Abstract
A direct regioselective C-H cyanation of purines was developed through a sequential triflic anhydride activation, nucleophilic cyanation with TMSCN, followed by a process of base-mediated elimination of triflous acid (CF3SO2H). In most cases, the direct C-H cyanation occurred on the electron-rich imidazole motif of purines, affording 8-cyanated purine derivatives in moderate to excellent yields. Various functional groups, including allyl, alkynyl, ketone, ester, nitro et al. were tolerated and acted as a C8 directing group. The electron-donating 6-diethylamino, as C2-directing group substituent, can switch the regioselectivity of purine from 8- to 2-position, enabling the synthesis of 8- and 2-cyano 6-dialkylaminopurines from corresponding 6-chloropurine in different reaction order. Further functional manipulations of the cyano group allow the conversions of 8-cyanopurines to corresponding purine amides, imidates, imidothioates, imidamides, oxazolines, and isothiazoles.
1. Introduction
Purine is a fundamental motif in DNA and RNA nucleic acids, and a primary heterocyclic framework in pharmaceuticals and medicinal chemistry. Purine derivatives bearing a cyano group on their framework have received a great deal of attention due to their biological activities such as antimalarial activity [1]. In addition, they also serve as T. brucei’s cysteine protease inhibitors to cure the Human African trypanosomiasis [2]. The cyanation of purines is generally derived from the corresponding purine halides, with 6-chloropurines as the most useful one, via either an SN2Ar process with KCN [3]/Bu4NCN [4] (Scheme 1a) or palladium catalyzed cross-coupling with Zn(CN)2 [5,6,7]. The cyanation of less reactive 2-chloropurines required harsh reaction conditions [2,8,9] or a transition-metal (TM) catalysis (Scheme 1b) [10,11]. Similarly, 8-cyanopurines were prepared from the corresponding 8-halopurines through a 3/4-step traditional protocol involving swapping the bromine to a more electronegative fluorine [12] or sulfonyl group [13,14], or a transition metal (TM)-catalyzed cross coupling (Scheme 1c) [15,16]. Generally, the cyanation of purines required multiple-step synthesis, with extremely toxic agent (KCN or Bu4CN) or the TM catalysis. Owing to the fact that 6- and 2-chloropurines are commercially available and easily obtained from adenine and guanine, we thus planned to develop an expedient and highly regioselective C8-H cyanation protocol through the triflic anhydride activation on the electron-rich imidazole motif of purines.
Scheme 1.
Cyanation of purines.
As is generally known, purine consists of an electron-deficient pyrimidine and an electron-rich imidazole partner (Scheme 1d). In other previous work and our own work, when purines were exposed to nucleophilic reagents such as Grignard reagents [17,18,19,20] and nucleophilic radical agents (Minisci reaction) [21,22,23,24,25,26], the regioselectivity of the reaction predominantly lay at the electron-deficient 6-position [12,13,14,15,16]. In contrast, when the electrophilic bromine was introduced, a C8-brominated purine derivative was obtained (Scheme 1c). To facilitate cyanide-type nucleophilic attack at the 8-position, the polarity of electron-rich imidazole motif should be reversed. In our previous work, we found that trifluoroacetic acid was critical to activate the pyrimidine core of the purine via protonation, facilitating the C6-arylation of purines with arylboronic acid through a Minisci-type reaction [24]. The research also revealed that with the equivalents of trifluoroacetic acid increased, a small amount of 2,8-biarylated purines was found in the transformation [24]. We thus envisaged that with a suitable activator, the polarity of the purine core would be reversed, that is the electrophilic site might switch from the originally electron-deficient pyrimidine to the electron-rich imidazole motif. After a series of investigations on Lewis acids, triflic anhydride (Tf2O) was found to be suitable for this polarity inversion. This interesting development was succeeded in the application of the C8 or C2-cyanation with trimethylsilyl cyanide (TMSCN) through a one-pot procedure (Scheme 1e).
2. Results and Discussion
In our model reaction, 9-benzylpurine (1a), holding three reactive sites at 2-, 6-, and 8-positions of the purine skeletal, was explored first in the presence of TFA (trifluoroacetic acid), a reagent used in our previous work [24], but no reaction occurred. A strong acid TfOH and its alternative TMSOTf cannot promote the reaction either (Scheme 1, entry 2). A typical Lewis acid BF3.OEt2 was investigated but the reaction was intact (entry 3). Tf2O is a useful electrophile to activate 6-membered pyridine compounds, as reported by Corey’s [27], McNally’s [28], and Dixon’s groups [29]. We were pleased to discover that the reaction proceeded smoothly to afford the desired 8-cyanoproduct 2a, whose structure was verified by both 1H NMR with the signal loss of 8-H at 8.0 ppm and single crystal XRD (CCDC: 2217776; Table 1). Further solvent evaluation showed that the reaction did not occur in THF or MeCN but reacted smoothly in toluene and haloalkane solvents (entries 5 and 6). Various bases, including pyridine (Py), Et3N, N-methylmorpholine (NMM), 1,5-diazabicyclo [4.3.0]non-5-ene (DBN), 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU), and 1,4-diazabicyclo [2.2.2]octane (DABCO), were investigated, but DBU provided the best result (Table 1, entries 11~15). The reaction time, temperature, and the equivalents of reactants were finally screened, with the detailed information listed in Supplementary Materials (SI, page 3).

Table 1.
Optimization of the cyanation of purine 1a.
Armed with the optimized conditions, we embarked on the task of investigating on the substrate scope. 6-Chloropurines are the most widely used substrates for the construction of various other substituted purine derivatives. With respect to this, various 9-alkyl 6-chloropurines (1) were investigated (Scheme 2). 9-Propyl and more sterically hindered isopropyl purines (1b and 1c) provided the desired 8-cyano products 2b and 2c in 88% and 75% of yields, respectively. Substrates bearing allyl (1d) and propynal (1e), were tolerated under current conditions. 9-Benzyl (1f) and its derivatives with electron-donating methyl (1g), methoxy (1i), and electron-withdrawing nitro (1h) all caused the corresponding 8-cyano products in satisfactory to excellent yields, but electron-rich substrates obviously displayed better reactivity than the electron-deficient one (2g, 2h vs. 2i). Moreover, the electrophilic ketone and ester substituents (1j and 1k) were also tolerated under current conditions, delivering the desired 2j and 2k in 79% and 63% of yields, respectively. N9-Arylpurines are highly suitable in this transformation. 6-Chloro-N-phenylpurine provided cyanated product 2l in 94% of the yield, while substituted purines, including N-para-methyl 1m, methoxy 1n, methylthio 1o, bromo 1p, and N-meta-chloro 1q all produced the desired products 2m~2q in 76~94% of yields.

Scheme 2.
Substrates scope with 6-chloropurines.
