Abstract
Monoterpenes and meroterpenes are two large classes of isoprene-based molecules produced by terrestrial plants and unicellular organisms as diverse secondary metabolites. The global rising incidence of cancer has led to a renewed interest in natural products. These monoterpenes and meroterpenes represent a novel source of molecular scaffolds that can serve as medicinal chemistry platforms for the development of potential preclinical leads. Furthermore, some of these natural products are either abundant, or their synthetic strategies are scalable as it will be indicated here, facilitating their derivatization to expand their scope in drug discovery. This review is a collection of representative updates (from 2016–2023) in biologically active monoterpene and meroterpenoid natural products and focuses on the recent findings of the pharmacological potential of these bioactive compounds as well as the newly developed synthetic strategies employed to access them. Particular emphasis will be placed on the anticancer and antioxidant potential of these compounds in order to raise knowledge for further investigations into the development of potential anti-cancer therapeutics. The mounting experimental evidence from various research groups across the globe regarding the use of these natural products at pre-clinical levels, renders them a fast-track research area worth of attention.
1. Background
1.1. History
Throughout history, herbal medicines have been used to treat various diseases in humans. The earliest documented incidence of patient treatment using these methods dates back to approximately 2600 BC in Ancient Egypt [1]. These medicinal practices utilized a variety of plant extracts to treat various maladies ranging from simple infections to cardiovascular diseases [1]. These naturally derived compounds are still utilized today in a medicinal context as molecular scaffolds or starting points to build new drugs and molecular probes.
In the past 30 years approximately 50% of all approved drugs for cancer treatments were either natural products or derived from a natural product scaffold [2]. Although modern medicine has overcome some of the historical reliance of physicians on natural therapeutic agents, it is estimated that 80% of people worldwide rely on natural-derived medicinal agents as some form of primary care [3]. These compounds still provide an invaluable opportunity to further explore human disease and treatment. Furthermore, as of 2005 less than 10% of the biodiversity on this planet had been biologically evaluated for their therapeutic potential [4] this underscoring the vast potential of these resources. Various recent reviews have highlighted the importance of natural product research in anticancer and antibiotic research for similar natural products not included here [5,6,7,8].
1.2. Classification
Terpenes are the largest classification of biologically produced organic molecules accounting for approximately one third of all characterized natural products. These compounds are structurally classified as combinations of monomeric isoprene units because are likely to follow the isoprene rule. In 1953, this rule was proposed to rationalize the biogenesis of terpenoid compounds. The isoprene rule not only clarified the biochemical origin of this class of molecules but also serves as the basis for their classification [9].
The subgroups of terpenes are defined by the number of isoprene units present in the molecule. Because all terpenes are composed of a number of isoprene units, they will share the hydrocarbon formula (C5H8)n. Activated isoprenoid precursors, isopentyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), polymerize in a variety of combinations through enzymatically catalyzed coordination-insertion, coordinative chain growth and radical ionic mechanisms. These polymerizations afford higher order terpenoid compounds (Figure 1). The various combinations of IPP and DMAPP as well as the potential for rearrangement mechanisms at each step yields a high degree of structural diversity in this class of molecules.
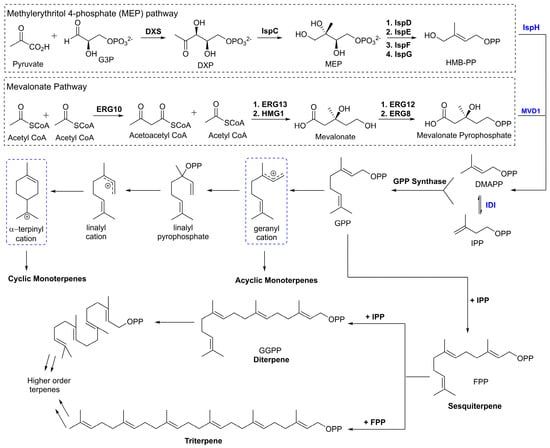
Figure 1.
Simplified detailing the convergent biosynthesis of acyclic, monocyclic, and bicyclic monoterpenes highlighting the mevalonate and MEP pathways.
Monoterpenes are the smallest molecules of the isoprenoids with a conserved hydrocarbon formula of C10H16. There are more than 400 known chemical structures that have been classified as monoterpenes [10]. There have been many monoterpenes isolated and elucidated from natural sources that contain unique and intricate structures each having their own properties in biological systems. These molecules can further be classified as a-, mono-, or bicyclic systems.
1.3. Biosynthesis of Monoterpenes
The methylerythritol 4-phosphate (MEP) pathway is a series of enzymatically mediated steps that was identified in bacteria that yields both of these activated derivatives as shown in Figure 1 [11]. The first committed step of this pathway is the decarboxylation reaction between glyceraldyhyde-3-phosphate (G3P) and pyruvate catalyzed by the enzyme deoxy-d-xylulose-5-phosphate synthase (DXS) [12]. This step results in the production of 1-deoxy-d-xylulose 5-phosphate (DXP). Subsequent enzymatic reactions will produce (E)-4-Hydroxy-3-methyl-but-2-enyl pyrophosphate (HMBPP) which then undergoes a catalytic dehydration to leave DMAPP and IPP. The mevalonate (MVA) pathway begins with the condensation of three molecules of acetyl CoA to produce mevalonate. Mevalonate is then phosphorylated in two catalytic steps to give mevalonate pyrophosphate. This compound is then decarboxylated to provide IPP which can be isomerized further to produce DMAPP [13].
Following the biosynthesis of the activated isoprene units, a key polymerization reaction takes place between DMAPP and IPP yielding geranyl pyrophosphate (GPP). The enzyme responsible for this reaction, GPP synthase (GPPS), is a highly regulated enzyme that is responsible for the first committed step towards production of all monoterpene compounds [14]. Through subsequent catalyzed reactions two important intermediates emerge as the starting point for further derivatization of monoterpenes. The first, geranyl cation, serves as the substrate for production of most acyclic monoterpenes. From this cationic intermediate a variety of reactions can occur leading to the monoterpenes. The second transition state, α-terpinyl cation, is thought to be the branch point for the synthesis of mono- and bi-cyclic monoterpenes [15].
1.4. Meroterpenes
Meroterpenoids are a large group of compounds characterized as being biosynthetically mixed, hybrid molecules. These compounds typically involve the combination of a terpene with polyketides, alkaloids, phenols, or amino acids (Figure 2). The large potential for combination affords this class of molecules with a high degree of structural diversity. Recently these compounds have been gaining attention from chemists and biologists alike for their complex scaffolds and intriguing bioactivities. While many of these compounds have been isolated and identified there is much more to be gleaned from these molecules. Biological evaluations of these compounds have shown their aptitude as potential therapeutic compounds.
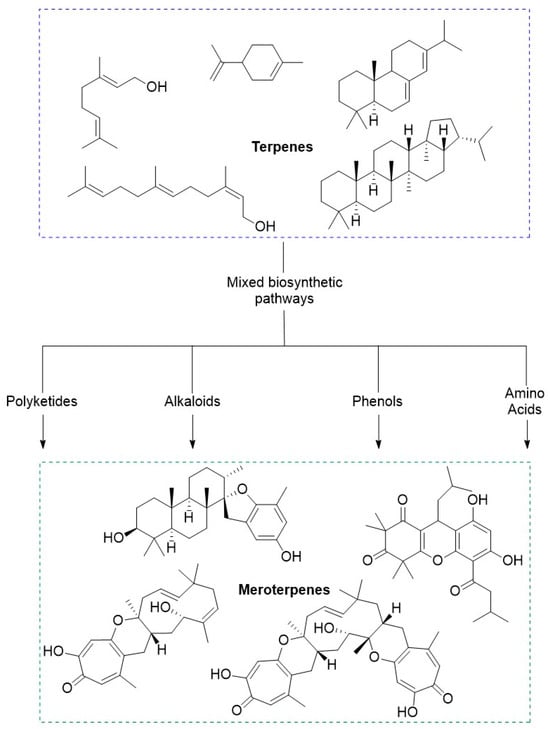
Figure 2.
Conceptual depiction of the relationship and biochemical origin of meroterpenoids.
2. Biological Activities of Monoterpenes
2.1. Acyclic Monoterpenes
Terpenes have been known to be responsible for crucial roles in their parent organism’s biochemical regulation and response to external stimuli. This diverse class of compounds are involved in many biological processes ranging from signaling, hormonal processes, and as biochemical defense mechanisms. In medicine, these compounds have been researched for their biological relevance as potential natural product therapeutics. These natural products have demonstrated antiviral, antifungal, antimicrobial, analgesic, anti-inflammatory, antioxidant, and antitumor properties [16]. This wide array of beneficial biological attributes increases the interest placed on these compounds by medicinal chemists and biologists.
The acyclic monoterpene subgroup is quite diverse and only representatives are shown in Figure 3 (1a-6). Currently, a few of these members have been identified for having antioxidant or anticancer properties, and it remains an active field with a promising future. As it is highlighted here, many of these identified compounds show both antioxidant and anti-cancer activity, but recent representatives are described.
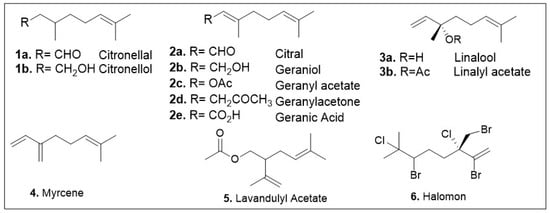
Figure 3.
Structures of acyclic monoterpenes that have been identified for their antioxidant or anticancer activities.
3,7-Dimethyl-2,6-octadienal or citral (2a, Figure 3), is an aldehyde-containing monoterpenoid that has been isolated as a volatile compound in essential oils from many different species disbursed across the kingdoms Eukaryota and Plantae [17]. Studies of essential oils containing this natural product have identified citral as having anticancer and antioxidant properties. A recent computational study of the effect of essential oils from Cymbopogon or lemongrass species on the protein network implicated citral as a chemo-preventative compound [18]. Another study utilized proteomic and molecular approaches to show that the essential oil from Ocimum × africanum induced apoptosis of human gastric cancer cell line by causing endoplasmic reticulum stress and impairment of the formation of the ribosome. GC-MS analysis of the essential oil showed that citral is a major component comprising 19% of this oils mass [19]. The potential of citral as a medicinal agent was demonstrated using citral-loaded micelles (CLM) as a treatment method for in vitro breast cancer models. This treatment method was tested in multiple breast cancer cell lines, and all showed IC50 values of 145, 152, 139, and 130 μM for MCF7, MDA-MB-321, MDA-MB-468, and HCC-1806 respectively [20]. Concerns with these findings are the high concentrations used as well as the lack of data evaluating the citrus compounds cytotoxicity to normal cell lines, therefore further validations studies are warranted.
A recent study conducted in murine models showed this monoterpene’s ability to stave off methotrexate induced lung injury. Treatment of damaged lung tissue with citral resulted in decreased congestion and hemorrhage while no necrotic tissue, alveolar dilation, or interstitial fibrosis was noted in comparison with the methotrexate-treated control [21]. The combined studies suggest citral can mitigate inflammation.
In addition, antioxidant properties of citral were reported in 2021 [22]. The study aimed to determine the mode of action of citral using computational and experimental studies including molecular biology assays such as MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, a colorimetric assay for assessing cell metabolic activity, and Chorioallantoic Membrane Assay (CAM) assay, which assess the antiangiogenic properties of the compounds. The studies indicated that citral might affect PPARγ and vascular endothelial growth factor receptors (VEGFR-1 and VEGFR-2). Furthermore, the results of the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assays conducted by this research team confirm this compound’s antioxidant properties [22].
Citronellal (1a, Figure 3), a prominent constituent of essential oils, has been reported to display anticancer and antioxidant properties [23]. In addition, citronellal was identified as a promising agent against triple negative breast cancer cell lines. Ho showed that a crude extracts of Citrus hystrix DC, citronellol, and citronellal significantly reduced cell proliferation and migration by inducing cell cycle arrest in the MDA-MB-231 cell line [23]. The IC50 values of the aforementioned extracts were in the millimolar range while those of compounds citronellol and citronellal were 1.16 and 1.41 nM respectively. However, the cytotoxicity of these compounds on normal tissue is of concern as the IC50 values of citronellol and citronellal were shown to be 1.96 and 1.81 nM respectively when tested against human monocyte derived macrophages. Independent antiproliferative studies of citronellal using Sulforhodamine B (SRB), MTT, and NRU (neutral red uptake) antiproliferative assays indicate its potential against a triple-negative breast cancer cell model (MDA-MB-231) [24]. The study also conducted in vitro experiments to evaluate citronellal’s activity against LOX-5 (arachidonate 5-lipoxygenase), finding the IC50 value to be in the low micromolar range [24]. Furthermore, the compounds efficacy was evaluated using the Ehrlich Ascites Carcinoma (EAC) model where it was found to inhibit tumor cell growth by 46% at 75 mg/kg, well below the lethal dose of 1000 mg/kg [25]. The anti-inflammatory and redox-protective effects of citronellal, indicated by in vitro and in vivo methods were also reported using leukocyte injected mouse models and carrageenan rat models [25]. The redox protective effects were demonstrated by reduced hepatic lipoperoxidation, as well as oxidation of certain protein. Citronellal administration via intraperitoneal route was able to inhibit the carrageenan and arachidonic acid- induced rat hind paw edema models (popular methods for evaluation of anti-inflammatory activity of lipoxygenase inhibitors) [25]. Citronellol (1b, Figure 3) has demonstrated notable anticancer properties as well as citronellol-containing essential oils have been reported to display anticancer potential in cell models [21,26].
Geranic acid (2e, Figure 3) isolated from Thymus × citriodorus was recently identified as an anti-inflammatory agent [27]. It is worth noting that in some instances of treatment the required concentrations to elicit a favorable biochemical response proved cytotoxic. While the findings of this study are promising, further work is required prior to the use of the relevant compounds as potential therapeutic agents. Geraniol (2b, Figure 3) has been identified as a main bioactive component in essential oils and validated as a pure compound. Geraniol is a major component of Palmarosa, lemon grass, and rose essential oils which have been documented for their beneficial attributes. Geraniol has been shown to protect against cyclophosphamide-induced hepatotoxicity in rat models by modulating expression levels of NFK-β, PPARγ, inducible nitric oxide synthase, and cyclo-oxygenase 2 [28]. Cyclophosphamide is a chemotherapeutic agent with liabilities due to its hepatotoxicity so identification of compounds that can prevent this unfavorable effect are highly desirable. Dosing of rats was done at a concentration of 25 mg/kg once daily one hour after their respective treatments of corn oil (vehicle), or geraniol (100 and 200 mg/kg). The groups treated with geraniol showed lowered expression levels of IL-1β, TNF-α, MDA and other hepatotoxicity markers. The only enzymes shown to be upregulated when treated with geraniol were GSH, and PPARγ. The data shows the promising ability of geraniol to curb some negative side effects caused by the treatment of patients with this effective and well-documented cancer drug.
Geranyl acetate (2c, Figure 3) is a major component of various essential oils and plant extracts that have shown biological activity. A recent study described the cytotoxicity of this compound from the essential oils of kaffir lime callus [29]. This study conducted MTT assays on the breast cancer cell line, T47D and non-cancerous cells Sel Vero. The results of these assays showed a low cytotoxicity in both cancerous and noncancerous cells. The determined IC50 values exceeded 100 μg/mL in both cases [29]. These findings suggest that the acetate group makes this compound much more cell permeable.
Geranylacetone (2d, Figure 3) has been identified in a number of essential oils and plant extracts that have demonstrated anticancer activities. Recently, geranylacetone was identified as a major component of Lithocarpus polystachyus Rehd. a plant with documented anticancer properties [30]. This monoterpene was shown as a major constituent of the volatile compound mixture, present at a concentration of about 283 μg/mL in the distilled sample [30]. An independent study identified geranylacetone as a major component of Dianella ensifolia (6% GC-MS) [31]. This study conducted a series of in vitro experiments to understand the antioxidant potential of this oil. A series of biological assays (DPPH, ABTS, and FRAP) were conducted to determine the antioxidant properties of this essential oil (EO) in comparison with positive control BHT (butylated hydroxytoluene). Geranylacetone is also able to react as a free radical scavenger. Both geraniol and geranylacetone have been demonstrated to serve as free radical scavengers, albeit at high concentrations of 4–8.0 mM. DPPH radical scavenging assay showed that geranylacetone after 15 min indicated a modest dose-dependent effect [32].
Halomon (6, Figure 3), a halogenated acyclic monoterpenoid, has been reported to be active against the non-small cell lung carcinoma cell model (NSCLCN6-L16) and colon cancer cell model (HCT-116) in vitro. Furthermore, in vivo studies have demonstrated the anticancer activity of this compound against U251 brain cancer. Halomon showed cytotoxicity against many cancer subtypes, including renal-, brain-, and colon-derived solid tumor cell lines. It also showed that this monoterpenoid acts as an inhibitor for DNA methyltransferase-1 with an IC50 value of 1.25 μM [33], which decreases the expression of tumor suppressor genes. Preliminary in vivo studies evaluated the biological effect of Halomon via intraperitoneal injections on U251 brain cancer xenograft model. This model involves the implantation of cancer cells into immunocompromised rats; the tumor is allowed to grow, and then the effects of a treatment are evaluated in vivo. It was demonstrated that five daily doses of 50 mg/kg had a 40% success rate in treatment [33].
Anticancer and antioxidant activity have also been demonstrated with lavandulyl acetate (5, Figure 3) which is found in essential oils and plant extracts such as Lavandula angustifolia (lavender oil). GC-MS indicated that lavandulyl acetate is found in 23 percent (w/w) of the overall mixture on average of this essential oil [34]. The essential oil was evaluated on several assays including cytokine cellular response, LPS-stimulated HaCaT Keratinocytes, and LPS-prestimulated human monocyte derived macrophages (hMDMs). A dose dependency was observed between lavandulyl acetate in the oil and a potent anti-inflammatory and pro-regenerative response [34]. The studies indicate that this compound might hit several targets as these biological processes involve different pathways. The pharmacological effects of lavender oil depend on the concentration of lavandulyl acetate as independent studies have also shown other biological activities for this essential oil [31].
Linalool (3a, Figure 3), a chiral acyclic monoterpenoid, has demonstrated anticancer and antioxidative properties [35,36]. The anticancer activity of the essential oil from Ocimum forskolei Benth with 28–10% of linalool has been reported [37]. The oil’s IC50 values were shown to be close to 17, 7, 5, and 25 μg/mL for MCF7, HT29, HCT116, and MRC5 respectively. A recent study done found that linalool displays antiproliferation effects on breast cancer cell lines MCF-7 and MDA-MB-231. This research demonstrated that at 100 μM concentration of linalool there was approximately 55 and 65 % cell viability for MCF7 and MDA-MB-231 respectively after 24 h [35]. The determined IC50 values of linalool were a mild 588 and 480 μM for MDA-MB-231 and MCF7 respectively [35]. Unfortunately, no data was provided for normal cells, which calls for further studies to experimentally determine therapeutic index and its potential as anticancer agent.
Linalool has demonstrated antioxidative properties in a study from Plumeria alba [38]. The essential oils from this plant have been shown to have antioxidant properties. Gas Chromatography-Mass Spectroscopy studies identified that linalool composes about 24% of the active essential oil. The samples prepared from the plants flowers gave IC50 values of 370, 1014, and 1943 μg/mL for the H2O2 scavenging assay, DPPH radical scavenging assay, and thiobarbituric acid reactive substances (TBARS) assay respectively [38]. In addition, in vivo studies in hemiparkinsonian rat models indicated neuroprotective effects. Linalool treatment demonstrated neuroprotectivity in the form of anti-inflammatory and antioxidative action in dosages as low as 50 mg/kg [39]. Through the observed behavioral and neurochemical effects linalool indicated neuroprotective activity presumably through its antioxidative properties [39]. Linalyl acetate (3b, Figure 3) has been well documented to have favorable bioactivities. Anticancer properties have been associated with essential oils from Lavandula angustifolia, Lavandula officinalis, Lavandula latifolia, and bergamot [40,41,42,43,44], all of which contain linalyl acetate in concentrations as high as 44%. A study conducted on the lavender essential oils from Lavandula angustifolia demonstrated an IC50 values of 0.36% (v/v) against HaCaT cell line in an MTT assay [43]. Additionally, there have been reports of linalyl acetate associated antioxidant activity in essential oils of Salvia sclarea, and Myrtus nivellei, across different regions [45,46]. The essential oil attained from Myrtus nivellei demonstrated an IC50 value of 2.25 mg/mL [46].
Myrcene (4, Figure 3) has been shown to display anticancer bioactivities when used as a mixture in the essential oils from Mentha spicata, Matricaria chamomilla, and Citrus pseudolimon [47,48]. The IC50 values of the essential oil from Matricaria chamomilla were demonstrated to be between 200 and 320 μg/mL for cancer cell lines A549, MCF7, and PC3. Meanwhile the IC50 values for the Mentha spicata essential oils were 672, 708, and 206 μg/mL for cell lines A549, MCF7, and PC3 respectively [47].
It has been documented that plant essential oils with high concentrations of myrcene have antioxidant potential in biological systems. Essential oils from Hedyosmum purpurascens, Pinus halepensis, Salvia officinalis, Cymbopogon citratus, Piper eriopodon, and Prangos trifida, all having high concentrations of myrcene, have demonstrated antioxidant activities [49,50,51,52,53]. Myrcene has been demonstrated to inhibit oxidative stress by down regulating pro-inflammatory cytokines and inflammatory mediators [54]. This compound’s ability to decrease the oxidative stress induced by rotenone was demonstrated by the significant reduction of pro-inflammatory cytokines upon myrcene-rotenone co-treatment. While monitoring expression levels of inflammatory markers a dramatic decrease in expression was noted in iNOS and COX-2 [54].
2.2. Cyclic Monoterpenes
The cyclic monoterpenes 7–12 (Figure 4) have a strong background in biological activities as they are common members of essential oils used in ethnopharmacology [55]. The natural product p-cymene (97, Figure 4) is an aromatic, cyclic monoterpene that has been demonstrated to have antioxidant properties in vivo. A study conducted in diabetes induced Wistar rats demonstrated this compound’s ability to upregulate mTOR, AKT, and phospho-AKT enzymes all of which being crucial regulators of oxidative stress [56]. The diabetes induced rats were dosed with either 55 mg/kg of metformin, an approved type II diabetes medication, or 25, 50, 100 mg/kg of p-cymene. All groups were successful in increasing the expression of these key oxidative stress regulatory enzymes [56]. It is worth noting that although the 25, and 50 mg/kg groups were less effective than metformin, the 100 mg/kg group was able to upregulate these enzymes to a comparable degree as the metformin group [56].
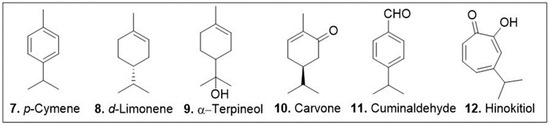
Figure 4.
Selected cyclic monoterpenes with reported antioxidant or anticancer activities.
d-Limonene (8, Figure 4) is a chiral, cyclic monoterpene that has been shown to have both anticancer and antioxidant properties. A study conducted in vivo animal models showed this compound’s ameliorative effect on chloroform induced cardiotoxicity by lowering oxidative stress, inflammatory and cardiac markers [57]. The study monitored the change in cardiac injury markers whose expression was increased by CCl4; following two weekly doses of 200 mg/kg of d-limonene approximately 50% reduction in serum levels of cardiac inflammatory markers IL-6, Hs-CRP, and TNFα was noted [57]. An independent study observed the effects of d-limonene in cancer therapy. This work used d-limonene for its antioxidant effects in a co-treatment study with doxorubicin [58]. The designed nanoparticle delivery system showed a strong decrease in cell viability against HepG2 human liver cancer cell line and a decrease in cytotoxicity of the LX2 normal cells [58]. Unfortunately, there was no further biological evaluation of this treatment system to further support the hypothesis of the mechanism of action with the doxorubicin-d-limonene nanoparticle delivery systems.
Carvone (10, Figure 4) is a chiral, cyclic monoterpenoid present in large quantities in caraway and spearmint essential oils that has been isolated identified for its anticancer and anti-oxidative properties. A study on the anticancer effects of carvone on myeloma cells (KMS-5) indicated that this meroterpenoid induces apoptosis through inhibition of the p38 MAPK signal transduction pathway [59]. It was found that the IC50 of carvone against this cell line was 20 μM [59]. Apoptosis was identified as the mode of action for this treatment by DAPI, AO/EB, and V/PI staining assays carried out at concentration of 20 μM carvone. Furthermore, western blot analysis demonstrated the decrease in expression of p-P38, a dual regulator of apoptosis, in response to carvone treatment at 20 μM leading to the conclusion that this compound may inhibit the expression of this regulatory enzyme [59]. An independent study investigated this compound’s effects in alleviation of doxorubicin induced cardiotoxicity. The IC50 value of carvone on the MCF7 cell line was determined to be 14 μM while in the healthy cell line H9C2 this monoterpenoid was nontoxic up to 200 μM [60]. In vivo analysis of male BALB/c mice was done to evaluate the synergistic effects of carvone during concomitant treatment with doxorubicin. These experiments showed that the myocardial disorganization and degeneration imposed by 20 mg/kg intraperitoneal (i.p) injection of doxorubicin was alleviated when i.p. injection of 75 mg/kg carvone was done one hour prior to doxorubicin treatment [60]. Identical injection of 150 mg/kg carvone resulted in no notable tissue generation upon histological analysis [60].
Cuminaldehyde (11, Figure 4) is an aromatic, cyclic monoterpenoid with anticancer biological properties. Research done on cuminaldehydes effects on human colorectal adenocarcinoma (COLO 205) cells showed that this compound induces cell death through topoisomerase I and II inhibition [61]. An in vitro evaluation of cuminaldehyde treatment of COLO 205 cells demonstrated a notable decrease in cell growth and increase in cytotoxicity at μM concentrations. After 24 h of cell treatment with 40 μM cuminaldehyde there was a 50% decrease in cell viability. This compound was further tested against topoisomerase I and II to show the inhibitory relationship between these molecules. The degradation of monomeric DNA in the presence of 5–20 μM cuminaldehyde indicates the interaction between this monoterpenoid and the nuclear proteins. Furthermore, tumor reduction capacity was demonstrated using a COLO 205 xenograft in a nude mice model. Daily subcutaneous injection of 10 and 20 mg/kg cuminaldehyde resulted in 48 and 69 % tumor reduction respectively [61] with no notable loss in body weight. Although these findings are promising the lack of comparative data with healthy cell lines makes it difficult to evaluate this compound for its efficacy as a potential therapeutic.
Hinokitiol (12, Figure 4) is a tropolone related, cyclic monoterpenoid that has been reported to show activity in anti-inflammatory and antioxidation processes while simultaneously promoting apoptosis and autophagy in cancer cells [62,63]. Western blot analysis of B16F10 murine melanoma cell line and 4T1 murine mammary carcinoma cell line showed this natural products potential of decreasing expression of heparanase, an enzyme known for its pro-metastasis effects in concentrations of 1 to 1250 nM. Furthermore, this research conducted in vivo studies on C57BL/6J and BALB/c mice. The results showed an inhibition of tumor metastasis and prolonged survival [62]. While these results are promising, the study did not analyze the effects of the monoterpene as a form of administered medication and instead injected B16F10 and 4T1 cells that had been pretreated in 1250 nM concentrations of hinokitiol into the mice. An independent study analyzed the cellular response of human osteosarcoma cell lines to treatment of hinokitiol in vitro.
Cell viability assays of U2OS and MG-63 human osteosarcoma cell lines showed a concentration dependent decrease in cell viability with hinokitiol treatments the 10–80 μM concentration range [63]. Furthermore, this research conducted western blot analysis of MG-63 cells showing that with treatment of hinokitiol results in PARP and caspase-3 cleavage both of which being known mediators of apoptosis. Cleavage of PARP was noted after treatment of hinokitiol in concentrations as low as 20 μM and demonstrated a concentration dependent increase through 80 μM [63] suggesting this compound induces programed cell death. Unfortunately, the study did not analyze any of the aforementioned phenomena on healthy cell lines.
3. Chemistry and Biological Activities of Selected Meroterpenoids
Meroterpenoids are a large group of compounds characterized as being biosynthetically hybrid molecules, which typically involve the combination of a terpene with polyketides, alkaloids, phenols, or amino acids. The large potential for variations in the combination of synthons provides a class of molecules with a high degree of structural diversity. Recently, these natural products have gained attention from chemists and biologists alike as there are new chemical applications and synthetic strategies to access these complex molecular scaffolds. While many of these compounds have been isolated and identified there is a new need for larger quantities to pursue biological studies. Biological evaluations of these compounds have shown potential for therapeutic development, many of these intriguing meroterpene structures have already been reviewed [64] and they are not included in this review. Specific synthetic lessons and innovative approaches are highlighted for the members of this meroterpene family, particularly natural products 100–188 (Figure 5). This diagram highlights the complexity and diversity of these natural products containing 3 to 6 rings, which also includes an increase in ring size within this family of natural products.
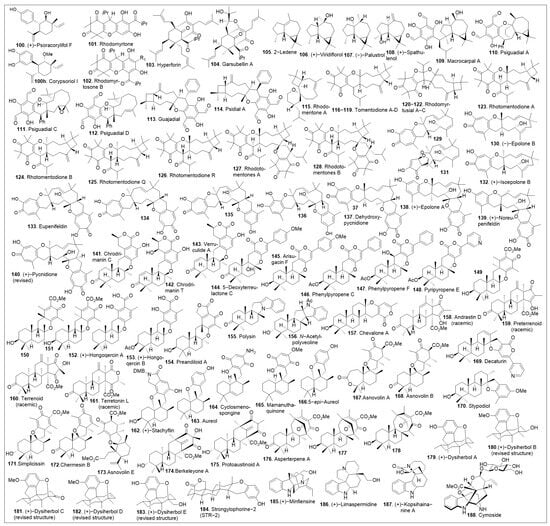
Figure 5.
Structures of representative meroterpenoids.
Total syntheses of small molecules can be challenging as insufficient functional groups are available to facilitate carbon-carbon bond formation and/or introduce chirality. Enantioselective divergent total syntheses for several meroterpenoids from Psoralea corylifolia have been recently reported [65]. The synthesis commenced with the readily available 2-methylcyclohexanone 16 in gram-scale, which was coupled with methyl acrylate mediated by thiourea catalyst (Cat. 1, Scheme 1) followed by treatment with 2-iodoxybenzoic acid (IBX) to oxidatively generate the corresponding α,β- system and subsequent saponification furnished acid 15. The newly formed compound 15 was subjected to catalytic PdCl2(PPh3)2 mediated intramolecular decarboxylative vinylation to establish the desired carbon quaternary center of enone 14. Compound 14 was then subjected to a 1,4-Michael addition reaction in the presence of copper and quenched with an aryl electrophile. This installs the α-arylation reaction sequence yielding diastereomers 13 and 13’, with compound 13 being predominant in 8:1 d.r. and can be separated by column chromatography. To improve the yield and favor a closer 1:1 ratio, Cesium fluoride was used instead of the copper acetate ligand which provided a 20% increase in yield. When applied properly, strategic conjugate addition reactions, when applied properly can expedite syntheses by forming carbon-carbon bonds efficiently, leading to improved overall yields. Compound 13 was subjected to reduction of the C-1 ketone with LiAlH4 or amine borane reagent to enrich one diastereomer over the other one providing alcohols 17 and 17’ in either 14:1 or 1:7 respectively. Compound 17 can be deprotected by BBr3 to yield the natural product (+)-psoracorylifol F (100a) in good yield, which was subjected to regioselective mCPBA epoxidation, and subsequent LiAlH4 reductive opening of the epoxide provided (−)-7β,8β-hydroxy-12β-cyclobakuchiol C (100b). Aromatic methoxy deprotection of compound 17’ by thermal treatment in the presence of the thio-anion led to the natural product (−)-8α-hydroxy-12β-psoracorylifol F (18). Compound 13 was exposed to a three-step synthetic sequence strategy, α-epimerization, carbonyl reduction via NaBH4 treatment, and heat-mediated thio-anion methyl deprotection of the phenol provided natural product (−)-7β,8α-hydroxy-12β-psoracorylifol F (100d). Alternatively, compound 17 was treated with MeI to protect the secondary hydroxyl group, and the aromatic methoxy group was revealed under thio-anion reaction conditions to yield compound 19 in excellent yields. The resultant compound 19 was oxidized at the alpha phenol with IBX followed by reduction with NaBH4 of the newly formed ortho-diketone to provide the natural product corypsoriol J (100c). In parallel efforts, compound 13’ was subjected to carbonyl-reduction followed by LiAlH4 or amine borane reagent to enhance the diastereoselectivity yielding alcohols 20 and 20’ in 6.5:1 or 18:1 ratio respectively. Deprotection of compound 14 mediated by thio-anion treatment under heat led to natural product (−)-corypsoriol H (100e), which upon two-step sequence epoxidation mediated by mCPBA and subsequent reductive opening of the epoxide with LiAlH4 provided (−)-8α-hydroxy-cyclobakuchiol C (100g). In a similar fashion, intermediate 20 was subjected to methyl-protecting group maneuver via intermediate 21, followed by one-pot oxidation mediated by IBX and subsequent reduction of the newly formed ortho-diketone. This, mediated by NaBH4 yielded corypsoriol I (100h). Finally, methyl deprotection of compound 20’ mediated by thermal treatment in the presence of thio-anion yielded (−)-psoracorylifol G (100f). The short and efficient synthetic strategy enables access to a large number of natural products, which can be further investigated for their biological properties.
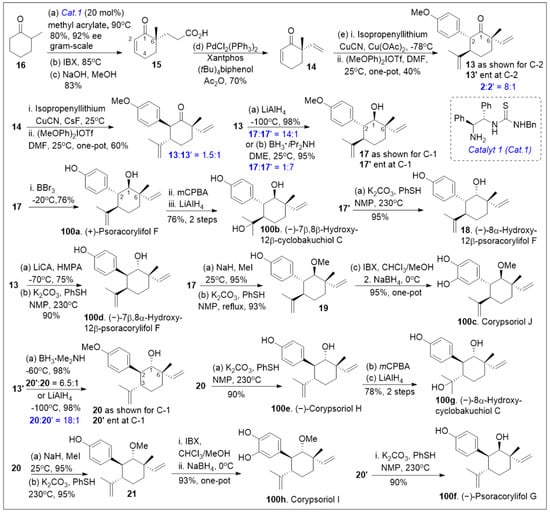
Scheme 1.
