
Synthesis, Computational, and Anticancer In Vitro Investigations of Aminobenzylnaphthols Derived from 2-Naphtol, Benzaldehydes, and α-Aminoacids via the Betti Reaction
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
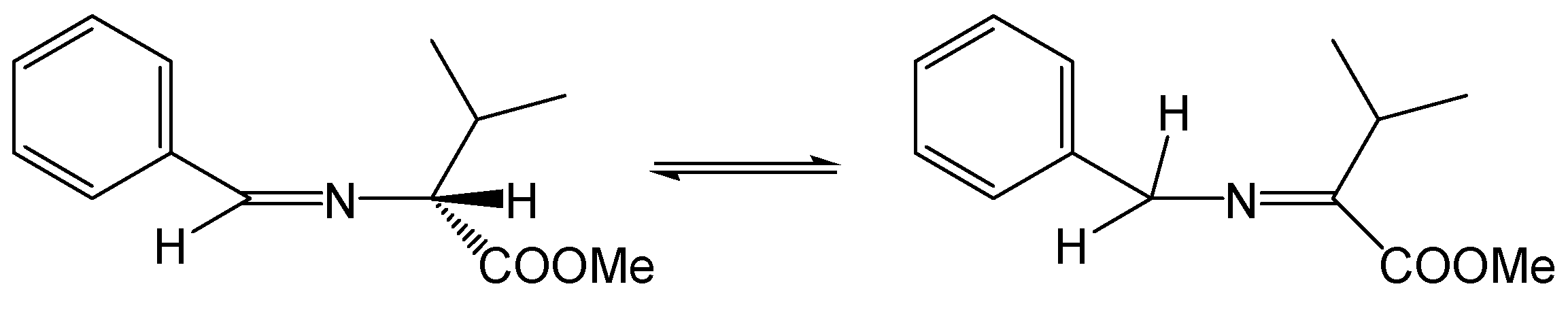