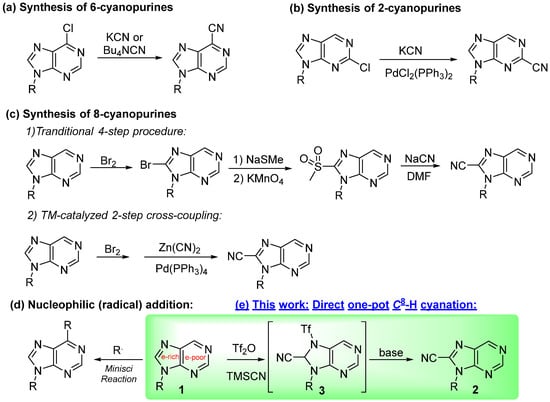
We also briefly investigated the purines bearing other substituents besides the chlorine (Scheme 3). It is worth noting that the reactive intermediates 3 were unstable, sensitive, and generally difficult to isolate. Nevertheless, the key intermediate 3r was isolated when 2,6-dichloropurine (1r) was exploited. Moreover, 3r was transformed into the desired 2r in 68% of the yield by treatment with 1 equiv. of DBU (Scheme 3a). The yield of 2r was increased to 72% when 1.5 equiv. of DBU was added. Moreover, 6-benzylthio purine 1s afforded the desired 8-cyanated product 2s in 97% of the yield, whereas 6-methoxy purine 1t caused the corresponding 2t in only 45% of the yield, with most of the substrate remaining unreactive (Scheme 3b,c). Another interesting result was obtained from the reaction of 6-p-tolyl purine 1u, in which a mixture of 8-monocyanated product 2u (23%) and 2,8-dicyanoated 4u (42%) were generated (Scheme 3d). The results demonstrated that the electron-donating alkoxy and aryl substituents might enhance the electron density of the pyrimidine motif of the purines, causing the decreasing regioselectivity over the imidazole motif. To verify our hypothesis, we introduced a strong electron-donating diethylamino (Et2N) on the 6-position of purine (1v) but the reaction of 1v was relatively complicated. After careful isolation and structural identification, a purine derivative 2v’ bearing the nitrile on its 2-position, rather than common 8-position, was obtained albeit in only 16% of the yield (Scheme 3e). None of the desired 8-cyanopurine 2v was traced by the TLC and 1H NMR of the reaction system. Obviously, the strong donating diethylamino completely switched the reactivity of the purine skeletal, enabling the pyrimidine motif to be more reactive than the imidazole counterpart. To obtain the desired 8-cyano-6-diethylaminopurine 2v, the direct amination reaction of 6-chloro-8-cyanopurine 2f and diethylamine (Scheme 3f) was performed in the presence of Et3N in EtOH, providing the desired 2v in 83% of the yield. The 1H NMR of 8-cyanopurine 2v and 2-cyanopurine 2v’ are totally different (Scheme 3g), as well as the 13C NMR shown in SI (page S25–S28). It is worth mentioning that the alkyl carbons in Et2N in both 2v and 2v’ are hard to emerge especially the 2v’ in which 1250 scans (~80 min) were tested while the peak appearances were broad and weak (much weaker than the ipso aromatic carbons). The HSQC test of 2v’ was thus performed for further verification of the alkyl carbons in Et2N (Figure 1). Interestingly, when heating the reaction system, an imidaminopurine derivative 5f was obtained in 33% of the yield from a Et2NH/EtOH solution at 90 °C through a base-mediated Pinner-type reaction [30]. This yield was further optimized to 91% with pure Et2NH as solution at 60 °C (Scheme 3h). The results demonstrated that besides the 6-chloro reactive site of purine, the nitrile group is also a highly potential electrophilic reactive position.
Scheme 3.
Substrates scope with other purines. The reactions in Scheme (a–h) were carried out in 0.2 mmol scale under standard condition. Scheme (g): the 1H NMR comparation of 2v and 2v’.
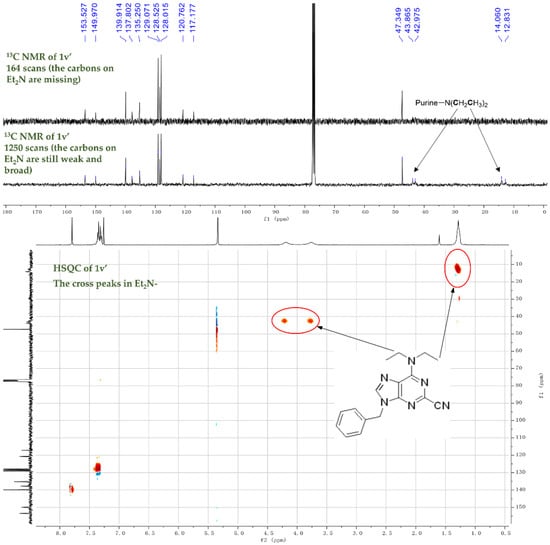
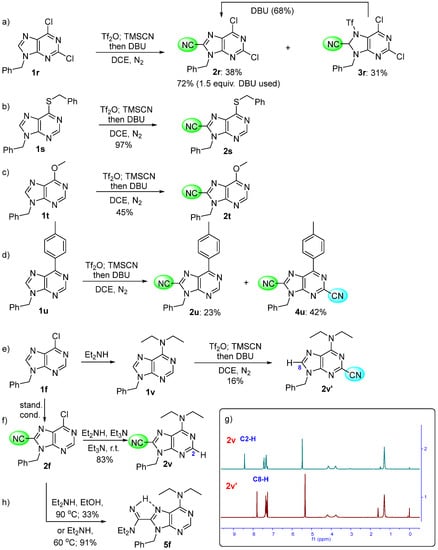
Figure 1.
The 13C NMR (101 MHz; 164 scans and 1250 scans) and the HSQC of 2v’ (the structure identification of the broad and weak carbons in Et2N). The sample was prepared from 10 mg of 2v in 0.5 mL CDCl3.
As building blocks in purine chemistry, 6-chloropurines can be converted into various purine derivatives (Scheme 4). Gram-scale reactions with 1f were carried out, affording 2f in 1.03 g (76%, 5 mmol scale) and 1.38 g (73%, 7 mmol scale) (Scheme 2-2f). A Sonogashira coupling product 6f was obtained in 52% of the yield from the reaction of 2f with p-methoxyphenylacetylene under the palladium catalysis [31]. Similarly, a Suzuki–Miyaura coupling with p-tolylboronic acid delivered 2u in 28% of the yield, along with 2f recovered in 26% of the yield [32]. The reaction of 1f with sodium methoxide provided a complicated mixture, but it reacted smoothly to cause the desired 6-methoxypurine 2t in the presence of CuBr (10 mol%) [33]. Moving forward, the alkylthiolation with benzyl thiol produced the desired 6-benzylthio purine 2s in 24% of the yield and an extra thiol to nitrile adduct, imidothioate 5s, similar to the adduct from Et2NH (Scheme 4) in 54% of the yield. The results are similar to our previous discovery, in which an interesting adduct was formed from the addition of potassium thioacetate (KSAc) to acetonitrile (MeCN) [34].

Scheme 4.
Gram-scale synthesis of 2f and its derivation.
The further derivation of the nitrile group was performed with 9-benzyl-6-benzylthio-8-cyano purine (1s) as an example. A 6 mmol Gram-scaled reaction was carried out, from which 1.60 g of desired 2s was obtained in 74% of the yield (Scheme 5). Treatment of 1s with benzylthiol (BnSH) at r.t. afforded the desired adduct 5s in 78% of the yield (Scheme 5). The result indicated that the cyano group on the purine skeleton was more reactive than the common arylnitriles. We thus explored its reaction with sodium methoxide (NaOMe), which produced the corresponding adduct methyl carbimidate 6s in 80% of the yield. Generally, carbimidate was commonly unstable but herein 6s along with 5s were generated and could be separated through the column chromatography, probably due to the stabilization effect by the intramolecular hydrogen bonding with the nitrogen in the purine scaffold. As that carbimidate was also a general intermediate in the synthesis of oxazoline [35], we thus reacted 6s with 2-amino-2-methylpropan-1-ol in toluene. After an overnight Dean–Stark reflux (~10 h), oxazoline 7s was obtained in 70% of the yield. The transformation of 2s to 7s was also carried out through a one-pot process, delivering a total yield of 75%. The hydrolysis of a nitrile to an amide is a classic transformation. However, the direct hydrolysis of 2s in aqueous NaOH solution failed due to its poor solubility in water, with all starting materials floating on the water surface. The addition of EtOH can disperse 2s into the solution, but only ethyl carbimidate homolog 6s’ was obtained with NaOH in EtOH/H2O (7:3). Finally, the desired amide 8s was attained in a solution of NaOH in THF/H2O. It is worth noting that the isolation of 5s, 6s, 6s’, and 8s were quite convenient because all adducts directly precipitated from their solutions and only filtration and drying were required in the work-up process. Very recently, Yang and coworkers reported a highly effective diastereoselective (3 + 2) transannulations of 1,2,3-thiadiazoles with cyanoepoxides to afford the corresponding isothiazoles under the catalysis of [Rh(COD)Cl]2 and DPPF [36]. We also developed the transannulation of 1,2,3-thiazoles with α,β-unsaturated nitriles [37]. Finally, referring to Yang’s conditions, the reaction of 2s with thiadiazole 9 caused the desired isothiazole 10s in a quantitative yield.
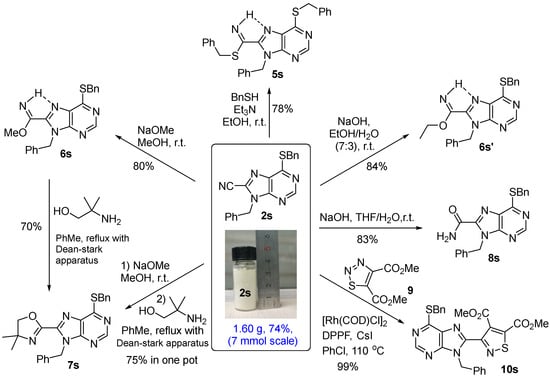
Scheme 5.
Gram-scale synthesis of 2s and its synthetical application.