Total synthesis of several meroterpenoids from Psoralea corylifolia.
Rhodomyrtone belongs to a large and diverse family of tricyclic meroterpenoid systems isolated from Rhodomyrtus tomentosa [66,67,68,69,70]. This set of compounds has been shown to have strong antibacterial activity, acting primarily against gram-positive strains. In addition, the combined extracts of Rhodomyrtus tomentosa have shown anti-cancer and anti-tumoral effects in various models [66,67]. The synthesis of the unusual caryophyllene-derived meroterpenoids (CDMTs) including rhodotomentone A (127) and rhodotomentone B (128) was reported via a biomimetic pathway [68,69,70], (Scheme 2). The synthesis began from the natural product leptospermone (22), a commonly occurring acylphloroglucinol, which was subjected to dehydration mediated by DIBAL-H, yielding the dearomatized ortho-quinone methide (o-QM) intermediate 23 in gram-scale. This maneuver was followed by a [4+2] Diels-Alder photo-cycloaddition reaction with singlet oxygen, generated under blue LED (455 nm) radiation to provide endoperoxide intermediates 24a and 24b in 1:1 ratio. The formed o-QM intermediate 23 was subsequently subjected to hetero-Diels-Alder (HDA) reaction with β-caryophyllene 25 to produce a mixture of rhodomentone A (115), and the related natural products, tomentodione A, B, C and D (116, 117, 118, and 119). Rhodomentone A (115) can be treated with endoperoxide 24b to undergo the key Kornblum-DeLamare rearrangement and Diels-Alder cycloaddition reaction sequence under thermal reaction conditions. This produces rhodotomentone A(127) and B(128), along with two other rhodotomentone diastereoisomers, compounds 27 and 28. Additionally, endoperoxide 24b was reacted with β-caryophyllene (25) to undergo the Kornblum-DeLamare rearrangement and Diels-Alder cycloaddition reaction sequence under thermal conditions to generate rhotomentodine A, B, Q and R (123, 124, 125 and 126), along with rhodomyrtusial A, B and C (120, 121 and 122) and compound 29 [70].
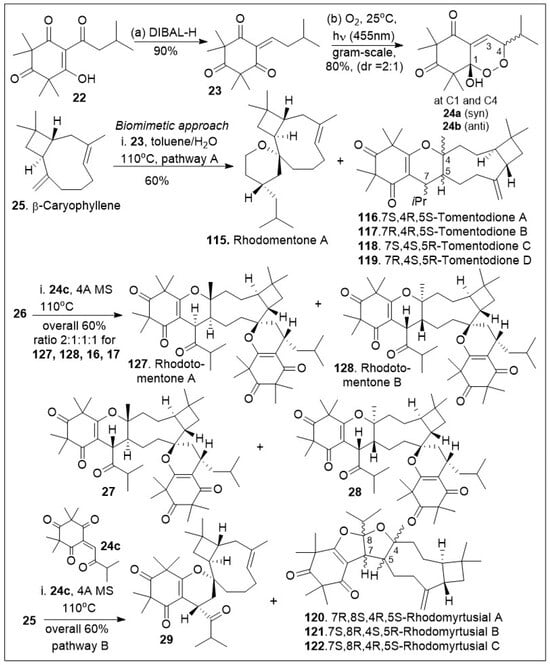
Scheme 2.
Biomimetic-inspired synthesis of caryophyllene-derived meroterpenoids (part 1).
In an efficient approach (Scheme 3), CDMTs 123, 124, 125 and 126 were easily converted to the corresponding natural products rhodomentone A (127), 28, rhodomentone B (128) and 27 respectively. This was carried out under hetero-Diels-Alder (HDA) reaction conditions, requiring Lewis acid, ZnI2 and heat, in relatively modest yields. The synthetic approach offers rapid access to impressively complex structures [70].
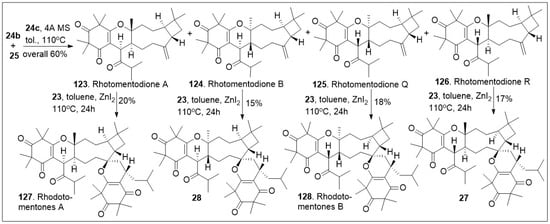
Scheme 3.
Biomimetic-inspired synthesis of caryophyllene-derived meroterpenoids (part 2).
The meroterpenoids macrocarpals and psigudials isolated from Psidium guajava a food crop widely abundant in tropical regions has been reported to display an array of medicinal properties [71,72]. These compounds have displayed significant biological properties as anti-HIV and anti-bacterial agents. To further evaluate their properties, access to sufficient quantities of these compounds is required. The application of a hetero-Diels-Alder (HDA) reaction in meroterpenoid synthesis is frequently utilized due to the likely probability that a similar pathway is taking place in nature to generate these natural products [72]. A short biomimetic-inspired synthesis to guajadial 113 and compound 30 was recently reported (Scheme 4). The synthesis commenced with a Knoevenagel condensation between benzaldehyde 31 and diformylphloroglucinol 32 providing compound 33 for in situ generated o-quinone methide 34. The HDA cycloaddition reaction between caryophyllene 30 and o-QM 34 led to guajadial 113, psidial A 114 and cycloadduct 36 [72].
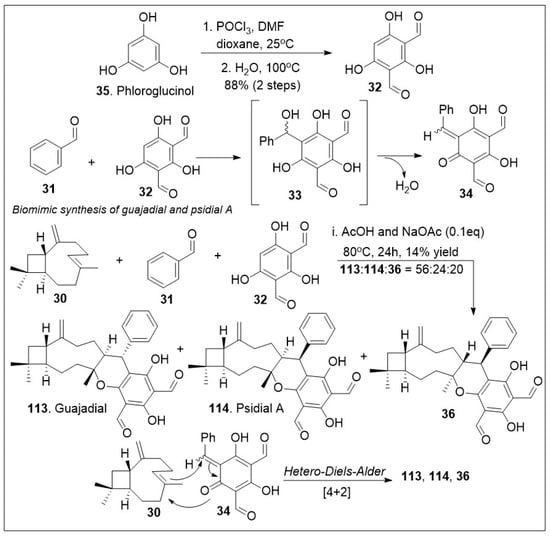
Scheme 4.
Biomimetic synthesis of guajadial and psidial A.
Hyperforin-like structures are a large family of meroterpenes that share a challenging molecular caged motif. A recent innovative synthetic strategy based on an annulative approach of polycyclic polyprenylated acylphloroglucinol (PPAP) meroterpenoids with 3,5-dimethylorsellinic (DMOA) was reported as shown in Scheme 5 [73]. The synthesis commenced with the readily available methylcyclopenenone synthon, which was subjected to sequential 1,4-copper-mediated conjugate addition reaction, namely formation of the lithium enolate followed by alkylative enolate trapping to generate the substituted cyclopetanone 37, which was subjected to further α-alkylated to furnish the highly substituted cyclopetanone 38. Next, cyclopetanone 38 was subjected to the key annulation reaction with β-lactone diketene, leading to diketone 39 as the major product, along with C-acylated product 40 and O-acylated product 41. Diketone 39 was further methylated with trimethylsilyldiazomethane to generate the vinylogous ester isomers 42 and 43. Compound 43 was subjected to oxidative rearrangement mediated by hypervalent iodine to provide product 45 with bicyclo[3.3.1]nonane ring system via intermediate 44. Compound 45 was subjected to a subsequent chlorination reaction to provide halogenated compound 46. Finally, a deprotonation/prenylation alkylation, acylation reaction sequence was used to introduce the last isoprenyl group generating the most challenging quaternary center. This was followed by deprotection of the methoxy group to yield hyperforin (103) in modest yields. The short strategy is flexible enough to generate a potential diverse library of derivatives for future biological evaluations.
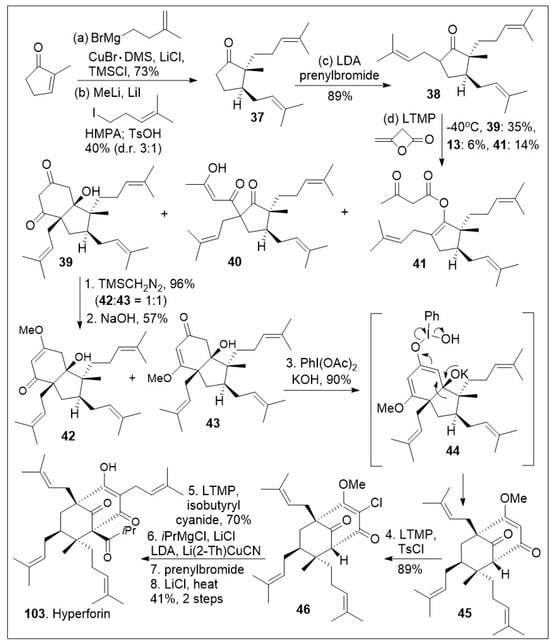
Scheme 5.
Total synthesis of hyperforin.
A similar approach has been applied to the caged meroterpenoid garsubellin A (104, Scheme 6) [73]. The total synthesis was initiated with methylcyclopenenone, which was α-prenylated to generate compound 47, followed by 1,2 Grignard reaction to access allylic alcohol 48. The resultant compound 48 was subjected to an anionic oxy-Cope rearrangement to provide bisprenylated cyclopentanone 49. Then, compound 49 underwent an annulation reaction with β-lactone diketene to produce diketone 50, followed by Pd(OAc)2/Cu(OAc)2 mediated unusual Wacker-type-intramolecular cyclization to provide tricycle system 51. Tricyclic system 51 was treated with PhI(OAc)2 under basic condition for an oxidative ring expansion leading to compound 52, the needed bicyclo[3.3.1]nonane ring system that upon cobalt catalyzed Mukaiyama hydration reaction provided tricycle 53. At this point in the synthesis compound 53 was treated with lithium tetramethylpiperidide (LTMP), and TsCl to introduce the α-vinylogous chlorine to generate compound 54, followed by acylation to introduce the congested quaternary center leading to compound 55. Various types of alkylation reactions are feasible for such compound, but alkylation with borane prenyl system mediated by [Pd(allyl)Cl2], and catalytic CPhos ligand under basic conditions introduce the prenyl group and remove the silyl protecting group to provide garsubellin A (104), as well as compound 56, which could be treated with acid or base to yield garsubellin A (104).
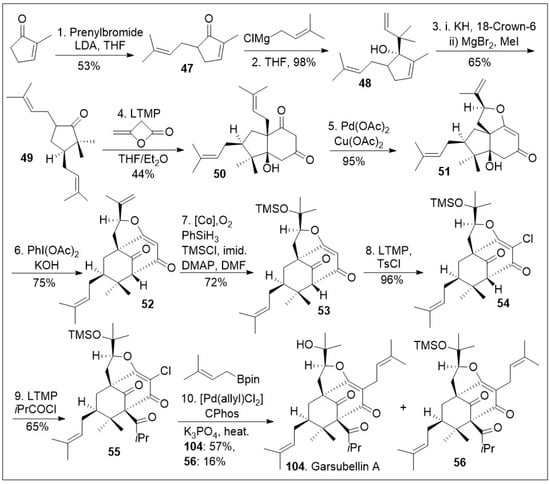
Scheme 6.
Total synthesis of garsubellin A.
The polycyclic fused ring meroterpenes such as berkeleyone A have displayed anti-inflammatory activity [74], while preandiloid A has shown antimicrobial properties. Asperterpene A was identified as a potent BACE-1 inhibitor with an IC50 value of 78 nM against this enzyme, out preforming commercial BACE-1 inhibitors. Furthermore, in vivo animal model studies were congruent with the in vitro efficacy studies of this natural product [74]. These findings indicate that these compounds require further studies and synthetic strategies to access them in large quantities.
A successful approach to these natural products involves the use of a cyclization reaction to set up the tricyclic system in a single step as shown in Scheme 7 [73]. The synthesis began with farnesyl bromide being subjected to alkylation with the anion of propionitrile, followed by one-pot bromohydrin formation/cyclization reaction sequence to generate epoxide 57, which upon reductive epoxide-opening radical cyclization furnished tricyclic ketone 58. Then, ketone intermediate 58 was subjected to annulation reaction with β-lactone diketene to produce diketone 59, which was O-methylated, followed by oxidative ring expansion mediated by PhI(OAc)2 under basic conditions to furnish bicyclo[3.3.1]nonane ring system 60. The newly formed tetracyclic 60 was exposed to Wittig olefination and subsequent chlorination reaction to yield halogenated polycyclic compound 61.
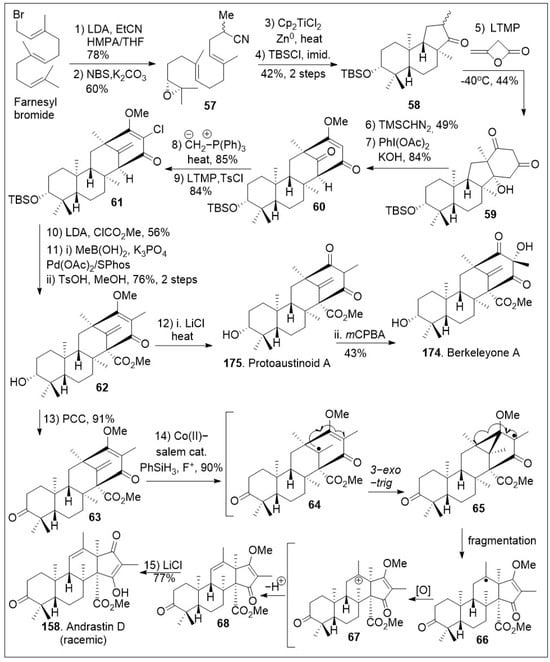
Scheme 7.
Synthesis of protoaustinoid A, berkeleyone A and andrastin D.
Upon further three-step α-alkylative methyl ester functionalization, methylation via Suzuki coupling reaction of the vinyl chloride with MeB(OH)2, and acid mediated deprotection yielded advanced intermediate 62. Then, compound 62 was subjected to the final chloride-mediated demethylation, leading to the DMOA-derived metabolite, protoaustinoid A (175). Compound 175 was further oxidized with mCPBA to provide berkeleyone A (174). In addition, advanced polycyclic compound 62 was treated with pyridinium chlorochromate (PCC) to generate compound 63, which was subjected to Shigehisa’s cobalt(II)-catalyzed hydroalkoxylation reaction leading to polycyclic intermediate 68. Finally, this compound was treated with LiCl to free the trapped enolate system, yielding the natural product andrastin D (158) in racemic form. Furthermore, as shown in Scheme 8, the tetracyclic compound 63 was subjected to Carreira’s alkene hydrochlorination conditions to provide the polycyclic compound 69 via hydrogen-atom-transfer (HAT) process involving radical intermediate 66. Subsequent demethylation of polycyclic compound 69 mediated by LiCl yielded the natural product preterrenoid (159), which underwent stereoselective α-oxidation mediated by magnesium monoperoxyphthalate (MMPP) to afford terrenoid (160). Upon ring-expansion via retro-Claisen/esterification cascade reaction under basic conditions, compound 160 was converted to racemic terretonin L (161). This synthetic strategy is efficient at enabling the generation of multiple members of this meroterpene family in good overall chemical yields [73]. This approach is not asymmetric, however, there are multiple opportunities to introduce asymmetry.
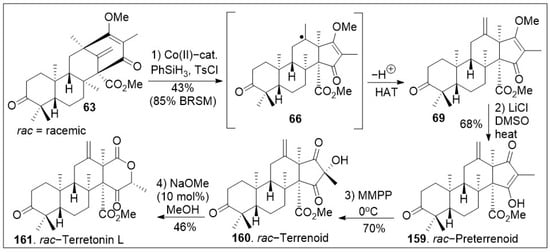
Scheme 8.
Synthesis of preterrenoid, terrenoid, and terretonin L natural products.
Forging complicated carbon-carbon bonds in complex natural products represents a challenge that requires groundbreaking synthetic endeavors or engaging in biomimetic synthesis, the process of imitating nature’s way to make molecules. Biomimetic synthesis of the psiguadial meroterpenoids featuring the key cationic cyclization reaction using terpene (+)-bicyclogermacrene 70 as the platform has been reported [75] and it is depicted in Scheme 9. The synthesis commenced with the generation of ketone-aldehyde 72, which was generated from (+)-2-carene (71) upon treatment with KMnO4 to mediate the dihydroxylation, and subsequent oxidative diol cleavage reaction. Compound 72 was further treated with Wittig reagent A under basic reaction conditions to provide compound 73, which was deprotected, and cyclized under samarium mediation to provide the 10-membered cyclic system 75, which was then converted to (+)-bicyclogermacrene 70 with the required E-olefin geometry using a modified Corey-Winter protocol. Compound 70 was then converted to 2-ledene (105) via acid facilitated intramolecular cationic cyclization. The natural product 105 was transformed to (+)-viridiflorol (106) upon treatment with hydroboration/hydroxylation conditions. Alternatively, compound 105 was subjected to Mukaiyama’s cobalt-catalyzed phenylsilane/O2 hydration conditions to provide natural product (−)-palustrol 107. Finally, epoxidation of compound 70 was facilitated upon treatment with mCPBA, followed by intramolecular cyclization provided (+)-spathulenol (108).
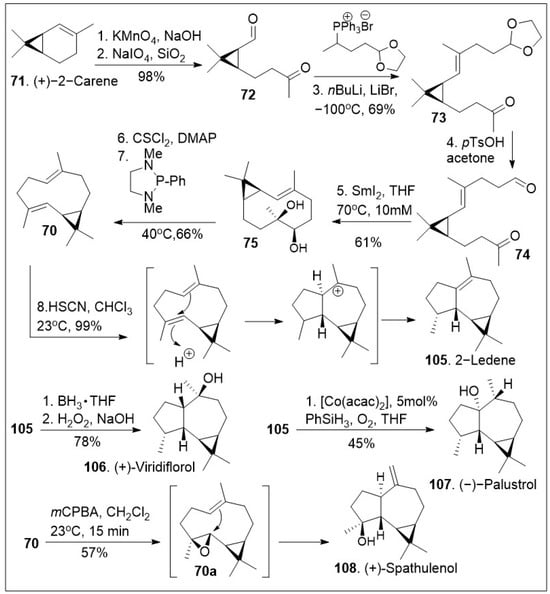
Scheme 9.
Biomimetic synthesis of several aromadendrene terpenoids.
As highlighted in Scheme 10, biomimetic approaches allow for flexible synthesis of various members of the same natural product family [75]. Thus, compound 70 was treated with intermediate o-QM 77 to enable the hetero-Diels-Alder (HDA) reaction, in order to yield the natural product psiguadial D (112). The compound o-QM 77 was synthesized in situ as the Knoevenagel-type condensation reaction product between bisformylated phloroglucinol 76 and phenyl aldehyde. Furthermore, 112 was converted to psigudial C (111) via epoxidation reaction. Under the similar reaction conditions, the o-QM 77 was unified with compound 4 via sequential cationic intermolecular coupling reaction followed by intramolecular cyclization cascade reactions. Briefly, intermediate 77 under acidic conditions generated the benzylic carbocation, which was able to react with compound 70 via addition reaction to yield intermediate 79. Compound 79 underwent an intramolecular cyclization, and a 1,2-hydride shift to establish the most stable cation 81. Finally, intramolecular cyclization from the phenolic system facilitated the installation of the last 7-member ring system of the natural product psiguadial A (110).
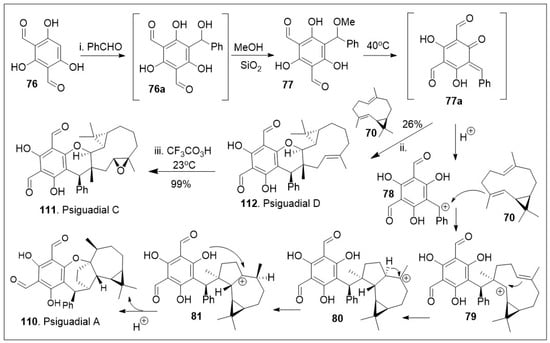
Scheme 10.
Biomimetic synthesis of psiguadial A and C.
The sesquiterpene-tropolones belong to a class of structurally distinctive meroterpenoids characterized as sesquiterpene mono- and bistropolones, which includes pycnidione and eupenifeldin, featuring a unique polycyclic ring system with rich stereochemistry. These compounds are isolated from Neosetophoma sp. a species of fungi. These compounds were tested against a series of cancer cell lines: human breast cancer (MDA-MB-231), two human ovarian cancer cell lines (OVCAR-3 and OVCAR-8), human mesothelioma (MSTO-211H), and human lung cancer (A549) to determine their IC50 values; the most potent compound, eupenifeldin (133, Scheme 11) had IC50 values of 2.83, 0.33, 0.02, 0.08, 1.33 μM for the above listed cell lines respectively [76]. To further evaluate the cytotoxicity of these compounds towards healthy tissues a mitochondrial toxicity assay was conducted for compounds 129, 131 and 133 (Figure 5). The results of this experiment showed no cytotoxicity up until a maximum dose of 12.5 μM, which was well above the majority of the IC50 values for the compounds tested [76]. Further study of this natural product was conducted by Maldonado et al. focusing on its effects on a panel of ovarian cancer cell lines. The antiproliferative effect (EC50) of eupeninfeldin on three ovarian cancer cell lines (OVCAR-3, OVCAR-5, OVCAR-8) and nontumorigenic fallopian tube cells (FTT3-Tag) were determined to be 10, 11, 12, and 170 nM respectively [77]. The results indicate the selectivity of the compound for cancer cell models over normal tissue with a greater than 10-fold difference. Additional mechanistic studies included cell migration assays, which provided favorable outcomes [77]. To validate their in vitro findings, hollow fiber studies were conducted in murine models. OVCAR-3 and OVCAR-8 hollow fibers were implanted in mice, which were treated with eupeninfeldin or vehicle daily for 4 days. A significant reduction of OVCAR-3 cell model was observed while no significant inhibition of OVCAR-8 cell model was recorded suggesting the compound is working via a specific pathway. To further investigate the mechanism of action specific protein evaluation was conducted for apoptosis pathway (caspase 3/7 activation, PARP cleavage, autophagy assays) and global protein analysis using tandem mass tag (TMT), a chemical label technique that facilitates sample multiplexing in mass spectrometry (MS)-based quantification and identification of the proteome. Bafilomycin A1, a member of macrolide antibiotics and an autophagy inhibitor, was used as a co-treatment with eupenifeldin on their selected ovarian cancers cell lines (OVCAR-3, OVCAR-5, and OVCAR-8) to determine if this compound is mediating cell death through autophagy as a primary cell death modality. The resultant data indicated that indeed bafilomycin was able to significantly rescue the cells from death. Their combined findings indicate that this natural product displays selectivity. It is likely mediating cell death through autophagy [77]. Pycnidione (140, Figure 5) is another example of bioactive sesquiterpene-bistropolone meroterpenoid natural products. First isolated from fungal species, Phoma sp., this compound has been studied extensively for its biological activities. Kaneko studied the effects of pycnidione on leukemia cell models [78]. This compound alone or in combination with bleomycin against a panel of human cancer cell lines had a significant anti-proliferative effect. Ongoing studies indicate that pycnidione inhibits of topoisomerase II at an IC50 value of 19 μM and modulates the cell cycle at the G2 phase [78]. These biological studies highlight the importance of having access to large quantities of these natural products for further derivatization to improve efficacy. The recent total synthesis of the structurally revised flagship members of the pycnidione family, namely (+)-pycnidione (140), dehydroxypycnidione (137) and (−)-epolone B (130) was elegantly designed on a modular [4+2] inverse electron-demand hetero-Diels-Alder (HDA) reaction between the in situ generated ortho-quinone methide (o-QM) 82 and the corresponding dienophile, hydroxyhumulene 83 or humulene 84 (Scheme 11). While the majority of [4+2] cycloaddition reactions require heat, microwave assistance can also promote the rection, and minimize decomposition of starting materials or intermediates. A unified synthetic endeavor was recently used to generate various members of these complex meroterpenes through the in situ generation of compound 82 (Scheme 11). The resultant transition state of either dienophile 83 or 84 can proceed either as a single monocyclic formation step, TS1 or a bis-cyclization formation, TS2. They indicated that the reaction can be modified to favor one or various products. However, the diastereomers of eupenifeldin series have yet to be synthesized and remain a highly active area of research [79].
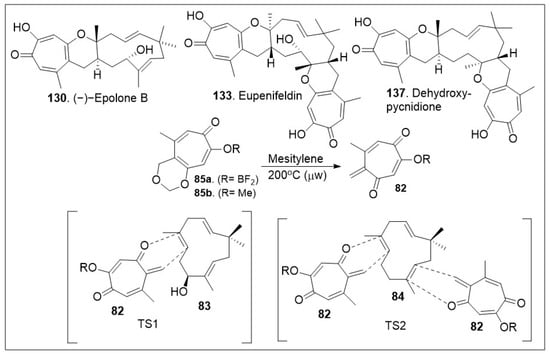
Scheme 11.
Hetero-Diels–Alder (HDA) strategy to access the tropolones.
The synthesis capitalizes on the advantages to access building blocks 83, o-quinone methide precursors 85a and 85b in sufficient quantities (Scheme 12) [79]. Their strategic synthesis began with 1,3-cyclopentanedione 90, followed by O-alkylation with 91, which is formed in situ with paraformaldehyde and chlorotrimethylsilane under basic conditions, providing the vinyl chloride 87 that can easily undergo a [2+2] intramolecular photocycloaddition furnishing tricycle 92. Light-driven [2+2] cycloaddition reactions are the most direct approach to build tetrasubstituted cyclobutanes that may lead to diverse structures. Upon one-pot iodination/Kornblum oxidation, the tricycle 92 was converted to hydroxy enone 86, followed by O-methylation provide intermediate 93. Use of either AgBF4 or UV light (254 nm) facilitated the ring expansion via fragmentation of compound 93 to provide the methoxy-tropolone 85 in good yields (Scheme 12A). Alternatively, treatment of compound 86 with Lewis acid, boron trifluoride diethyl etherate, resulted in tropolone 84. Thus, the use of simple transformations easily generates the large ring-containing molecule in sufficient quantities. (−)-Kobusone 89 was converted to the corresponding α-hydroxylated product 95 via Davis’ oxiziridine treatment as shown in Scheme 12B. The resultant α-hydroxyl group was protected with a silyl group, followed by 1-carbon Wittig reaction yielding compound 88, setting the stage for the ring opening. This reaction previously discovered by the Shenvi group [80] highlights the strategic use of Cobalt/Sal/silane catalyst systems to mediate a Hydrogen-Atom Transfer (HAT) retrocycloisomerization of compound 88 to afford compound 96 in excellent yields. Rhenium catalyzed deoxygenation and silyl group removal with TBAF provided the desired compound 83 as the starting material for the cycloaddition reaction.
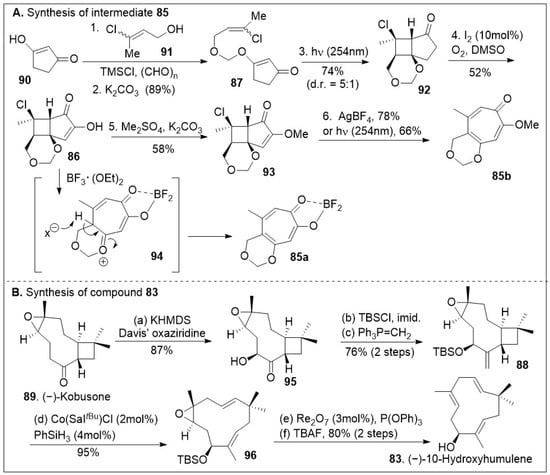
Scheme 12.
Synthesis of quinone methide precursor and (−)-10-hydroxyhumulene.
The application of this HDA reaction to various ene-systems is an efficient approach to generate natural products with high order architecture as shown in Scheme 13. Furthermore, these types of cyclization reactions tend to be selective and can be applied to large scale settings, enabling carbon-carbon bond formations in an economical manner. With the in situ generated o-QM 82 from tropolone 85a or 85b and dienophile 83, the platform was established for the [4+2] inverse electron demand HDA reaction [79]. The thermal microwave assisted HDA reaction between (−)-hydroxyhumulene 83 and o-QM (generated in situ from precursor 85) provided cycloadducts 99 and 189. Note that mesitylene was used rather than toluene to reach higher boiling points and provide the desired products in modest yields. Next deprotection of methoxy group of compounds 99 or 189 via base, led to the natural product epimers 132 or 130a respectively. Then, compound 99 was subjected to HDA reaction again with compound 85b to yield 191, which was deprotected under basic reaction conditions to afford the natural product epimer compound 140a. The same treatment could be provided to compound 189 to yield the natural product epimer 140b (8,9-epi, epi-(−)-pycnidione) in good yields. Finally, the HDA reaction of between humulene 84 (no chirality) and in situ generated o-QM (from 85a or 85b) was conducted to produce compounds 97 or 98 respectively. Deprotection of these compounds led to the natural product 130b (dehydroxyepolone B) as a racemate in good yields. Compound 98 was further treated with dienophile for a second HDA reaction under the same reaction conditions to provide the adduct precursor of 137 in poor yields indicating the second HDA reaction is more difficult for 83 than for the allylic system 84. Perhaps, the presence of the hydroxyl group lowers the barrier slightly in favor of that second reaction. Deprotection of the methoxy group provided natural product 137 [79].
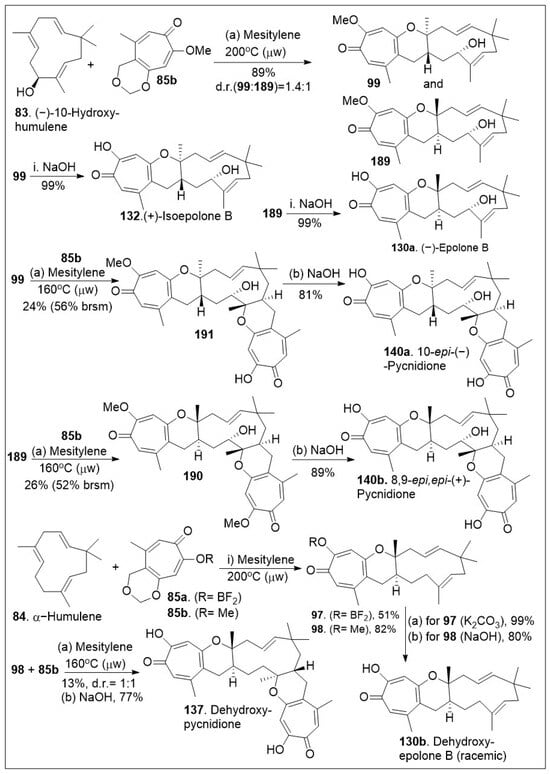
Scheme 13.
Synthesis of (−)-epolone B and family members.
Recent synthetic approaches towards the trans-syn-fused drimane meroterpenoids have capitalized in advancements in chemoenzymatic reactions [81] to facilitate carbon economic and diversity oriented synthetic strategies. The total synthesis application using a chiral-pool natural product is highlighted in Scheme 14. The synthesis commenced with the commercially and readily available sclareolide 192, which was treated with acid to epimerize the β-hydrogen, and re-formation of the syn-lactone (199, Scheme 15). Then, stereoselective enzymatic hydroxylation of the methylene group adjacent to the quaternary center using P450BM3 variant KSA15 provided compound 193 in respectful yields in gram scale. Compound 193 was treated with nBuLi, trapped with a silyl group, and coupled with 2-methyl aniline anion to provide compounds 194 and 195. This indole reaction known as Smith-modified Madelung indole synthesis protocol is usually high yielding and relatively compatible with other functional groups. This mixture was converted to 196 under acid treatment to facilitate the dehydration. System 196 was then exposed to Albright-Goldman oxidation to generate the corresponding ketone, which was further subjected to two-step microwave-promoted intramolecular Friedel-Crafts cyclization reaction and subsequent in situ trapping of the enolate with acetic anhydride, affording enol ether adducts 197 and 198. Next palladium catalyzed hydrogenation and subsequent K-selectride mediated diastereoselective reduction of enol ether adduct 197, provided N-acetyl-polyveoline (156). In parallel, compound 198 was treated with potassium carbonate to release the acetate group, resulting in the natural product polysin (155) in good yield.
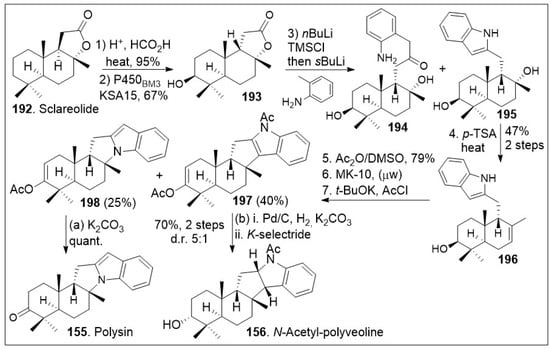
Scheme 14.
Chemoenzymatic synthesis of drimane meroterpenoid polysin.
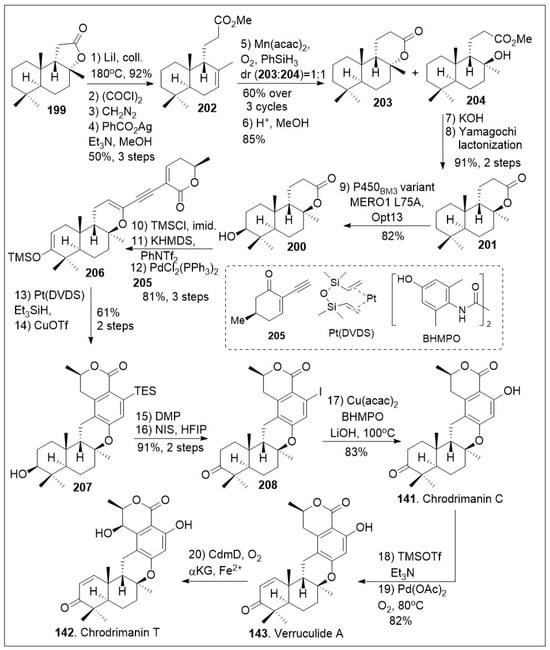
Scheme 15.