2.1. Chemistry
2.2. Biological Studies
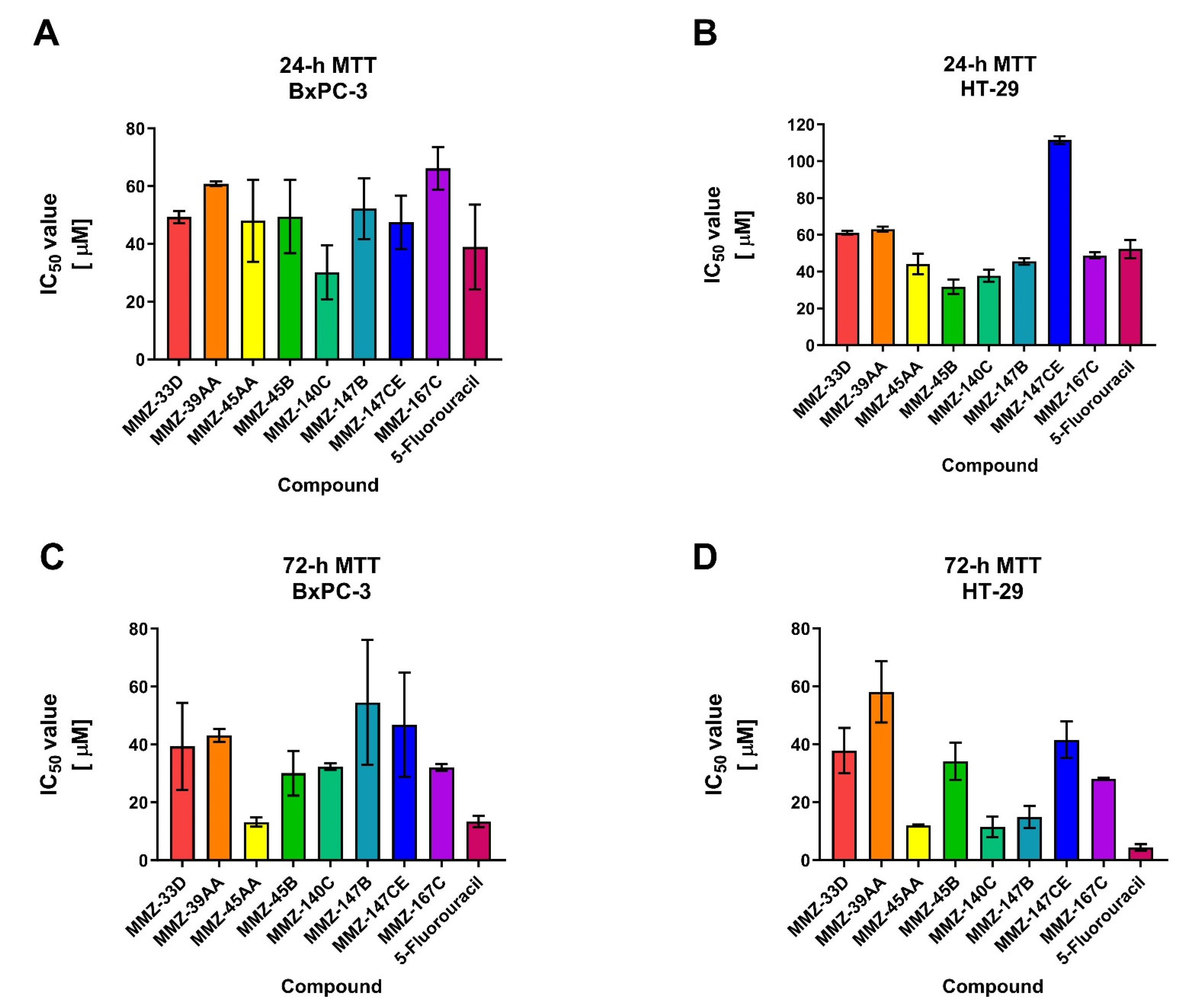
2.2.1. MTT Assay
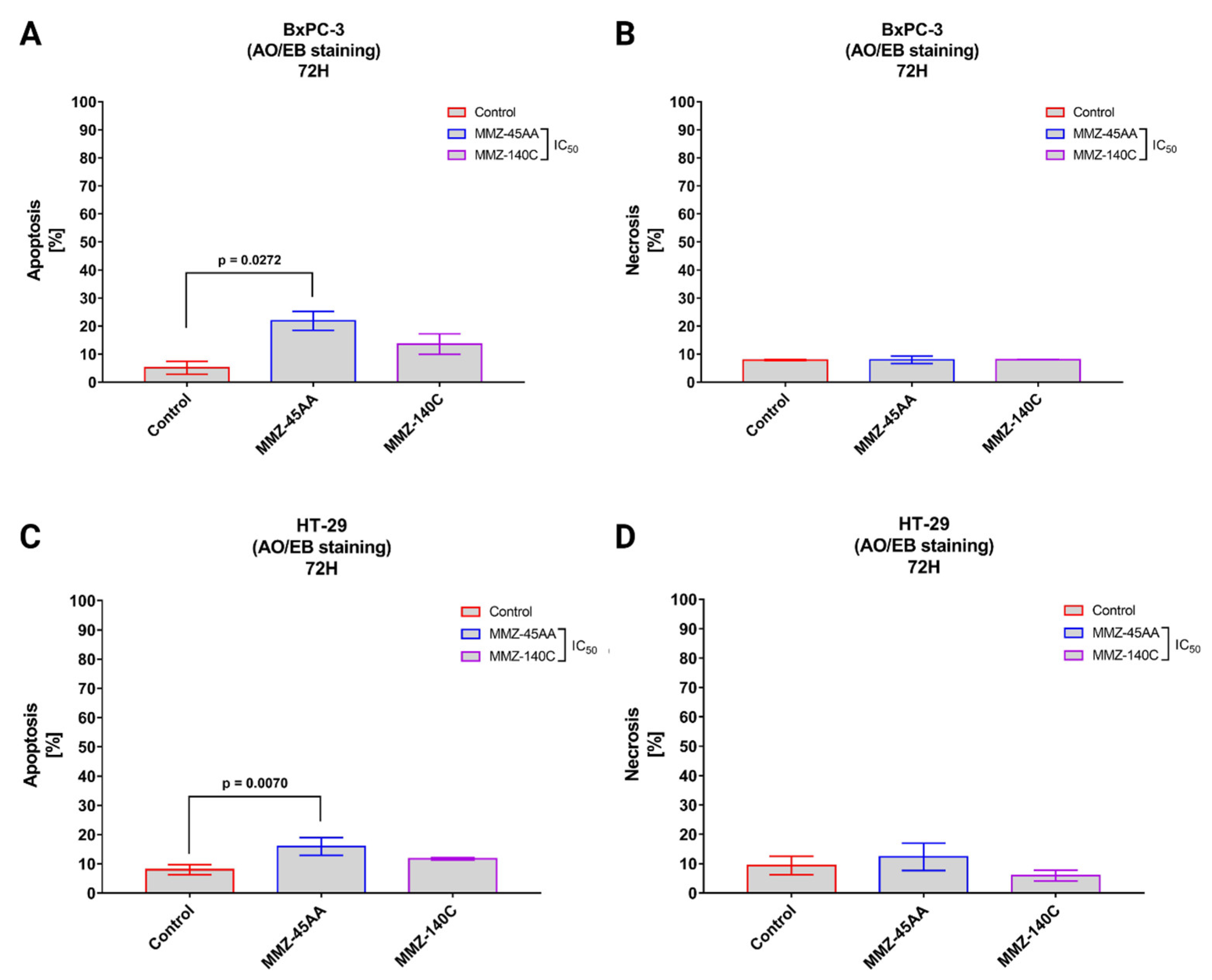
