Chatsungnoen, T.; Dangtungee, R.; Bhuyar, P. Phytochemistry of Medicinal Herbs Belongs to Asclepiadaceae Family for Therapeutic Applications: A Critical Review. Mol. Biotechnol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.G.; Zhao, T.T.; Lu, N.; Yang, Y.A.; Zhu, H.L. Research Progress of Glycyrrhizic Acid on Antiviral Activity. Mol. Biotechnol. 2019, 19, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Yuan, S.; Li, L.; Zheng, J.; Zhao, D.; Wang, C.; Wang, H.; Liu, X.; Liu, J. Application of Terpenoid Compounds in Food and Pharmaceutical Products. Fermentation 2023, 9, 119. [Google Scholar] [CrossRef]
- Caputi, L.; Aprea, E. Use of terpenoids as natural flavouring compounds in food industry. Recent Patents Food Nutr. Agric. 2011, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lipeeva, A.V.; Dolgikh, M.P.; Tolstikova, T.G.; Shults, E.E. A Study of Plant Coumarins. 18. Conjugates of Coumarins with Lupane Triterpenoids and 1,2,3-Triazoles: Synthesis and Anti-Inflammatory Activity. Russ. J. Bioorg. Chem. 2020, 46, 125–132. [Google Scholar] [CrossRef]
- Ticona, L.A.A.; Serban, A.M.; Madorrán, M.J.P.; Fernández-Grifol, M.; Sánchez, Á.R. Anti-Melanogenic and Anti-Inflammatory Activities of Triterpenoids from Jatropha macrantha. Rev. Bras. Farmacogn. 2021, 31, 40–50. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Petrova, A.V.; Kazakova, O.B. Antiviral potency of lupane and oleanane alkynyl-derivatives against human cytomegalovirus and papillomavirus. J. Antibiot. 2024, 77, 50–56. [Google Scholar] [CrossRef]
- Smirnova, I.; Petrova, A.; Lobov, A.; Minnibaeva, E.; Phoung, T.T.T.; Van, L.T.; Khine, M.M.; Esaulkova, I.; Slita, A.; Zarubaev, V.; et al. Azepanodipterocarpol is potential candidate for inhibits influenza H1N1 type among other lupane, oleanane, and dammarane A-ring amino-triterpenoids. J. Antibiot. 2022, 75, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Ayodele, O.A.; Awotuya, I.O.; Taiwo, B.J.; Osungunna, O.M.; Vuyisa, M.; Kasim, S.L. Two New Triterpenoids from the Leaf of Ficus vogelii and Their Antibacterial Activities. Chem. Afr. 2024, 7, 63–70. [Google Scholar] [CrossRef]
- Salimova, E.V.; Magafurova, A.A.; Tretyakova, E.V.; Kukovinets, O.S.; Parfenova, L.V. Indole Derivatives of Fusidane Triterpenoids: Synthesis and the Antibacterial Activity. Chem. Heterocycl. Compd. 2020, 56, 800–804. [Google Scholar] [CrossRef]
- Chinthanom, P.; Vichai, V.; Dokladda, K.; Sappan, M.; Thongpanchang, C.; Isaka, M. Semisynthetic modifications of antitubercular lanostane triterpenoids from Ganoderma. J. Antibiot. 2021, 74, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Sajid, A.R.; Javeed, A.; Aslam, M.; Ahsan, T.; Hussain, D.; Mateen, A.; Li, X.; Qin, P.; Ji, M. Antioxidant, antifungal, and aphicidal activity of the triterpenoids spinasterol and 22,23-dihydrospinasterol from leaves of Citrullus colocynthis L. Sci. Rep. 2022, 12, 4910. [Google Scholar] [CrossRef] [PubMed]