Chemoenzymatic synthesis of verruculide A, chrodrimanin C, and T.
To generate other meroterpenoid family members (Scheme 15), lactone 199 was converted to methyl ester 202 through a series of four linear steps [81]. The lactone was first opened using heat, followed by three-step Arndt-Eistert homologation to furnish the corresponding methyl ester 202 in excellent yields. Then, compound 202 was treated with Mukaiyama hydration protocol which led to a mixture of lactone 203 and tertiary alcohol 204 in relatively good yields. In addition, lactone 203 was converted back to methyl ester 202 and recycled to increase the overall yield. Upon saponification and subsequent Yamaguchi lactonization of tertiary alcohol 204, the trans-syn-trans-fused lactone 201 was generated. At this point, the application on enzymatic transformation at the methylene group adjacent to quaternary center was conducted. The enzymatic hydroxylation took place under P450BM3 variant MERO1 L75A on a preparative scale to produce alcohol 200. Next vinyl triflate formation was conducted upon treatment of alcohol 200 with silyl group protection and trapping of the enolate to provide the corresponding triflate, which underwent Sonogashira coupling reaction with alkyne 205, to generate dienyne 206. Subsequent hydrosilylation of the newly formed dienyne 206 catalyzed by Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex known as Pt(DVDS) in the presence of Et3SiH, afforded the desired hydrosilylated triene intermediate, which could readily undergo 6π electrocyclization/aromatization reaction under copper(II) triflate to yield arene 207. The resultant compound was treated with Dess-Martin periodinane (DMP) to mediate the oxidation of the secondary hydroxyl group followed by desilylative iodinization under NIS mediation to provide iodo-arene 208. Compound 208 was subjected to copper-catalyzed hydroxylation of aryl halides under mild conditions, namely Cu(acac)2 in the presence of N,N′-bis(4-hydroxyl-2,6-dimethylphenyl)oxalamide (BHMPO) and base to yield chrodrimanin C (141). The natural product 141 can be converted to verruculide A (143) upon treatment with Saegusa 2-step protocol to install the α,β-unsaturated system. Furthermore, this natural product can be converted to chrodrimanin T (142) under Fe(II)/α-ketoglutarate-dependent oxygenase treatment to introduce the hydroxyl group stereoselectively at the γ-position of the lactone ring. Overall, the Renata group provided an impressive use of enzymatic and modern organic chemistry reactions [81].
Another effective synthetic approach using natural products as a chiral pool is the strategy towards the arisugacin natural products. These drimane meroterpenoids have been isolated from Penicillium sp. and other sources. Their members have demonstrated acetylcholinesterase inhibitory activity in the nanomolar range [82]. Access to both libraries and large quantities of these compounds is desirable to interrogate their mode of action. Stypodiol is a pentacyclic meroterpenoid isolated from the brown algae S. flabelliforme, which displays a broad range of biological activity. Stypodiol has shown anti-proliferative effects against a number of cancer cell lines and also has antimicrobial capacity. This compound showed promising anti-proliferative activity in the low micromolar range against a human neuroblastoma cell line (SH-SY5Y) [83]. While multiple cancer cell lines were tested most of them showed weak anti-proliferative effects, indicating the compound is likely to have selectivity towards a specific cancer subtype. Therefore, efficient synthetic approaches to this family of compounds are desirable. A recent rapid and modular approach using modern organic chemistry, such as radical-based reactions combined with chemoenzymatic reactions has been reported [81]. As shown in Scheme 16, the synthesis commenced with natural product sclareolide 192, which underwent enzymatic hydroxylation using P450BM3 variant BM3 MERO1 in the presence of thermostabilized phosphate dehydrogenase (Opt13) at the methylene group adjacent to the quaternary center. The corresponding compound 210 was protected with a silyl group followed by stereoselective α-hydroxylation, conducted via trapping of the enolate with oxygen to efficiently generate compound 211. Next, lactone 211 was reduced to the triol system followed by oxidative cleavage of the diol moiety, rapidly furnishing the key aldehyde 209. Aldehyde 209 was coupled with pyrone 213 via a formal [3+3] cycloaddition/hydrogen atom transfer (HAT) reaction mediated by phosphoric acid 215 to generate cyclized product 217, which was acetylated in a one-pot procedure with acetic anhydride. Phosphoric acid catalyst promoted in situ alcohol dehydration and formal [3+3] cycloaddition to provide the shown stereocenter as the major product. Then, it was treated with Mn(dpm)3 in the presence of TBHP (tert-butyl hydrogen peroxide) and PhSiH3 to facilitate a chemoselective HAT reduction reaction to provide the natural product phenylpyropene C (146). This Mukaiyama reduction protocol has high stereospecificity as it favored the hydride addition from the bottom face, yielding the product in a 20 to 1 ratio with respect to its diastereoisomer.
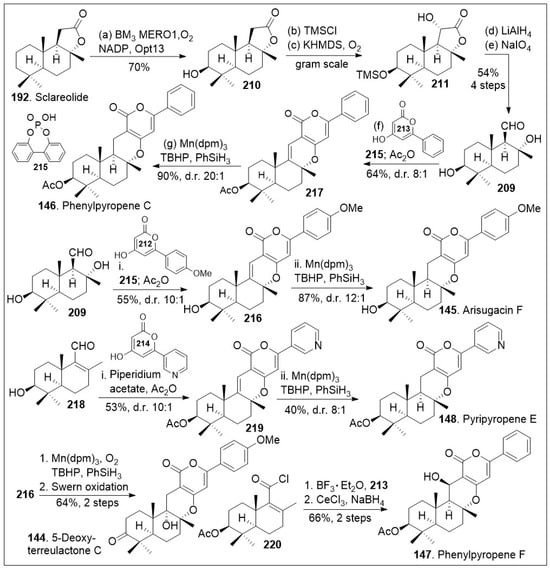
Scheme 16.
Modular total syntheses to access phenylpyropene C, and related family members.
Alternatively, aldehyde 209 was coupled with pyrone 212 via a formal [3+3] cycloaddition reaction mediated by phosphoric acid 215 to generate cyclized product 216, which was converted to arisugacin F (145) via chemoselective HAT reduction reaction mediated by Mn(dpm)3 in the presence of TBHP and PhSiH3. Compound 209 was dehydrated to provided enal 218, which was coupled with the shown pyrone via formal [3+3] cycloaddition mediated by piperidium acetate to generate compound 219. The corresponding compound can be then converted to the natural product pyripyropene E (148) via chemoselective HAT reduction reaction using their established reaction conditions for these systems in modest yields and good diastereomeric ratio. In addition, compound 216 was treated with Mukaiyama hydration chemoselective HAT reaction mediated by Mn(dpm)3 in the presence of oxygen gas and PhSiH3, followed by Swern oxidation to provide 5-deoxyterreulactone C (144) in good yields. Finally, the acyl chloride 220 that arises from aldehyde 209 was subjected to Fredel-Crafts acylation reaction with pyrone 213 mediated by BF3 etherate followed by cyclization reaction. Then, reduction of the ketone system under Luche reduction conditions provided the natural product phenylpyropene F (147). The total syntheses of various members of this drimane meroterpenoid family was achieved through the creative use of strategic enzymatic and chemoselective reactions to provide flexible, modular, and a generalized synthetic route that may facilitate biological studies of these natural products and their derivatives.
Another modular synthetic effort is highlighted in Scheme 17 to access these natural products, but mainly to stypodiol [84]. The synthesis commenced with the available natural product sclareol 222, which was converted to intermediate 223 via a 3-step sequential protocol: acylation with acryloyl chloride, γ-lactone ring formation via intramolecular olefin metathesis mediated by Grubbs second generation catalyst, and finally dehydration reaction mediated by base treatment.
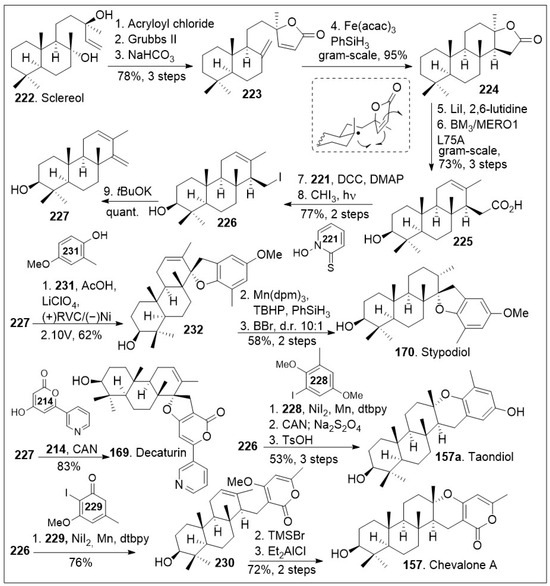
Scheme 17.
Total Syntheses of stypodiol and chevalone A.
Next, gram-scale HAT-based intramolecular Giese type coupling reaction was conducted on compound 223 with Fe(acac)3 in the presence of PhSiH3, which goes through the radical transition state shown to preferentially provide the tetracyclic compound 224. Then, a two-step sequence mediated by lithium iodide resulted in the opening of the lactone ring and generation of the corresponding carboxylic acid. This was subjected to enzymatic hydroxylation using P450BM3 variant BM3 L75A, leading to regioselective hydroxylated compound 225. The resultant compound could be treated with Barton reaction conditions to undergo decarboxylation, followed by photocatalyzed halogenation reaction to generate the corresponding iodide 226, which was then converted to diene 227 under basic conditions. Then, electrochemical SET-based [3+2] coupling reaction of diene 227 with phenol 231 via constant potential electrolysis (using reticulated vitreous carbon as cathode and Ni as the anode) generated advanced intermediate 232. Compound 232 was treated with Mn(dpm)3 in the presence of TBHP and PhSiH3 to induce diastereoselective HAT reduction followed by deprotection to generate the natural product stypodiol (170) in good yield. Using a similar synthetic protocol, diene 227 was coupled with excess pyrone 214 via single-electron-transfer (SET) to promote a formal [3+2] cycloaddition mediated by cerium ammonium nitrate (CAN) to yield the natural product decaturin (169). The iodide intermediate 226 was subjected to nickel-catalyzed cross-coupling with arene iodide 228, followed by the redox maneuver mediated by CAN and subsequent acid catalyzed intramolecular cyclization provided natural product taondiol (157a). Furthermore, iodide intermediate 226 was subjected to nickel-catalyzed cross-coupling with pyrone iodide 229 to afford compound 230, which upon Lewis acid catalyzed intramolecular cyclization afforded the desired natural product chevalone A (157) in good yields [84]. This synthetic strategy offers the opportunity to access many derivatives of these meroterpene natural product derivatives by fine-tuning the coupling partner.
The hongoquercins are tetracyclic meroterpenoid natural products with the trans-transoid decalin-dihydrobenzopyran ring system, which display a range of promising bioactivities. This subgroup of the meroterpenoids natural products incorporates a sesquiterpene unit to form the unique hongoquercins. These natural products have attracted attention because they pose a synthetic challenge with four continuous stereocenters and a highly substituted arene scaffold. Access to a modular synthesis can enable the establishment of a medicinal chemistry platform to investigate their exact mode of action. Over the past 25 years multiple synthetic endeavors have been reported towards meroterpenoid (+)-hongoquercin A (152, Scheme 18) and (+)-hongoquercin B (153, Scheme 19), but a recent simplified approach was disclosed by the Barrett group [85], featuring a sequential polyketide aromatization and cationic polyene cyclization reaction. The synthesis commenced with thermolysis of dioxane-4,6-dione dioxanone 236 in the presence of trans, trans-farnesyl 234 or geraniol 235 to provide dioxinone β-keto ester 238 or 237 respectively, which upon C-acylation generated the corresponding ester systems. The resultant products underwent highly regioselective decarboxylative allylic rearrangement mediated by Pd2dba3, followed by in situ aromatization to provide farnesyl resorcylate 233 or the geranyl substituted analogue 239 respectively. Compound 233 was then treated with SnCl4 complex with ligand 240 as a dual Bronsted and Lewis acid, in the presence of SnCl4 and trifluoroacetic acid, which facilitated the enantioselective protonation of the substrate, followed by facile cationic intramolecular polyene cyclization reaction, leading to meroterpenoid 241. Compound 241 upon saponification provided the natural product (+)-hongoquercin A (152). Alternatively, O-allylation of farnesyl resorcylate 233 with allylbromide under basic condition provided allyl ether 242, which upon asymmetric dihydroxylation generated diol 244. This step is followed by mesylating the secondary hydroxyl group, which enables the epoxide formation under a classic SN2 reaction to provide epoxide 245. Compound 245 was then deprotected, followed by biomimetic cationic cyclization of epoxide mediated by Lewis acid, Iron (III) to generate the advanced meroterpenoid derivative 247 in good enantioselective excess.
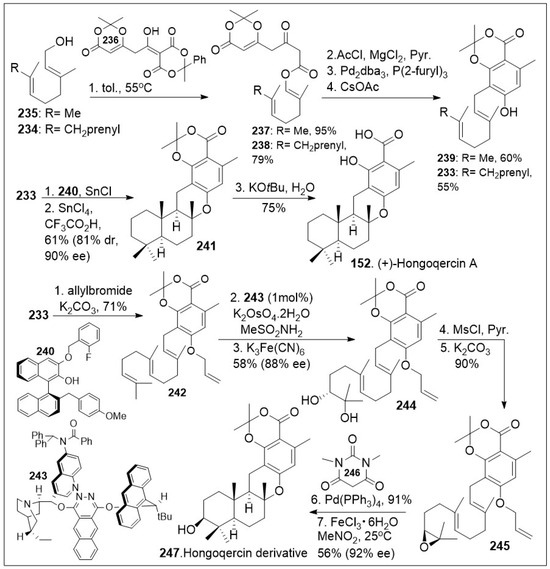
Scheme 18.
Total synthesis of (+)-Hongoqercin A.
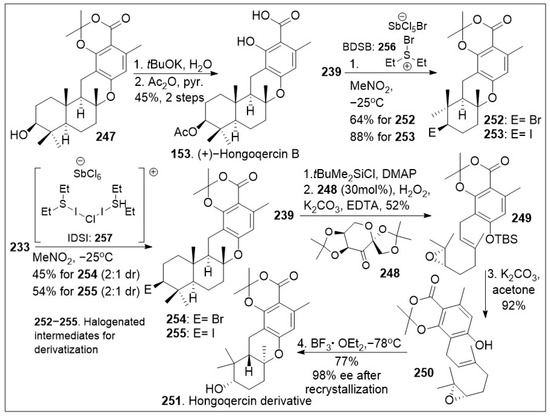
Scheme 19.
Total synthesis of (+)-Hongoqercin B and halogenated derivatives.
As shown in Scheme 19, compound 247 was deprotected to yield the corresponding acid, followed by selective acetylation of the secondary hydroxyl group to provide natural product 153. Furthermore, halocyclization of farnesyl resorcylate 233 or geranyl-substituted analogue 239 (Scheme 18) in the presence of halogenating reagents 256 (Bromodiethylsulfonium bromopentachloroantimonate V) or 257 (iodonium reagent), provided halogenated meroterpenoid 254/255, or 252/253 respectively in good yields [85]. These important intermediates will allow the generation of several derivatives that can further provide insight into the biological mode of action of these natural products. An interesting meroterpenoid derivative of hongoquercin was generated geranyl-substituted resorcylate 239. This was protected with a silyl group followed by regioselective asymmetric epoxidation with Shi chiral ketone to generate epoxide 249. The resultant system was deprotected under basic conditions to yield epoxide 250, which underwent cationic polyene cyclization reaction mediated by boron trifluoride etherate to provide the advanced meroterpenoid derivative 251. This compound should serve as a chemical tool to interrogate the biological properties of these natural products.
A modular synthetic approach to the tetracyclic meroterpenoids was recently reported [86] and it is as illustrated in Scheme 20. The synthesis featuring sterically demanding sp2-sp3 cross-coupling and acid mediated cyclization/isomerization for installation of the required cis or trans-decalin stereochemistry of the tetracyclic meroterpenoid scaffold is highlighted. The total synthesis commenced with a solvent-free Alder-Rickert reaction between the dimedone-derived bis-trimethylsilyl enol ether 258 and dimethyl 2-butynedioate (DMAD) provided resorcinol, followed by mono-methylation yielded phenol, which upon thermal treatment in the presence of 3,4-dimethoxybenzylamine (DMBNH2) yielded imide 259. Regioselective halogenation of imide 259 generated intermediate 259a, which upon regioselective imide reduction provided key isoindolinone 260. Next diene 263 was generated from β-hydroxyketone 261 via three step protocol, involving the formation of benzyl-protected ketone, subsequent vinyltriflate formation, followed by Stille coupling with vinyl(nBu3)Sn mediated by Pd(PPh3)4. Then, a Lewis acid catalyzed Diels-Alder [4+2] cycloaddition reaction between diene 263 and dienophile 264 provided intermediate 265 as the major product due to the shown transition state. Compound 265, following a two-step sequential reduction reaction, generated alcohol via thioester intermediate 266, and it was converted to iodide 267 via two linear steps (mesylation of the alcohol followed by SN2 reaction). Then, the key step was conducted, namely sp2-sp3 Negishi coupling of isoindolinone 260 with iodide 267 to provided 5,6-dehydrodecalin intermediate 268. Next compound 268 underwent deprotection, intramolecular cation trapping of the phenol group, followed by hydrogenation to yield the cyclized product 272. This established the required meroterpenoid scaffold. Lastly, global deprotection of intermediate 272 provided the natural product (+)-stachyflin (162).
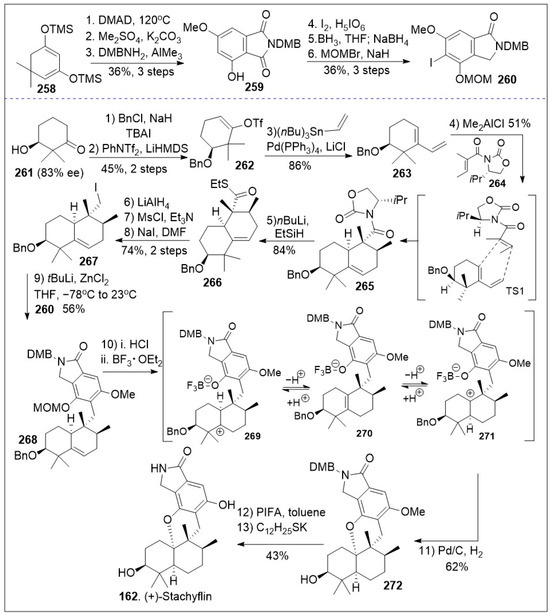
Scheme 20.
Modular synthesis of tetracyclic meroterpenoids.
Next as shown in Scheme 21, thioester 273 was reduced to the corresponding aldehyde 274 via Pd(II) in the presence of triethylsilane hydride in good yields. Then, aldehyde was coupled with the anion of arene 275 or 277, followed by 2-step Barton-McCombie deoxygenation to produce intermediate 276 or 278 respectively. Then, compound 278 was converted to mamanuthaquinone (165) via global acid deprotection and salcomine (a coordination complex of salen ligand and cobalt) mediated oxidation reaction sequence in moderate yields. In addition, compound 276 was subjected to deprotection and intramolecular cation trapping with the free phenol group to provide natural product aureol (279), which was subsequently converted to 5-epi-aureol (166) via epimerization of the decalin bridge carbon, using HI. Intermediate 166 is known to generate natural product cyclosmenospongine 164 [86]. The efficient synthesis provides rapid access to various family members and the established platform will enable the generation of derivatives for further biological studies.
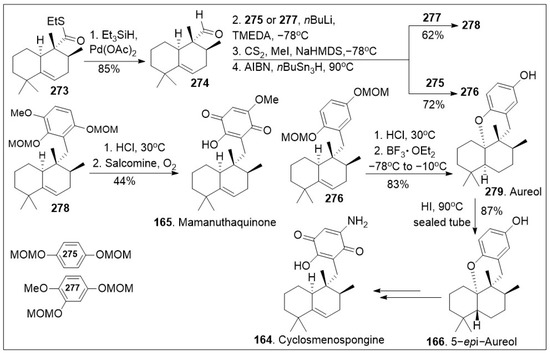
Scheme 21.
Synthesis of (+)-stachyflin, aureol, cyclosmenospongine, and mamanuthaquinone.
These unique spiromeroterpenoids share a common biosynthetic pathway that presumably arises from the terpenoid precursor farnesyldiphosphate and display promising biological properties [87]. The total synthesis of asnovolins and related spiromeroterpenoids feature a key lithium mediated coupling reaction to establish the carbon-carbon bond formation that giving rise to the spirocenter, was recently reported by Porco [87], shown in Scheme 22. The synthesis began with known chiral hydroxy enone 283, which was converted to the corresponding neopentyl iodide via one-pot triflation, followed by an SN2 reaction, which upon subsequent Luche reduction, provided allylic alcohol 282. Coupling reaction between the decalin system 281 and the lithium exchanged form of the iodide 282 via 1,2-addition reaction afforded compound 284. Next, treatment of the resultant product 284 with MnO2 was converted to spirocycle 280 via tandem allylic oxidation, followed by oxa-Michael addition reaction sequence. Spirocycle 280 was then transformed to spirocyclic intermediate 285 via a metal hydride atom transfer (MHAT) process mediated by Fe(acac)3 in the presence of hydride source PhSiH3, establishing the desired stereochemistry. Enolization of spirocyclic intermediate 285 with TMSOTf followed by treatment with Eschenmoser’s salt, generated exocyclic enone 286. Under various reactions conditions compound 286 resisted isomerization to the endocyclic enone 287. Therefore, compound 285 was oxidized with 2-iodoxybenzoic acid (IBX) to the spirocyclic enone system, which was iodinated at the α-carbon to provide the halogenated spirocyclic intermediate 288. This system was then subjected to Suzuki coupling reaction to generate the desired endocyclic enone 287, followed by final desaturation, and protecting group removal to generate the spiromeroterpenoid chermesin B (172). Compound 288 could be further functionalized via Pd mediated methylation to produce 287 again, which was treated with base to conduct an enolate acylation and deprotection to access the natural product asnovolin B (168). The natural product 168 was then subjected to Baeyer-Villiger oxidation in the presence of Sc(OTf)3 to yield asnovolin A (167). The lactone ring opening of asnovolin A (167) was conducted using methanolysis conditions to provide natural product asnovolin E (173). In addition, compound 287 was acylated to generate compound 289, which was desaturated and deprotected to afford advanced spirocyclic intermediate 290. Finally, compound 290 was subjected to Meerwein-Ponndorf-Verley reduction conditions to produce simplicissin (171) as the major product. The powerful linear synthesis enables access to many members of these natural products
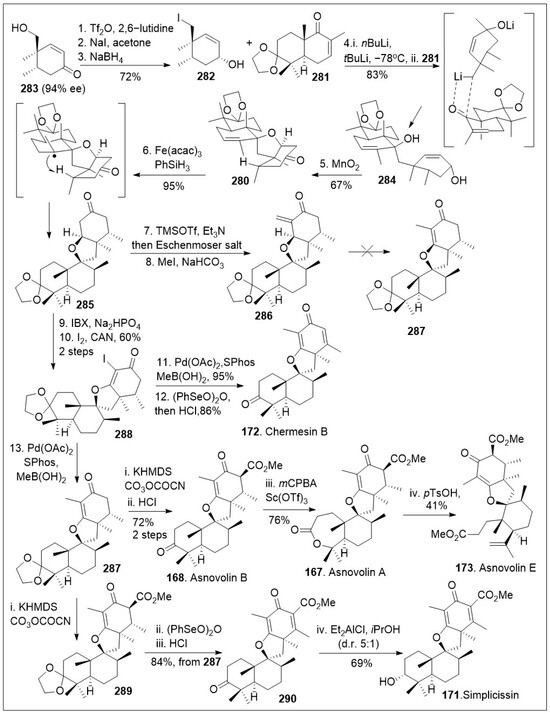
Scheme 22.
Synthesis of asnovolin A, asnovolin B, asnovolin E, chermesn B and simplicissin.
Asperterpene A and its related family are interesting structures with unique chemistry and biology. Asperterpene meroterpenoids displays an unprecedented 1,2,5-trimethyl-4,9-dioxobicyclo [3.3.1]non-2-ene-3-carboxylic acid moiety that is distinct from other similar meroterpenoids such as berkeleytrione and preaustinoid A. These natural products have exhibited promising BACE1 inhibitory activities in cell-based assays, with IC50 values of 60–80 nM. This is more active than LY2811376 (IC50 of 260 nM), a potent clinical BACE1 inhibitor produced by Eli Lilly. Additional in vivo studies of one of these natural products showed significant decrease of BACE1 activity and Ab42 levels in 3xTg AD mice, similar to LY2811376 [74]. Therefore, the synthesis of these compounds has clinical relevance, and more potent derivatives could be generated once a streamlined synthetic endeavor has been established. An impressive biomimetic synthesis towards these natural products was recently reported as shown in Scheme 23 [88]. The synthesis began with the coupling reaction of 3,5-dimethylorsellinic acid 291 with (2E,6E)-10,11-epoxyfarnesyl mesylate 292 as the electrophile under basic condition, followed by in situ methyl esterification to generate (2E,6E)-10,11-epoxyfarnesyl-5-dimethylorsellinic acid methyl ester 293. Then, a dearomatization-driven polycyclization reaction was mediated by treatment with alkyl aluminum-pair as a Lewis acid system to promote the intramolecular polyene cyclization cascade reaction to afford asperterpene scaffold 177 and related meroterpenoid 149 via intermediate 297 in a 1 to 3 ratio favoring compound 294. Alternatively, compound 291 was coupled with electrophile (2Z,6E)-10,11-epoxyfarnesyl mesylate 295 (the stereoisomer of 292) using base treatment, followed by in situ methyl esterification to generate compound 296. The resultant product was treated with alkyl aluminum Lewis acid system to mediate the intramolecular polyene cyclization cascade reaction, which afforded meroterpenoid scaffolds 150, 151, and 178. In addition, it was found that compound 149 could be treated with formic acid to provide natural product 178 in good yields. While the chemical yields are modest, this synthetic approach is remarkable as several centers are established in a single step, generating high complexity rapidly.
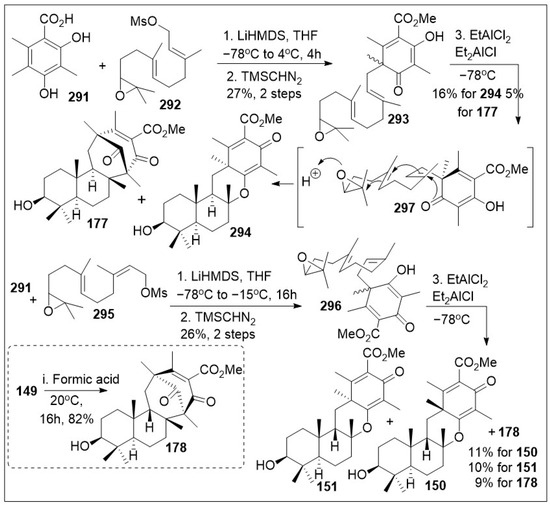
Scheme 23.
Biomimetic synthesis of asperterpene A derivative and related meroterpenoids.
Dysiherbols A-C are a set of sesquiterpene hydroquinone meroterpenoids first isolated and characterized from Dysidea sp., a species of marine sponge found in the South China Sea first isolated by Lin and co-workers [89]. These natural products are structurally unified by a sterically compact 6/6/5/6/6 pentacyclic skeleton embracing an impressive oxabicyclo[3.3.1] ring system. There are 5−6 contiguous stereogenic centers in their molecular scaffold, four of which are quaternary centers. These natural products as extract mixtures were identified to inhibit NF-κB [89]. Isolation and purification of these compounds demonstrated that the pure compounds had potent inhibitory values in the low micromolar range against NF-κB, finding IC50 values of 0.49 to 6 μM for dysiherbols A, B, and C respectively. Additionally, these compounds were tested against a human myeloma cancer cell line, NCI H-929, and showed antiproliferation effects in the low micromolar range, with dysiherbols A being the most active with an EC50 in the 500 nanomolar range. Recent divergent total syntheses of the revised structures of these sesquiterpene hydroquinone meroterpenoids (+)-dysiherbols A-E (179–183, Figure 5) was reported as shown in Scheme 24 and Scheme 25 [90]. The modular synthetic strategy began with compound 299 which had been reported to arise from the readily available dimethyl predysiherbol. Compound 299 was treated with gold to catalyze the tandem intramolecular cyclization reactions providing known Schmalz’s intermediate 300. Hydroboration/oxidative hydroxylation and subsequent Dess Martin Periodinane (DMP) oxidation reaction of compound 300 was converted to ketone 301. Then, compound 301 was followed by regioselective deprotonation/o-methylation under lithium hydride, α,α,α’-tris(4-hydroxyphenyl)-1-ethyl-4-isopropylbenzene (TPPA) followed by MeI, and subsequent Simmons-Smith cyclopropanation reaction sequence generated cyclopropane 302, which upon further proteolytic cleavage generated ketone 303. Then, enol triflate was formed to enable a Stille coupling reaction to afford compound 298. This compound was subjected to Lewis acid treatment at low temperatures to provide intermediate 304 through an intramolecular oxy-cyclization reaction, which upon increasing of the temperature under the same Lewis acid removed the additional methoxy group to yield the natural product (+)-dysiherbol A (179). Alternatively, compound 298 was treated with a 2-step allylic oxidation and reduction reaction sequence, followed by Lewis acid mono deprotection and subsequent intramolecular oxy-cyclization to provide compound 305. Finally, compound 305 was deprotected to provide natural product (+)-dysherbol E (183).
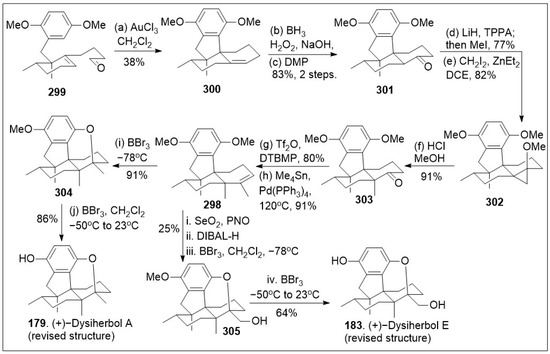
Scheme 24.
Divergent syntheses of (+)-dysiherbols A and (+)-dysiherbols E.
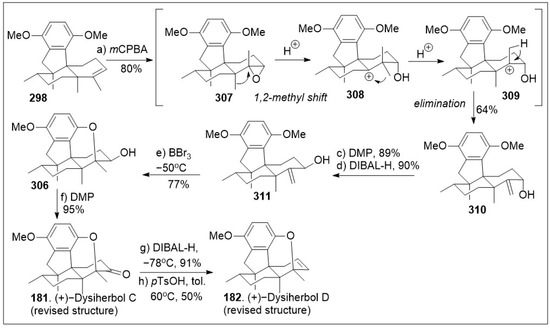
Scheme 25.
Divergent synthesis of marine meroterpenoids (+)-dysiherbols C and (+)-dysiherbols D.
In parallel, as shown in Scheme 25, compound 298 was treated with mCPBA to undergo cationic 1,2-methyl shift and elimination reaction sequence to provide allylic alcohol 310. The hydroxyl group geometry in 310 was inverted via a 2 step-reaction transformation. Namely, oxidation of the hydroxyl group using DMP treatment, followed by DIBAL-H reduction, which provided allylic alcohol 311. Then, intramolecular oxy-cyclization of 311 mediated by BBr3 generated cyclized ether 306, which was oxidized with DMP to yield (+)-dysiherbol C (181). Reduction of this enone with DIBAL-H, followed by acid mediated dehydration reaction provided (+)-dysiherbol D (182). In summary, this synthetic strategy of the tetracyclic core of dysiherbols through the use of critical acid mediated ring construction sequences [90]. This unique synthetic strategy serves as the foundation to access the natural products and their derivatives.
The strongylophorines meroditerpenoids have been isolated from various sources including the marine sponge Petrosia corticata, and they have been demonstrated to be promising proteasome inhibitors [91]. Access to these natural products to develop compound libraries is desirable. A recent streamlined total synthesis of strongylophorine-2 (STR-2, 184) is highlighted in Scheme 26 [92]. The synthesis capitalizes on a novel iron (III)-mediated rearrangement-cyclization cascade reaction. The synthesis commenced with the esterification of natural product isocuppressic acid 312, followed by treatment with 1,1'-(Azodicarbonyl)dipiperidine (ADDP) under Mitsunobu reaction conditions to etherify the allylic alcohol, generating compound 314. Next, compound 314 was exposed to Lewis acid Iron trichloride, which mediated the key acid catalyzed reaction that facilitated the C-O to C-C [3,3]-Claisen rearrangement, followed by ring closure to afford compound 316 as the major product as shown in Scheme 26. Benzyl deprotection with Pd/C provided STR-1 (184a), and further deprotection of the methoxy group led to STR-3 (184b). Alternatively, 184a could be subjected to amination conditions to yield compound 317, which was further functionalized with lead tetraacetate (LTA) and iodine-promoted hypoiodite reaction under photolysis to provide the di-iodo compound 318. Compound 318 was set for an intramolecular cyclization under basic conditions with elimination of the other iodo-group to provide 1,2-didehydro-STR-9 (184c) as the unique intermediate, which was reduced under Pd/C to provide STR-9 (184d). Next removal of the methoxy-protection group resulted in the natural product STR-2 (184) in good yields.
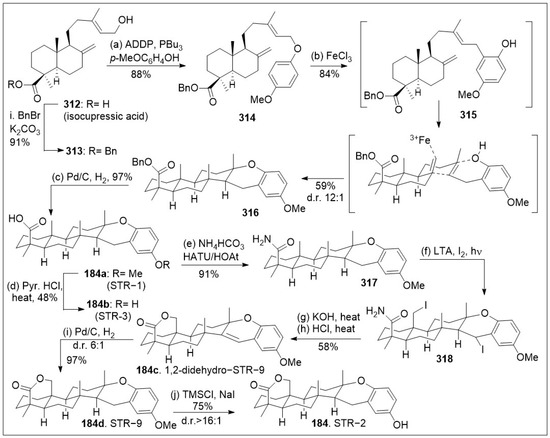
Scheme 26.