2.2.2. Dual Acridine Orange/Ethidium Bromide (AO/EB) Fluorescent Staining
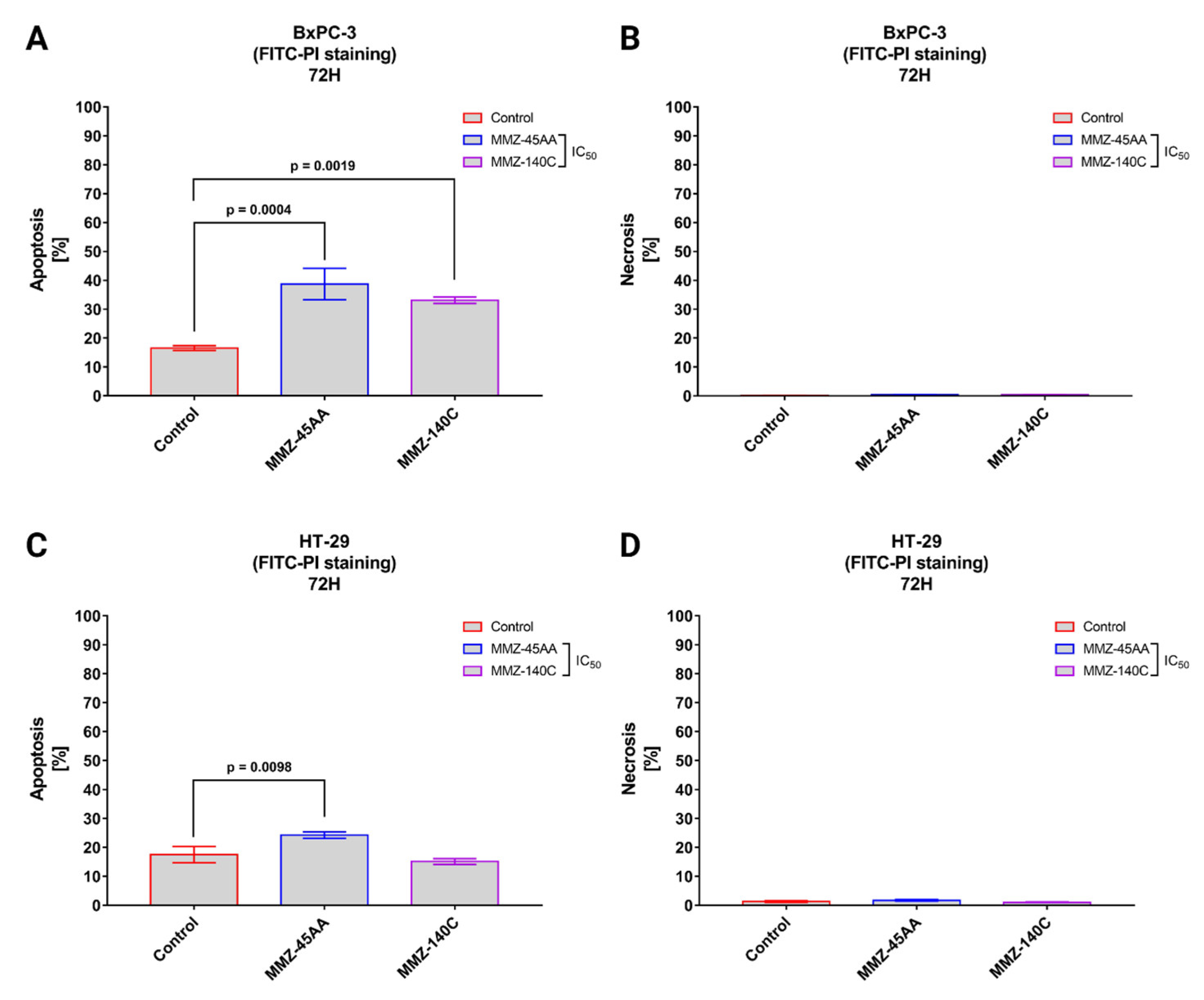