- Hang, T.X.H.; Jarupinthusophon, S.; Hairani, R.; Nguyen, V.-K.; Chavasiri, W. Cycloartane-type triterpenoids from the leaves of Sandoricum koetjape and their efficacy on α-glucosidase inhibition activity. J. Nat. Med. 2024, 78, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Desmiaty, Y.; Hanafi, M.; Saputri, F.C.; Elya, B.; Rifai, E.A.; Syahdi, R.R. Two triterpenoids from Rubus fraxinifolius leaves and their tyrosinase and elastase inhibitory activities. Sci. Rep. 2021, 11, 20452. [Google Scholar] [CrossRef] [PubMed]
- Aly, S.H.; Elbadry, A.M.M.; Doghish, A.S.; El-Nashar, H.A.S. Unveiling the pharmacological potential of plant triterpenoids in breast cancer management: An updated review. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024. [Google Scholar] [CrossRef]
- Irungu, B.N.; Nyangi, M.; Ndombera, F.T. Anticancer potential of four triterpenoids against NCI-60 human tumor cell lines. Beni-Suef Univ. J. Basic Appl. Sci. 2024, 13, 50. [Google Scholar] [CrossRef]
- Nazarov, M.A.; Tolmacheva, I.A.; Gagarskih, O.N.; Grishko, V.V. Synthesis and cytotoxic activity of triterpenoids with N,O-heterocyclic fragments based on 2-formyl-1(2)-ene derivative of methyldihydrobetulonate. Chem. Pap. 2023, 77, 2219–2227. [Google Scholar] [CrossRef]
- Giniyatullina, G.V. Synthesis and Antitumor Activity of O- and N-Propylamino-Derivatives of Betulin. Chem. Nat. Compd. 2022, 58, 684–692. [Google Scholar] [CrossRef]
- Govdi, A.I.; Sorokina, I.V.; Baev, D.S.; Bryzgalov, A.O.; Tolstikova, T.G.; Tolstikov, G.A.; Vasilevsky, S.F. Acetylenic derivatives of betulonic acid amide as a new type of compounds possessing spasmolytic activity. Russ. Chem. Bull. 2015, 64, 1327–1334. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Medvedeva, N.I.; Lopatina, T.V.; Apryshko, G.N.; Pugacheva, R.B.; Yavorskaya, N.P.; Golubeva, I.S.; Tolstikov, G.A. Synthesis and the antineoplastic activity of imidazolides of betulonic acid. Russ. J. Bioorg. Chem. 2015, 41, 305–314. [Google Scholar] [CrossRef]
- Giniyatullina, G.V.; Kazakova, O.B. Synthesis and Cytotoxicity of Lupane Mono- and Bis-Piperazinylamides. Chem. Nat. Compd. 2021, 57, 698–705. [Google Scholar] [CrossRef]
- Chue, K.T.; Chang, M.S.; Ten, L.N. Synthesis and antibacterial activity of betulonic acid amides with piperazine derivatives. Chem. Nat. Compd. 2011, 47, 759–763. [Google Scholar] [CrossRef]
- Sorokina, I.V.; Baev, D.S.; Zhukova, N.A.; Tolstikova, T.G.; Antimonova, A.N.; Petrenko, N.I.; Shults, E.E.; Grigor’ev, I.A. Hepatoprotective activity of betulonic acid amides containing piperidine or pyrrolidine nitroxide moieties. Russ. J. Bioorg. Chem. 2013, 39, 668–670. [Google Scholar] [CrossRef]
- Sunitha, V.; Kumar, A.K.; Saikrishna, B. Synthesis of Novel Benzofuran Based 1,2,3-Triazoles, Their Antimicrobial and Cytotoxic Activities, and Molecular Docking Studies. Russ. J. Gen. Chem. 2022, 92, 1348–1359. [Google Scholar] [CrossRef]
- Prasad, C.; Nagesh, P.; Kishan, C.; Krishna, V.M.; Balaswamy, A.; Manga, V.; Prashanth, B.; Aparna, Y. Synthesis, Antimicrobial Evaluation, and In Silico Molecular Docking Studies of Chalcone-Based 1,2,3-Triazoles. Russ. J. Gen. Chem. 2023, 93, 1162–1170. [Google Scholar] [CrossRef]
- Kaushik, C.P.; Chahal, M. Synthesis and antibacterial activity of benzothiazole and benzoxazole-appended substituted 1,2,3-triazoles. J. Chem. Sci. 2020, 132, 142. [Google Scholar] [CrossRef]