Synthesis of strongylophorine-2 (STR-2).
Finally, a selected number of monoterpene indole alkaloids, which represent a large group of more than 3000 different compounds and are mainly present in three families: Rubiaceae, Apocynaceae, and Loganiaceae will be included in this review. Numerous phytochemical studies have led to the discovery of potent bioactive molecules as exemplified by the anti-cancer natural product vinblastine. Minfiensine, a secoiridoid indole alkaloid-type, which was isolated from the African plant Strychnos minfiensis by Massiot and co-workers in 1989 with no structural precedence at the time has since attracted various synthetic chemists. Extracts containing these meroterpene alkaloids are used throughout the world in the practice of traditional medicine, with some heightened interest in the medicinal potential of the pure natural products. A recent short total synthesis of monoterpene alkaloid (+)-minfiensine (185) via an asymmetric palladium mediated cyclization reaction was reported [93]. The total synthesis is depicted in Scheme 27, and it commenced with the coupling reaction of readily available allylic halide 319 and methyltryptamine 320. The 2-step transformation took place via a N-bromosuccinimide (NBS) mediated radical methyl-bromination of 320, followed by subsequent coupling with the allyl indium intermediate derived from the allylic halide 319 to provide compound 321, which upon DIBAL-H reduction and subsequent esterification produced allyl carbonate 322. Protecting group maneuver of compound 322 provided intermediate 323, which is subjected to an indole dearomative asymmetric allylic alkylation reaction mediated by Palladium (II) and the shown chiral ligand. The tetracyclic product 324 was generated in acceptable enantiomeric excess and good yields, and was N-protected, followed by epoxidation with mCPBA to afford a mixture of diastereomers of epoxide 326. Allylic alcohol 327 was subsequently generated from epoxide 326 via an epoxide ring-opening mediated by 2,2,6,6-Tetramethylpiperidine-dimethyl Aluminum complex (TMP-AlMe2). Protection of the resultant alcohol with a silyl group followed by coupling with vinyl iodide 328 under basic condition provided N-alkylated intermediate 329. Use of a Pd(OAc)2-catalyzed intramolecular Heck-type reaction developed by Overman and Wrobleski for this molecule, followed by a spontaneous β-elimination reaction generated a polycyclic intermediate. This intermediate was deprotected under basic conditions provided (+)-minfiensine (185). The synthesis is efficient at generating high complexity.
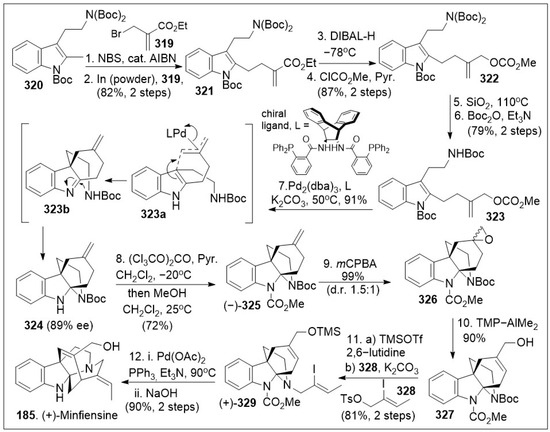
Scheme 27.
Total synthesis of monoterpene indole alkaloid (+)-minfiensine.
The total synthesis of meroterpene indole alkaloid (+)-limaspermidine (186, Scheme 28) was completed in a unified approach to demonstrate the power of palladium mediated cyclization reactions [94]. Scheme 28 illustrates an efficient approach to this natural product from β-amidoester 332, which was generated from sequential double C-alkylation of dihydropyrido[1,2-a]indolone 330 first alkylation with allyl cyanoformate in the presence of LiHMDS, and second alkylation using (2-benzyloxy)ethyl iodide 331 under basic condition reactions. Then, decarboxylative asymmetric allylation of compound 332 was mediated by Pd2(pmdba)3 with a chiral ligand to provide compound 333 featuring the required α-quaternary all-carbon stereocenter in good enantioselective excess and chemical yields. Next, compound 333 was subjected to a formal anti-Markovnikov hydroamination reaction to generate the corresponding primary amine, which was directly treated with LiAlH4 and quenched with aqueous acetic acid to promote intramolecular indole-iminium cyclization reaction. This furnished the cis-fused tetracyclic system followed by chemoselective piperidine N-alkylation to afford compound 335. Compound 335 was converted to polycyclic imine 336 via 2-step pyrrolidine annulation by generating the leaving group, followed by base treatment. Subsequent NaBH4 mediated reduction of polycyclic imine 336 and benzyl deprotection provided the natural product (+)-limaspermidine 186 in overall good yields.
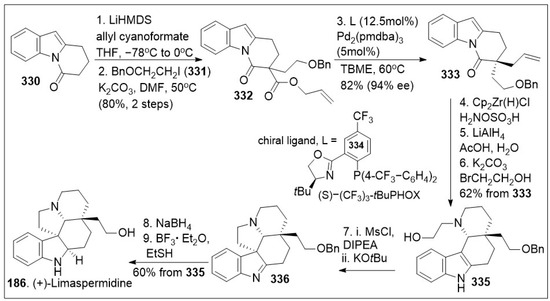
Scheme 28.
Total synthesis of (+)-limaspermidine.
In parallel, other similar synthetic strategies to this family of meroterpenoids with alkaloid features have been reported [94]. The total synthesis of (+)-kopsihainanine A (187) is shown in Scheme 29 and begins with β-amidoester 337, which was generated via sequential double α-C-alkylation of dihydropyrido[1,2-a]indolone 330, using allyl cyanoformate in the presence of LiHMDS. This was followed by treatment with methyl acrylate 331 under basic conditions. Further decarboxylative asymmetric allylation of compound 337 in the presence of Pd2(pmdba)3 with a chiral ligand provided an intermediate with the α-quaternary all-carbon stereocenter, which underwent Rh-catalyzed hydroboration to yield alcohol 338. Compound 338 was mesylated and displaced by the azide group to generate compound 339, followed by Staudinger reduction mediated by polymer-bond PPh3 with concomitant translactamization, which provided δ-lactam 340. Subsequent Bischler-Napieralski cyclization of δ-lactam 340 furnished trans-fused tetracycle 341. This was subjected to intramolecular lactamization between the piperidine nitrogen, and the pendant methyl ester group mediated by bicyclic guanidine base triazabicyclodecene (TBD) and generated pentacycle 342. Upon α-hydroxylation mediated by lithium dimethylamide (LDMA) in the presence of HMPA, using bis(trimethylsilyl) peroxide as the oxidant successfully afforded the natural product (+)-kopsihainanine A (187). The overall synthetic maneuvers maximize the chemical yields, and a distinctive feature of this concise synthetic endeavors is the use of palladium-catalyzed reactions to form all the carbon-carbon bonds in the transformation of simple precursors to highly elaborate natural products with promising biological properties.
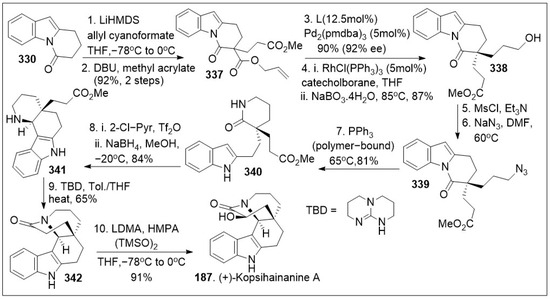
Scheme 29.
Total synthesis of (+)-kopsihainanine A.
The total synthesis of cymoside, an oxidized derivative of strictosidine with an unprecedented hexacyclic skeleton which includes a double bridge linking the indole and the monoterpene moieties was isolated from the leaves of Chimarrhis cymose and provides a fertile ground for organic chemists as shown in Scheme 30. The total synthesis of cymoside (188) via biomimetic oxidative cyclization was reported [95]. The synthesis commenced with Knoevenagel condensation reaction between aldehyde 345 and 3-formyl-1,1,1-trichloroacetone 346 which generated enone intermediate, which upon inverse-demand hetero-Diels-Alder (HDA) reaction with enol ether 347 furnished cyclic intermediate 348 together with other diastereoisomers. Upon methanolysis and sulfoxide elimination, intermediate 348 was further converted to olefin intermediate, followed by deprotection furnished Tietze secologanin aglycon 349. Subsequent Pictet-Spengler reaction between the secologanin aglycon 349 and tryptamine 356, followed by protection of the secondary amine furnished the mixture of the protected strictosidine aglycon ethyl ether 350a and its epimer 350b. The biomimetic cascade reaction sequence of oxidation with oxaziridine 351 and acid catalyzed cyclization reaction of strictosidine aglycon ethyl ether 350a furnished the complete framework 352 of cymoside via intermediate 352a, 352b, and 352c, which was deprotected to generate ethyl ether aglycon 353 of cymoside. Hydrolysis of acetal 352 provided hemiacetal 354, which upon Schmit glycosylation with glycosyl trichloroacetimidate, generated glycosylated product 355a with its diastereoisomer, together with glycosylated product 355b. Global deprotection of intermediate 355a furnished the natural product cymoside 188, global deprotection of compound 355b provided product 188a as the diastereoisomer of cymoside 188.