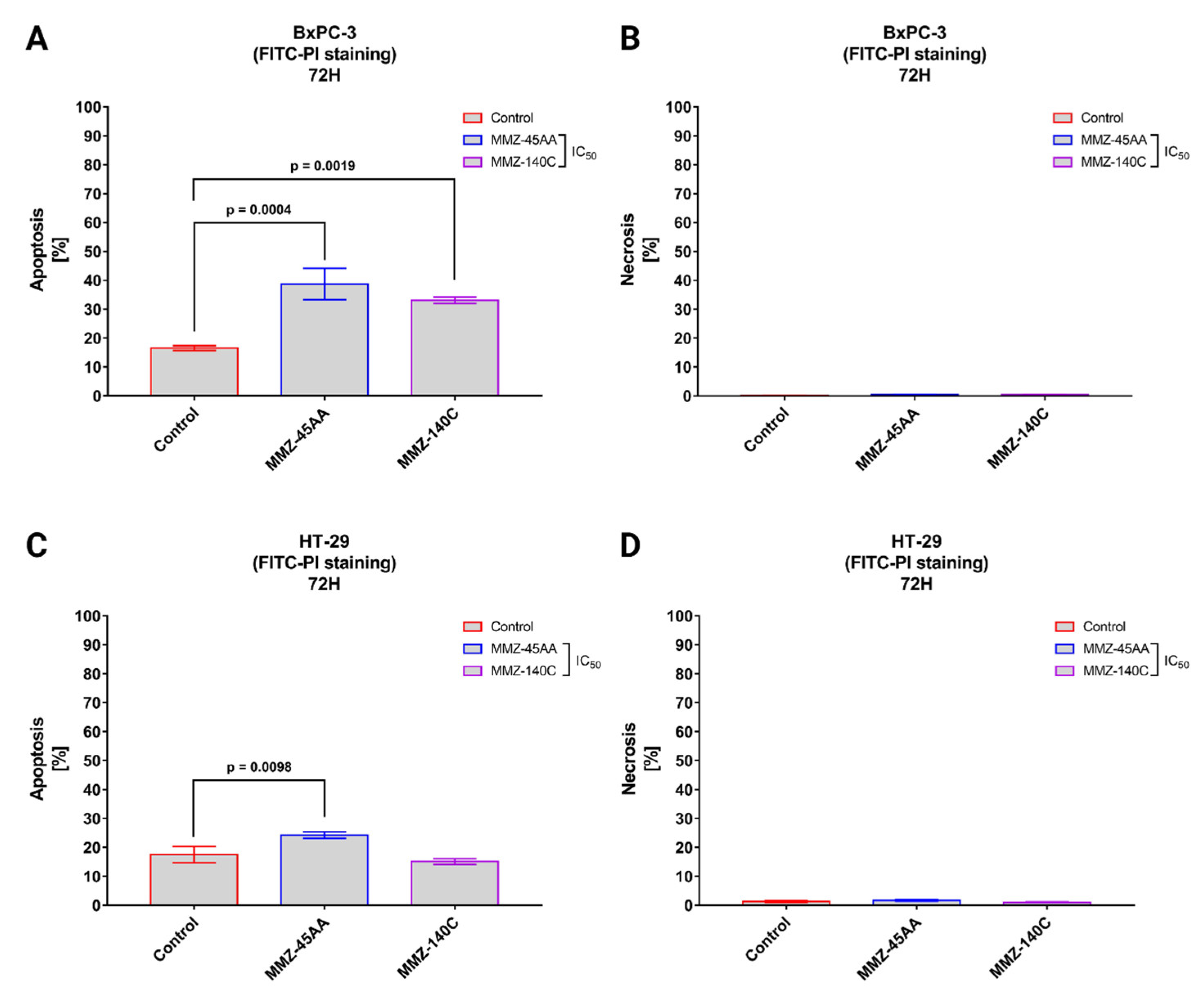
2.2.3. Annexin V-FITC and Propidium Iodide Flow Cytometry Analysis
2.3. Computational Studies