The proposed reaction mechanism is portrayed in Scheme 6. Triflylation of the most nucleophilic nitrogen (N-7) of the purines 1 activates the electron-rich imidazole motif and enables it to be subjected to the subsequent nucleophilic attack by TMSCN, in this way the sensitive and unstable key adducts 3 are formed. We believe that the base-mediated elimination of triflous acid (CF3SO2H) from 3 furnishes the formal C-H cyanation products 2, and the transformation of intermediate 3r in Scheme 3 solidly supports this suggested mechanistic insight.
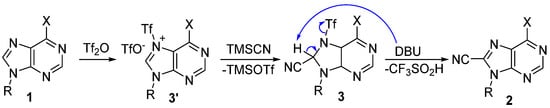
Scheme 6.
Reaction mechanism of the 8-cyanation of purines.
3. Materials and Methods
3.1. General Information
Moisture sensitive reactions were carried out under the nitrogen atmosphere with flame-dried apparatus. The solvents, THF, PhCl, PhMe, CHCl3, used in Table 1 and the DCE used under the standard conditions, were freshly distilled by referring the standard dehydration process. Other solvents used in Scheme 4, Scheme 5 and Scheme 6, besides DCE, were used directly from the commercial source. Melting points were obtained on a MP-500 melting point apparatus and are uncorrected. TLC were performed on silica gel GF254 plates, the plates of which were visualized under UV light. 1H and 13C-NMR spectra were recorded on a 400 MHz Bruker spectrometer in CDCl3 with TMS as an internal standard and the chemical shifts (δ) are reported in parts per million (ppm). HRMS measurements were carried out on an Agilent LC/MSD ESI-TOF mass spectrometer. Petroleum ether (PE, 60–90 °C) and ethyl acetate (EtOAc) were used for column chromatography.
All purine substrates were synthesized according to our previous work [24,38].
3.2. General Procedure for the 8-Cyanation of N-Benzylpurine, 6-Chloropurines and 6-Alkoxy, Alkylthio, and Aryl Purines 2
To a frame-dried 10 mL vial was added solid purine (0.2 mmol), and the vial was capped with a septum followed by the N2 protection. Anhydrous 1,2-dichloroethane (2.0 mL) was then injected and the reaction was cooled in an ice-water bath. Triflic anhydride (0.24 mmol, 40 μL) was then injected dropwise, and the reaction system was stirred at 0 °C for 15 min followed by the injection of trimethylsilyl cyanide (TMSCN; 0.4 mmol, 50 μL). The system was stirred at 84 °C for 3 h. After addition of 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU, 39 μL, 1.3 mmol) the mixture was stirred at 84 °C for another 6 h. After cooling to r.t., the reaction was quenched with 1.0 mL of saturated NaHCO3 solution and the organic phase was separated. The aqueous phase was then extracted with 3.0 mL of DCE. The combined organic phase was dried over anhydrous Na2SO4. After removing the solvent in vacuo, the residue was purified by silica gel column chromatography with a mixture PE and EtOAc as the eluent.
The Gram-scale reactions of 2f and 2s were scaled up proportionally.
3.2.1. 9-Benzyl-9H-purine-8-carbonitrile (2a)
Colorless crystals, mp: 178–180 °C. [CCDC: 2217776] yield: 26 mg, 54%. Rf = 0.12 (PE/EtOAc = 5/1, v/v). 1H NMR (400 MHz, CDCl3) δ 9.26 (s, 1H, C6-H in purine), 9.17 (s, 2H, C2-H in purine), 7.48–7.45 (m, 2H, ArH), 7.37–7.32 (m, 3H, ArH), and 5.61 (s, 2H, NCH2). 13C NMR (101 MHz, CDCl3) δ 155.4, 151.3, 150.2, 133.8, 133.0, 129.2, 129.1, 128.7, 128.4, 110.2, and 47.8. HRMS (ESI): m/z [M + H]+ calcd. for C13H10N5+, 236.0931; found: 236.0923.
3.2.2. 6-Chloro-9-propyl-9H-purine-8-carbonitrile (2b)
Colorless crystals, mp: 81–83 °C, yield: 39 mg, 88%. Rf = 0.75 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.89 (s, 1H, C2-H in purine), 4.46 (t, J = 7.2 Hz, 2H, NCH2), 2.03 (sext, J = 7.2 Hz, 2H, CH2), and 1.00 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 154.4, 153.9, 150.9, 131.1, 129.1, 109.7, 46.9, 23.2, and 11.0. HRMS (ESI): m/z [M + H]+ calcd. for C9H9ClN5+, 222.0541; found: 222.0548.
3.2.3. 6-Chloro-9-isopropyl-9H-purine-8-carbonitrile (2c)
Colorless crystals, mp: 112–113 °C, yield: 33 mg, 75%, Rf = 0.65 (PE/EtOAc = 3/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.86 (s, 1H, C2-H in purine), 5.15 (sept, J = 6.8 Hz, 1H, NCH2), and 1.80 (d, J = 6.8 Hz, 6H, 2CH3). 13C NMR (101 MHz, CDCl3) δ 153.9, 153.8, 150.6, 131.4, 127.9, 110.2, 51.3, and 21.6. HRMS (ESI): m/z [M + H]+ calcd. for C9H9ClN5+, 222.0541; found: 222.0537.
3.2.4. 9-Allyl-6-chloro-9h-purine-8-carbonitrile (2d)
Colorless crystals, mp: 108–110 °C, yield: 36 mg, 82%. Rf = 0.50 (PE/EtOAc = 3/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.90 (s, 1H, C2-H in purine), 6.03 (ddt, J = 17.2, 10.4, 6.0, 17.2 Hz, 1H, CH), 5.42 (d, J = 10.0 Hz, 1H, 1H in CH2), 5.32 (d, J = 17.2 Hz, 1H, 1H in CH2), and 5.09 (d, J = 6.0 Hz, 2H, NCH2). 13C NMR (101 MHz, CDCl3) δ 154.6, 153.9, 150.7, 131.0, 129.4, 128.8, 121.2, 109.5, and 46.9. HRMS (ESI): m/z [M + H]+ calcd. for C9H7ClN5+, 220.0384; found: 222.0368.
3.2.5. 6-Chloro-9-(propargyl)-9H-purine-8-carbonitrile (2e)
Colorless crystals, mp: 134–136 °C, yield: 38 mg, 87%, Rf = 0.77 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.93 (s, 1H, C2-H in purine), 5.25 (d, J = 2.4 Hz, 2H, NCH2), and 2.57 (t, J = 2.4 Hz, 1H, CH). 13C NMR (101 MHz, CDCl3) δ 154.9, 154.1, 150.3, 130.8, 128.3, 109.1, 76.1, 74.0, and 33.9. HRMS (ESI): m/z [M + H]+ calcd. for C9H5ClN5+, 218.0228; found: 218.0235.
3.2.6. 9-Benzyl-6-chloro-9H-purine-8-carbonitrile (2f)
Colorless crystals, mp: 102–104 °C, yield: 46 mg, 85%. Rf = 0.51 (PE/EtOAc = 3/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.94 (s, 1H, C2-H in purine), 7.49–7.46 (m, 2H, ArH), 7.39–7.36 (m, 3H, ArH), and 5.62 (s, 2H, NCH2). 13C NMR (101 MHz, CDCl3) δ 154.7, 153.9, 150.8, 133.4, 131.0, 129.35, 129.33, 128.6, 128.5, 109.9, and 48.6. HRMS (ESI): m/z [M + H]+ calcd. for C13H9ClN5+, 270.0541; found: 270.0550.
3.2.7. 6-Chloro-9-(4-methylbenzyl)-9h-purine-8-carbonitrile (2g)
Colorless crystals, mp: 91–92 °C. Yield: 49 mg, 86%. Rf = 0.62 (PE/EtOAc = 3/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.93 (s, 1H, C2-H in purine), 7.36 (d, J = 8.0 Hz, 2H, ArH), 7.16 (d, J = 8.0 Hz, 2H, ArH), 5.58 (s, 2H, NCH2), and 2.32 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 154.6, 153.8, 150.7, 139.4, 131.0, 130.5, 129.9, 128.6, 128.5, 109.9, 48.4, and 21.1. HRMS (ESI): m/z [M + H]+ calcd. for C14H11ClN5+, 284.0697; found: 284.0707.