- Ashram, M.; Habashneh, A.Y.; Bardaweel, S.; Taha, M.O. A Click Synthesis, Molecular Docking and Biological Evaluation of 1,2,3-triazoles-benzoxazepine hybrid as potential anticancer agents. Med. Chem. Res. 2023, 32, 271–287. [Google Scholar] [CrossRef]
- Seck, I.; Ciss, I.; Diédhiou, A.; Baldé, M.; Ka, S.; Ba, L.A.; Ndoye, F.S.; Figadère, B.; Seon-Meniel, B.; Gomez, G.; et al. 1,2,3-triazenes and 1,2,3-triazoles as antileishmanial, antitrypanosomal, and antiplasmodial agents. Med. Chem. Res. 2023, 32, 158–164. [Google Scholar] [CrossRef]
- Kaushik, C.P.; Chahal, M. Synthesis, antimalarial and antioxidant activity of coumarin appended 1,4-disubstituted 1,2,3-triazoles. Monatsh. Chem. 2021, 152, 1001–1012. [Google Scholar] [CrossRef]
- Denisov, M.S. Synthesis of 2-Heteroylidene Triterpenoids: Complex Formation with Palladium and in vitro Cytotoxic Activity. Russ. J. Gen. Chem. 2023, 93, 37–42. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Salikhova, T.I.; Abdullin, T.I.; Eva, L.R.G.; Khozyainova, S.A.; Mironov, V.F. Synthesis, Anticancer, and Antibacterial Activity of Betulinic and Betulonic Acid C-28-Triphenylphosphonium Conjugates with Variable Alkyl Linker Length. Anticancer Agents Med. Chem. 2020, 20, 286–300. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Mustafin, A.G.; Babkov, D.A.; Sokolova, E.V.; Spasov, A.A. Evaluation of Cytotoxicity and α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids. Molecules 2020, 25, 4833. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Govdi, A.I.; Shults, E.E.; Shakirov, M.M.; Sorokina, I.V.; Tolstikova, T.G.; Baev, D.S.; Tolstikov, G.A.; Alabugin, I.V. Efficient synthesis of the first betulonic acid-acetylene hybrids and their hepatoprotective and anti-inflammatory activity. Bioorg. Med. Chem. 2009, 17, 5164–5169. [Google Scholar] [CrossRef]
- Yang, S.-J.; Liu, M.-C.; Zhao, Q.; Hu, D.-Y.; Xue, W.; Yang, S. Synthesis and biological evaluation of betulonic acid derivatives as antitumor agents. Eur. J. Med. Chem. 2015, 96, 58–65. [Google Scholar] [CrossRef]
- Prakash, R.; Opsomer, T.; Dehaen, W. Triazolization of Enolizable Ketones with Primary Amines: A General Strategy toward Multifunctional 1,2,3-Triazoles. Chem. Rec. 2021, 21, 376–385. [Google Scholar] [CrossRef]
- Tulegenova, A.U. State Pharmacopoeia of the Republic of Kazakhstan; Publishing House “Zhibek Zholy”: Almaty, Kazakhstan, 2015; Volume I, 720p. [Google Scholar]
- Mironov, A.N. (Ed.) Guidelines for Conducting Preclinical Studies of Drugs—Part 1; GRIF-K: Moscow, Russia, 2012; p. 206. [Google Scholar]
- Meyer, B.N.; Ferrigni, N.R.; Putnam, J.E.; Jacobsen, L.B.; Nicholsand, D.E.; McLaughlin, J.L. Brine Shrimp: A Convenient General Bioassay for Active Plant Constituents. Planta Medica 1982, 45, 31–34. [Google Scholar] [CrossRef]
- McLaughlin, L. Crown Gall Tumors on Potato Discs and Brine Shrimp Lethality: Two Simple Bioassays for Higher Plant Screening and Fractionation. Methods Plant Boichem. 1991, 6, 1–32. [Google Scholar]