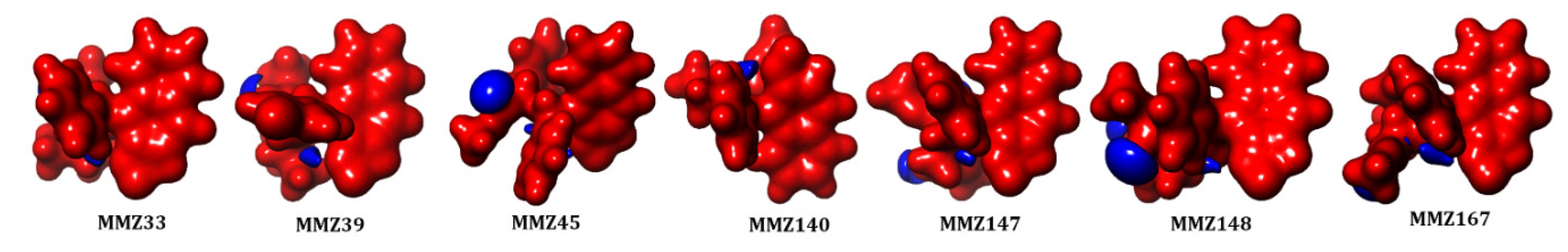
2.3.1. Drug Likeness and ADMET
2.3.2. Density Functional Theory (DFT) Calculations