3.2.8. 6-Chloro-9-(4-nitrobenzyl)-9H-purine-8-carbonitrile (2h)
Light yellow crystals, mp: 217–220 °C. Yield: 40 mg, 64%. Rf = 0.60 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H, C2-H in purine), 8.22 (d, J = 8.8 Hz, 2H, ArH), 7.65 (d, J = 8.8 Hz, 2H, ArH), and 5.85 (s, 2H, NCH2). 13C NMR (101 MHz, DMSO-d6) δ 154.7, 152.0, 151.1, 147.4, 141.7, 130.4 129.4, 129.0, 123.9, 110.2, and 47.1. HRMS (ESI): m/z [M + H]+ calcd. for C13H8ClN5+, 315.0392; found: 315.0401.
3.2.9. 6-Chloro-9-(4-methoxybenzyl)-9H-purine-8-carbonitrile (2i)
Colorless crystals, mp: 158–160 °C. Yield: 55 mg, 92%, Rf = 0.81 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.94 (s, 1H, C2-H in purine), 7.44 (d, J = 8.8 Hz, 2H, ArH), 6.88 (d, J = 8.8 Hz, 2H, ArH), 5.56 (s, 2H, NCH2), and 3.78 (s, 3H, OCH3). 13C NMR (101 MHz, CDCl3) δ 160.3, 154.6, 153.9, 150.7, 131.1, 130.2, 128.6, 125.6, 114.6, 110.0, 55.3, and 48.2. HRMS (ESI): m/z [M + H]+ calcd. for C14H11ClN5O+, 300.0647; found: 300.0640.
3.2.10. Ethyl-2-(6-chloro-8-cyano-9H-purin-9-yl)acetate (2j)
Colorless crystals, mp: 108–109 °C. Yield: 42 mg, 79%. Rf = 0.40 (PE/EtOAc = 3/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H, C2-H in purine), 5.20 (s, 2H, NCH2), 4.31 (q, J = 7.2 Hz, 2H, OCH2), and 1.33 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 165.0, 154.8, 154.0, 150.9, 130.8, 129.3, 109.2, 63.3, 44.7, and 14.0. HRMS (ESI): m/z [M + H]+ calcd. for C10H9ClN5O2+, 266.0439; found: 266.0432.
3.2.11. 6-Chloro-9-(2-(4-chlorophenyl)-2-oxoethyl)-9H-purine-8-carbonitrile (2k)
Colorless crystals, mp: 204–207 °C. Yield: 42 mg, 63%. Rf = 0.65 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H, C2-H in purine), 8.01 (d, J = 8.8 Hz, 2H, ArH), 7.59 (d, J = 8.8 Hz, 2H, ArH), and 5.87 (s, 2H, NCH2). 13C NMR (101 MHz, CDCl3) δ 187.5, 154.7, 154.1, 151.1, 142.0, 131.5, 131.0, 129.8, 129.7, 109.4, and 49.6. HRMS (ESI): m/z [M + H]+ calcd. for C14H8Cl2N5O+, 332.0100; found: 332.0097.
3.2.12. 6-Chloro-9-phenyl-9H-purine-8-carbonitrile (2l)
Colorless crystals, mp: 158–161 °C. Yield: 48 mg, 94%. Rf = 0.85 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.91 (s, 1H, C2-H in purine), 7.65–7.70 (m, 3H, ArH), and 7.59–7.62 (m, 2H, ArH). 13C NMR (101 MHz, CDCl3) δ 155.2, 154.2, 151.1, 131.4, 131.1, 130.9, 130.3, 128.7, 125.9, and 109.7. HRMS (ESI): m/z [M+H]+ calcd. for C12H7ClN5+, 256.0384; found: 256.0371.
3.2.13. 6-Chloro-9-(p-tolyl)-9H-purine-8-carbonitrile (2m)
Colorless crystals, mp: 120–123 °C, yield: 46 mg, 85%. Rf = 0.80 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.90 (s, 1H, C2-H in purine), 7.47 (s, 4H, ArH), and 2.50 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 155.2, 154.2, 151.2, 141.4, 131.1, 130.9, 128.9, 128.8, 125.7, 109.7, and 21.3. HRMS (ESI): m/z [M + H]+ calcd. for C13H9ClN5+, 270.0541; found: 270.0544.
3.2.14. 6-Chloro-9-(4-methoxyphenyl)-9H-purine-8-carbonitrile (2n)
Colorless crystals, mp: 122–125 °C. Yield: 48 mg, 84%. Rf = 0.75 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H, C2-H in purine), 7.49 (d, J = 8.8 Hz, 2H, ArH), 7.14 (d, J = 8.8 Hz, 2H, ArH), and 3.91 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 161.2, 155.1, 154.1, 151.3, 130.9, 129.0, 127.3, 123.8, 115.4, 109.7, and 55.7. HRMS (ESI): m/z [M+H]+ calcd. for C13H9ClN5O+, 286.0490; found: 286.0499.
3.2.15. 6-Chloro-9-(4-(methylthio) phenyl)-9H-purine-8-carbonitrile (2o)
Yellow crystals, mp: 149–151 °C. Yield: 51 mg, 85%. Rf = 0.80 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.90 (s, 1H, C2-H in purine), 7.51 (d, J = 8.8 Hz, 2H, ArH), 7.47 (d, J = 8.8 Hz, 2H, ArH), and 2.57 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 155.2, 154.3, 151.1, 143.3, 131.1, 128.7, 127.7, 127.0, 126.0, 109.7, and 15.2. HRMS (ESI): m/z [M + H]+ calcd. for C13H9ClN5S+, 302.0262; found: 302.0281.
3.2.16. 9-(4-bromophenyl)-6-chloro-9H-purine-8-carbonitrile (2p)
Colorless crystals, mp: 165–167 °C, yield: 51 mg, 76%. Rf = 0.85 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.92 (s, 1H, C2-H in purine), 7.84–7.81 (m, 2H, ArH), and 7.52–7.26 (m, 2H, ArH). 13C NMR (101 MHz, CDCl3) δ 155.4, 154.5, 150.9, 133.6, 131.1, 130.3, 128.3, 127.3, 125.2, and 109.5. HRMS (ESI): m/z [M + H]+ calcd. for C12H6BrClN5+, 333.9490; found: 333.9504.
3.2.17. 6-Chloro-9-(3-chlorophenyl)-9H-purine-8-carbonitrile (2q)
Colorless crystals, mp: 139–141 °C. Yield: 48 mg, 83%. Rf = 0.85 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.93 (s, 1H, C2-H in purine), 7.65–7.61 (m, 3H, ArH), and 7.54–7.51 (m, 1H, ArH). 13C NMR (101 MHz, CDCl3) δ 155.4, 154.5, 151.0, 136.1, 132.3, 131.3, 131.2, 131.1, 128.3, 126.3, 124.1, and 109.5. HRMS (ESI): m/z [M + H]+ calcd. for C12H6Cl2N5+, 289.9995; found: 290.0001.
3.2.18. 9-Benzyl-2,6-dichloro-9h-purine-8-carbonitrile (2r) and 9-Benzyl-2,6-dichloro-7-((trifluoromethyl)sulfonyl)-8,9-dihydro-7H-purine-8-carbonitrile (3r)
2r: Colorless crystals, mp: 141–143 °C. Yield: 116 mg, 38%. Rf = 0.65 (PE/EtOAc = 5/1, v/v). 1H NMR (400 MHz, CDCl3) δ 7.49–7.38 (m, 5H, ArH), and 5.58 (s, 2H, NCH2). 13C NMR (101 MHz, CDCl3) δ 156.3, 154.8, 151.9, 133.0, 130.1, 129.5, 129.4, 129.1, 128.6, 109.6, and 48.8. HRMS (ESI): m/z [M+H]+ calcd. for C13H8Cl2N5+, [M + H]+ 304.0152, found: 304.0155.
3r: Colorless oil, yield: 93 mg, 31%. Rf = 0.72 (PE/EtOAc = 5/1, v/v). 1H NMR (400 MHz, CDCl3) δ 7.50–7.33 (m, 5H, ArH), 6.12 (s, 1H, CH), 5.32 (d, J = 15.2 Hz, 1H, NCH), and 4.29 (d, J = 15.2 Hz, 1H, NCH). 13C NMR (101 MHz, CDCl3) δ 161.2, 158.1, 145.7, 131.3, 129.74, 129.67, 128.47, 118.9 (q, J = 319.7 Hz, CF3), 117.3, 110.6, 68.4, and 47.1. 19F NMR (376 MHz, CDCl3) δ -74.13. HRMS (ESI) calcd. for C14H9Cl2F3N5O2S+ [M + H]+ 437.9801, found: 437.9796.