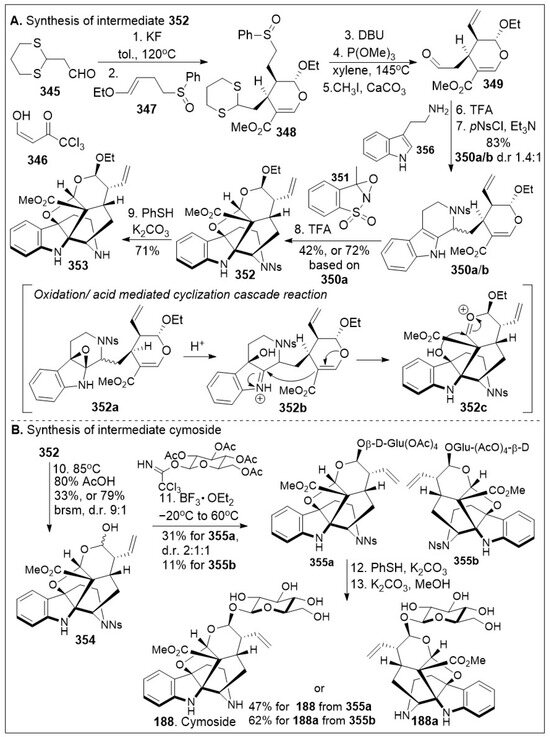
Scheme 30.
Total synthesis of cymoside via biomimetic oxidative cyclization strategy.
4. Conclusions
The described monoterpene and meroterpenoid families featured herein display an array of impressive biological activities, including antioxidant/anticancer properties. Natural resources have been an essential part of human health development for centuries and it continues to serve as an invaluable source of inspiration. The medical community has made great advances in small molecule development, but there is still a heavy reliance on natural products for their pharmaceutical applications. Biological evaluation of these natural products provide the scientific community with a larger selection of tools to understand biological and chemical systems as well as a strong starting point for the production and refinement of these chemical entities as potential therapeutic agents. Additionally, the development of synthetic methodology to access these compounds is a crucial process in the effort to conduct biological studies and large-scale production and derivatization. In order to meet the challenges of generating these complex meroterpenoids, several innovative synthetic strategies were developed including modern chemoenzymatic transformations, cationic polyene cyclization reaction, dearomatization-driven polycyclization reaction, metal mediated rearrangement cyclization and photochemical transformations.
Author Contributions
Conceptualization, F.R. and T.L.; writing—original draft preparation, B.J.B.; F.R. and T.L.; writing—review and editing, T.L., B.J.B. and F.R. project administration, F.R. and T.L.; funding acquisition, F.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health grant number R15GM148983, LSU-2022-CCRI-9, and Board of Regents Support Fund Award LEQSF-RD-A-05 to F.R.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Metwaly, A.M.; Ghoneim, M.M.; Eissa, I.H.; Elsehemy, I.A.; Mostafa, A.E.; Hegazy, M.M.; Afifi, W.M.; Dou, D. Traditional Ancient Egyptian Medicine: A Review. Saudi J. Biol. Sci. 2021, 28, 5823–5832. [Google Scholar] [CrossRef] [PubMed]
- Veeresham, C. Natural Products Derived from Plants as a Source of Drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Ekor, M. The Growing Use of Herbal Medicines: Issues Relating to Adverse Reactions and Challenges in Monitoring Safety. Front Pharmacol. 2014, 4, 177. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Biodiversity: A Continuing Source of Novel Drug Leads. Pure Appl. Chem. 2005, 77, 7–24. [Google Scholar] [CrossRef]
- Kamran, S.; Sinniah, A.; Abdulghani, M.A.M.; Alshawsh, M.A. Therapeutic Potential of Certain Terpenoids as Anticancer Agents: A Scoping Review. Cancers 2022, 14, 1100. [Google Scholar] [CrossRef]
- Tomko, A.M.; Whynot, E.G.; Ellis, L.D.; Dupré, D.J. Anti-Cancer Potential of Cannabinoids, Terpenes, and Flavonoids Present in Cannabis. Cancers 2020, 12, 1985. [Google Scholar] [CrossRef]
- Wróblewska-Łuczka, P.; Cabaj, J.; Bargieł, J.; Łuszczki, J.J. Anticancer Effect of Terpenes: Focus on Malignant Melanoma. Pharmacol. Rep. 2023, 75, 1115–1125. [Google Scholar] [CrossRef]
- Martignago, C.C.S.; Soares-Silva, B.; Parisi, J.R.; Silva, L.C.S.e.; Granito, R.N.; Ribeiro, A.M.; Renno, A.C.M.; de Sousa, L.R.F.; Aguiar, A.C.C. Terpenes Extracted from Marine Sponges with Antioxidant Activity: A Systematic Review. Nat. Prod. Bioprospect. 2023, 13, 23. [Google Scholar] [CrossRef]
- Ruzicka, L. The Isoprene Rule and the Biogenesis of Terpenic Compounds. Experientia 1953, 9, 357–367. [Google Scholar] [CrossRef]
- Alcántara, A.R.; Hernaiz, M.-J.; Sinisterra, J.-V. Biocatalyzed Production of Fine Chemicals. In Comprehensive Biotechnology; Elsevier: Amsterdam, The Netherlands, 2011; pp. 309–331. ISBN 978-0-08-088504-9. [Google Scholar]
- Ganjewala, D.; Kumar, S.; Luthra, R. An Account of Cloned Genes of Methyl-Erythritol-4-Phosphate Pathway of Isoprenoid Biosynthesis in Plants. Curr. Issues Mol. Biol. 2009, 11 (Suppl. S1), i35–i45. [Google Scholar]
- Banerjee, A.; Wu, Y.; Banerjee, R.; Li, Y.; Yan, H.; Sharkey, T.D. Feedback Inhibition of Deoxy-D-Xylulose-5-Phosphate Synthase Regulates the Methylerythritol 4-Phosphate Pathway. J. Biol. Chem. 2013, 288, 16926–16936. [Google Scholar] [CrossRef] [PubMed]
- Kuzuyama, T.; Seto, H. Two Distinct Pathways for Essential Metabolic Precursors for Isoprenoid Biosynthesis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, C.; Yang, L.; Choi, E.-S.; Kim, S.-W. Geranyl Diphosphate Synthase: An Important Regulation Point in Balancing a Recombinant Monoterpene Pathway in Escherichia Coli. Enzyme Microb. Technol. 2015, 68, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Leferink, N.G.H.; Dunstan, M.S.; Hollywood, K.A.; Swainston, N.; Currin, A.; Jervis, A.J.; Takano, E.; Scrutton, N.S. An Automated Pipeline for the Screening of Diverse Monoterpene Synthase Libraries. Sci. Rep. 2019, 9, 11936. [Google Scholar] [CrossRef]
- Sharma, E.; Anand, G.; Kapoor, R. Terpenoids in Plant and Arbuscular Mycorrhiza-Reinforced Defence against Herbivorous Insects. Ann. Bot. 2017, 119, 791–801. [Google Scholar] [CrossRef] [PubMed]
- LOTUS: Natural Products Online. Available online: https://lotus.naturalproducts.net/compound/lotus_id/LTS0246122 (accessed on 12 November 2023).
- Bansal, H. Bioactivity Assessment of Essential Oils of Cymbopogon Species Using a Network Pharmacology Approach. Biol. Futur. 2022, 73, 107–118. [Google Scholar] [CrossRef]
- Boonyanugomol, W.; Rukseree, K.; Prapatpong, P.; Reamtong, O.; Baik, S.C.; Jung, M.; Shin, M.K.; Kang, H.; Lee, W.K. Endo-plasmic Reticulum Stress and Impairment of Ribosome Biogenesis Mediate the Apoptosis Induced by Ocimum x africanum Essential Oil in a Human Gastric Cancer Cell Line. Medicina 2022, 58, 799. [Google Scholar] [CrossRef]
- Abu-Serie, M.M.; Andrade, F.; Cámara-Sánchez, P.; Seras-Franzoso, J.; Rafael, D.; Díaz-Riascos, Z.V.; Gener, P.; Abasolo, I.; Schwartz, S., Jr. Pluronic F127 micelles improve the stability and enhance the anticancer stem cell efficacy of citral in breast cancer. Nanomedicine 2021, 16, 1471–1485. [Google Scholar] [CrossRef]
- Sakineh, A.; Noorbakhsh, M.F.; Ahmadi, N.; Saeed, N.; Behdokht, B. Evaluation of the Protective Effect of Citral, Si-lymarin, and Thymoquinone on Methotrexate-Induced Lung Injury in Rats. J. Pharmacopunct. 2023, 26, 184–191. [Google Scholar] [CrossRef]