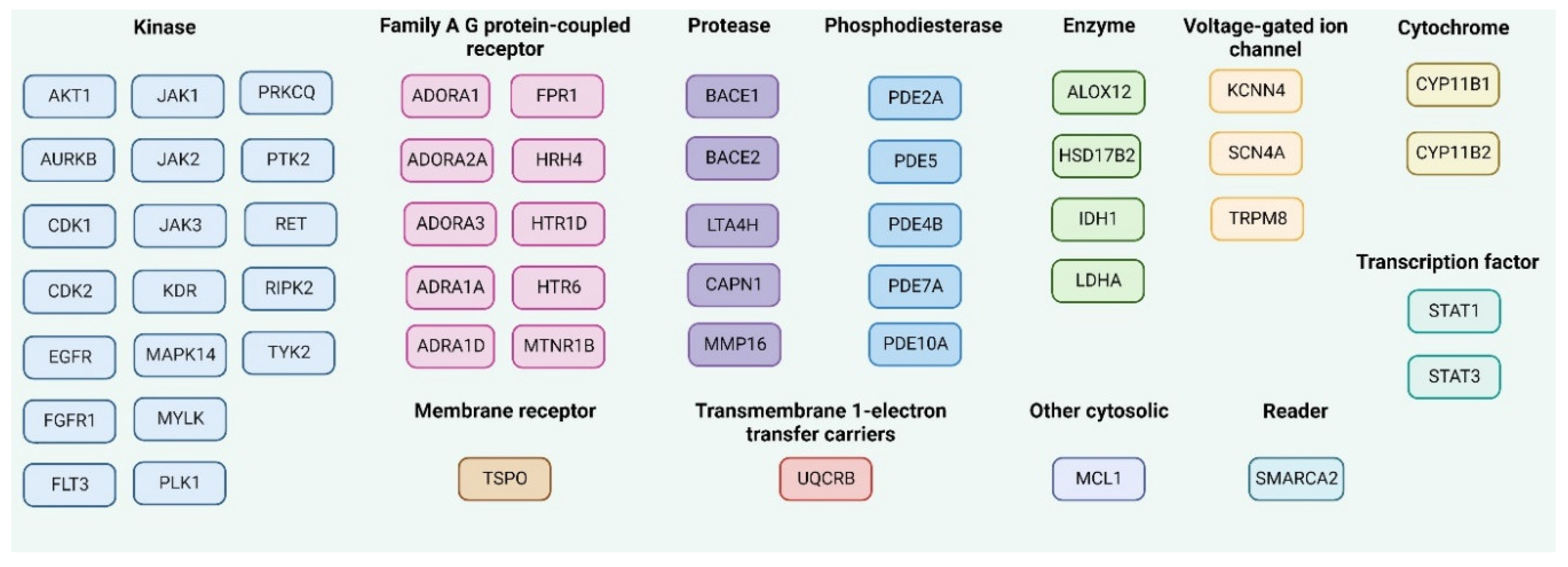
2.3.3. Molecular Target Prediction
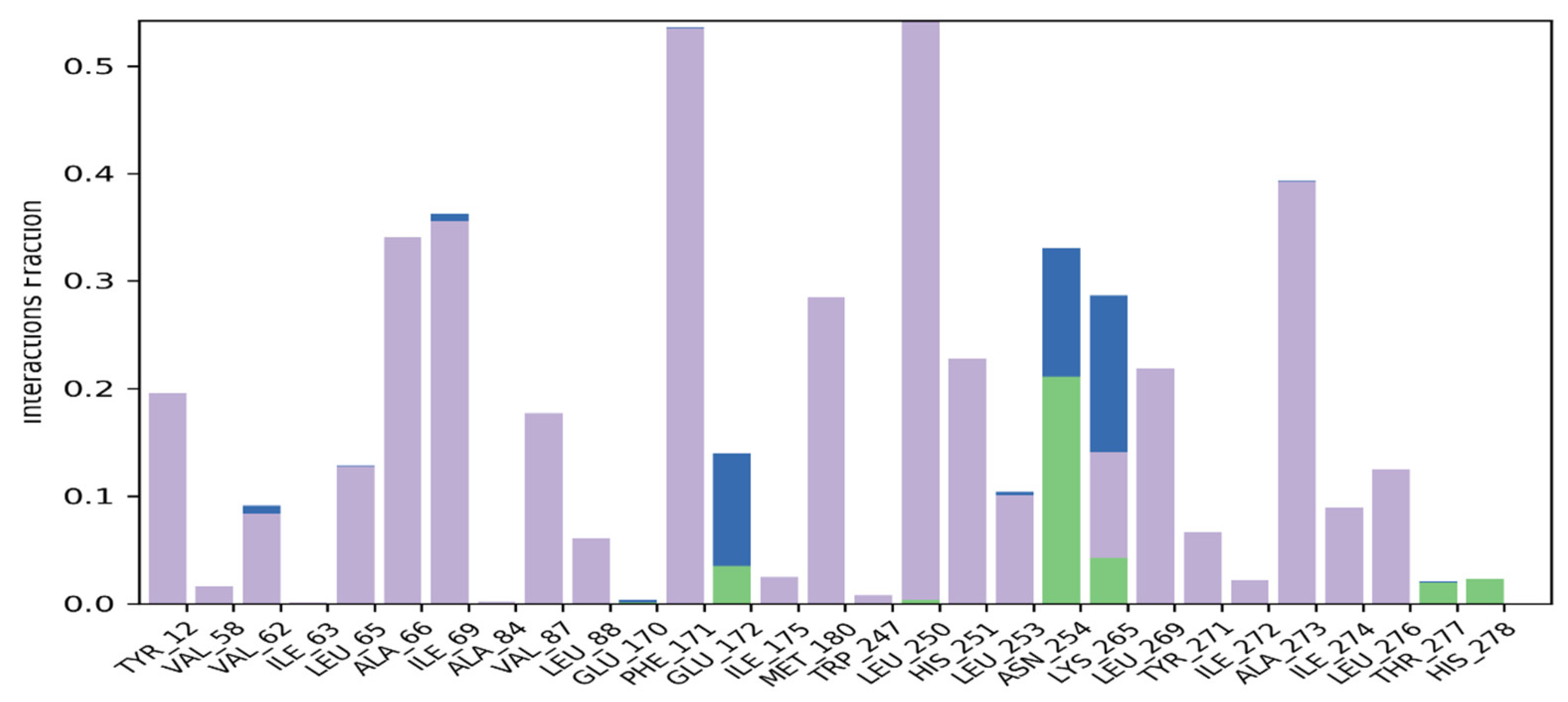