3.2.19. 9-Benzyl-6-(benzylthio)-9H-purine-8-carbonitrile (2s)
Colorless crystals, mp: 111–113 °C. Yield: 69 mg, 97%. Rf = 0.90 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H, C2-H in purine), 7.45–7.43 (m, 4H, ArH), 7.27–7.35 (m, 6H, ArH), 5.55 (s, 2H, NCH2), and 4.66 (s, 2H, SCH2). 13C NMR (101 MHz, CDCl3) δ 164.3, 154.4, 148.0, 136.7, 134.0, 130.6, 129.2, 129.1, 129.0, 128.6, 128.4, 127.5, 125.9, 110.4, 48.0, and 33.0. HRMS (ESI) calcd. for C20H16N5S+ [M+H]+, 358.1121, found: 358.1131.
3.2.20. 9-Benzyl-6-methoxy-9H-purine-8-carbonitrile (2t)
Colorless crystals, mp: 138–141 °C. Yield: 24 mg, 45%. Rf = 0.80 (PE/EtOAc = 1/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H, C2-H in purine), 7.46–7.44 (m, 2H, ArH), 7.38–7.33 (m, 3H, ArH), 5.57 (s, 2H, NCH2), and 4.22 (s, 3H, OCH3). 13C NMR (101 MHz, CDCl3) δ 162.2, 155.2, 151.2, 134.1, 129.2, 129.0, 128.4, 125.8, 121.8, 110.5, 54.8, and 48.1. HRMS (ESI) calcd. for C14H12N5O+ [M+H]+, 266.1036, found: 266.1039.
3.2.21. 9-Benzyl-6-(p-tolyl)-9H-purine-8-carbonitrile (2u) and 9-Benzyl-6-(p-tolyl)-9H-purine-2,8-dicarbonitrile (4u)
2u: Colorless crystals, mp: 138–140 °C. Yield: 25 mg, 23%. Rf = 0.19 (PE/EtOAc = 30/1, v/v). 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H, C2-H in purine), 8.70 (d, J = 8.0 Hz, 2H, ArH), 7.51–7.48 (m, 2H, ArH), 7.38–7.35 (m, 5H, ArH), 5.63 (s, 2H, NCH2), and 2.46 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 157.5, 155.2, 151.4, 142.8, 134.2, 131.9, 130.2, 130.1, 129.6, 129.2, 129.0, 128.5, 127.5, 110.7, 47.8, and 21.7. HRMS (ESI) calcd. for C20H16N5+ [M + H]+ 326.1400, found: 326.1397.
4u: Colorless crystals, mp: 38–40 °C. Yield: 46 mg, 42%. Rf = 0.25 (PE/EtOAc = 30/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.73 (d, J = 8.4 Hz, 2H, ArH), 7.53–7.51 (m, 2H, ArH), 7.41–7.36 (m, 5H, PhH), and 5.63 (s, 2H, NCH2), 2.47 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 158.4, 151.2, 144.4, 140.0, 133.4, 130.9, 130.6, 130.3, 129.9, 129.8, 129.5, 129.4, 128.7, 116.1, 110.0, 48.5, and 21.8. HRMS (ESI) calcd. for C21H15N6+ [M + H]+ 351.1353, found 351.1349.
3.2.22. 9-Benzyl-6-(diethylamino)-9H-purine-2-carbonitrile (2v’)
2v’: Colorless crystals, mp: 88–90 °C. Yield: 10 mg, 16%, Rf = 0.25 (PE/EtOAc = 5/1, v/v). 1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H, C8-H in purine), 7.39–7.32 (m, 5H, ArH), 5.34 (s, 2H, ArCH2), 4.19 (br s, 2H, NCH2), 3.76 (br s, 2H, NCH2), and 1.27 (t, J = 7.2 Hz, 6H, CH3). 13C NMR (101 MHz, CDCl3) δ 153.5, 150.0, 139.9, 137.8, 135.2, 129.1, 128.5, 128.0, 120.8, 117.2, 47.3, 43.9, 43.0, 14.1, and 12.8. HRMS (ESI) calcd. for C17H19N6+ [M + H]+ 307.1666, found 307.1670. [Note: The CH2 near the nitrogen atom was shown as a broad singlet in 1H NMR and 13C NMR because of both part double bond character in the amidine functionality and thus restricted rotation and the electric quadrupole of nitrogen-14. This structure was further verified by HSQC in Figure 1].
3.3. Synthesis of 9-Benzyl-6-(diethylamino)-9H-purine-8-carbonitrile (2v) and 9-Benzyl-6-(diethylamino)-N,N-diethyl-9H-purine-8-carboximidamide (5f)
Synthesis of 2v: To a 10 mL vial were added purine 2f (53.9 mg, 0.2 mmol), ethanol (2.0 mL), Et2NH (62 μL, 0.6 mmol), and Et3N (82 μL, 0.6 mmol) in order. The reaction mixture was stirred at room temperature for 2 h. The reaction system was filtered, and the filter residue was washed with EtOH/H2O followed by the infrared lamp drying to provide 2v directly as white solids (51 mg, 83%). Mp: 94–96 °C. Rf = 0.60 (PE/EtOAc = 3/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H, C2-H in purine), 7.45–7.31 (m, 5H, PhH), 5.48 (s, 2H, NCH2), 4.15 (br s, 2H, 1 CH2 in NEt2), 3.78 (br s, 2H, 1 CH2 in NEt2), and 1.28 (t, J = 6.9 Hz, 6H, 2CH3).13C NMR (101 MHz, CDCl3) δ 155.4, 154.1, 150.4, 134.8, 129.0, 128.6, 128.3, 121.7, 120.2, 111.4, 47.3, 44.1, 43.0, 13.8, and 12.6. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H, C2-H in purine), 7.36–7.30 (m, 5H, ArH), 5.52 (s, 2H, ArCH2), 4.09 (br s, 2H, NCH2), 3.73 (br s, 2H, NCH2), and 1.20 (br s, 6H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 155.7, 153.9, 150.7, 135.9, 129.4, 128.7, 128.0, 122.4, 119.8, 111.9, 47.2, 44.00, 42.8, 14.2, and 12.9. HRMS (ESI) calcd. for C17H19N6+ [M + H]+ 307.1666, found: 367.1687. [Note: The CH2 near the nitrogen atom was shown as a broad singlet in 1H NMR and 13C NMR because of both part double bond character in the amidine functionality and thus restricted rotation and the electric quadrupole of nitrogen-14].
Synthesis of 5f: To a 10 mL vial were added purine 2f (53.9 mg, 0.2 mmol) and Et2NH (1 mL). The reaction mixture was then heated at 60 °C for 3 h under stirring. After cooling to room temperature, the solvent was removed in vacuo and the residue was purified through column chromatography with DCM/MeOH (25:1, v/v) as the eluent to obtain 5f as colorless crystals (69 mg, 91%). Mp: 138–140 °C. Rf = 0.20 (DCM/MeOH = 25/1, v/v). 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H, C2-H in purine), 7.28–6.92 (m, 5H, ArH), 6.92 (br s, 1H, NH), 5.46 (s, 2H, NCH2), 3.97 (br s, 4H, NCH2), 3.09 (br s, 4H, NCH2), 1.28 (t, J = 7.2 Hz, 6H, CH3), and 1.08 (t, J = 7.2 Hz, 6H, CH3). 13C NMR (101 MHz, CDCl3) δ 157.2, 153.9, 153.3, 151.0, 141.6, 136.3, 128.6, 128.2, 128.0, 118.4, 46.3, 43.0, 42.3, 13.5, and 12.8. HRMS (ESI) calcd. for C21H30N7+ [M + H]+ 380.2557, found: 380.2590. [Note: The CH2 near the nitrogen atom was shown as a broad singlet in 1H NMR and 13C NMR because of both part double bond character in the amidine functionality and thus restricted rotation and the electric quadrupole of nitrogen-14].