- Habib, S.; Gupta, P.; Bhat, S.S.; Gupta, J. In silico, in-vitro and in vivo screening of biological activities of citral. Int. J. Vitam Nutr. Res. 2021, 91, 251–260. [Google Scholar] [CrossRef]
- Ho, Y.; Suphrom, N.; Daowtak, K.; Potup, P.; Thongsri, Y.; Usuwanthim, K. Anticancer Effect of Citrus hystrix DC. Leaf Extract and Its Bioactive Constituents Citronellol and, Citronellal on the Triple Negative Breast Cancer MDA-MB-231 Cell Line. Pharmaceuticals 2020, 13, 476. [Google Scholar] [CrossRef]
- Fatima, K.; Luqman, S. Suppression of Molecular Targets and Antiproliferative Effect of Citronellal on Triple-Negative Breast Cancer Cells. Curr. Mol. Pharmacol. 2021, 14, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.S.; Guimarães, A.G.; Santana, M.F.; Siqueira, R.S.; De Lima Ado, C.; Dias, A.S.; Santos, M.R.; Onofre, A.S.; Quintans, J.S.; De Sousa, D.P.; et al. Anti-inflammatory and redox-protective activities of citronellal. Biol. Res. 2011, 44, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Bordoloi, M.; Saikia, S.; Kolita, B.; Sarmah, R.; Roy, S.; Narzary, B. Volatile Inhibitors of Phosphatidylinositol-3-Kinase (PI3K) Pathway: Anticancer Potential of Aroma Compounds of Plant Essential Oils. ACAMC 2018, 18, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.S.; Rolo, J.; Gaspar, C.; Cavaleiro, C.; Salgueiro, L.; Palmeira-de-Oliveira, R.; Ferraz, C.; Coelho, S.; Pastorinho, M.R.; Sousa, A.C.; et al. Chemical characterization and bioactive potential of Thymus×citriodorus (Pers.) Schreb. preparations for anti-acne applications: Antimicrobial, anti-biofilm, anti-inflammatory and safety profiles. J. Ethnopharmacol. 2022, 287, 114935. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.J.; Tadros, M.G.; Michel, H.E. Geraniol protects against cyclophosphamide-induced hepatotoxicity in rats: Possible role of MAPK and PPAR-γ signaling pathways. Food Chem. Toxicol. 2020, 139, 111251. [Google Scholar] [CrossRef] [PubMed]
- Tunjung, W.A.S.; Fajarina, S.; Prabowo, B.H.; Damayanti, F.; Widyasari, A.; Sasongko, A.B.; Indrianto, A.; Semi-arti, E.; Hidayati, L. Evaluation of Anticancer Bioactive Compounds and Cytotoxicity of Kaffir Lime (Citrus hystrix Dc.) Callus Extract Post Preservation. Indones. J. Pharm. 2021, 32, 179–192. [Google Scholar] [CrossRef]
- Yan, N.; Wang, Z.; Gao, X. Analysis of the Volatile Components of Lithocarpus polystachyus Rehd. and a Preliminary Study of Their Anticancer Activity. Mod. Food Sci. Technol. 2017, 33, 35–39. [Google Scholar] [CrossRef]
- He, Z.; Shen, X.; Cheng, Z.; Wang, R.; Lai, P.; Xing, X. Chemical Composition, Antibacterial, Antioxidant and Cytotoxic Activities of the Essential Oil of Dianella ensifolia. Rec. Nat. Prod. 2019, 14, 160–165. [Google Scholar] [CrossRef]
- Agnieszka, S. Comparative study on the free radical scavenging mechanism exerted by geraniol and geranylacetone using the combined experimental and theoretical approach: Anti-radical activity of geraniol and geranylacetone. Flavour Fragr. J. 2015, 30, 399–409. [Google Scholar] [CrossRef]
- Rocha, D.; Seca, A.; Pinto, D. Seaweed Secondary Metabolites In Vitro and In Vivo Anticancer Activity. Mar. Drugs 2018, 16, 410. [Google Scholar] [CrossRef]
- Miastkowska, M.; Kantyka, T.; Bielecka, E.; Kałucka, U.; Kamińska, M.; Kucharska, M.; Kilanowicz, A.; Cudzik, D.; Cudzik, K. Enhanced Biological Activity of a Novel Preparation of Lavandula Angustifolia Essential Oil. Molecules 2021, 26, 2458. [Google Scholar] [CrossRef]
- Elbe, H.; Ozturk, F.; Yigitturk, G.; Baygar, T.; Cavusoglu, T. Anticancer activity of linalool: Comparative investigation of ultrastructural changes and apoptosis in breast cancer cells. Ultrastruct. Pathol. 2022, 46, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, S.; Deng, W. Dual responsive linalool capsules with high loading ratio for excellent antioxidant and anti-bacterial efficiency. Colloids Surf. B Biointerfaces 2020, 190, 110978. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.; Abdalla, A.N.; Obaid, N.A.; Youssef, L.; Naffadi, H.M.; Elzubier, M.E.; Almaimani, R.A.; Flamini, G.; Pieracci, Y.; El-Readi, M.Z. In Vitro Anticancer and Antibacterial Activities of the Essential Oil of Forsskal's Basil Growing in Ex-treme Environmental Conditions. Life 2023, 13, 651. [Google Scholar] [CrossRef]
- Mary Mawumenyo Mamattah, K.; Kusiwaa Adomako, A.; Nketia Mensah, C.; Borquaye, L.S. Chemical Characteriza-tion, Antioxidant, Antimicrobial, and Antibiofilm Activities of Essential Oils of Plumeria alba (Forget-Me-Not). Biochem. Res. Int. 2023, 1040478. [Google Scholar] [CrossRef] [PubMed]
- de Lucena, J.D.; Gadelha-Filho, C.V.J.; da Costa, R.O.; de Araújo, D.P.; Lima, F.A.V.; Neves, K.R.T.; de Barros-Viana, G.S. L-linalool exerts a neuroprotective action on hemiparkinsonian rats. Naunyn. Schmiedebergs Arch. Pharmacol. 2020, 393, 1077–1088. [Google Scholar] [CrossRef]
- Marchese, E.; D’onofrio, N.; Balestrieri, M.L.; Castaldo, D.; Ferrari, G.; Donsì, F. Bergamot essential oil nanoemulsions: Antimicrobial and cytotoxic activity. Z. Naturforsch C J. Biosci. 2020, 75, 279–290. [Google Scholar] [CrossRef]
- Omurtag Özgen, P.S.; Biltekin, S.N.; İduğ, T.; Macit, Ç.; Ayran, İ.; Çelik, S.A.; Kultur, Ş.; Yüksel, K.A.; Omurtag, G. Chemical composition and in vitro mutagenic, antioxidant and anti-inflammatory activities of Lavandula angustifolia Mill. essential oil from Turkey. J. Res. Pharm. 2022, 26, 781–791. [Google Scholar] [CrossRef]
- Ovidi, E.; Masci, V.L.; Taddei, A.R.; Paolicelli, P.; Petralito, S.; Trilli, J.; Mastrogiovanni, F.; Tiezzi, A.; Casadei, M.A.; Giacomello, P.; et al. Chemical Investigation and Screening of Anti-Proliferative Activity on Human Cell Lines of Pure and Nano-Formulated Lavandin Essential Oil. Pharmaceuticals 2020, 13, 352. [Google Scholar] [CrossRef]
- Caprari, C.; Fantasma, F.; Monaco, P.; Divino, F.; Iorizzi, M.; Ranalli, G.; Fasano, F.; Saviano, G. Chemical Profiles, In Vitro Antioxidant and Antifungal Activity of Four Different Lavandula angustifolia L. EOs. Molecules 2023, 28, 392. [Google Scholar] [CrossRef]
- Fahmy, M.; Farghaly, A.; Hassan, E.; Hassan, E.; Hassan, Z.; Mahmoud, K.; Omara, E. Evaluation of the Anti-Cancer/Anti-Mutagenic Efficiency of Lavandula Officinalis Essential Oil. Asian Pac. J. Cancer Prev. 2022, 23, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Al-Harrasi, A.; Shah, Y.A.; Jawad, M.; Al-Azri, M.S.; Ullah, S.; Anwer, M.K.; Aldawsari, M.F.; Koca, E.; Aydemir, L.Y. The Effect of Sage (Salvia sclarea) Essential Oil on the Physiochemical and Antioxidant Properties of Sodium Algi-nate and Casein-Based Composite Edible Films. Gels 2023, 9, 233. [Google Scholar] [CrossRef]
- Ramdane, F.; Medjour, O.; Ben, A.; Hadjadj, S.; Guemouda, M.; Kadri, M.; Hadj, M. Essential Oil and Ethanolic Extract Composition from Myrtus Nivellei Batt. & Trab. and Their Biological Evaluations. Acta Period. Technol. 2021, 52, 111–121. [Google Scholar] [CrossRef]
- Biltekin, S.N.; Karadağ, A.E.; Demirci, F.; Demirci, B. In Vitro Anti-Inflammatory and Anticancer Evaluation of Mentha spicata L. and Matricaria chamomilla L. Essential Oils. ACS Omega 2023, 8, 17143–17150. [Google Scholar] [CrossRef] [PubMed]