- Genet, C.; Strehle, A.; Schmidt, C.; Boudjelal, G.; Lobstein, A.; Schoonjans, K.; Souchet, M.; Auwerx, J.; Saladin, R.; Wagner, A. Structure-Activity Relationship Study of Betulinic Acid, A Novel and Selective TGR5 Agonist, and Its Synthetic Derivatives: Potential Impact in Diabetes. J. Med. Chem. 2010, 53, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Samoshina, N.F.; Denisenko, M.V.; Denisenko, V.A.; Uvarova, N.I. Synthesis of glycosides of lupane-type triterpene acids. Chem. Nat. Compd. 2003, 39, 575–582. [Google Scholar] [CrossRef]
- Barthel, A.; Stark, S.; Csuk, R. Oxidative transformations of betulinol. Tetrahedron 2008, 64, 9225–9229. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Staphylococcus aureus ATCC 6538 | Bacillus subtilis ATCC 6633 | Escherichia coli ATCC 25922 | Pseudomonas aeruginosa ATCC 27853 | Candida albicans ATCC 10231 |
|---|---|---|---|---|---|
| Betulonic acid 2 | 50 | 25 | - | - | 50 |
| 3 | 25 | 50 | 12.5 | - | 50 |
| 4 | 25 | 50 | 50 | - | - |
| 5 | 50 | 25 | 25 | - | 50 |
| 6 | 6.3 | - | 25 | - | - |
| 7 | 25 | 25 | 6.3 | 50 | 25 |
| 8 | 25 | 50 | - | - | - |
| 9 | 50 | - | - | - | - |
| 10 | 50 | 50 | - | - | - |
| 13 | 25 | - | 50 | - | - |
| 14 | 50 | - | - | - | - |
| 15 | 12.5 | 50 | 25 | - | - |
| 16 | 50 | - | 50 | - | - |
| 17 | 50 | 50 | 50 | - | - |
| 18 | 25 | 50 | 12.5 | - | - |
| 19 | 25 | 25 | 50 | - | 25 |
| 20 | 50 | 25 | 25 | - | 50 |
| 21 | 25 | - | 50 | - | - |
| 22 | 50 | - | - | - | - |
| Ceftriaxone | 6.3 | 12.5 | 6.3 | 6.3 | - |
| Benzylpenicillin sodium salt | 12.5 | 25 | 25 | 50 | - |
| Nystatin | - | - | - | - | 12.5 |
| Compounds | LD50, µg/mL |
|---|---|
| Betulonic acid 2 | 88.7 |
| 3 | 82.5 |
| 4 | 105.7 |
| 5 | - |
| 6 | 82.8 |
| 7 | 89.4 |
| 8 | 107.2 |
| 9 | 95.6 |
| 10 | 79.4 |
| 13 | 97.3 |
| 14 | 90.5 |
| 15 | 105.9 |
| 16 | 85.1 |
| 17 | - |
| 18 | 66.2 |
| 19 | 74.1 |
| 20 | 63.5 |
| 21 | - |
| 22 | 69.0 |
| Comparison drug: dactinomycin (actinomycin D) | 46.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalmakhanbetova, R.I.; Mukusheva, G.K.; Abdugalimov, A.S.; Zhumagalieva, Z.Z.; Dehaen, W.; Anthonissen, S.; Suleimen, Y.M.; Seidakhmetova, R.B. Synthesis and Investigation of Biological Activity of New Betulonic Acid Derivatives Containing 1,2,3-Triazole Fragments. Molecules 2024, 29, 3149. https://doi.org/10.3390/molecules29133149
Jalmakhanbetova RI, Mukusheva GK, Abdugalimov AS, Zhumagalieva ZZ, Dehaen W, Anthonissen S, Suleimen YM, Seidakhmetova RB. Synthesis and Investigation of Biological Activity of New Betulonic Acid Derivatives Containing 1,2,3-Triazole Fragments. Molecules. 2024; 29(13):3149. https://doi.org/10.3390/molecules29133149
Chicago/Turabian StyleJalmakhanbetova, Roza I., Gulim K. Mukusheva, Alisher Sh. Abdugalimov, Zharkyn Zh. Zhumagalieva, Wim Dehaen, Stijn Anthonissen, Yerlan M. Suleimen, and Roza B. Seidakhmetova. 2024. "Synthesis and Investigation of Biological Activity of New Betulonic Acid Derivatives Containing 1,2,3-Triazole Fragments" Molecules 29, no. 13: 3149. https://doi.org/10.3390/molecules29133149
APA StyleJalmakhanbetova, R. I., Mukusheva, G. K., Abdugalimov, A. S., Zhumagalieva, Z. Z., Dehaen, W., Anthonissen, S., Suleimen, Y. M., & Seidakhmetova, R. B. (2024). Synthesis and Investigation of Biological Activity of New Betulonic Acid Derivatives Containing 1,2,3-Triazole Fragments. Molecules, 29(13), 3149. https://doi.org/10.3390/molecules29133149