3.4. Synthesis of 9-Benzyl-6-((4-methoxyphenyl)ethynyl)-9H-purine-8-carbonitrile (6f)
To a 10 mL vial were added 2f (53.94 mg, 0.2 mmol), CuI (3.44 mg, 0.018 mmol), and (PPh3)2PdCl2 (4.2 mg, 0.006 mmol) and the whole system was exchanged with N2. Toluene (1 mL) and Et3N (0.028 mL, 0.2 mmol) were then injected in 5 min at below 25 °C. The whole system was then stirred overnight (~11 h) at room temperature. After filtering with the diatomite, the filtrate was dissolved in 5 mL of EtOAc, then the resulting solution was washed with saturated NH4Cl and NaCl solution. The organic phase was collected, dried over anhydrous Na2SO4, and filtered. After removing the solvent in vacuo, the residue was purified by silica gel column chromatography with PE and EtOAc (5:1, v/v) as the eluent to afford the product 6f as yellow crystals. Yield: 38 mg, 52%, mp: 141–143 °C. Rf = 0.41 (PE/EtOAc = 3/1, v/v). 1H NMR (400 MHz, CDCl3) δ 9.10 (s, 1H, C2-H in purine), 7.70 (d, J = 8.8 Hz, 2H, ArH), 7.47 (dd, J = 7.5, 2.2 Hz, 2H, ArH), 7.41–7.33 (m, 3H, ArH), 6.93 (d, J = 8.8 Hz, 2H, ArH), 5.62 (s, 2H, NCH2), and 3.86 (s, 3H, OCH3). 13C NMR (101 MHz, CDCl3) δ 161.5, 155.4, 150.4, 145.1, 134.8, 133.9, 133.1, 129.3, 129.1, 128.4, 128.3, 114.3, 112.7, 110.3, 102.2, 83.8, 55.4, and 48.0. HRMS (ESI) calcd. for C22H16N5O+ [M + H]+ 366.1350, found: 366.1356.
3.5. Synthesis of 2u from 2f
To a 10 mL reaction tube were added 2f (53.94 mg, 0.2 mmol), p-tolylboronic acid (33 mg, 0.24 mmol), Pd(PPh3)4 (15 mg, 0.01 mmol) and K3PO4 (212 mg, 1 mmol) and then a mixed solvent of DME:H2O = 10:1 (2 mL) was injected. The reaction system was stirred at 90 °C for 5 h. After cooling to room temperature, the reaction system was filtered, and the filtrate was dried over anhydrous Na2SO4. After removing the solvent in vacuo, the residue was purified by silica gel column chromatography with PE and EtOAc (10:1, v/v) as the eluent, to yield the 2u as colorless crystals, 19 mg, 28%.
3.6. Synthesis of 2t from 2f
To a 10 mL vial were added 2f (53.9 mg, 0.2 mmol), NaOMe (8.0 mg, 0.3 mmol), and CuBr (3.0 mg, 0.02 mmol) in DMF (2 mL). The vial was capped and heated at 110 °C for 5 h. After cooling to room temperature, EtOAc (5 mL) was added and the whole system was washed with brine (5 mL × 3). The aqueous phase was then extracted with EtOAc (5 mL). The organic phase was collected and dried over anhydrous Na2SO4. After removing the solvent in vacuo, the residue was purified by silica gel column chromatography with PE and EtOAc (5:1, v/v) as the eluent to afford the 2t as colorless crystals, 17 mg, 32%.
3.7. Synthesis of 2s and Benzyl 9-Benzyl-6-(benzylthio)-2-cyano-9H-purine-8-carbimidothioate (5s) from 2f
In a 10 mL vial, 2f (53.94 mg, 0.2 mmol) was dissolved into 2 mL of ethanol, and then benzylthiol (35 μL, 0.3 mmol) and Et3N (22 μL, 0.22 mol) were added in order. The resulting mixture was stirred at room temperature for 30 min. After removal of the solvent in vacuo, the residue was separated by column chromatography with PE and EtOAc (10:1, v/v) as the eluent to provide 2s as colorless crystals (18 mg, 24%) and 5s as colorless crystals (39 mg, 54%). 5s: Rf = 0.45 (PE/EtOAc = 5/1, v/v), mp: 110–112 °C. 1H NMR (400 MHz, CDCl3) δ 10.23 (s, 1H, NH), 8.78 (s, 1H, C2-H in purine), 7.46–7.44 (m, 2H, ArH), 7.37–7.17 (m, 13H, ArH), 5.94 (s, 2H, NCH2), 4.67 (s, 2H, SCH2), and 4.04 (s, 2H, SCH2). 13C NMR (101 MHz, CDCl3) δ 163.8, 162.0, 153.0, 150.4, 146.5, 137.2, 136.5, 134.1, 130.0, 129.1, 128.9, 128.6, 128.4, 127.8, 127.6, 127.5, 127.2, 47.3, 34.0, and 33.0. HRMS (ESI) calcd. for C27H24N5S2+ [M + H]+ 482.1468, found: 482.1468.
3.8. Synthesis of 5s from 2s
To a 10 mL vial were added purine 2s (71.49 mg, 0.2 mmol), ethanol (2.0 mL), benzyl thiol (27 μL, 0.24 mmol), and Et3N (23 μL, 0.24 mmol) in order. The reaction mixture was then stirred at room temperature for 1 h. After filtration and washing with H2O, the residue was dried under infrared lamp to provide 5s (75 mg, 78%) as a white solid.
3.9. Synthesis of Methyl 9-Benzyl-6-(benzylthio)-9H-purine-8-carbimidate (6s) from 2s
In a dry flask 2s (178.73 mg, 0.5 mmol) and NaOMe (2.7 mg, 0.05 mmol) were dissolved in anhydrous methanol (5 mL). The reaction mixture was stirred at room temperature for 4 h. The reaction mixture was filtered, and the filter residue was washed with H2O followed by the infrared lamp drying to provide 6s directly as white solids (155 mg, 80%). Rf = 0.15 (PE/EtOAc = 5/1, v/v), mp: 105–106 °C. 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H, NH), 8.81 (s, 1H, C2-H in purine), 7.48–7.46 (m, 2H, ArH), 7.33–7.14 (m, 8H, ArH), 5.74 (s, 2H, NCH2), 4.68 (s, 2H, SCH2), and 3.95 (s, 3H, Ome). 13C NMR (101 MHz, CDCl3) δ 161.9, 159.1, 153.1, 150.1, 142.8, 137.1, 136.4, 129.9, 129.1, 128.7, 128.5, 127.9, 127.4, 127.0, 53.8, 48.0, and 32.9. HRMS (ESI) calcd. for C21H20N5OS+ [M + H]+ 390.1383, found: 390.1383.
3.10. Synthesis of 2-(9-Benzyl-6-(benzylthio)-9H-purin-8-yl)-4,4-dimethyl-4,5-dihydrooxazole (7s) from 6s, and One-Pot Procedure from 2s
Procedure from 6s: 6s (155 mg,0.4 mmol), 2-amino-2-methylpropan-1-ol (38 μL, 0.4 mmol), TsOH·H2O (7.6 mg, 0.04 mmol) were dissolved in toluene (5 mL). The reaction mixture was refluxed with a Dean–Stark apparatus for 12 h. After the reaction was cooled to room temperature, the solvent was removed in vacuo and the residue was purified through a silica gel flash column chromatography (PE/EtOAc = 6/1, v/v) to afford 7s as a white solid (120 mg, 70%). Rf = 0.35 (PE/EtOAc = 5/1, v/v), mp: 105–107 °C. 1H NMR (400 MHz, CDCl3) δ 8.81 (s, 1H, C2-H in purine), 7.46–7.44 (m, 2H, ArH), 7.33–7.23 (m, 8H, ArH), 6.01 (s, 2H, NCH2), 4.65 (s, 2H, SCH2), 4.12 (s, 2H, OCH2), and 1.36 (s, 6H, 2CH3). 13C NMR (101 MHz, CDCl3) δ 162.4, 153.8, 153.0, 150.0, 140.3, 137.3, 136.4, 130.3, 129.1, 128.43, 128.37, 128.1, 127.8, 127.2, 78.7, 69.2, 47.2, 32.8, and 28.2. HRMS (ESI) calcd. for C24H24N5OS+ [M + H]+ 430.1696, found: 430.1696.
Procedure from 2s: 2s (178.73 mg, 0.5 mmol) was dissolved in anhydrous methanol (5 mL) in a flame-dried flask equipped with a magnetic stir bar. NaOMe (2.7 mg, 0.05 mmol) was added, and the reaction mixture was stirred at room temperature for 4 h. The solvent was removed in vacuo and toluene (5 mL) was added along with 2-amino-2-methylpropan-1-ol (47.5 μL, 0.5 mmol) and TsOH·H2O (9.5 mg, 0.05 mmol). The reaction system was refluxed with a Dean–Stark apparatus overnight (~12 h). After cooling to r. t., the solvent was removed in vacuo and the residue was purified through silica gel column chromatography (PE/EtOAc = 6/1, v/v) to afford 7s as a white solid (162 mg, 75%).