- Sajid, A.; Manzoor, Q.; Sajid, A.; Nazir, A.; Mumtaz, M.A.; Fatima, N.; Alshawwa, S.Z.; Iqbal, M.; Younas, U. Downregulation of NF-κB Activation Pathways Using Essential Oil Derived from Citrus Pseudolimon: Anticancer and Anti-Inflammatory Potential. Biocatal. Agric. Biotechnol. 2023, 47, 102599. [Google Scholar] [CrossRef]
- Calva, J.; Cartuche, L.; Castillo, L.N.; Morocho, V. Biological Activities and Chemical Composition of Essential Oil from Hedyosmum purpurascens (Todzia)-An Endemic Plant in Ecuador. Molecules 2023, 28, 2366. [Google Scholar] [CrossRef]
- Cortes-Torres, A.G.; López-Castillo, G.N.; Marín-Torres, J.L.; Portillo-Reyes, R.; Luna, F.; Baca, B.E.; Sandoval-Ramírez, J.; Carrasco-Carballo, A. Cymbopogon citratus Essential Oil: Extraction, GC-MS, Phytochemical Analysis, Antioxidant Activity, and In Silico Molecular Docking for Protein Targets Related to CNS. Curr. Issues Mol. Biol. 2023, 45, 5164–5179. [Google Scholar] [CrossRef]
- Dakhlaoui, S.; Bourgou, S.; Bachkouel, S.; Ben Mansour, R.; Ben-Jemaa, M.; Jallouli, S.; Megdiche-Ksouri, W.; Hessini, K.; Msaada, K. Essential oil composition and biological activities of Aleppo pine (Pinus halepensis Miller) needles collected from different Tunisian regions. Int. J. Environ. Health Res. 2023, 33, 83–97. [Google Scholar] [CrossRef]
- Maresca, V.; Badalamenti, N.; Ilardi, V.; Bruno, M.; Basile, A. The Antioxidant Properties and Protective Capacity of Prangos trifida and Cachrys cristata Essential Oils against Cd Stress in Lunularia cruciata and Brassica napus. Antioxidants 2023, 12, 793. [Google Scholar] [CrossRef]
- Muñoz-Acevedo, A.; González, M.C.; De Moya, Y.S.; Rodríguez, J.D. Volatile Metabolites of Piper eriopodon (Miq.) C.DC. from Northern Region of Colombia and Assessment of In Vitro Bioactivities of the Leaf Essential Oil. Molecules 2023, 28, 2594. [Google Scholar] [CrossRef]
- Azimullah, S.; Jayaraj, R.L.; Meeran, M.F.N.; Jalal, F.Y.; Adem, A.; Ojha, S.; Beiram, R. Myrcene Salvages Rotenone-Induced Loss of Dopaminergic Neurons by Inhibiting Oxidative Stress, Inflammation, Apoptosis, and Autophagy. Molecules 2023, 28, 685. [Google Scholar] [CrossRef]
- Zielińska-Błajet, M.; Feder-Kubis, J. Monoterpenes and Their Derivatives—Recent Development in Biological and Medical Applications. Int. J. Mol. Sci. 2020, 21, 7078. [Google Scholar] [CrossRef]
- Arabloei Sani, M.; Yaghmaei, P.; Hajebrahimi, Z.; Hayati Roodbari, N. Therapeutic Effect of P-Cymene on Lipid Profile, Liver Enzyme, and Akt/Mtor Pathway in Streptozotocin-Induced Diabetes Mellitus in Wistar Rats. J. Obes. 2022, 1015669. [Google Scholar] [CrossRef] [PubMed]
- AlSaffar, R.M.; Rashid, S.; Ahmad, S.B.; Rehman, M.U.; Hussain, I.; Parvaiz-Ahmad, S.; Ganaie, M.A. D-Limonene (5 (One-Methyl-Four-[1-Methylethenyl]) Cyclohexane) Diminishes CCl4-Induced Cardiac Toxicity by Alleviating Oxidative Stress, Inflammatory and Cardiac Markers. Redox Rep. 2022, 27, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Assali, M.; Jaradat, N.; Maqboul, L. The Formation of Self-Assembled Nanoparticles Loaded with Doxorubicin and d-Limonene for Cancer Therapy. ACS Omega 2022, 7, 42096–42104. [Google Scholar] [CrossRef]
- Ding, X.; Chen, H. Anticancer effects of Carvone in myeloma cells is mediated through the inhibition of p38 MAPK signalling pathway, apoptosis induction and inhibition of cell invasion. J. Buon. 2018, 23, 747–751. [Google Scholar]
- Abbas, M.M.; Kandil, Y.İ.; Abbas, M.A. R-(-)-Carvone Attenuated Doxorubicin Induced Cardiotoxicity In Vivo and Potentiated Its Anticancer Toxicity In Vitro. Balkan Med. J. 2020, 37, 98–103. [Google Scholar] [CrossRef]
- Tsai, K.; Liu, Y.-H.; Chen, T.-W.; Yang, S.-M.; Wong, H.-Y.; Cherng, J.; Chou, K.-S.; Cherng, J.-M. Cuminaldehyde from Cinnamomum Verum Induces Cell Death through Targeting Topoisomerase 1 and 2 in Human Colorectal Adenocarcinoma COLO 205 Cells. Nutrients 2016, 8, 318. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Hsu, W.-J.; Wu, L.-H.; Liou, H.-P.; Pangilinan, C.R.; Tyan, Y.-C.; Lee, C.-H. Hinokitiol Reduces Tumor Metastasis by Inhibiting Heparanase via Extracellular Signal-Regulated Kinase and Protein Kinase B Pathway. Int. J. Med. Sci. 2020, 17, 403–413. [Google Scholar] [CrossRef]
- Yang, S.-C.; Chen, H.-Y.; Chuang, W.-L.; Wang, H.-C.; Hsieh, C.-P.; Huang, Y.-F. Different Cell Responses to Hinokitiol Treatment Result in Senescence or Apoptosis in Human Osteosarcoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 1632. [Google Scholar] [CrossRef]
- Nazir, M.; Saleem, M.; Tousif, M.I.; Anwar, M.A.; Surup, F.; Ali, I.; Wang, D.; Mamadalieva, N.Z.; Alshammari, E.; Ashour, M.L.; et al. Meroterpenoids: A Comprehensive Update Insight on Structural Diversity and Biology. Biomolecules 2021, 11, 957. [Google Scholar] [CrossRef]
- Chen, X.-W.; Hou, Z.-C.; Chen, C.; Zhang, L.-H.; Chen, M.-E.; Zhang, F.-M. Enantioselective Total Syntheses of Six Natural and Two Proposed Meroterpenoids from Psoralea Corylifolia. Chem. Sci. 2023, 14, 5699–5704. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.M.; Hu, L.J.; Bai, Y.T.; Wang, J.; Qin, G.Q.; Song, Q.Y.; Su, J.C.; Huang, X.J.; Jiang, R.W.; Tang, W.; et al. Rhodomentosones A and B: Two Pairs of Enantiomeric Phloroglucinol Trimers from Rhodomyrtus tomentosa and Their Asymmetric Biomimetic Synthesis. Org. Lett. 2021, 23, 4499–4504. [Google Scholar] [CrossRef] [PubMed]
- Wenninger, M.; Maier, M.E.; Ammanath, A.V.; Huang, L.; Götz, F. Synthesis of Rhodomyrtone Analogs Modified at C7. Eur. J. Org. Chem. 2023, 26, e202300259. [Google Scholar] [CrossRef]
- Qin, X.J.; Rauwolf, T.J.; Li, P.P.; Liu, H.; McNeely, J.; Hua, Y.; Liu, H.Y.; Porco, J.A., Jr. Isolation and Synthesis of Novel Meroterpenoids from Rhodomyrtus tomentosa: Investigation of a Reactive Enetrione Intermediate. Angew. Chem. Int. Ed. Engl. 2019, 58, 4291–4296. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.M.; Hu, L.-H.; Tang, W.; Liu, J.-X.; Huang, X.-J.; Li, Y.-Y.; Li, Y.-L.; Ye, W.-C.; Wang, Y. A biomimetic synthesis-enabled stereochemical assignment of rhodotomentones A and B, two unusual caryophyllene-derived meroterpenoids from Rhodomyrtus tomentosa. Org. Chem. Front. 2021, 8, 5728–5735. [Google Scholar] [CrossRef]
- Deng, L.-M.; Tang, W.; Wang, S.-Q.; Song, J.-G.; Huang, X.-J.; Zhu, H.-Y.; Li, Y.-L.; Ye, W.-C.; Hu, L.-J.; Wang, Y. Discovery and Biomimetic Synthesis of a Polycyclic Polymethylated Phloroglucinol Collection from Rhodomyrtus Tomentosa. J. Org. Chem. 2022, 87, 4788–4800. [Google Scholar] [CrossRef]
- Tang, G.H.; Dong, Z.; Guo, Y.Q.; Cheng, Z.-B.; Zhou, C.-J.; Yin, S. Psiguajadials A–K: Unusual Psidium Meroterpenoids as Phosphodiesterase-4 Inhibitors from the Leaves of Psidium guajava. Sci. Rep. 2017, 7, 1047. [Google Scholar] [CrossRef]
- Lawrence, A.L.; Adlington, R.M.; Baldwin, J.E.; Lee, V.; Kershaw, J.A.; Thompson, A.L. A Short Biomimetic Synthesis of the Meroterpenoids Guajadial and Psidial A. Org. Lett. 2010, 12, 1676–1679. [Google Scholar] [CrossRef]
- Shen, X.; Thach, D.Q.; Ting, C.P.; Maimone, T.J. Annulative Methods in the Synthesis of Complex Meroterpene Natural Products. Acc. Chem. Res. 2021, 54, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Bao, J.; Wang, J.; Zhu, H.; Xue, Y.; Wang, X.; Li, H.; Sun, W.; Gao, W.; Lai, Y.; et al. Asperterpenes A and B, Two Unprecedented Meroterpenoids from Aspergillus Terreus with BACE1 Inhibitory Activities. Chem. Sci. 2016, 7, 6563–6572. [Google Scholar] [CrossRef]