3.11. Synthesis of Ethyl 9-Benzyl-6-(benzylthio)-9H-purine-8-carbimidate (6s’) from 2s
In a vial 2s (71.49 mg, 0.2 mmol) and NaOH (1.0 mg, 0.02 mmol) were dissolved in a mixture of EtOH/H2O = 7/3 (2 mL). The resulting solution was stirred at room temperature for 4 h. After filtration and washing with water, the solid was dried under the infrared lamp to afford 6s’ as a white solid (68 mg, 84%). Rf = 0.2 (PE/EtOAc = 5/1, v/v), mp: 56–58 °C. 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H, NH), 8.79 (s, 1H, C2-H in purine), 7.48–7.46 (m, 2H, ArH), 7.33–7.09 (m, 8H, ArH), 5.78 (s, 2H, NCH2), 4.68 (s, 2H, SCH2), 4.38 (q, J = 7.2 Hz, 2H, OCH2), and 1.30 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 161.8, 159.4, 153.0, 150.2, 143.0, 137.1, 136.4, 129.9, 129.1, 128.7, 128.5, 127.8, 127.3, 126.6, 63.0, 47.9, 33.0, and 13.9. HRMS (ESI) calcd. for C22H22N5OS+ [M + H]+ 404.1540, found: 404.1546.
3.12. Synthesis of 9-Benzyl-6-(benzylthio)-9H-purine-8-carboxamide (8s) from 2s
In a vial 2s (71.49 mg, 0.2 mmol) and NaOH (1.0 mg, 0.02 mmol) were dissolved in a mixture of THF/H2O = 7/3 (2 mL) and the reaction mixture was stirred at room temperature for 4 h. The reaction system was filtered, and filter residue was washed with H2O followed by the drying under infrared lamp to provide 8s as white solids (62 mg, 83%). Rf = 0.15 (PE/EtOAc = 5/1, v/v), mp: 184–186 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 1H, C2-H in purine), 8.37 (s, 1H in NH2), 8.04 (s, 1H in NH2), 7.48–7.46 (m, 2H, ArH), 7.33–7.23 (m, 8H, ArH), 5.86 (s, 2H, NCH2), and 4.70 (s, 2H, SCH2). 13C NMR (101 MHz, DMSO-d6) δ 161.4, 160.7, 153.3, 150.3, 144.3, 138.1, 137.4, 129.5, 129.4, 129.00, 128.97, 128.1, 127.8, 127.7, 47.4, and 32.34. HRMS (ESI) calcd. for C20H18N5OS+ [M + H]+ 376.1227, found: 376.1237.
3.13. Synthesis of Dimethyl 3-(9-Benzyl-6-(benzylthio)-9H-purin-8-yl)isothiazole-4,5-dicarboxylate (10s) from 2s
To a pre-dried reaction tube were sequentially added 2s (143 mg, 0.4 mmol), dimethyl 1,2,3-thiadiazole-4,5-dicarboxylate (40.4 mg, 0.2 mmol), [Rh(COD)Cl]2 (5 mg, 0.01 mmol), DPPF (13 mg, 0.024 mmol), and CsI (6 mg, 0.01 mmol). The reaction tube was evacuated (<1 mmHg) and refilled with nitrogen three times. Anhydrous chlorobenzene (2 mL) was injected via a syringe. The reaction tube was placed in a metal module pre-heated to 130 °C and stirred at 130 °C for 2 h. After cooling to r. t., the solution was purified by column chromatography on silica gel (PE/EtOAc = 8/1, v/v) to provide 10s as colorless crystals (106 mg, 99%). Rf = 0.35 (PE/EtOAc = 5/1, v/v), mp: 119–120 °C. 1H NMR (400 MHz, CDCl3) δ 8.79 (s, 1H, C2-H in purine), 7.48–7.46 (m, 2H, ArH), 7.34–7.20 (m, 8H, ArH), 6.07 (s, 2H, NCH2), 4.67 (s, 2H, SCH2), 4.09 (s, 3H, CH3), and 3.95 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 163.8, 161.7, 158.7, 155.6, 154.5, 152.8, 150.2, 143.7, 137.2, 136.1, 136.0, 130.3, 129.2, 128.5, 127.8, 127.4, 127.3, 53.7, 53.3, 47.4, and 32.9. HRMS (ESI) calcd. for C26H22N5O4S2+ [M + H]+ 532.1108, found: 532.1108.
4. Conclusions
A direct C-H cyanation of purines was developed from corresponding purines with TMSCN via sequential Tf2O activation, nucleophilic addition, and a base-mediated detrifluoromethanesulfination. The current transformation showed a good tolerance of variety of functional groups, including allyl, alkynyl, ketone, ester, nitro, etc. The cyanation occurred highly regioselectively at the 8-position of purines with various substituents, including electron-withdrawing 6-chloro, aryl, and electron-donating alkoxy and alkylthio groups. Strong electron-donating diethylamino can reverse the regioselectivity of purine, and 2-cyano-6-diethylamino purine is obtained. Therefore, both 8- and 2-cyano 6-diethylaminopurines can be obtained from corresponding 6-chloropurine from differently substituted purines. Moreover, 6-chloro-8-cyanopurines are also versatile substrates for the further transformations because both of 6-chloro and nitrile are good electrophilic sites for various nucleophiles. For the further synthetic applications of nitriles, cyanopurines were easily converted into numerous derivatives, such as amide, oxazolines, isothiazoles, imidates, imidothioates, and imidamides.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28030914/s1, X-ray crystal data, and copies of 1H-NMR and 13C-NMR spectra of unknown compounds are included in the Supporting Information.
Author Contributions
Conceptualization, N.C.; methodology, L.L., J.H., N.C. and J.X.; investigation, L.L. (Table 1, Scheme 2, Scheme 3a–c), J.H. (Scheme 3d–h, Scheme 4, Scheme 5), X.S. and Y.F. (raw materials); validation, N.C., J.H. and L.L.; data curation, J.H. and L.L.; resources, H.D., J.X. and N.C.; writing—original draft preparation, N.C.; writing—review and editing, J.X.; visualization, L.L., J.H. and N.C.; supervision, N.C.; project administration, N.C.; funding acquisition, N.C. and J.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 21772010.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We sincerely thanks Zhanhui Yang for his valuable suggestion.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Sample Availability
Not applicable.
References
- Mallari, J.P.; Guiguemde, W.A.; Guy, R.K. Antimalarial activity of thiosemicarbazones and purine derived nitriles. Bioorg. Med. Chem. Lett. 2009, 19, 3546–3549. [Google Scholar] [CrossRef] [PubMed]
- Mallari, J.P.; Shelat, A.A.; Obrien, T.; Caffrey, C.R.; Kosinsk, A.; Connelly, M.; Harbut, M.; Creenbaum, D.; McKerrow, J.H.; Guy, R.K. Development of potent purine-perived nitrile inhibitors of the Trypanosomal Protease TbcatB. J. Med. Chem. 2008, 51, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, A.; Suzuki, Y.; Ohta, K.; Higashino, T. Preparation of heteroarenecarbonitriles by reaction of haloheteroarenes with potassium cyanide with sodium p-toluenesulfinate as catalyst. Heterocycles 1994, 39, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Herdewijn, P.; Van Aerschot, A.; Pfleiderer, W. Synthesis of 2-amino-6-(acetamidomethyl)-9-(β-D-ribofuranosyl)purine. Synthesis 1989, 961–962. [Google Scholar] [CrossRef]
- Gundersen, L.-L. Synthesis of purinecarbonitriles by Pd(0)-catalyzed coupling of halopurines with zinc cyanide. Acta Chem. Scand. 1996, 50, 58–63. [Google Scholar] [CrossRef]
- Ding, Y.; Girardet, J.-L.; Hong, Z.; Lai, V.C.H.; An, H.; Koh, Y.-h.; Shaw, S.Z.; Zhong, W. Synthesis of 9-(2-β-C-methyl-β-D-ribofuranosyl)-6-substituted purine derivatives as inhibitors of HCV RNA replication. Bioorg. Med. Chem. Lett. 2005, 15, 709–713. [Google Scholar] [CrossRef]
- Ai, C.; Zhang, W.; Zhou, L.; Cai, X.; Zheng, Z. Molecular modeling of three-dimensional structure of hTRPV4 protein and experimental verification of its antagonist binding sites. J. Mol. Struct. 2021, 1227, 129421. [Google Scholar] [CrossRef]
- Tanji, K.; Higashino, T. Purines. IX. Reaction of 9-phenyl-9H-purine-2-carbonitriles with Grignard reagents. Heterocycles 1990, 30, 435–440. [Google Scholar] [CrossRef]
- Altmann, E.; Cowan-Jacob, S.W.; Missbach, M. Novel purine nitrile derived inhibitors of the Cysteine Protease Cathepsin K. J. Med. Chem. 2004, 47, 5833–5836. [Google Scholar] [CrossRef]
- Zulfiqar, F.; Kojima, H.; Nakanishi, M.; Ando, T.; Kitade, Y. Synthesis of carbocyclic 2-substituted adenine nucleoside and related Analogs. Nucleosides Nucleotides Nucleic Acids 2008, 27, 1153–1157. [Google Scholar] [CrossRef]
- Braendvang, M.; Gundersen, L.-L. Synthesis, biological activity, and SAR of antimycobacterial 2- and 8-substituted 6-(2-furyl)-9-(p-methoxybenzyl)purines. Bioorg. Med. Chem. 2007, 15, 7144–7165. [Google Scholar] [CrossRef] [PubMed]
- Butora, G.; Schmitt, C.; Levorse, D.A.; Streckfuss, E.; Doss, G.A.; MacCoss, M. The elusive 8-fluoroadenosine: A simple non-enzymatic synthesis and characterization. Tetrahedron 2007, 63, 3782–3789. [Google Scholar] [CrossRef]
- El Safadi, Y.; Marquet, R.; Aubertin, A.-M.; Vivet-Boudou, V. Synthesis and primary evaluation of novel HIV-1 Inhibitors. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Vivet-Boudou, V.; Isel, C.; Sleiman, M.; Smyth, R.; Ben Gaied, N.; Barhoum, P.; Laumond, G.; Bec, G.; Gotte, M.; Mak, J.; et al. 8-Modified-2’-deoxyadenosine analogues induce delayed polymerization arrest during HIV-1 reverse transcription. PLoS ONE 2011, 6, e27456. [Google Scholar] [CrossRef]
- Butler, R.S.; Myers, A.K.; Bellarmine, P.; Abboud, K.A.; Castellano, R.K. Highly fluorescent donor-acceptor purines. J. Mater. Chem. 2007, 17, 1863–1865. [Google Scholar] [CrossRef]
- Butler, R.S.; Cohn, P.; Tenzel, P.; Abboud, K.A.; Castellano, R.K. Synthesis, photophysical behavior, and electronic structure of push-pull purines. J. Am. Chem. Soc. 2009, 131, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, E.; Shimada, N.; Matsuoka, Y. Purines. 1. Reaction of 9-phenyl-9H-purine and 7-phenyl-7H-purine with Grignard reagents. Yakugaku Zasshi 1979, 99, 114–119. [Google Scholar] [CrossRef]
- D’Errico, S.; Piccialli, V.; Oliviero, G.; Borbone, N.; Amato, J.; D’Atri, V.; Piccialli, G. Probing the reactivity of nebularine N1-oxide. A novel approach to C-6 C-substituted purine nucleosides. Tetrahedron 2011, 67, 6138–6144. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Amato, J.; Borbone, N.; Cerullo, V.; Hemminki, A.; Piccialli, V.; Zaccaria, S.; Mayol, L.; Piccialli, G. Synthesis and biological evaluation of unprecedented ring-expanded nucleosides (RENs) containing the imidazo[4,5-d][1,2,6]oxadiazepine ring system. Chem. Commun. 2012, 48, 9310–9312. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Piccialli, V.; D’Atri, V.; Mayol, L.; Piccialli, G. Synthesis of 2,6-dialkyl(aryl)purine nucleosides by exploiting the reactivity of Nebularine N1-oxide towards Grignard reagents. Eur. J. Org. Chem. 2013, 2013, 6948–6954. [Google Scholar] [CrossRef]
- Xia, R.; Xie, M.-S.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. Radical route for the alkylation of purine nucleosides at C6 via Minisci reaction. Org. Lett. 2014, 16, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-C.; Xia, R.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M. Synthesis of cycloalkyl substituted purine nucleosides via a metal-free radical route. Org. Biomol. Chem. 2016, 14, 4189–4193. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-M.; Shang, R.; Fu, M.-C.; Fu, Y. Photoredox-catalysed decarboxylative alkylation of N-heteroarenes with N-(acyloxy)phthalimides. Chem. Eur. J. 2017, 23, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Yu, M.; Shi, T.; Hu, J.; Li, S.; Xu, J.X.; Chen, N.; Du, H.G. Silver-catalyzed direct C6-H arylation of purines and purine nucleosides with arylboronic acids. Eur. J. Org. Chem. 2017, 2017, 3415–3420. [Google Scholar] [CrossRef]
- Yu, M.; Zhou, Z.; Zou, C.; Wang, Z.; Wang, W.; Sun, K. Traceless proton aided regioselective C(sp2)–C(sp2) construction to synthesize C6-acylated purines and purine nucleosides without metal catalysts. Org. Chem. Front. 2022, 9, 4460–4465. [Google Scholar] [CrossRef]
- Yu, M.; Zhou, Z.; Chen, Y.; Wang, Z.; Wang, W.; Sun, K. Regioselective C6–H hydroxyalkylation of purines and purine nucleosides via α-C–H functionalization of alcohols at room temperature. Org. Lett. 2022, 24, 4886–4891. [Google Scholar] [CrossRef]
- Corey, E.J.; Tian, Y. Selective 4-arylation of pyridines by a nonmetalloorganic process. Org. Lett. 2005, 7, 5535–5537. [Google Scholar] [CrossRef]
- Hilton, M.C.; Dolewski, R.D.; McNally, A. Selective functionalization of pyridines via heterocyclic phosphonium salts. J. Am. Chem. Soc. 2016, 138, 13806–13809. [Google Scholar] [CrossRef]
- Elbert, B.L.; Farley, A.J.M.; Gorman, T.W.; Johnson, T.C.; Genicot, C.; Lallemand, B.; Pasau, P.; Flasz, J.; Castro, J.L.; MacCoss, M.; et al. C–H cyanation of 6-ring N-containing heteroaromatics. Chem. Eur. J. 2017, 23, 14733–14737. [Google Scholar] [CrossRef]
- Aly, A.A.; Brase, S.; Gomaa, A.A.-M. Amidines: Their synthesis, reactivity, and applications in heterocycle synthesis. Arkivoc 2018, vi, 85–138. [Google Scholar] [CrossRef]
- Faizi, D.J.; Nava, N.A.; Al-Amin, M.; Blum, S.A. Oxyboration: Synthesis of borylated benzofurans. Org. Synth. 2016, 93, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, M.-P.; Li, L.-J.; Huang, Q.; Hu, M.-Y.; Zhu, S.-F. Phenanthroline-imine ligands for iron-catalyzed alkene hydrosilylation. Chem. Sci. 2022, 13, 2721–2728. [Google Scholar] [CrossRef] [PubMed]
- Keegstra, M.A.; Peters, T.H.A.; Brandsma, L. Copper(I) halide catalyzed synthesis of alkyl aryl and alkyl heteroaryl ethers. Tetrahedron 1992, 48, 3633–3652. [Google Scholar] [CrossRef]
- Hu, L.B.; Zhu, H.; Du, D.-M.; Xu, J.X. Efficient Synthesis of taurine and structurally diverse substituted taurines from aziridines. J. Org. Chem. 2007, 72, 4543–4546. [Google Scholar] [CrossRef]
- Lu, D.-F.; Zhu, C.-L.; Jia, Z.-X.; Xu, H. Iron(II)-catalyzed intermolecular amino-oxygenation of olefins through the N–O bond cleavage of functionalized hydroxylamines. J. Am. Chem. Soc. 2014, 136, 13186–13189. [Google Scholar] [CrossRef]
- Dong, Z.Y.; Chen, C.Z.; Wang, J.; Xu, J.X.; Yang, Z.H. Dual roles of bisphosphines and epoxides: Rh-catalyzed highly chemoselective and diastereoselective (3 + 2) transannulations of 1,2,3-thiadiazoles with cyanoepoxides. Org. Chem. Front. 2021, 8, 6687–6698. [Google Scholar] [CrossRef]
- Wu, Q.Y.; Chen, N.; Xu, J.X. Chemoselectivity in the transannulaction of 1,2,3-thiadiazoles and alk-2-enenitriles: Specific synthesis of 3-(Alk-1-enyl)isothiazoles. Chemistryselect 2022, 7, e202103943. [Google Scholar] [CrossRef]
- Hu, J.B.; Wang, C.X.; Yu, M.W.; Zhang, S.J.; Chen, N.; Du, H.G. Palladium-catalyzed N3-directed C–H halogenation of N9-arylpurines and azapurines. Eur. J. Org. Chem. 2022, 2022, e202101266. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).