- Tran, D.N.; Cramer, N. Biomimetic Synthesis of (+)-Ledene, (+)-Viridiflorol, (−)-Palustrol, (+)-Spathulenol, and Psiguadial A, C, and D via the Platform Terpene (+)-Bicyclogermacrene. Chem.-A Eur. J. 2014, 20, 10654–10660. [Google Scholar] [CrossRef] [PubMed]
- El-Elimat, T.; Raja, H.A.; Ayers, S.; Kurina, S.J.; Burdette, J.E.; Mattes, Z.; Sabatelle, R.; Bacon, J.W.; Colby, A.H.; Grinstaff, M.W.; et al. Meroterpenoids from Neosetophoma sp.: A Dioxa[4.3.3]Propellane Ring System, Potent Cytotoxicity, and Prolific Expression. Org. Lett. 2019, 21, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, A.C.; Haughan, M.A.; Khin, M.; Ekiert, J.; Zhang, Z.; Lantvit, D.; Al Subeh, Z.Y.; Pierre, H.C.; Salkovski, M.; Hirschhorn, T.; et al. Probing the Cytotoxic Signaling Induced by Eupenifeldin in Ovarian Cancer Models. J. Nat. Prod. 2023, 86, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Matsuda, D.; Ohtawa, M.; Fukuda, T.; Nagamitsu, T.; Yamori, T.; Tomoda, H. Potentiation of Bleomycin in Jurkat Cells by Fungal Pycnidione. Biol. Pharm. Bull. 2012, 35, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Bemis, C.Y.; Ungarean, C.N.; Shved, A.S.; Jamieson, C.S.; Hwang, T.; Lee, K.S.; Houk, K.N.; Sarlah, D. Total Synthesis and Computational Investigations of Sesquiterpene-Tropolones Ameliorate Stereochemical Inconsistencies and Resolve an Ambiguous Biosynthetic Relationship. J. Am. Chem. Soc. 2021, 143, 6006–6017. [Google Scholar] [CrossRef] [PubMed]
- Crossley, S.W.M.; Barabé, F.; Shenvi, R.A. Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc. 2014, 136, 16788–16791. [Google Scholar] [CrossRef]
- Li, J.; Li, F.; King-Smith, E.; Renata, H. Merging Chemoenzymatic and Radical-Based Retrosynthetic Logic for Rapid and Modular Synthesis of Oxidized Meroterpenoids. Nat. Chem. 2020, 12, 173–179. [Google Scholar] [CrossRef]
- Otoguro, K.; Kuno, F.; Omura, S. Arisugacins, Selective Acetylcholinesterase Inhibitors of Microbial Origin. Pharmacol. Ther. 1997, 76, 45–54. [Google Scholar] [CrossRef]
- Pereira, D.M.; Cheel, J.; Areche, C.; San-Martin, A.; Rovirosa, J.; Silva, L.R.; Valentao, P.; Andrade, P.B. Anti-Proliferative Activity of Meroditerpenoids Isolated from the Brown Alga Stypopodium Flabelliforme against Several Cancer Cell Lines. Mar. Drugs 2011, 9, 852–862. [Google Scholar] [CrossRef]
- Li, F.; Renata, H. A Chiral-Pool-Based Strategy to Access trans-syn-Fused Drimane Meroterpenoids: Chemoenzymatic Total Syntheses of Polysin, N-Acetyl-polyveoline and the Chrodrimanins. J. Am. Chem. Soc. 2021, 143, 18280–18286. [Google Scholar] [CrossRef]
- Ma, T.-K.; Elliott, D.C.; Reid, S.; White, A.J.P.; Parsons, P.J.; Barrett, A.G.M. Meroterpenoid Synthesis via Sequential Polyketide Aromatization and Cationic Polyene Cyclization: Total Syntheses of (+)-Hongoquercin A and B and Related Meroterpenoids. J. Org. Chem. 2018, 83, 13276–13286. [Google Scholar] [CrossRef] [PubMed]
- Wildermuth, R.; Speck, K.; Haut, F.L.; Mayer, P.; Karge, B.; Brönstrup, M.; Magauer, T. A modular synthesis of tetracyclic meroterpenoid antibiotics. Nat. Commun. 2017, 8, 2083. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Porco, J.A. Unified, Asymmetric Total Synthesis of the Asnovolins and Related Spiromeroterpenoids: A Fragment Coupling Approach. J. Am. Chem. Soc. 2022, 144, 12970–12978. [Google Scholar] [CrossRef] [PubMed]
- Powers, Z.; Scharf, A.; Cheng, A.; Yang, F.; Himmelbauer, M.; Mitsuhashi, T.; Barra, L.; Taniguchi, Y.; Kikuchi, T.; Fujita, M.; et al. Biomimetic Synthesis of Meroterpenoids by Dearomatization-Driven Polycyclization. Angew. Chem. Int. Ed. Engl. 2019, 58, 16141–16146. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.-H.; Shi, G.-H.; Xu, T.-T.; Chen, G.-D.; Gu, B.-B.; Wang, Z.; Peng, S.; Wang, S.P.; Li, J.; Han, B.N.; et al. Dysiherbols A-C and Dysideanone E, Cytotoxic and NF-κB Inhibitory Tetracyclic Meroterpenes from a Dysidea sp. Marine Sponge. J. Nat. Prod. 2016, 79, 406–411. [Google Scholar] [CrossRef]
- Chong, C.; Chang, L.; Grimm, I.; Zhang, Q.; Kuang, Y.; Wang, B.; Kang, J.; Liu, W.; Baars, J.; Guo, Y.; et al. Divergent total synthesis of the revised structures of marine anti-cancer meroterpenoids (+)-dysiherbols A-E. Chem. Sci. 2023, 14, 3302–3310. [Google Scholar] [CrossRef]
- Noda, A.; Sakai, E.; Kato, H.; Losung, F.; Mangindaan, R.E.; de Voogd, N.J.; Yokosawa, H.; Tsukamoto, S. Strongylophorines, meroditerpenoids from the marine sponge Petrosia corticata, function as proteasome inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 2650–2653. [Google Scholar] [CrossRef]
- Yu, W.; Hjerrild, P.; Overgaard, J.; Poulsen, T.B. A Concise Route to the Strongylophorines. Angew. Chem. Int. Ed. Engl. 2016, 55, 8294–8298. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Chen, S.-C.; Jiao, L. Total Synthesis of (+)-Minfiensine: Construction of the Tetracyclic Core Structure by an Asymmetric Cascade Cyclization. Angew. Chem. Int. Ed. Engl. 2016, 55, 8090–8094. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, B.P.; Donckele, E.J.; Stoltz, B.M. Enantioselective Catalysis Coupled with Stereodivergent Cyclization Strategies Enables Rapid Syntheses of (+)-Limaspermidine and (+)-Kopsihainanine A. Angew. Chem. Int. Ed. Engl. 2017, 56, 12624–12627. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Kouklovsky, C.; Gandon, V.; Vincent, G. Enantioselective Total Synthesis of Cymoside through a Bioinspired Oxidative Cyclization of a Strictosidine Derivative. Angew. Chem. Int. Ed. Engl. 2020, 59, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
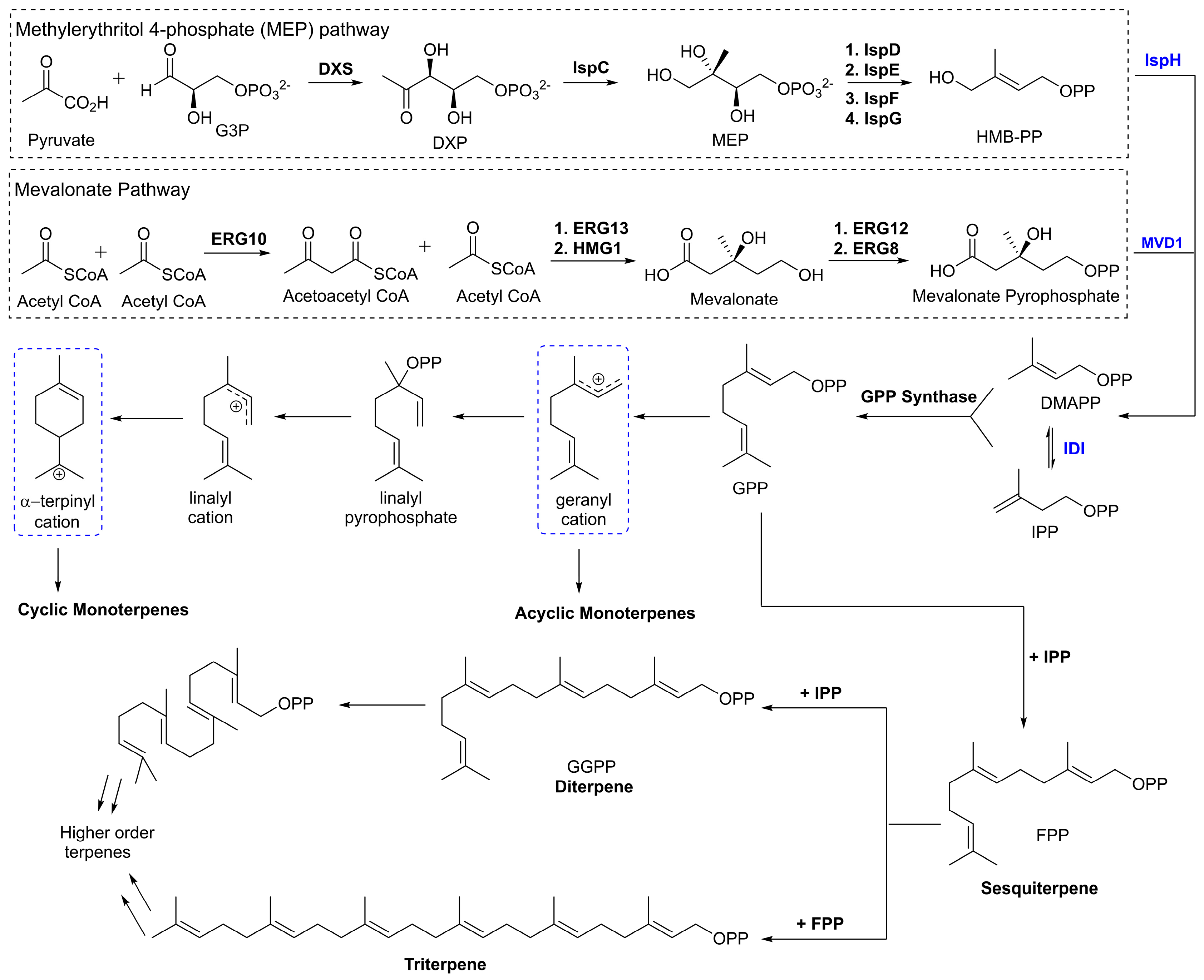
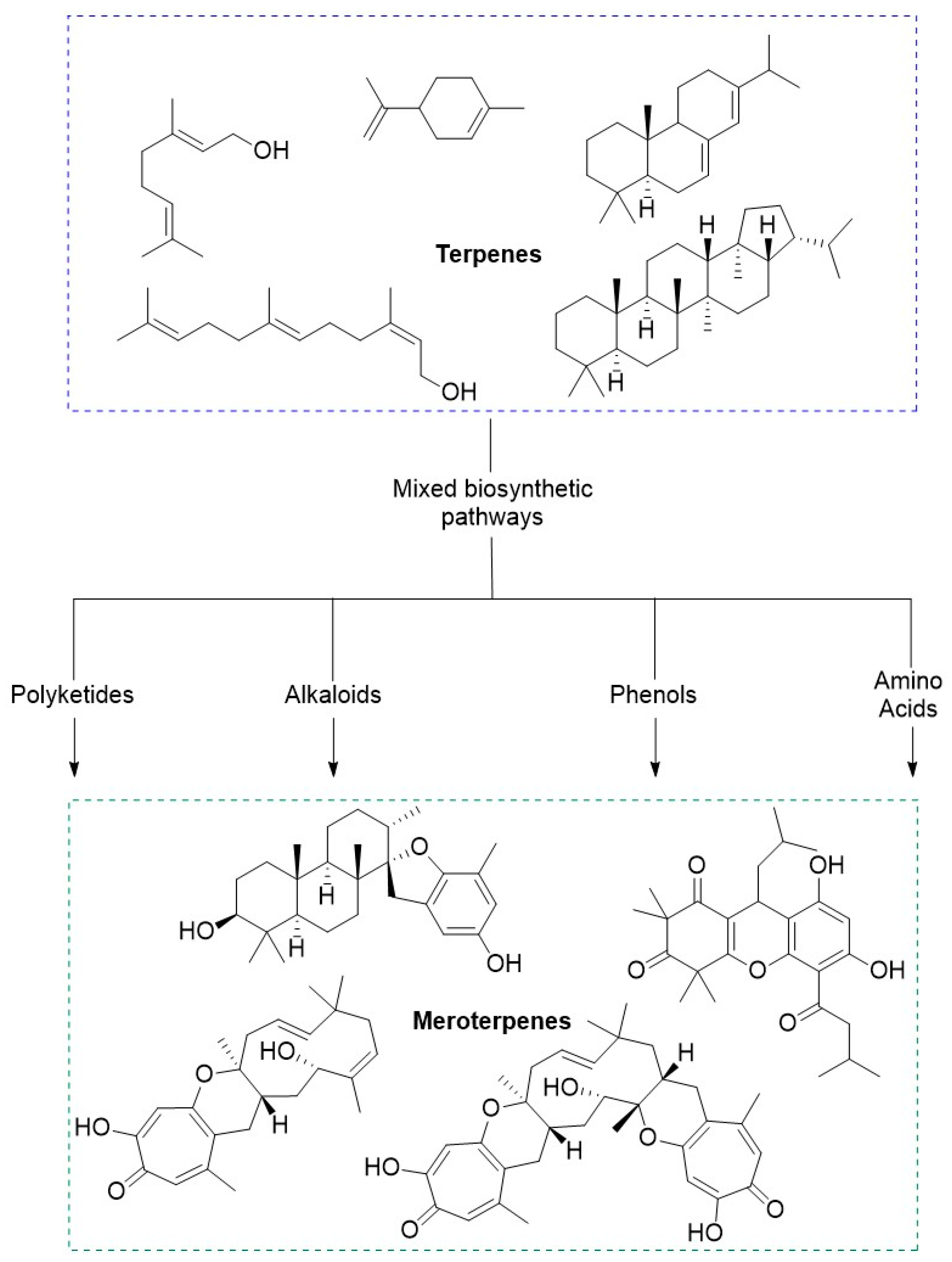
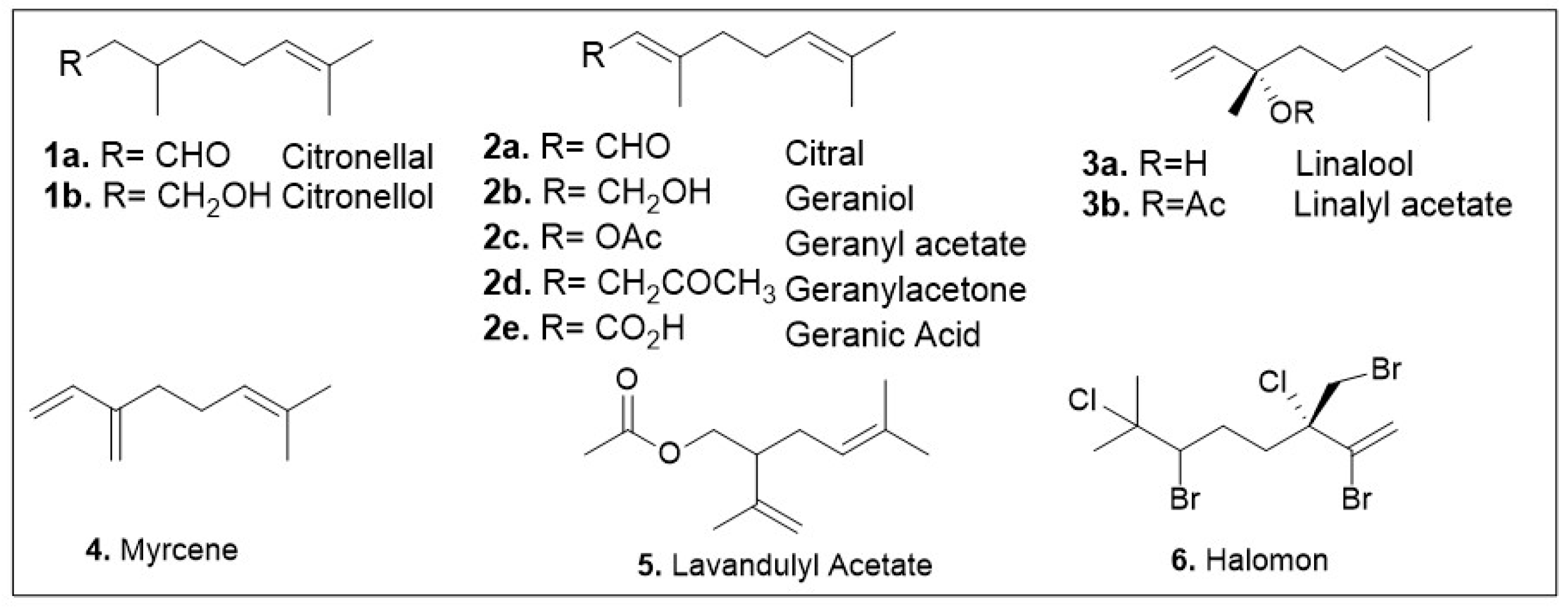
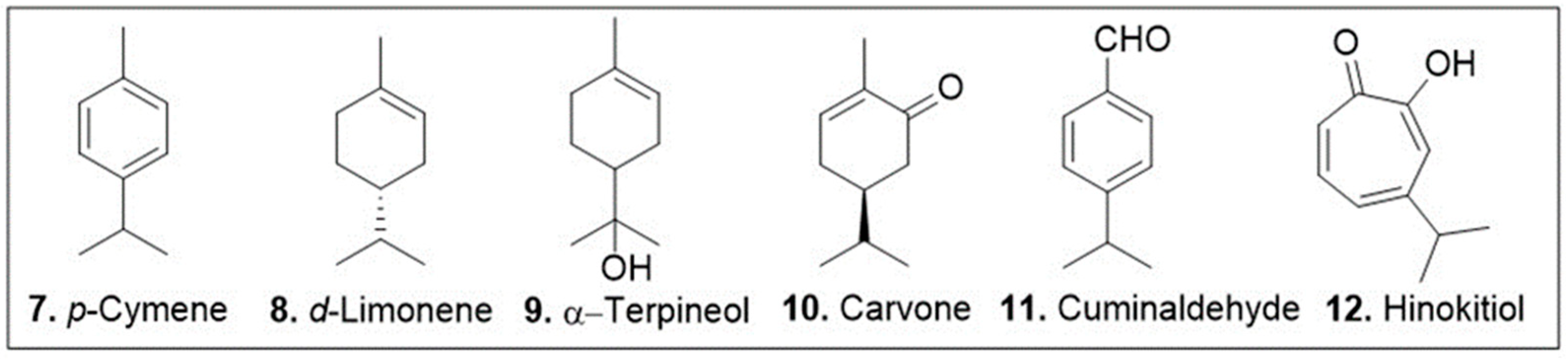
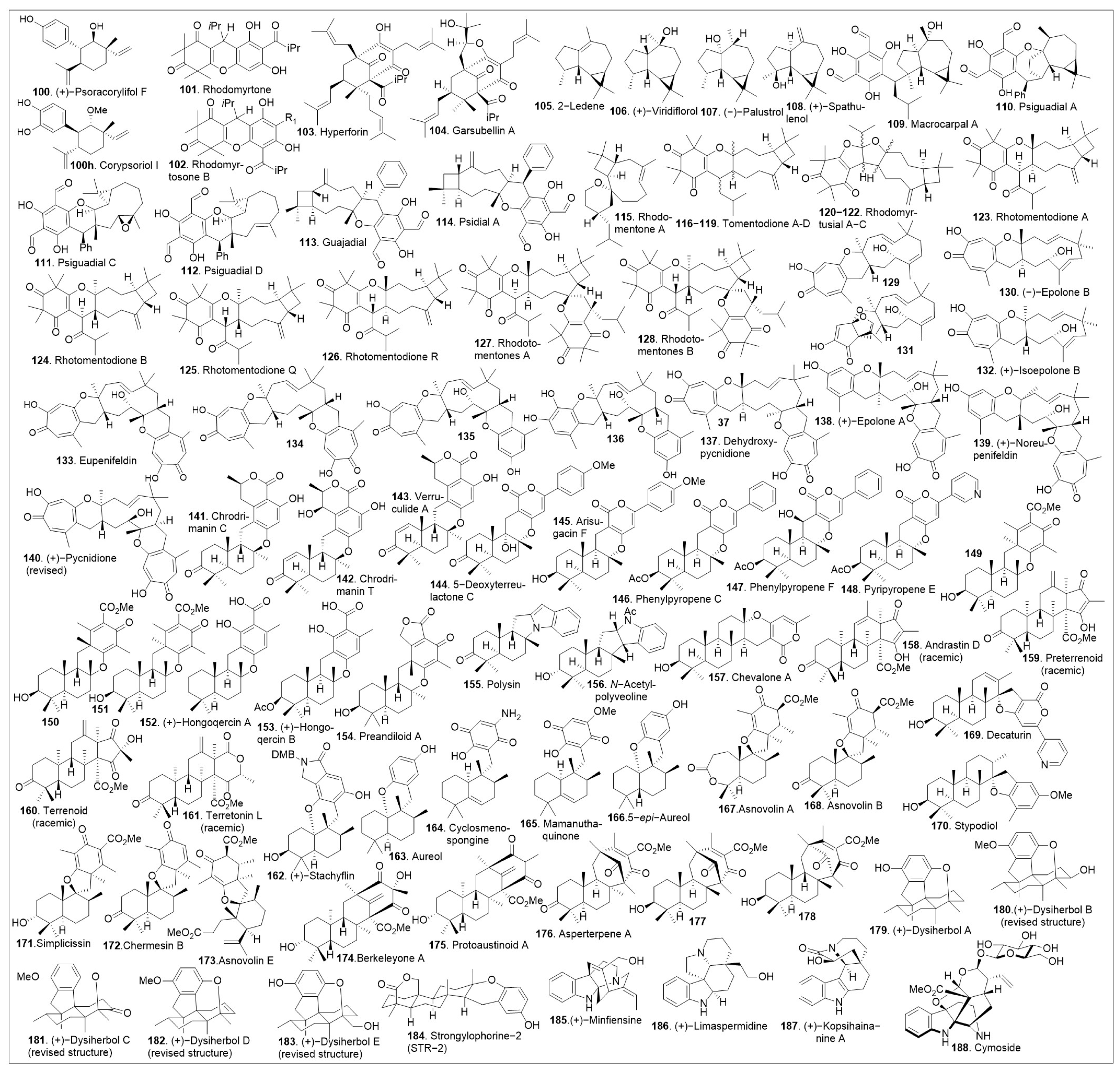
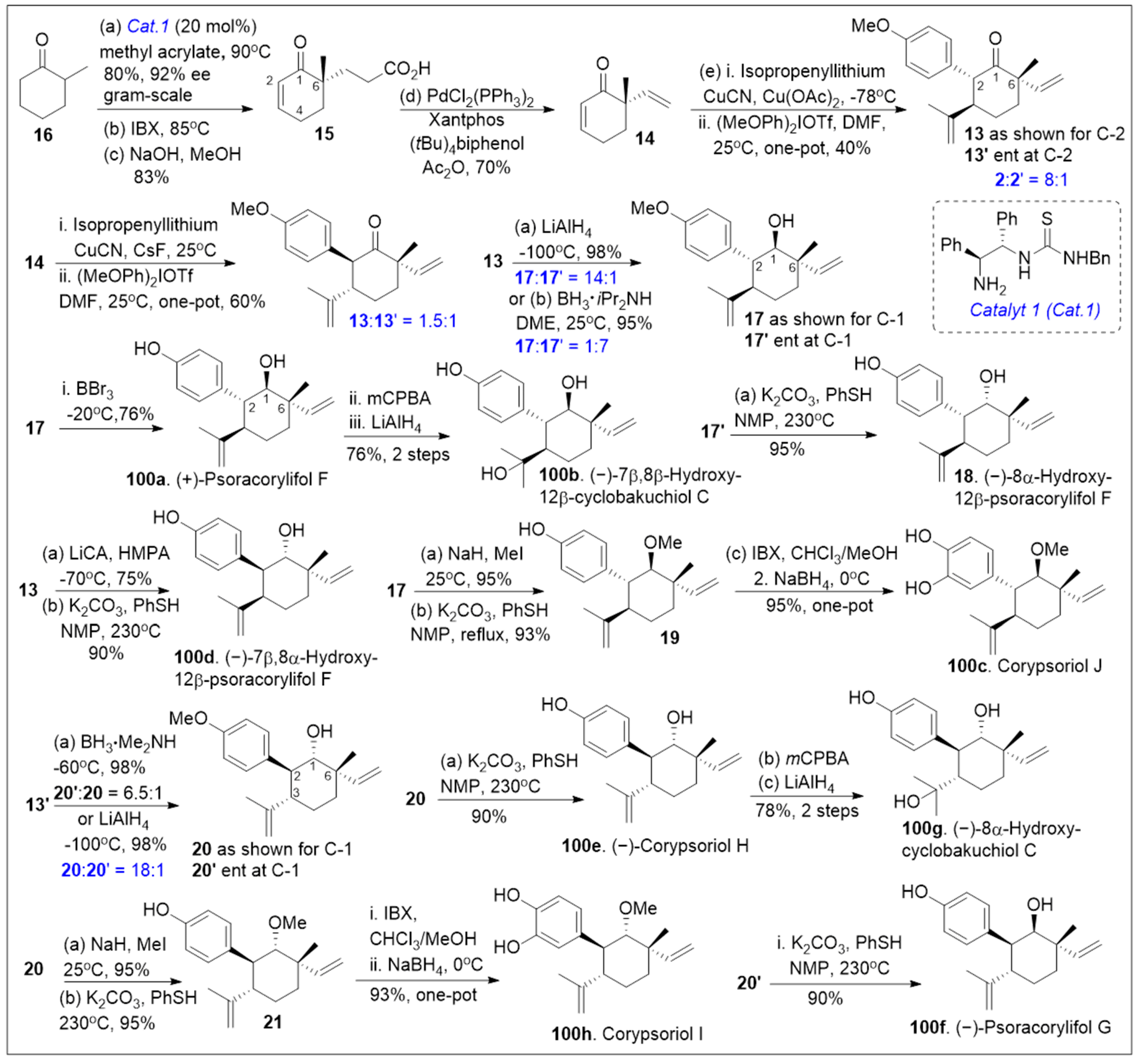
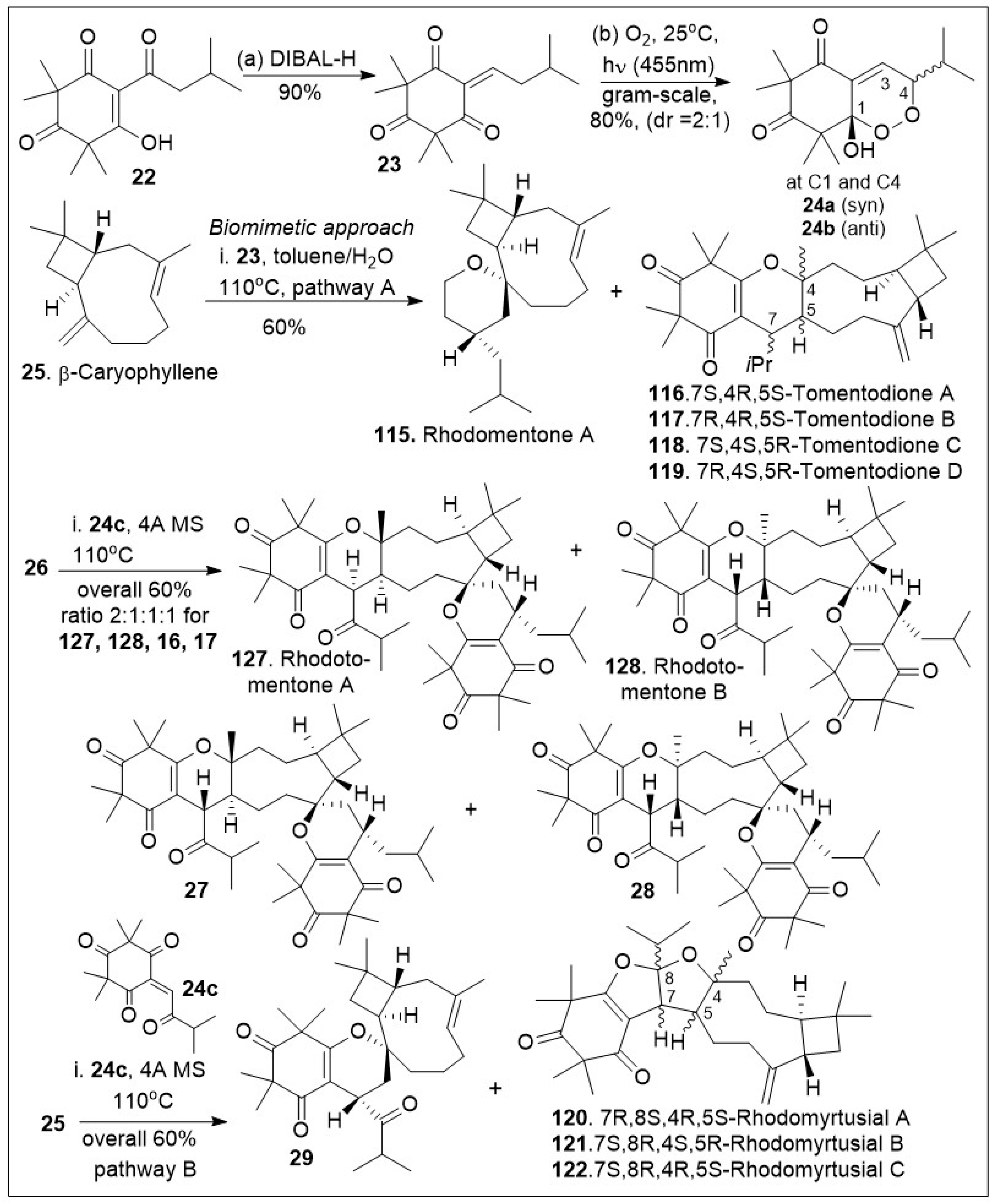
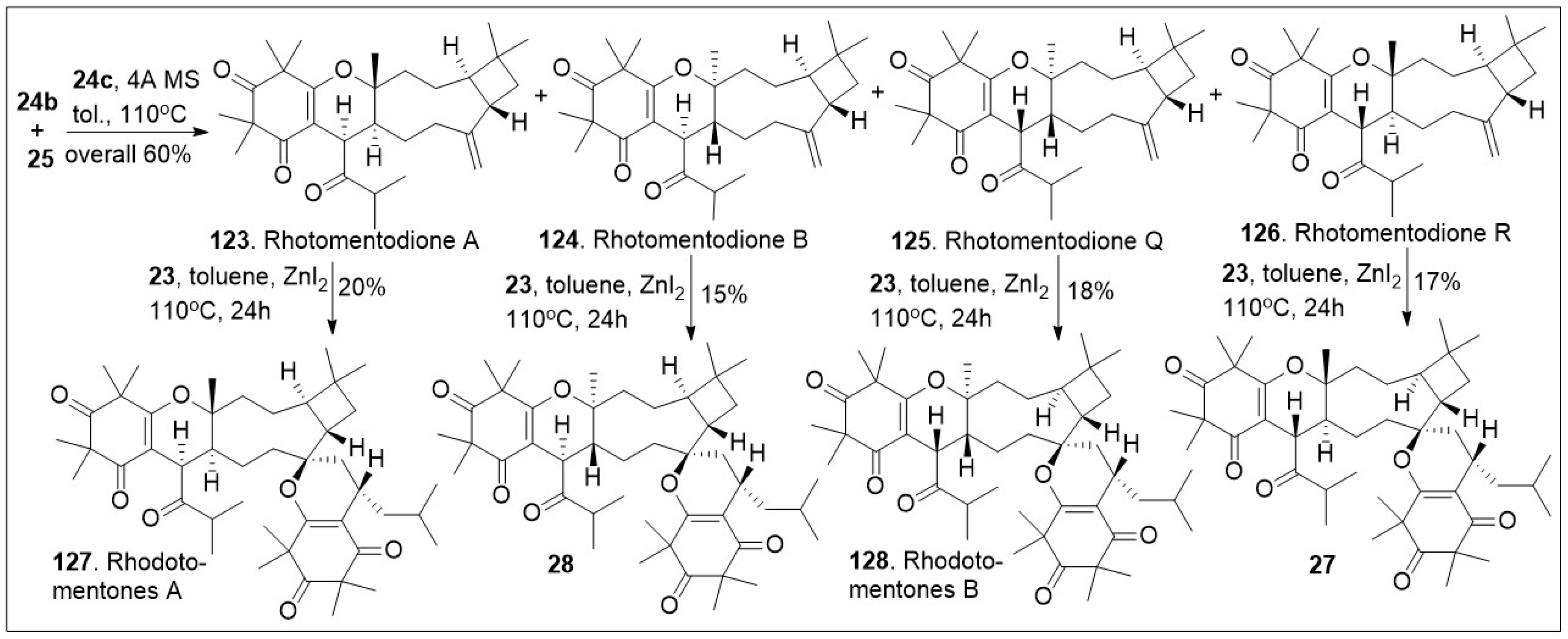
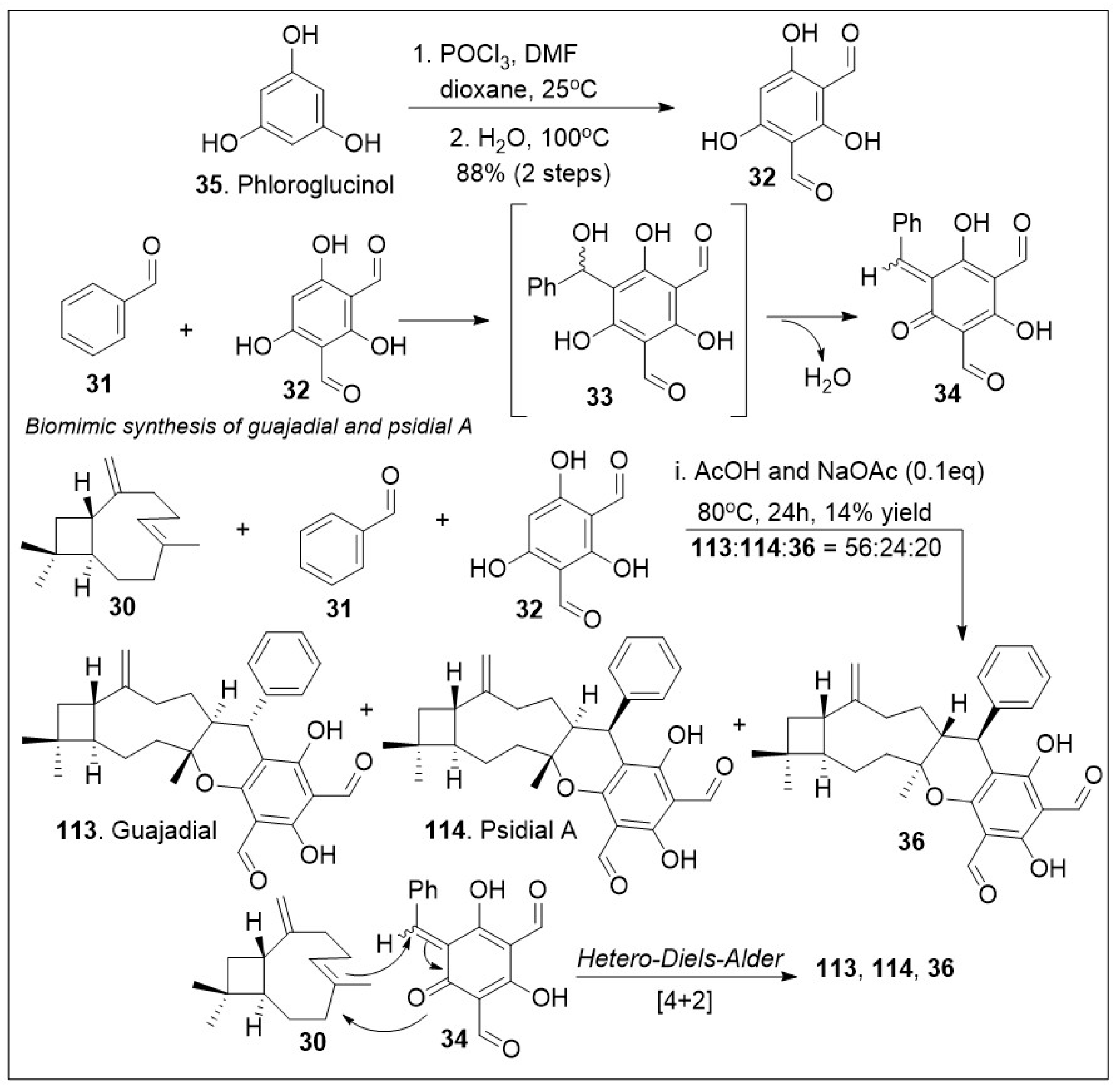
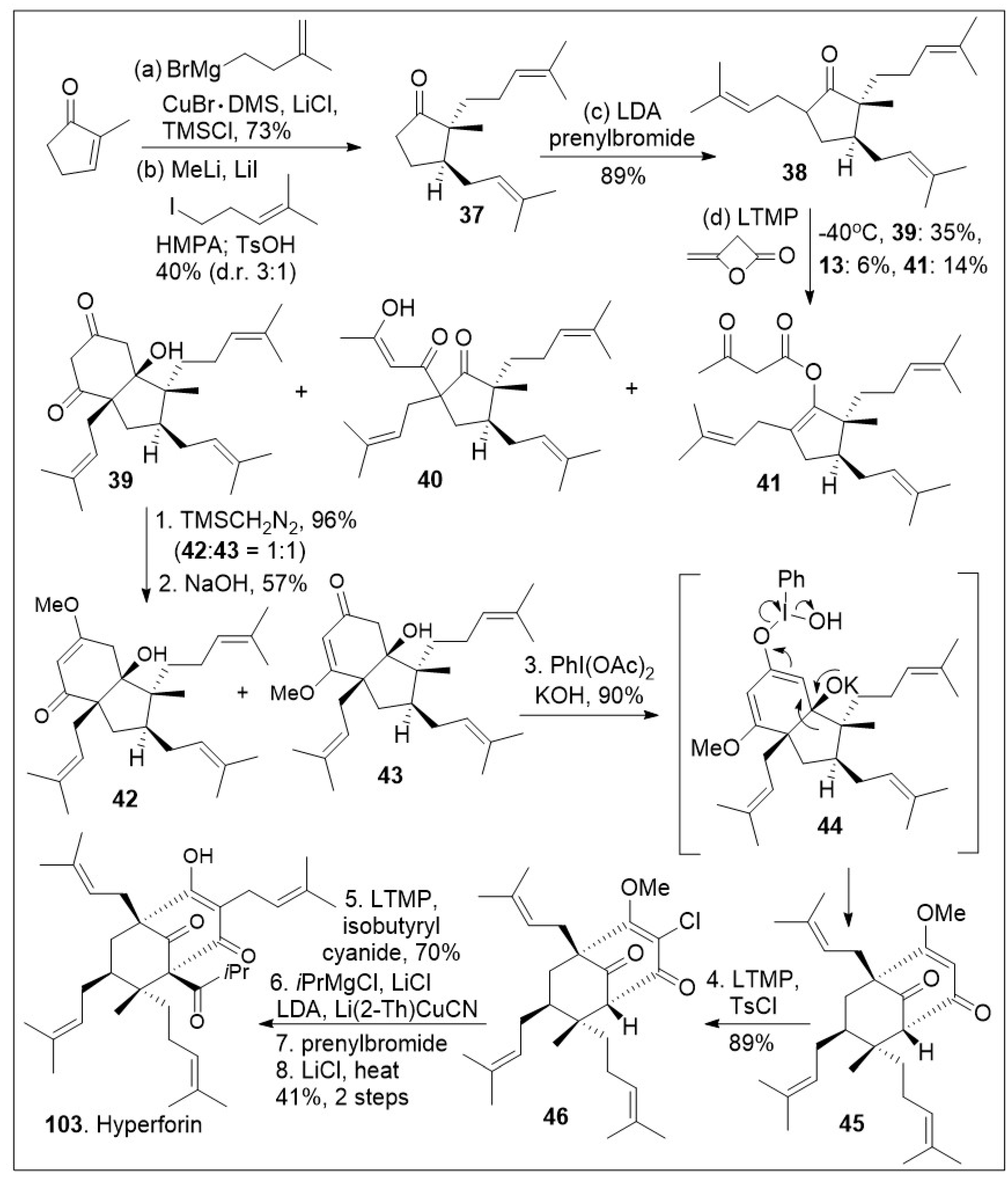
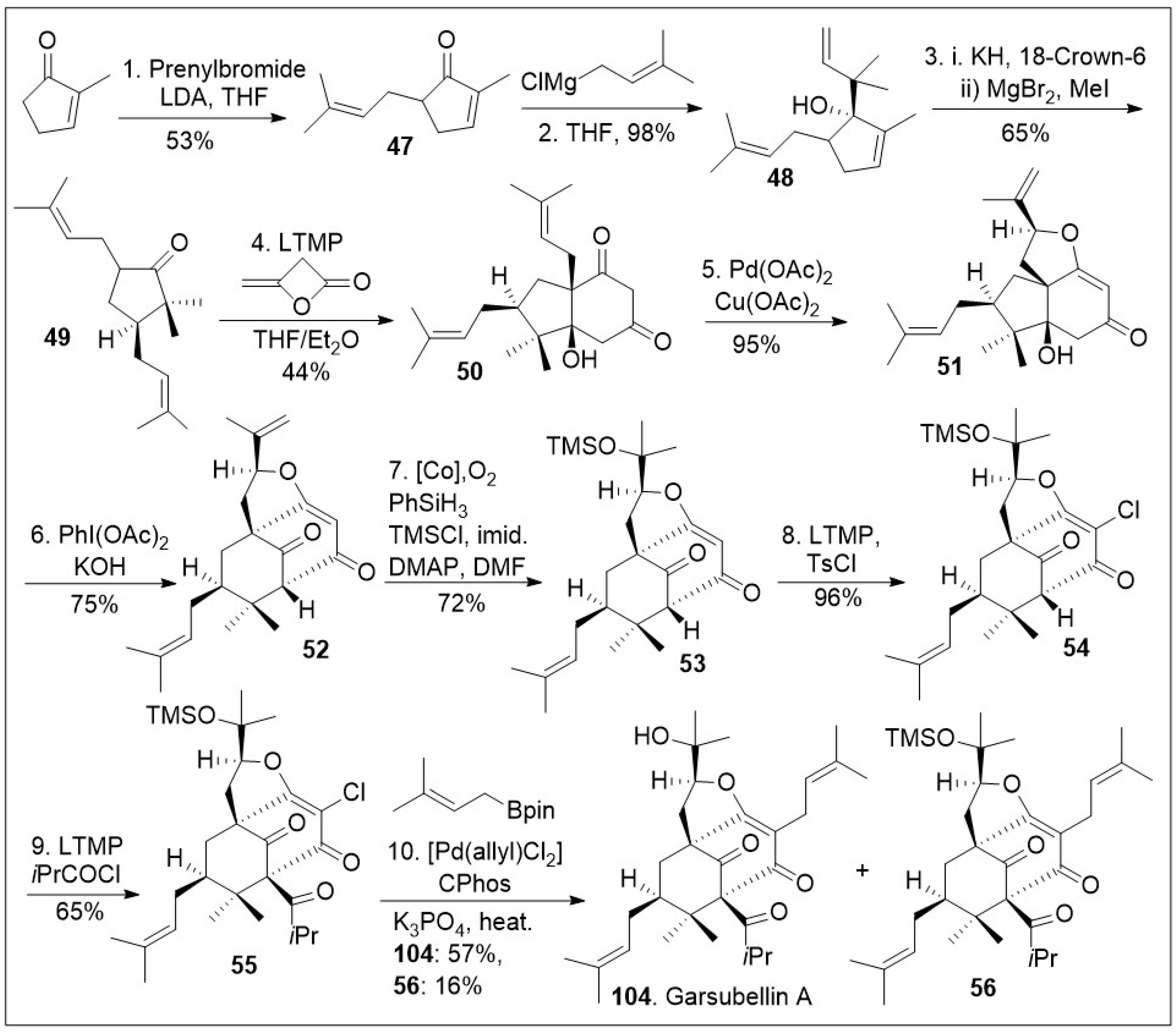
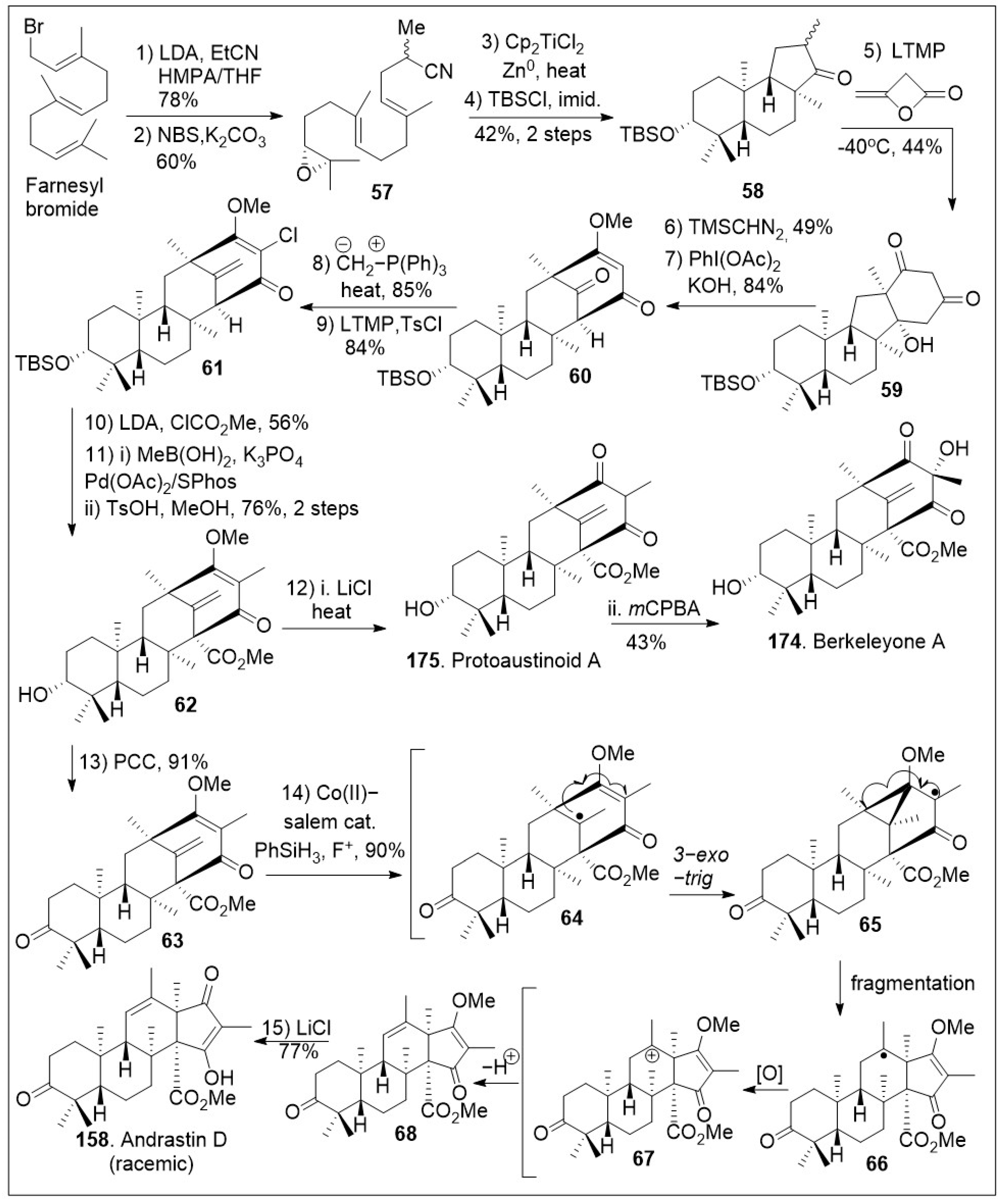
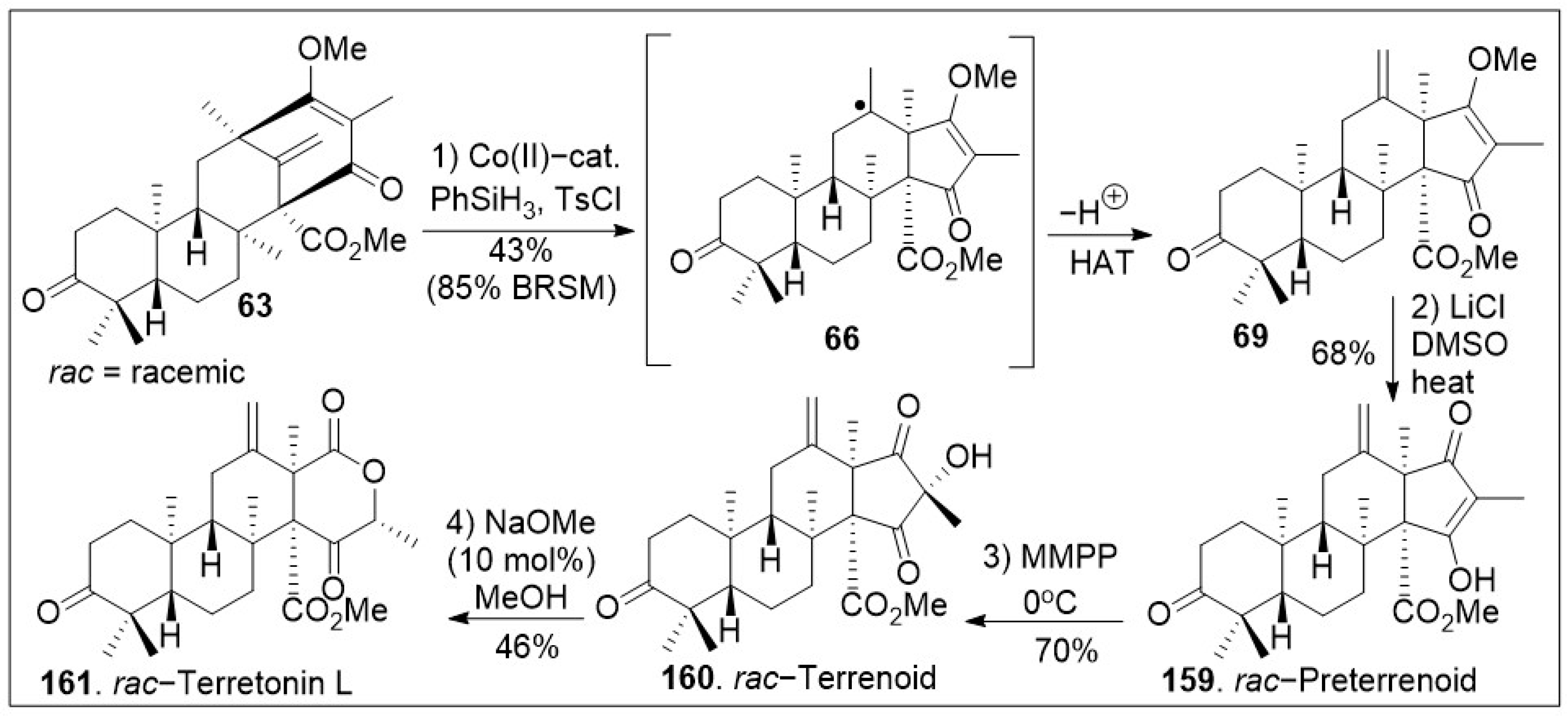
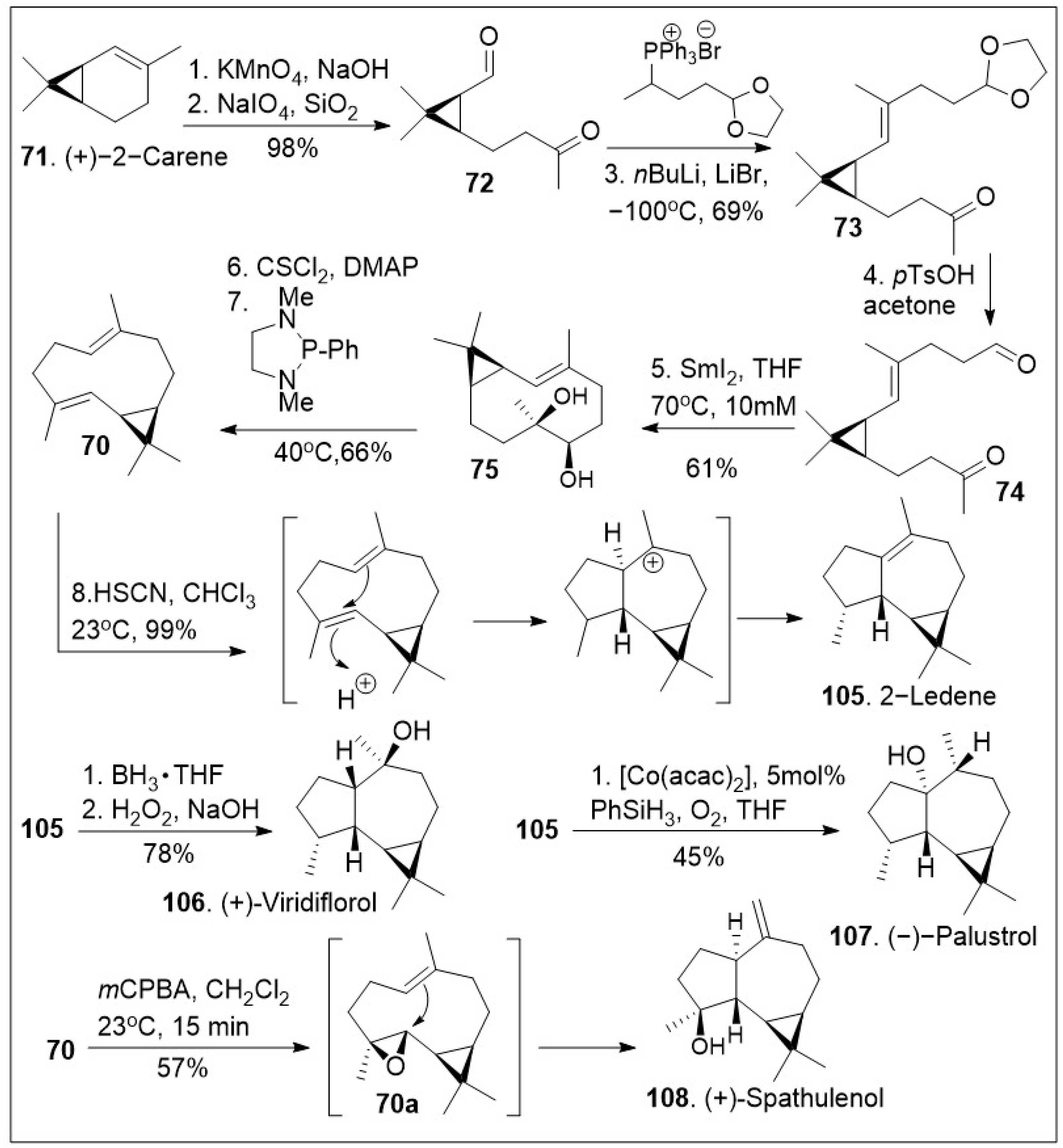
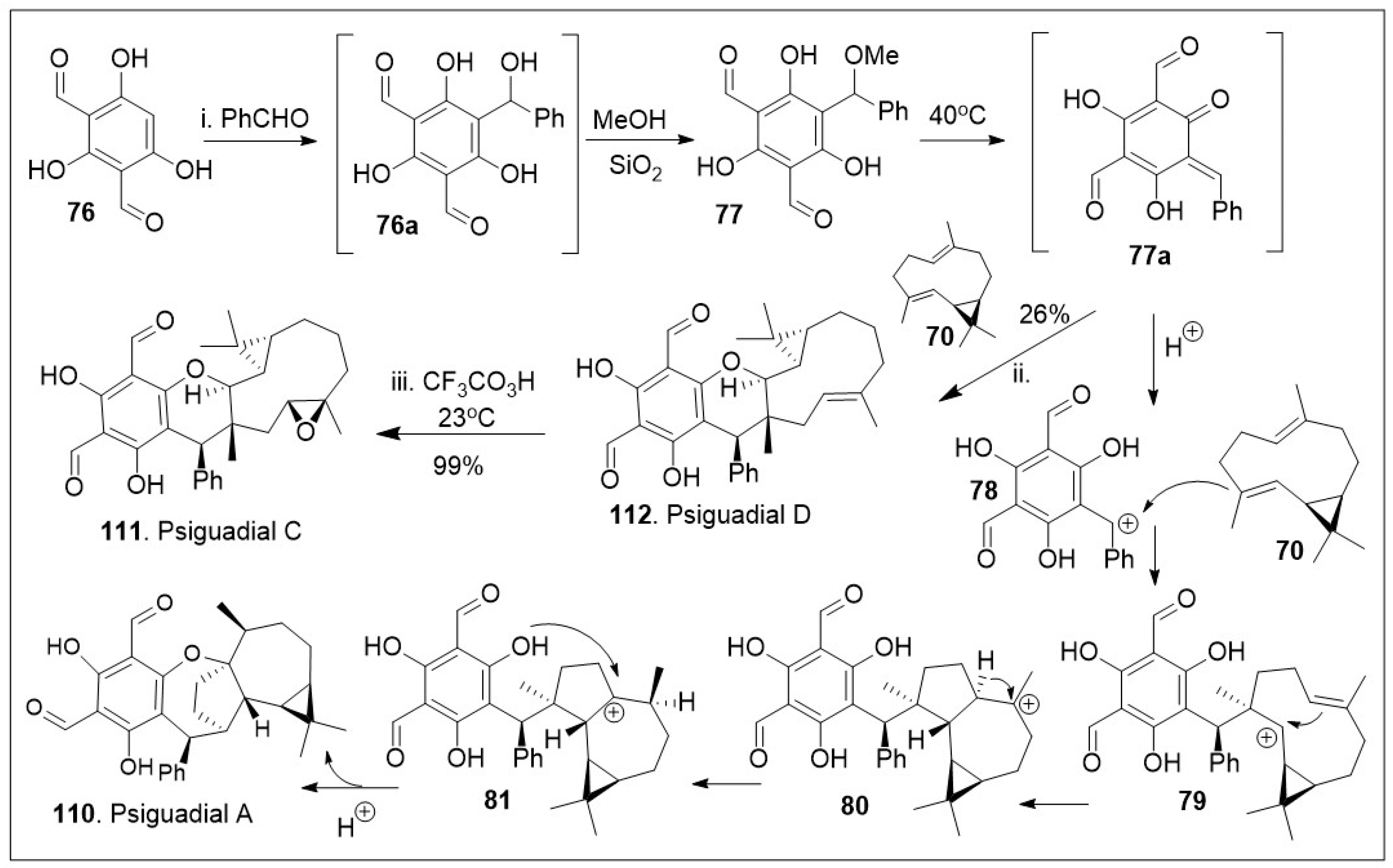
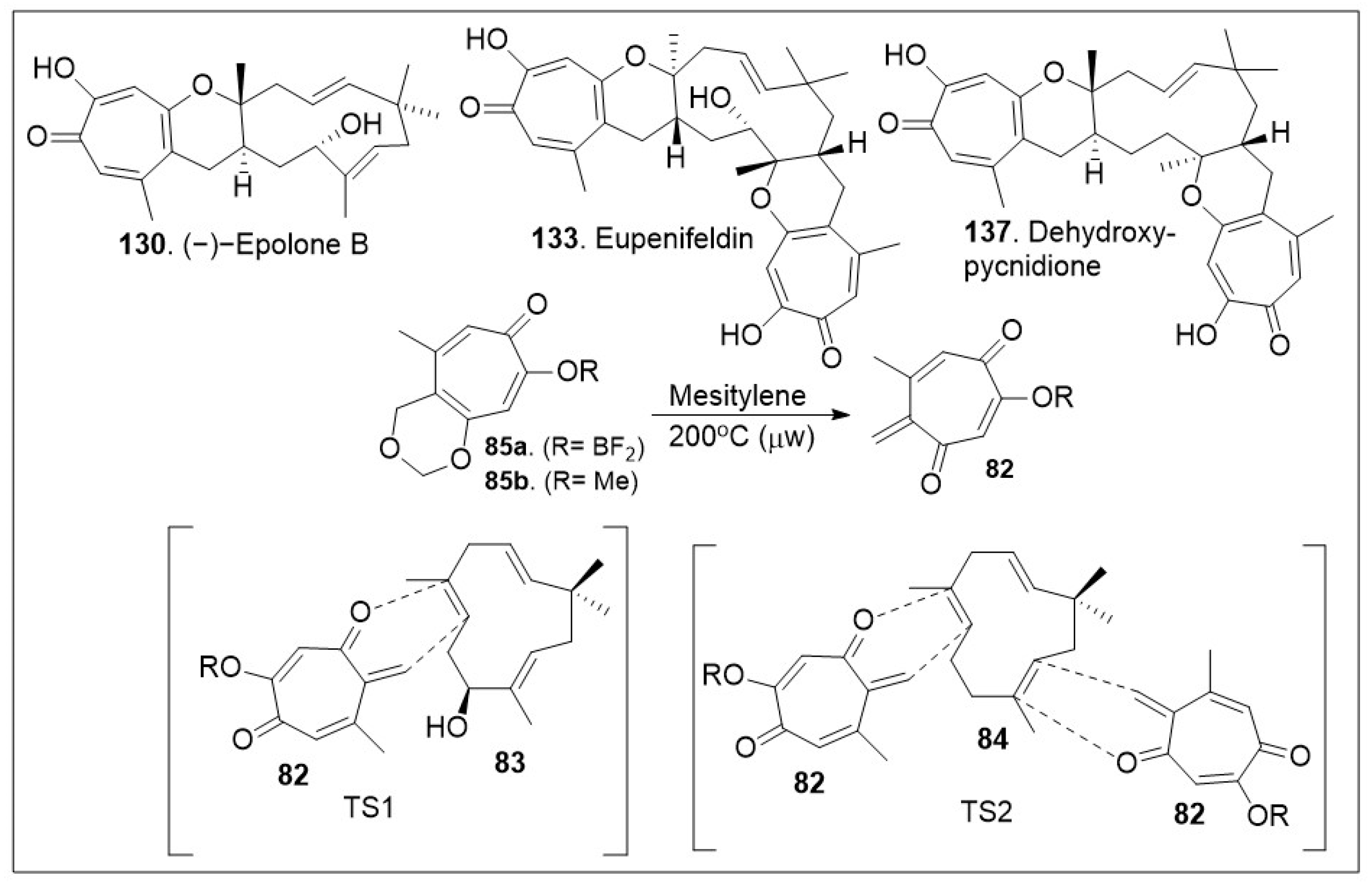
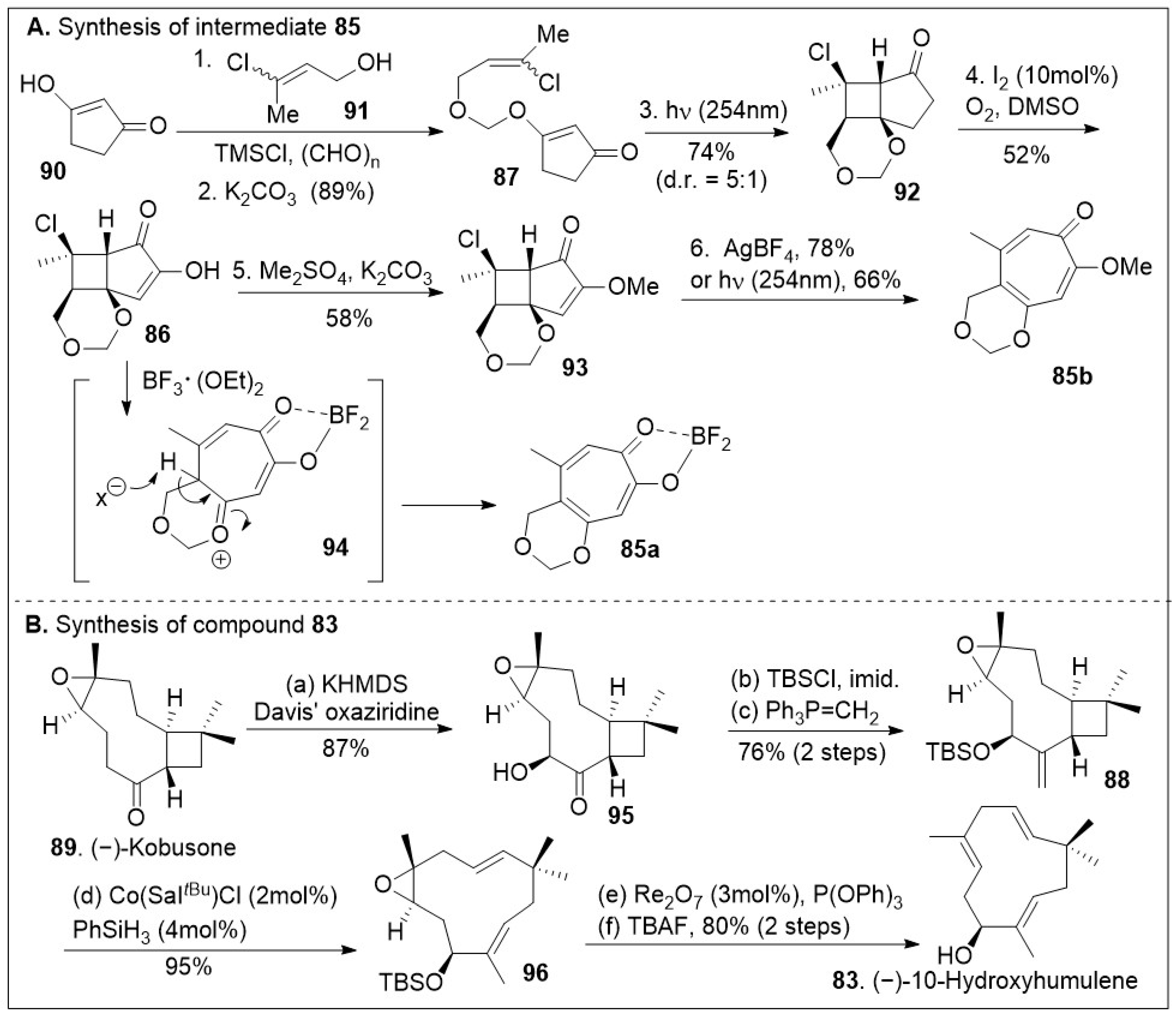
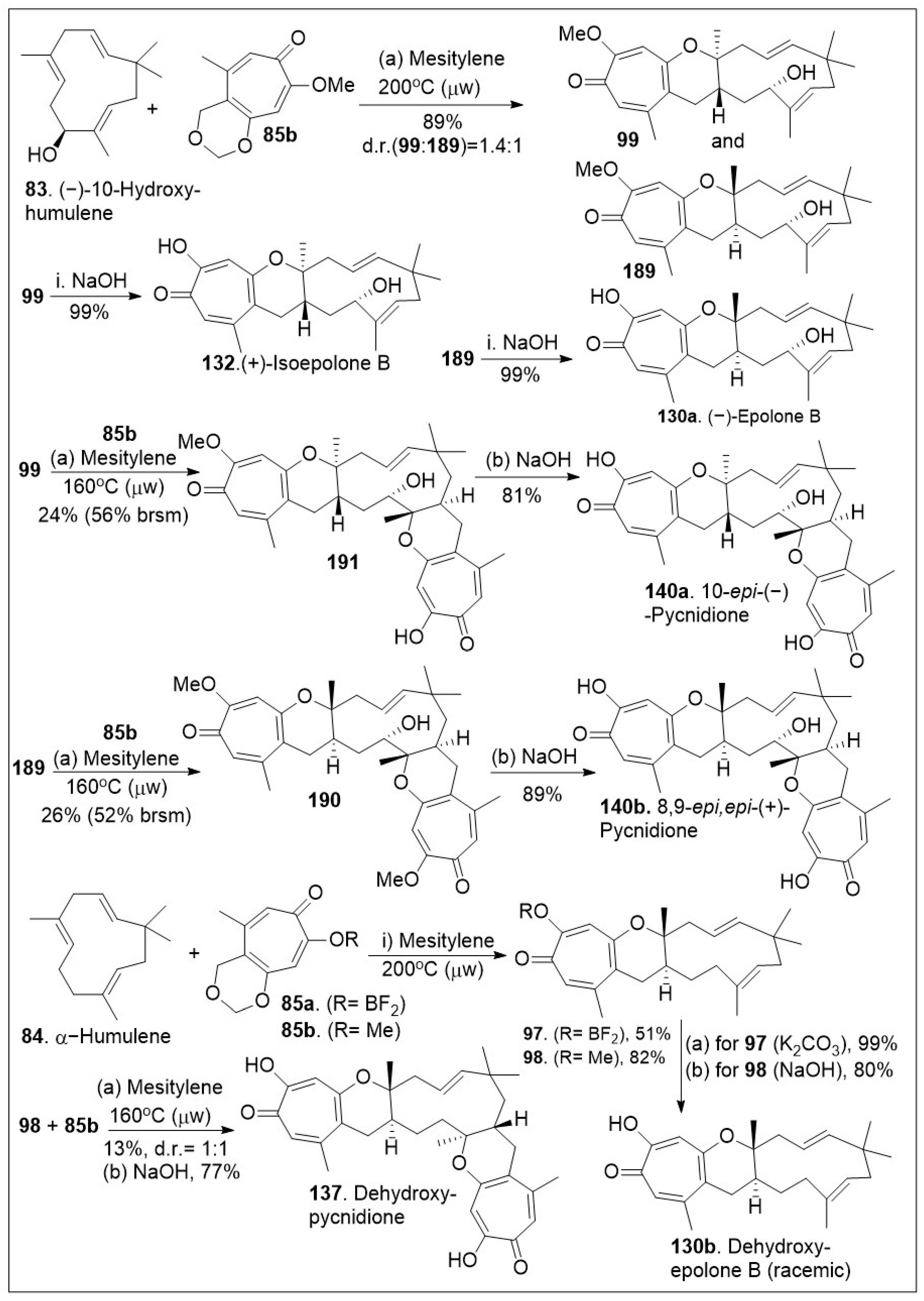
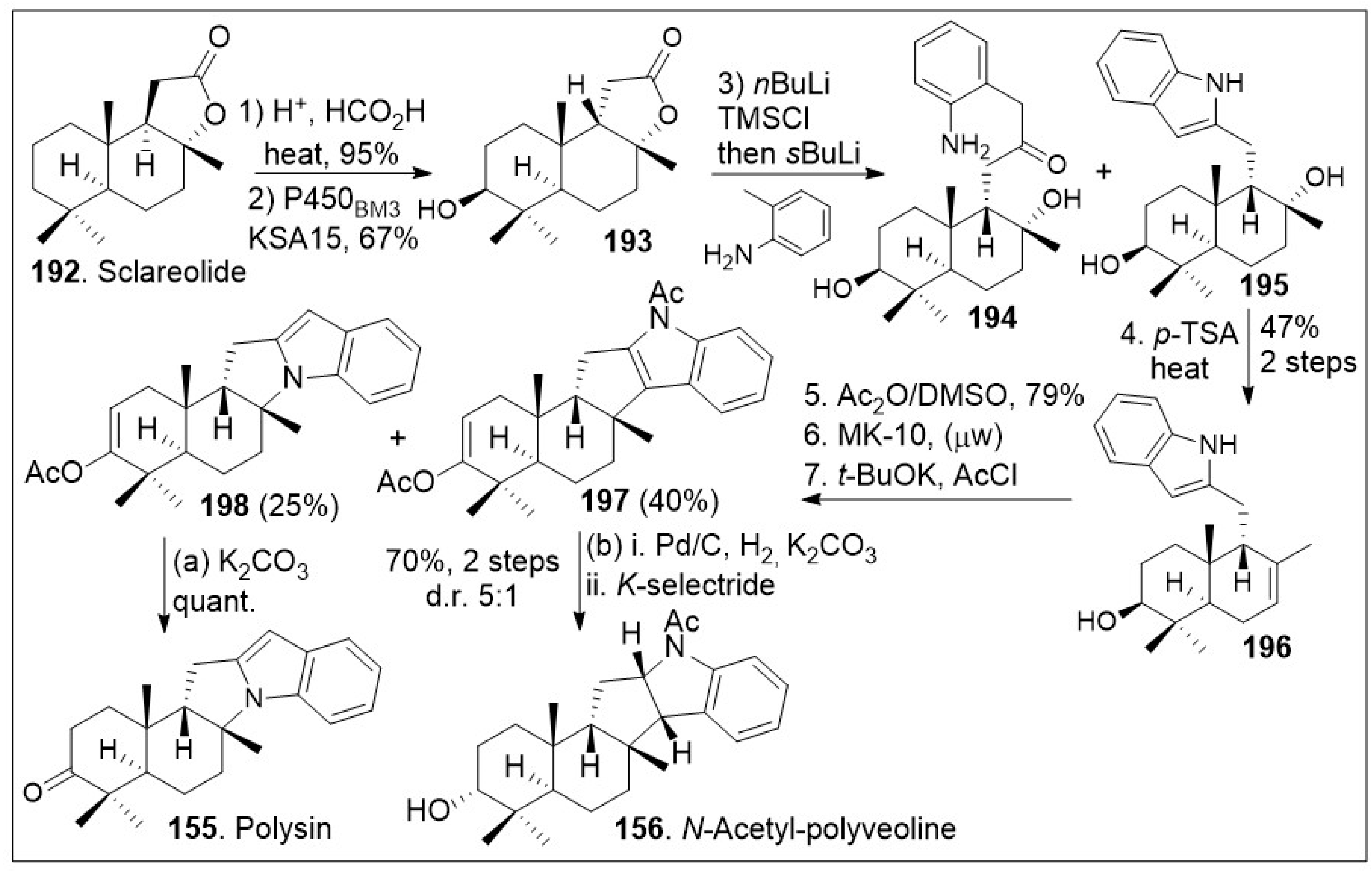
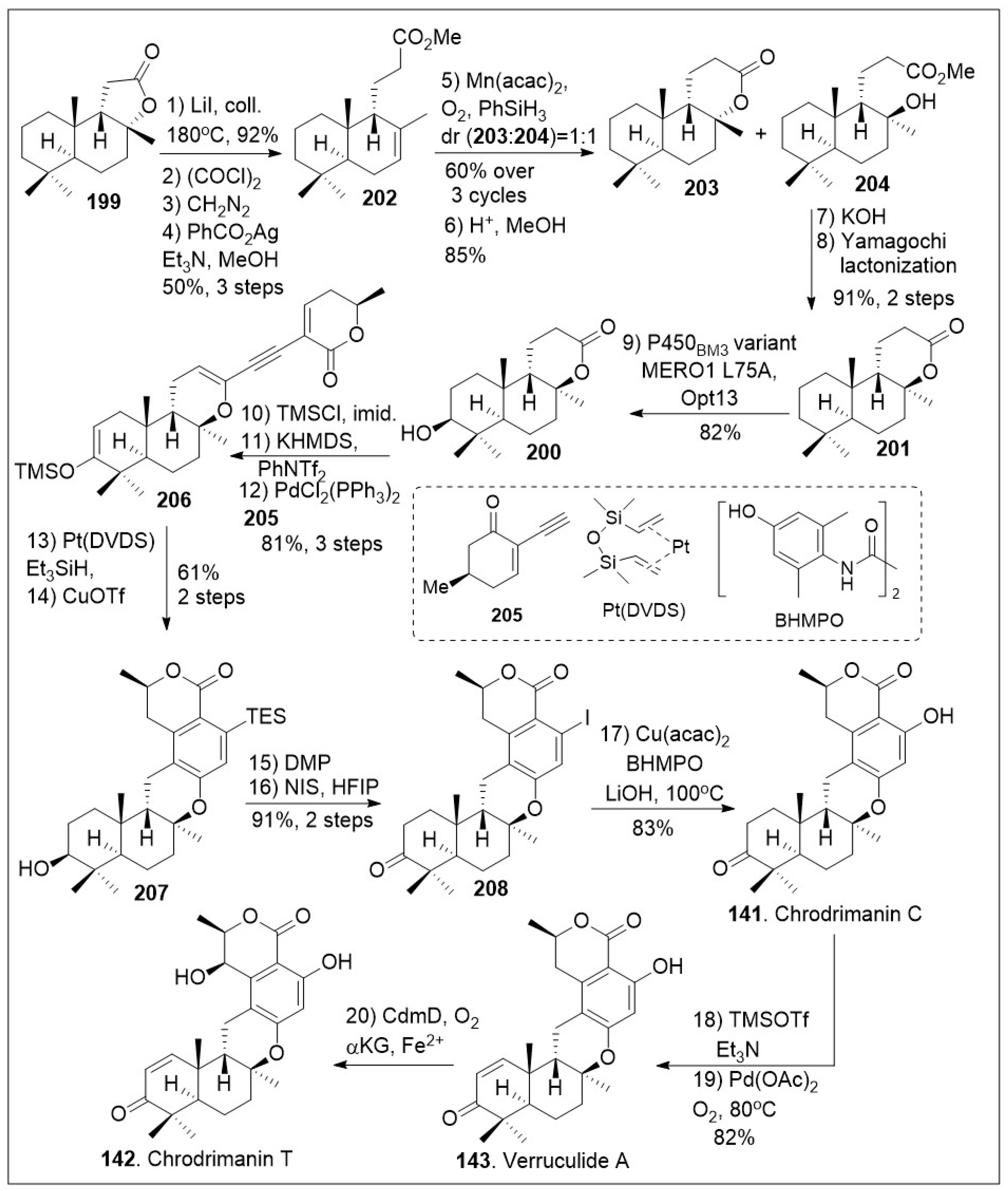
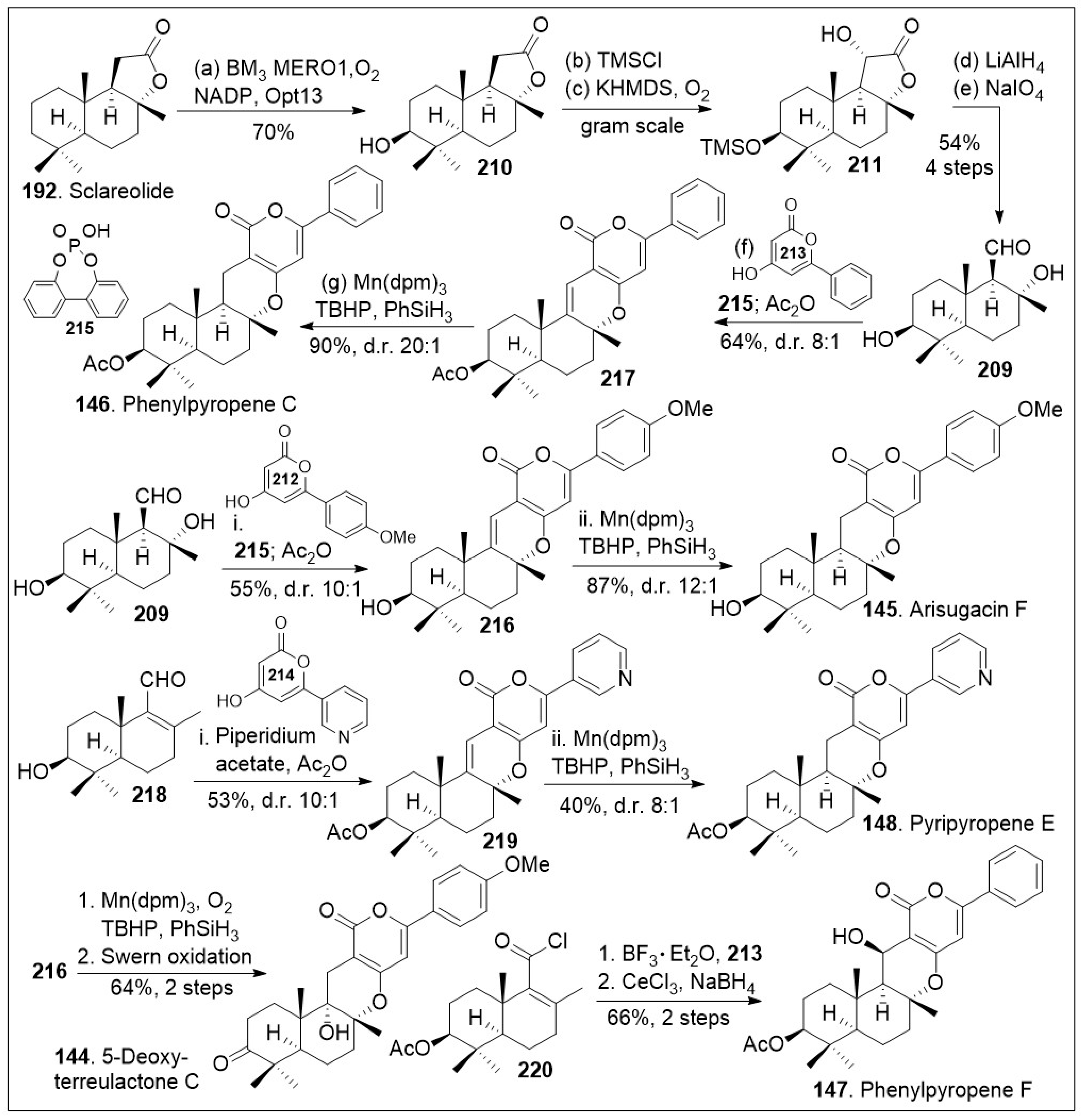
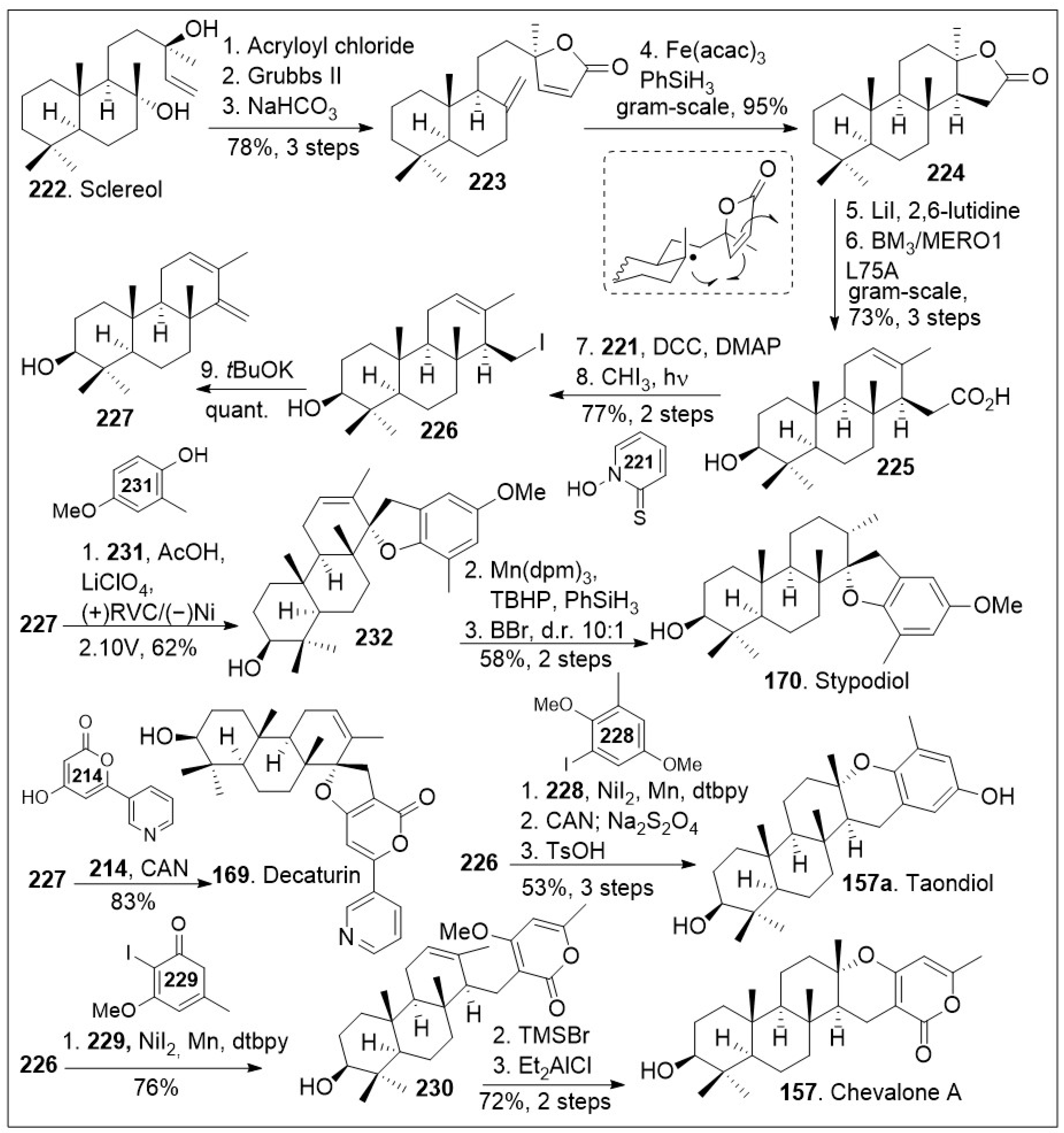
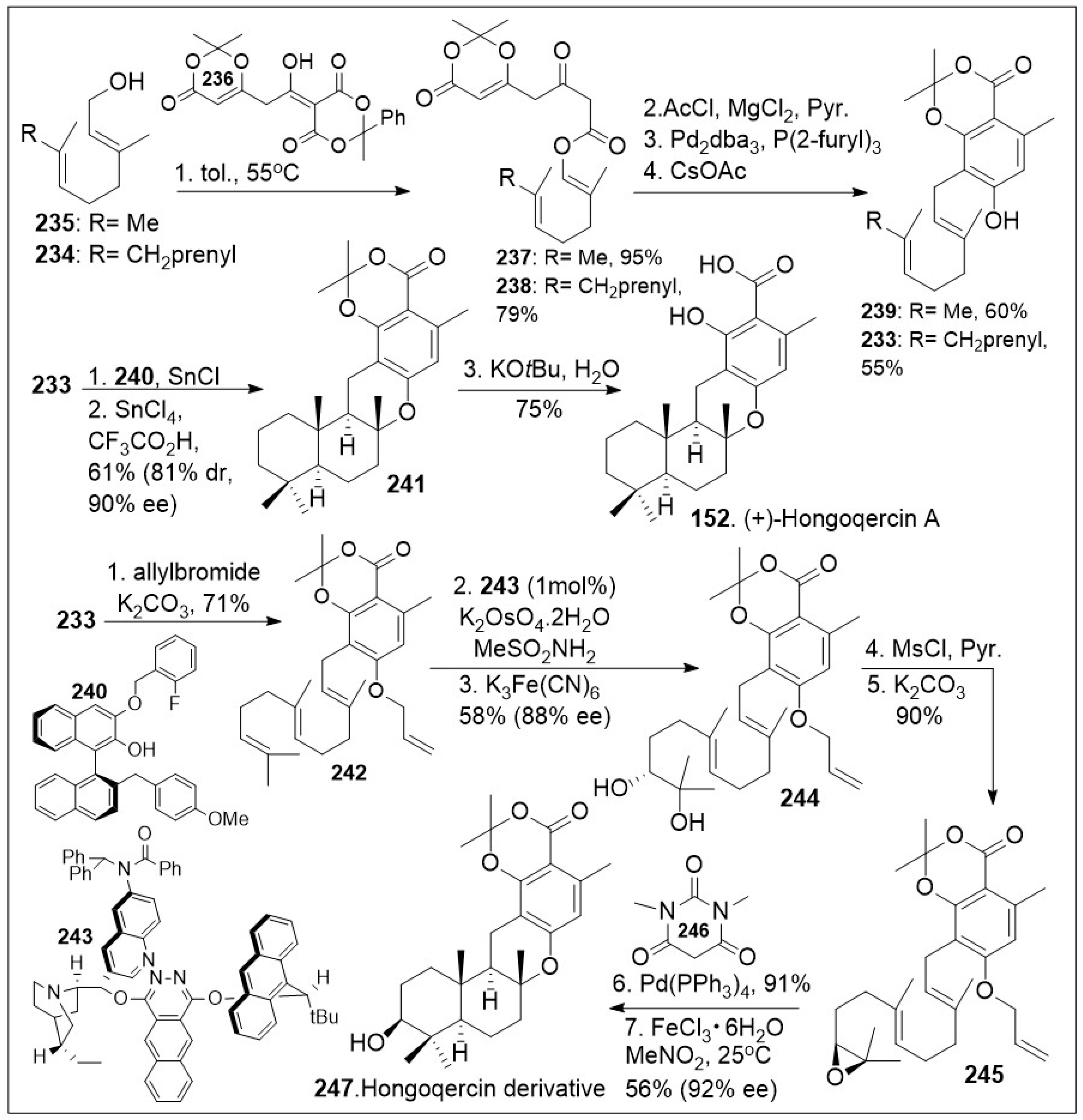
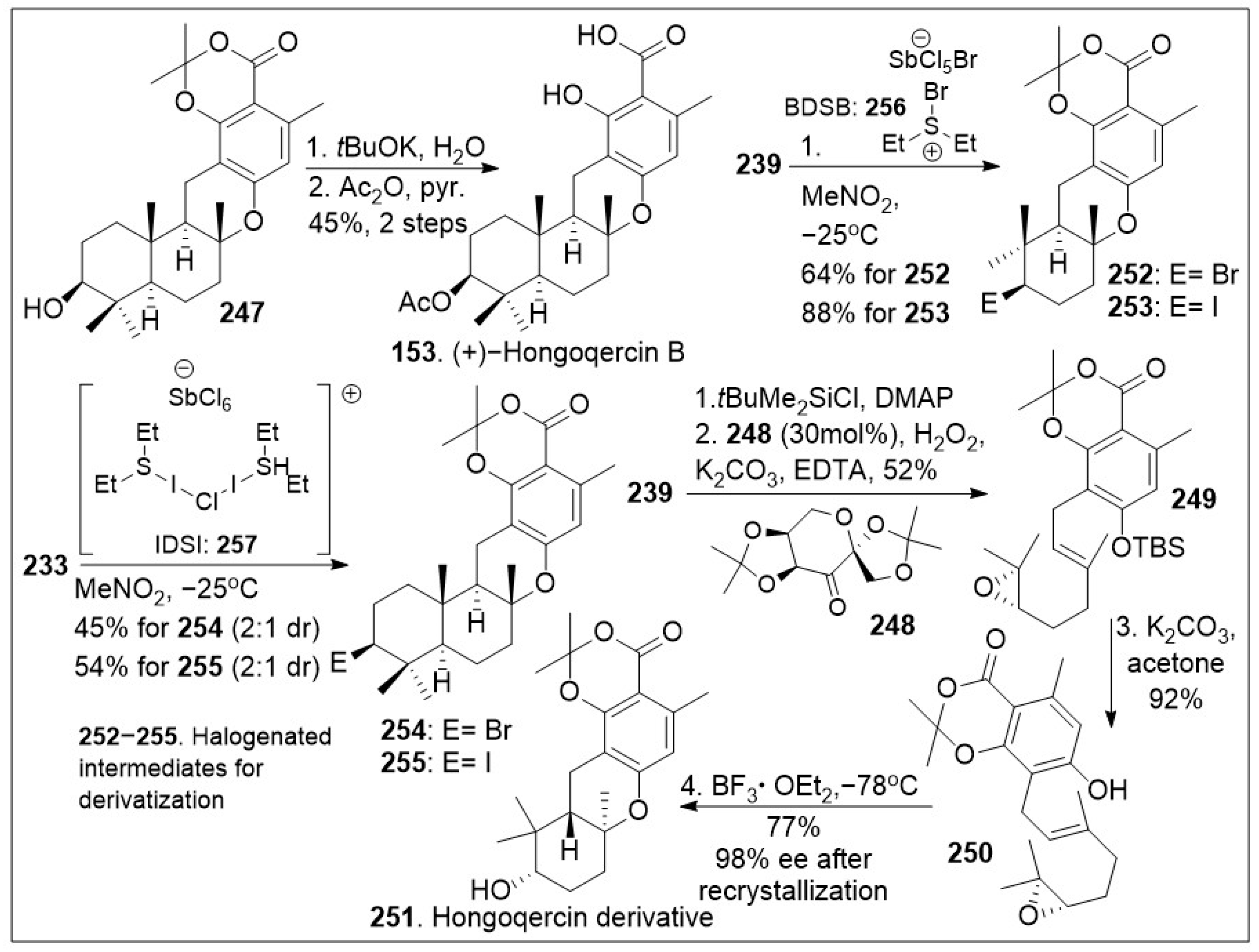
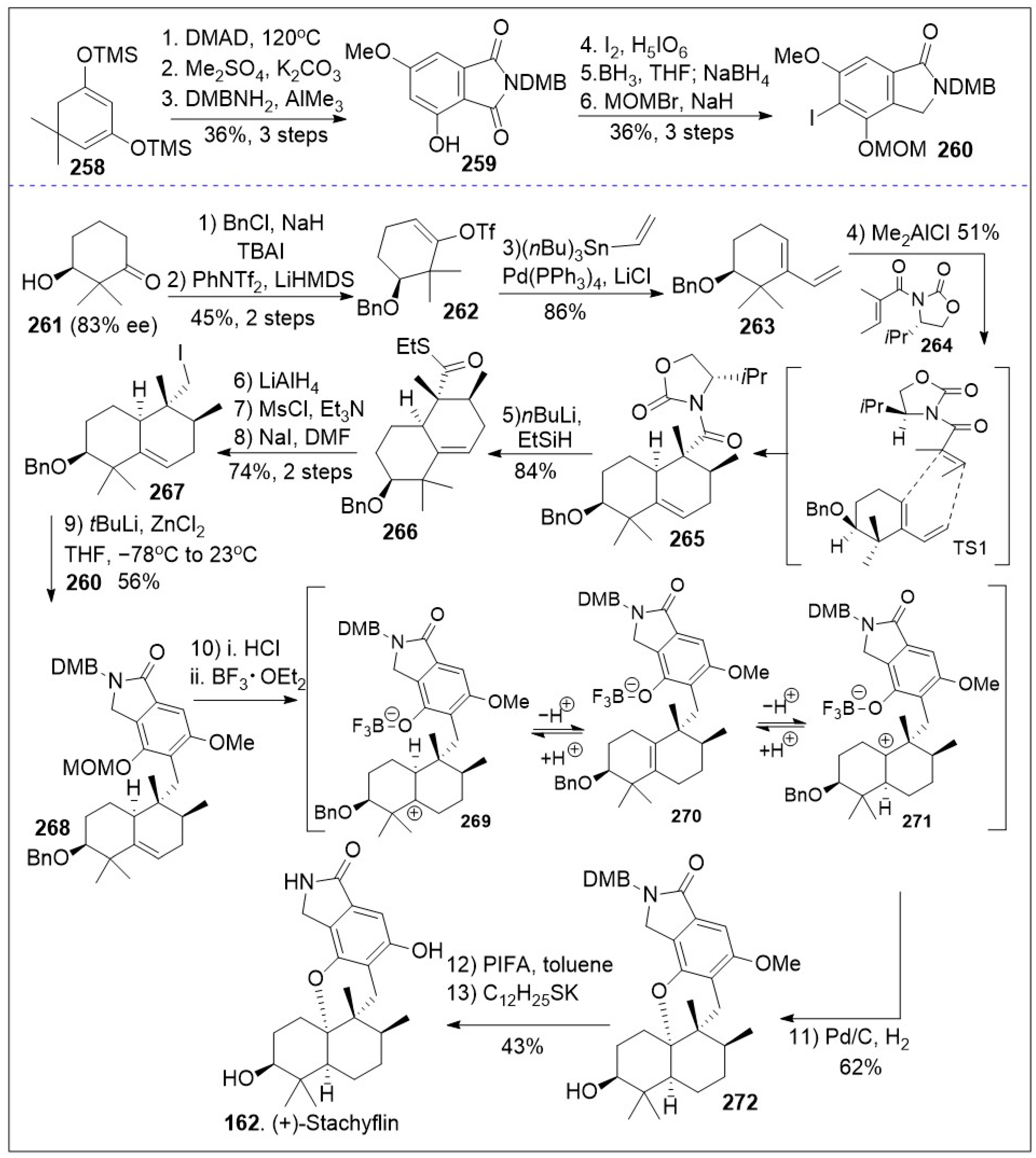
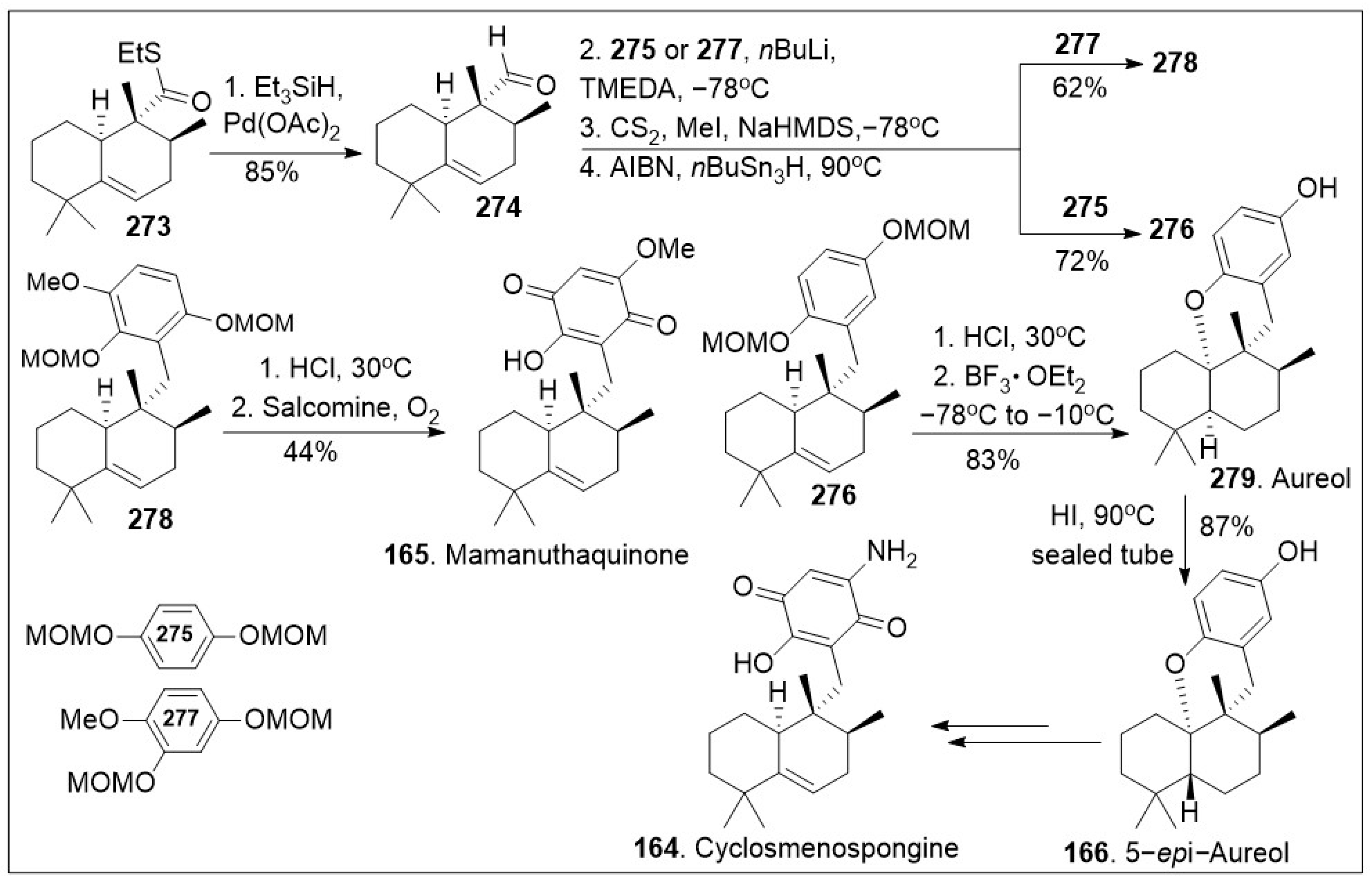
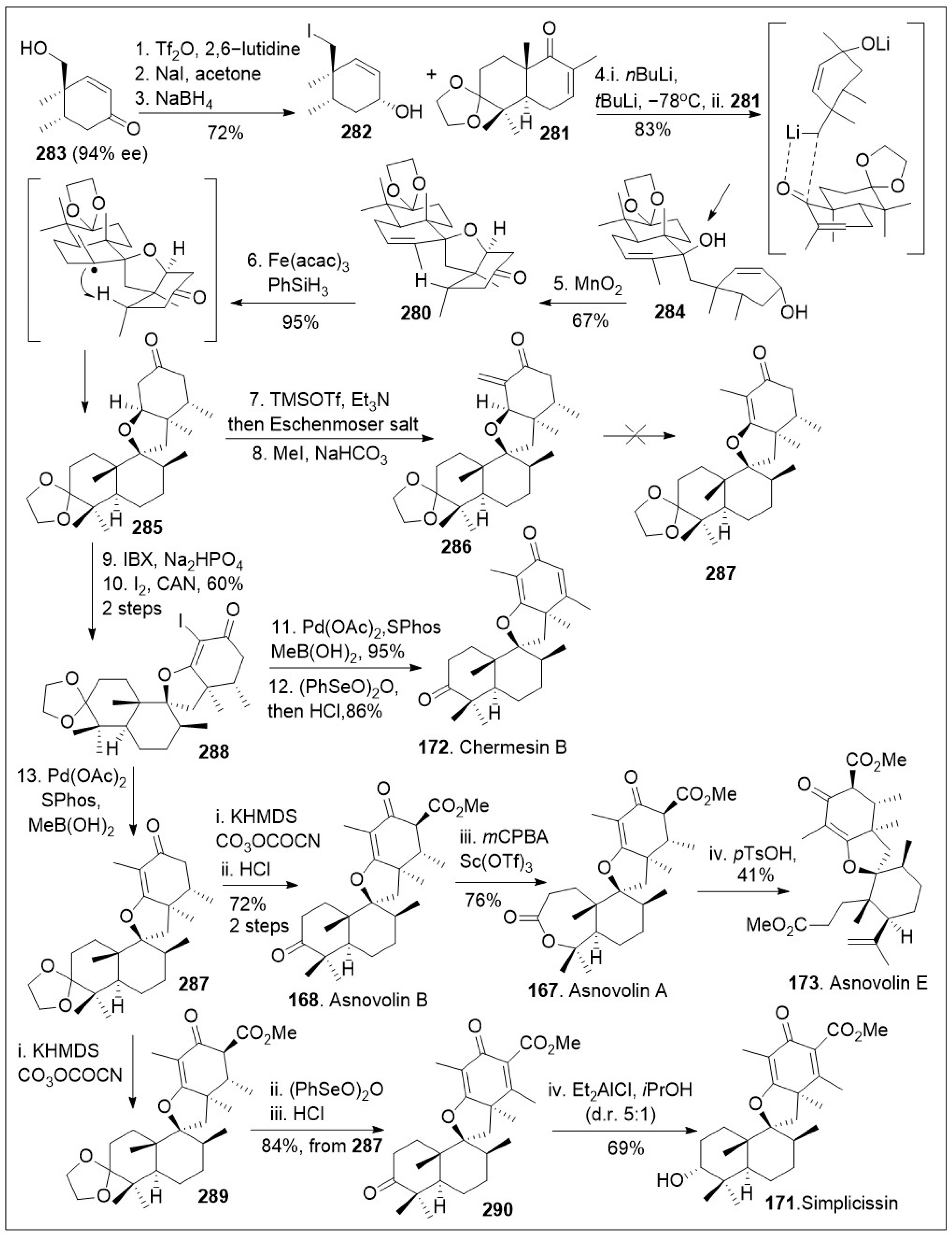
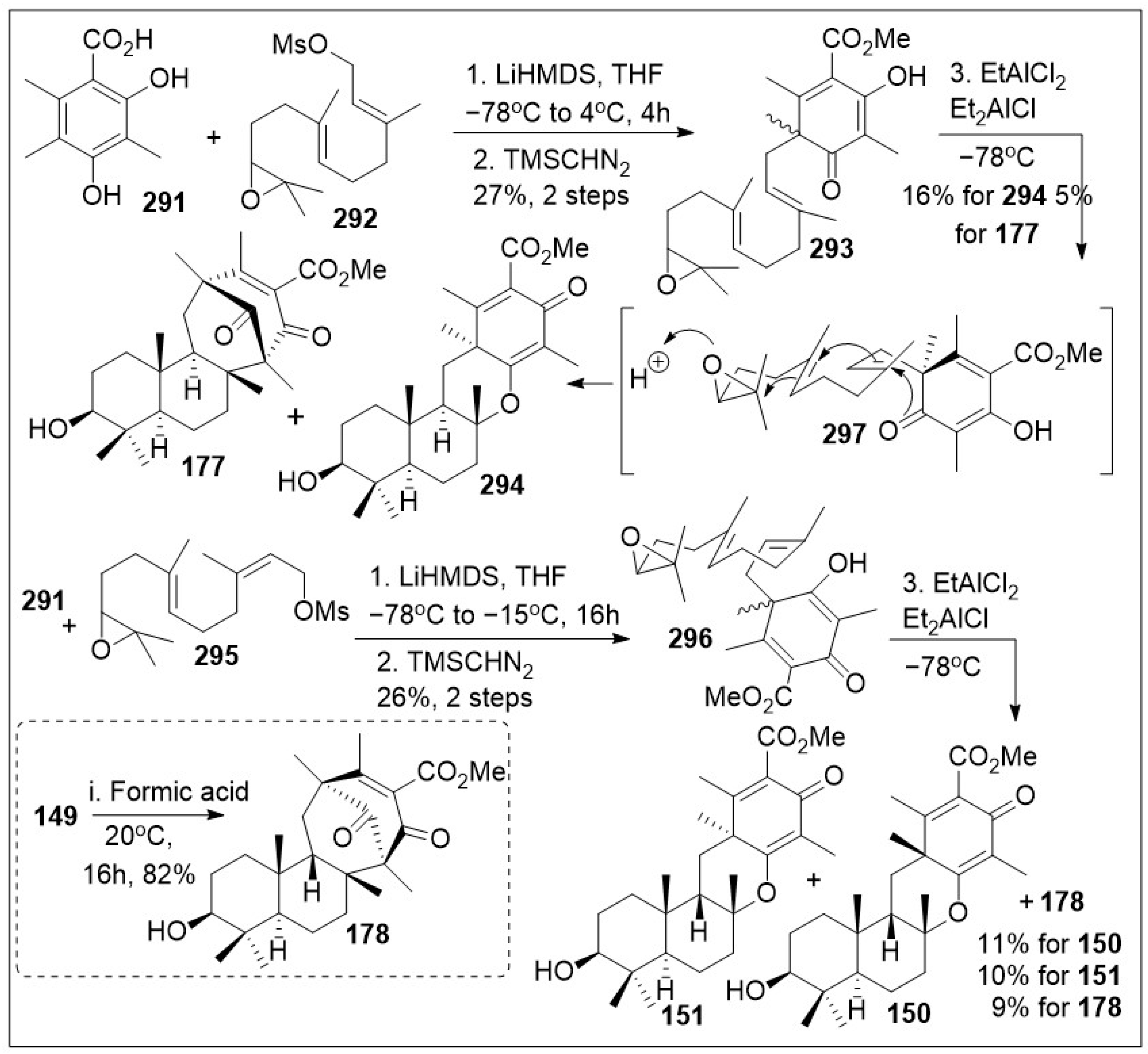
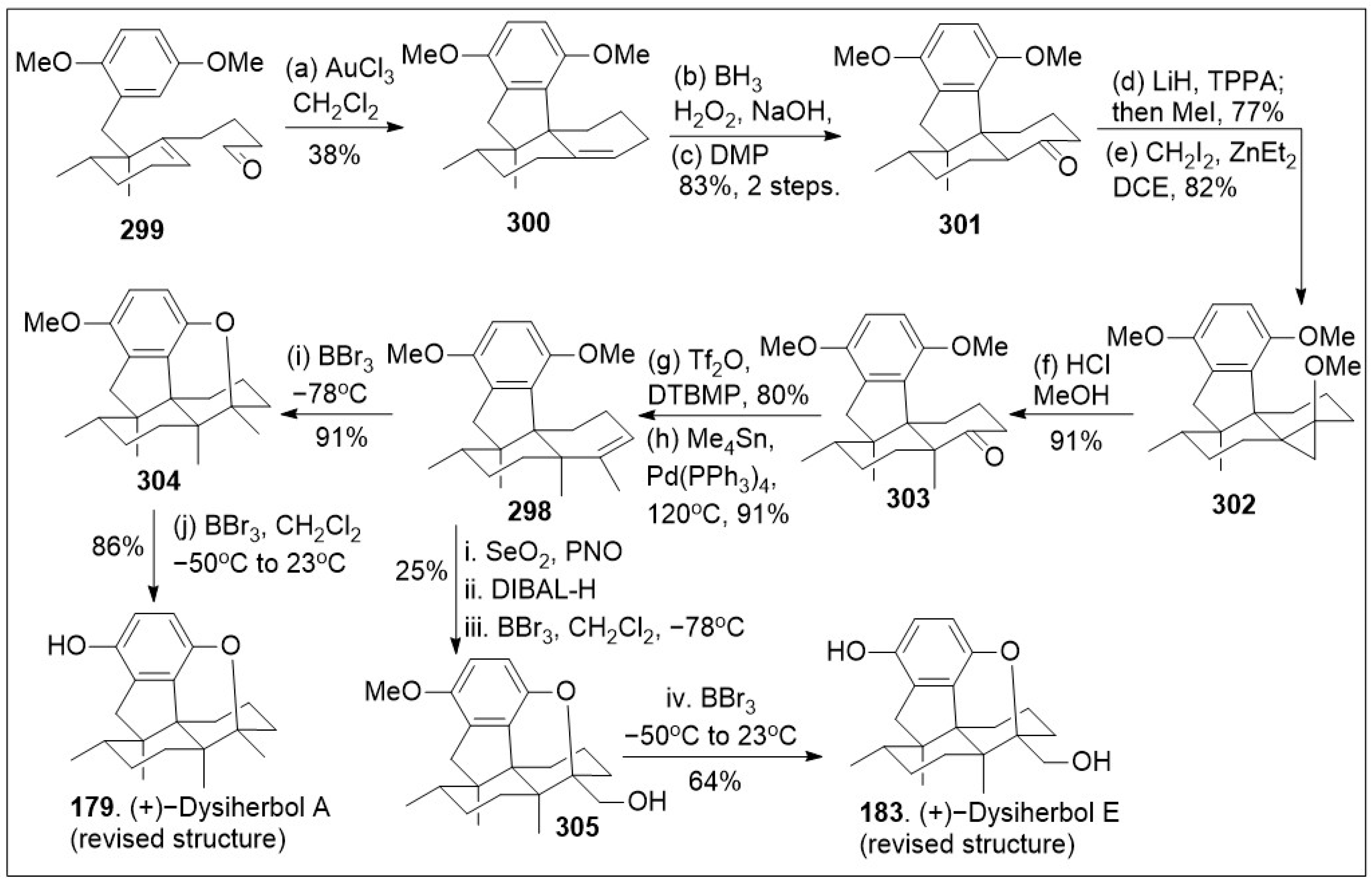
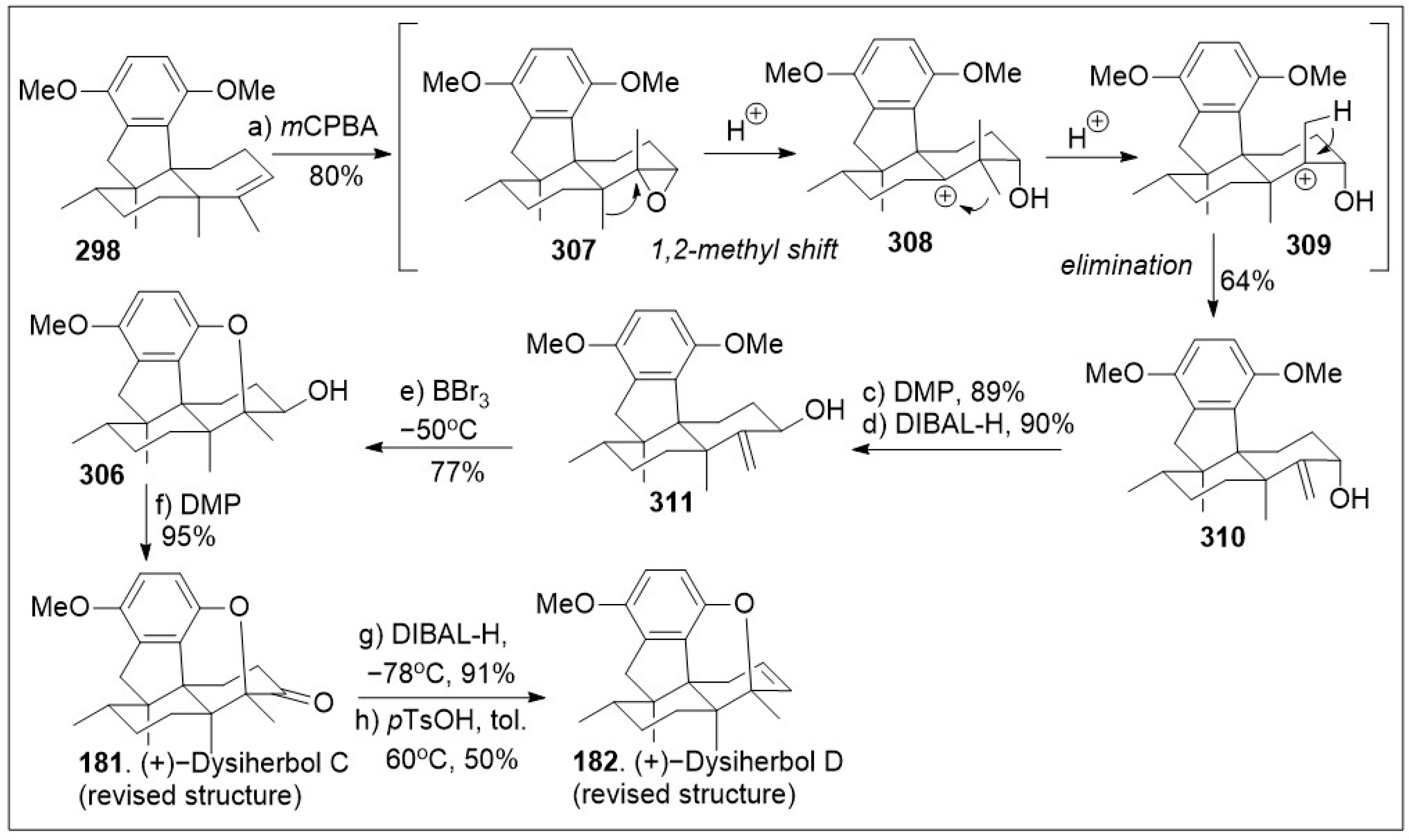
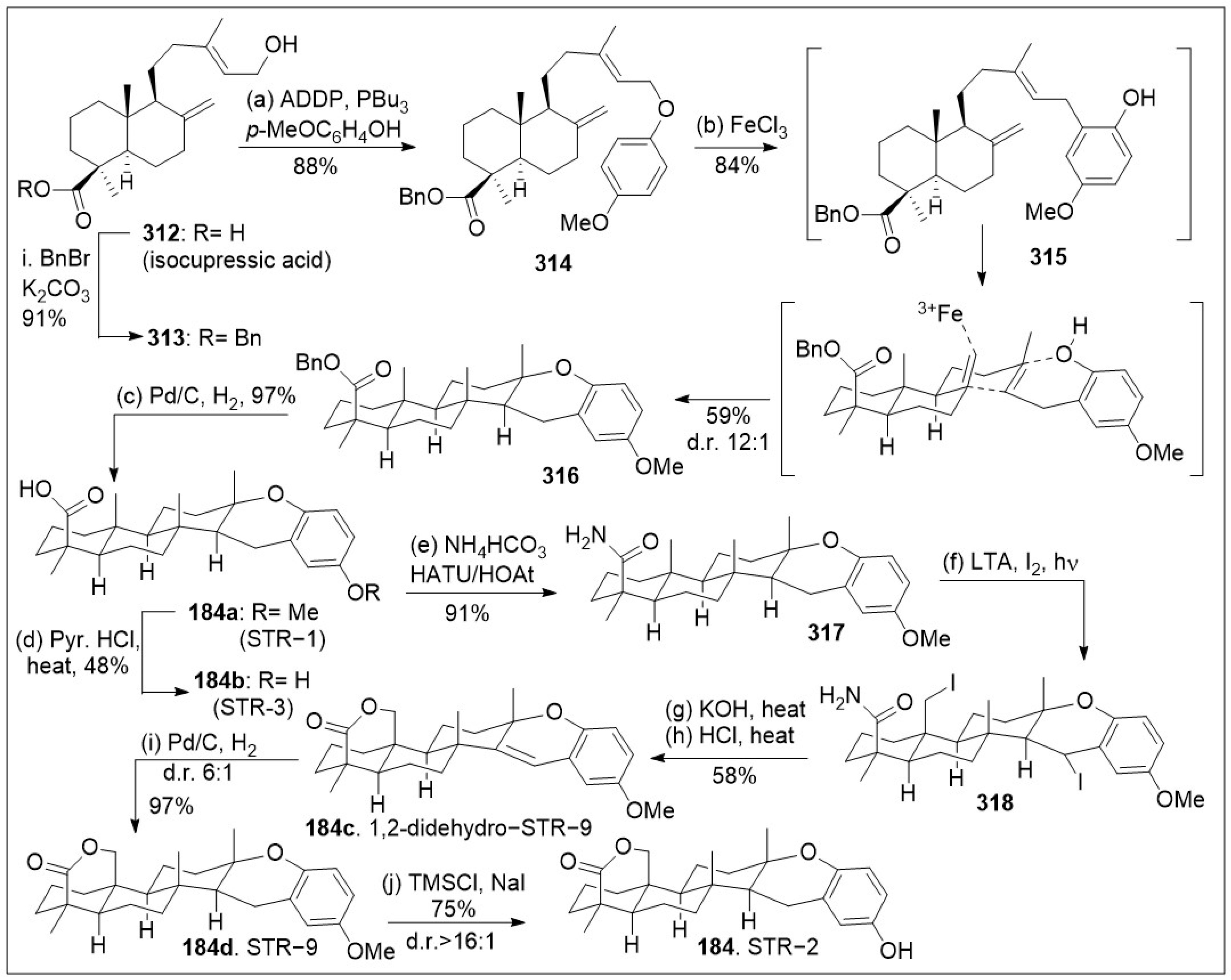
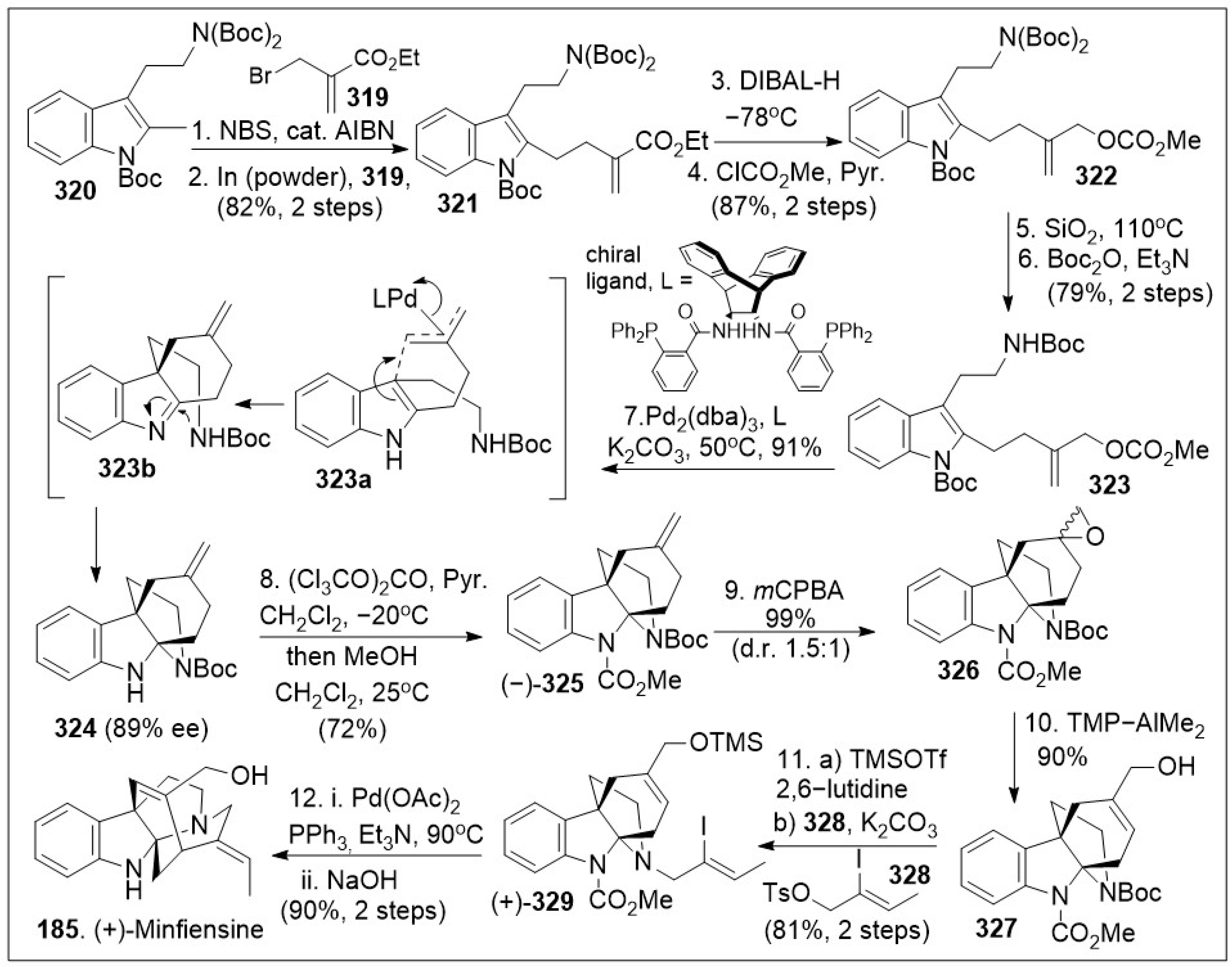
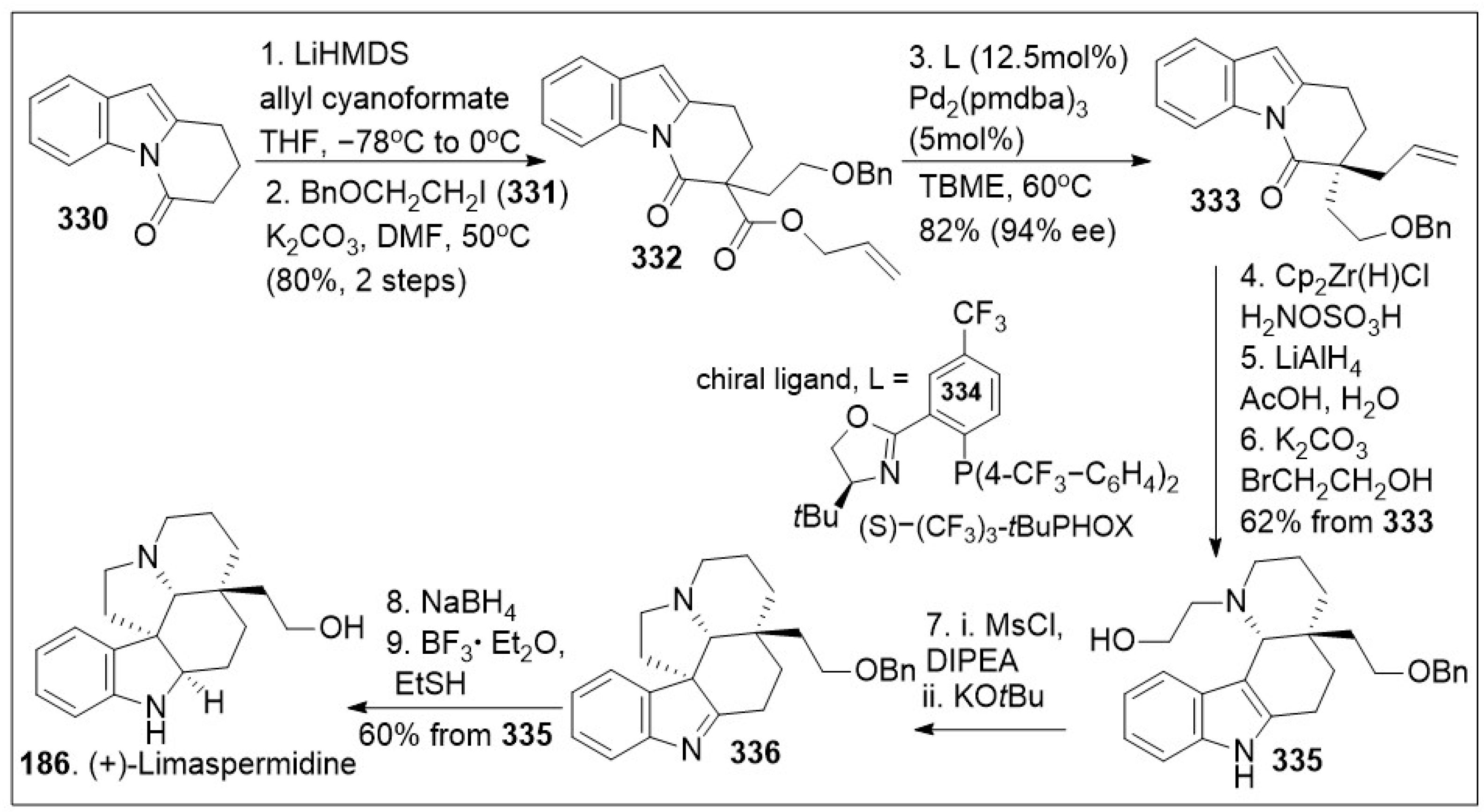
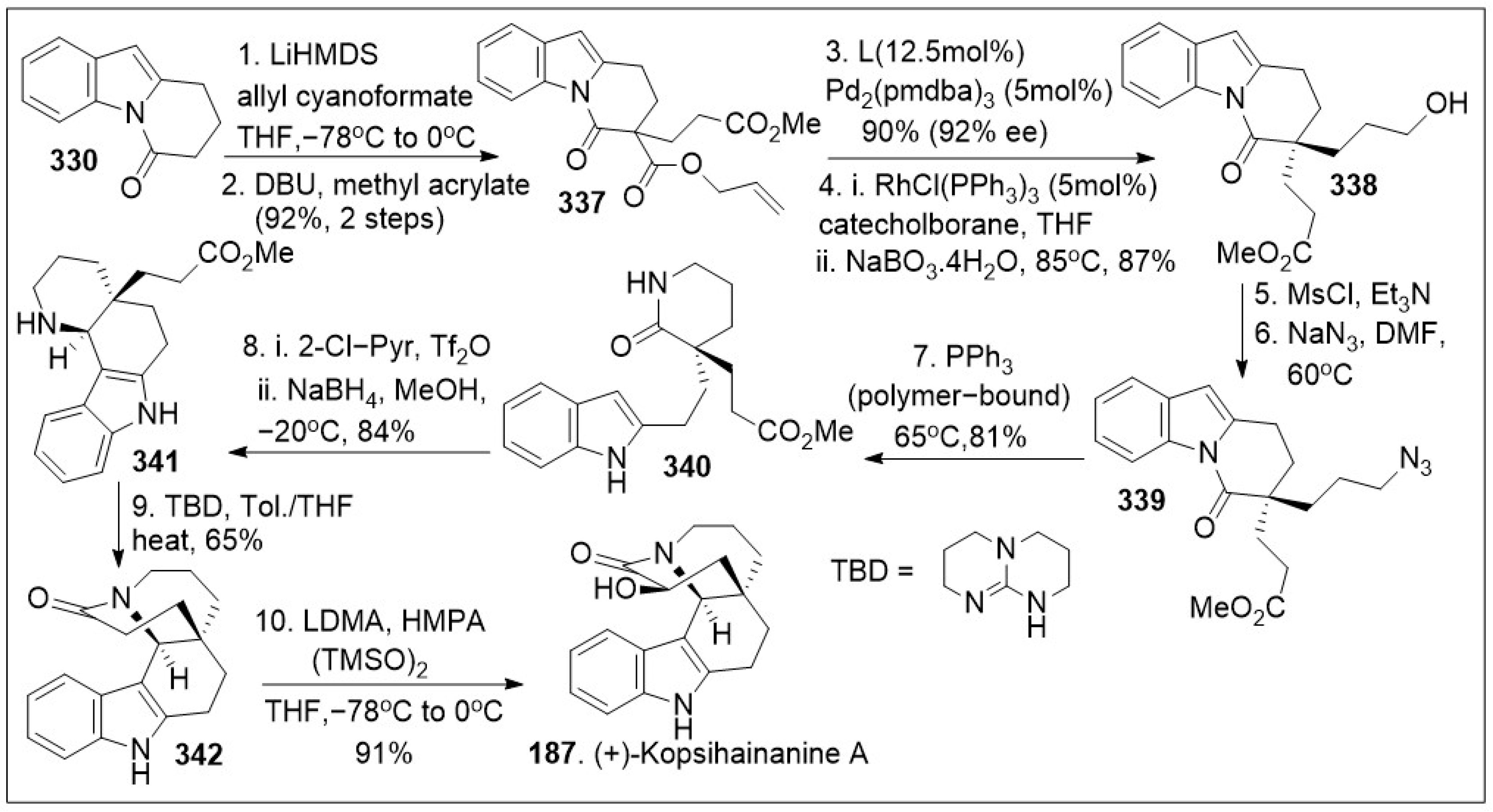
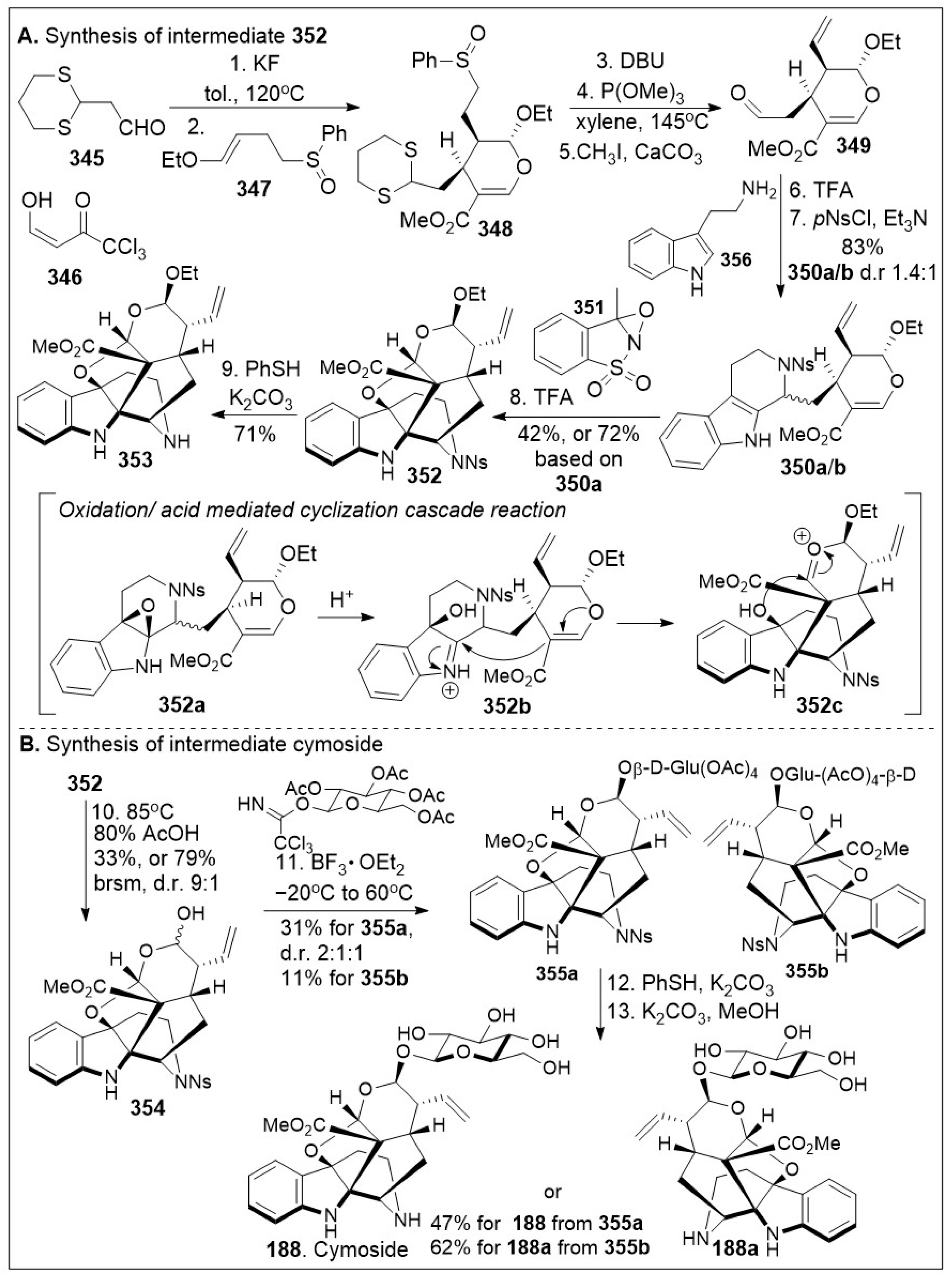
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).