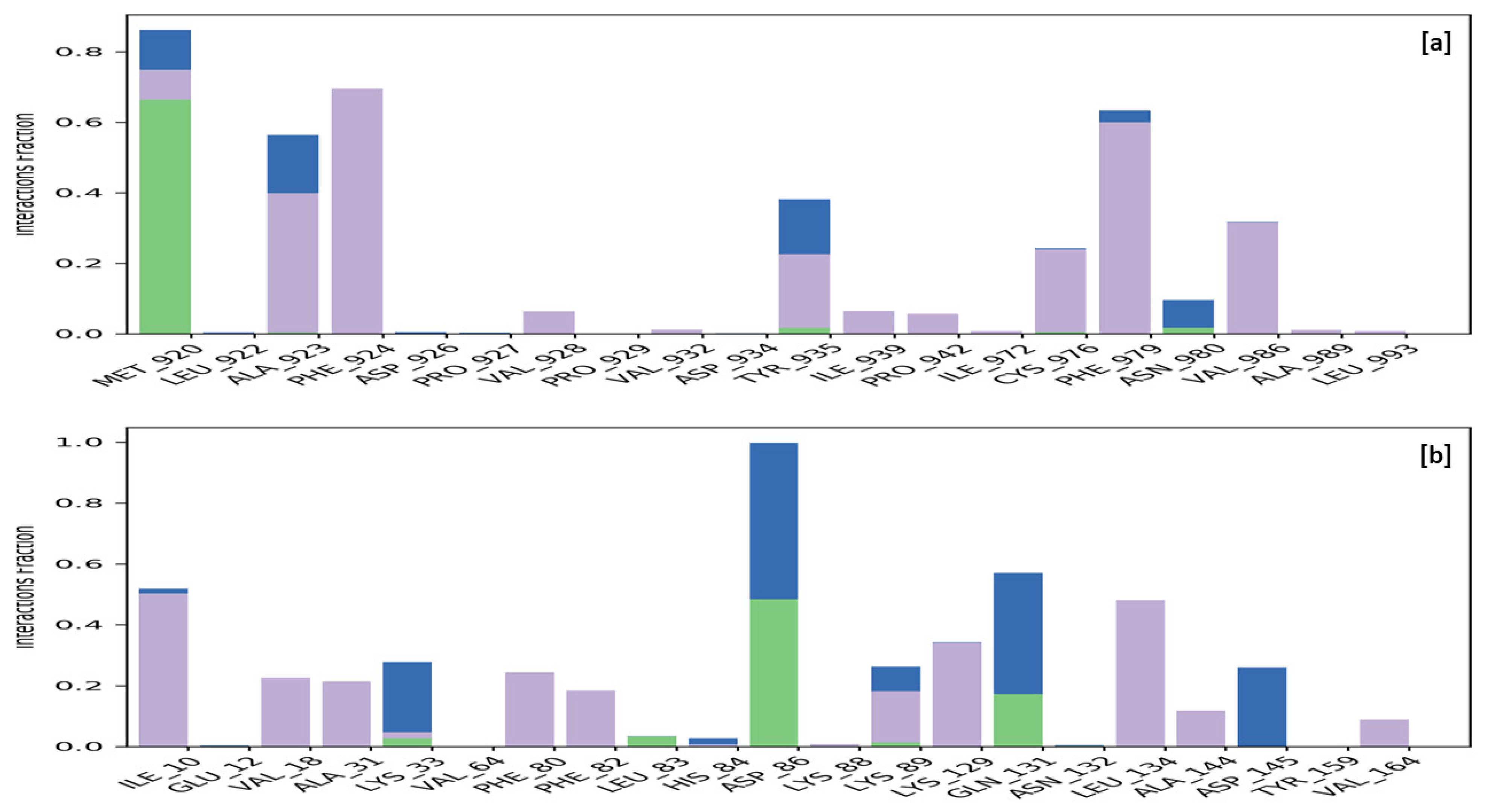
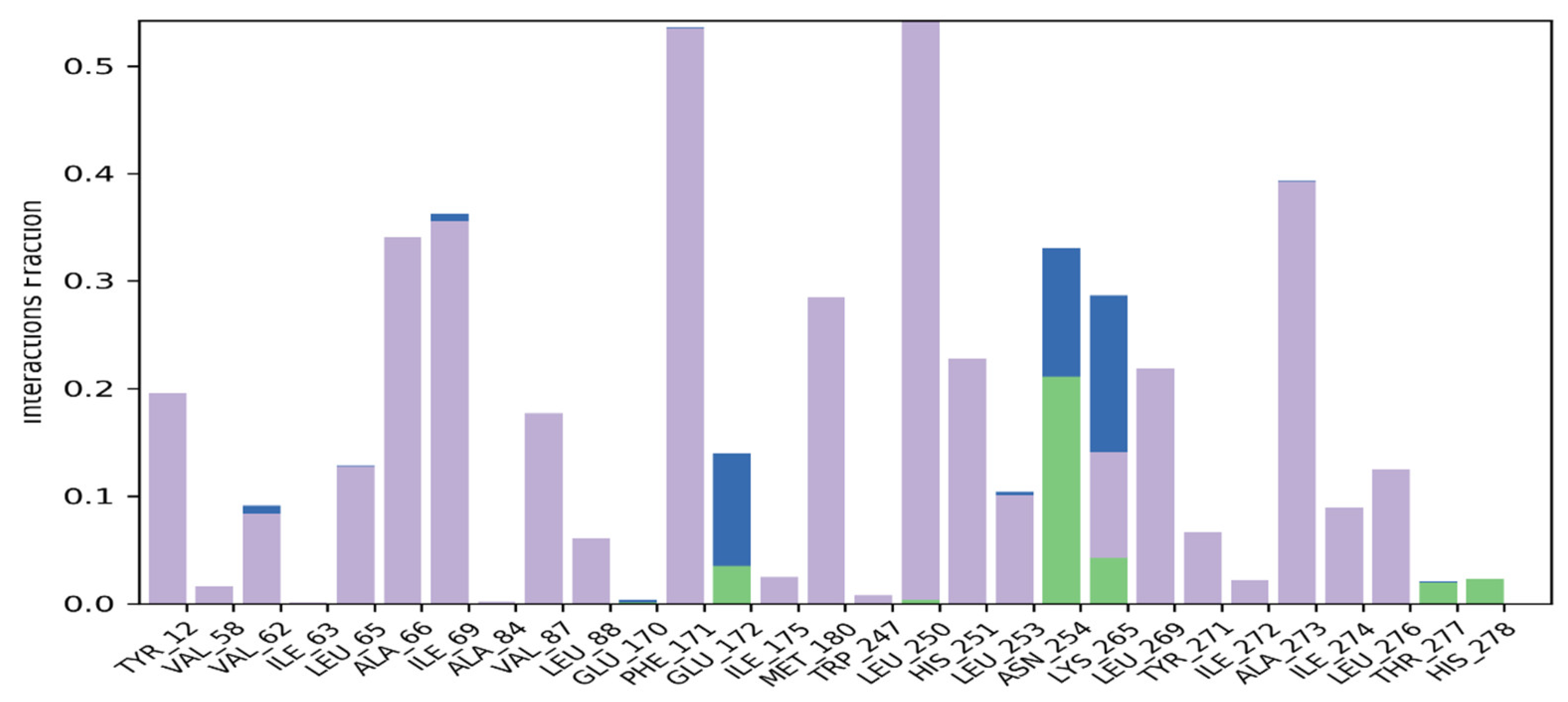
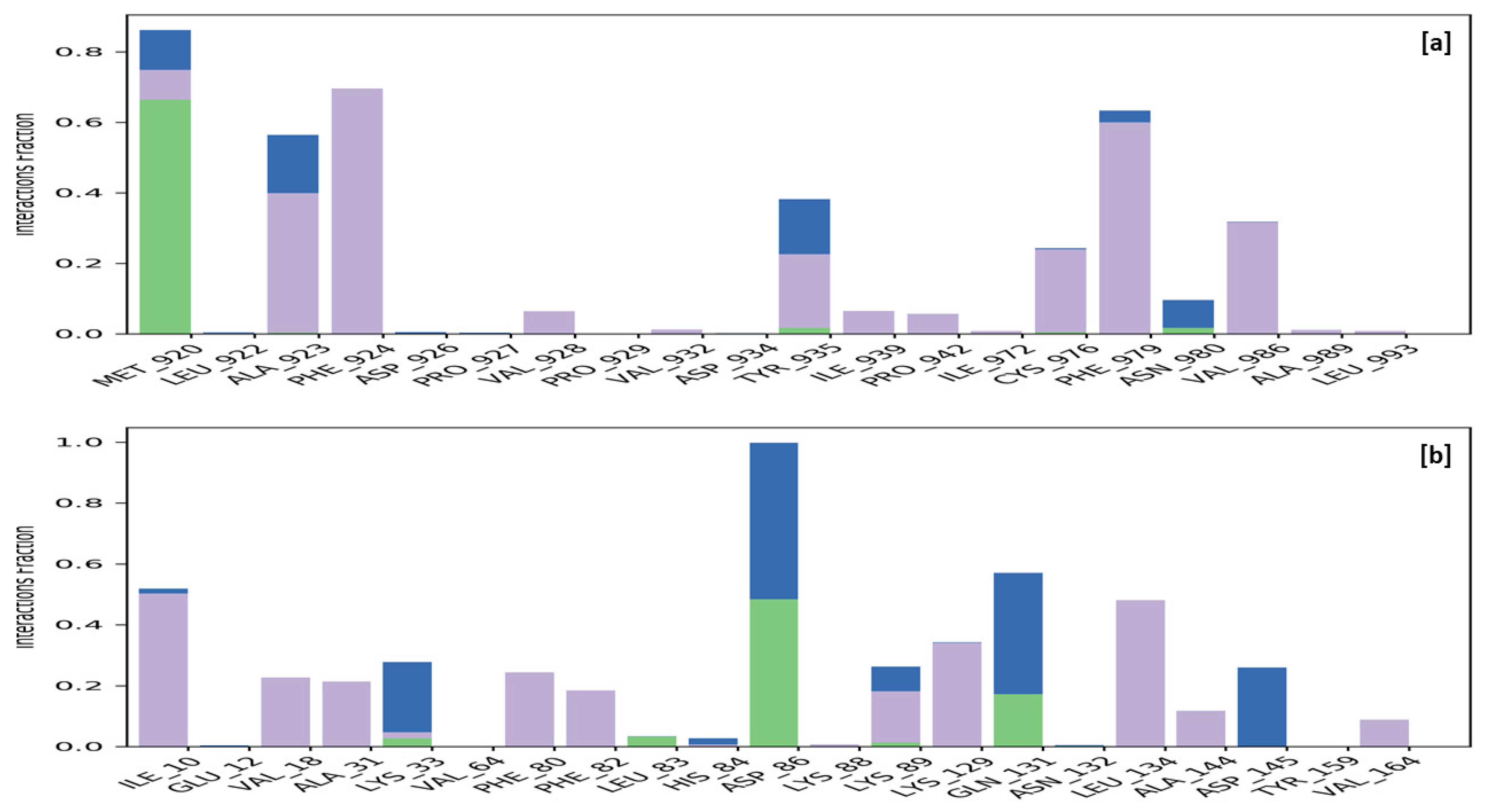
2.3.4. Molecular Docking
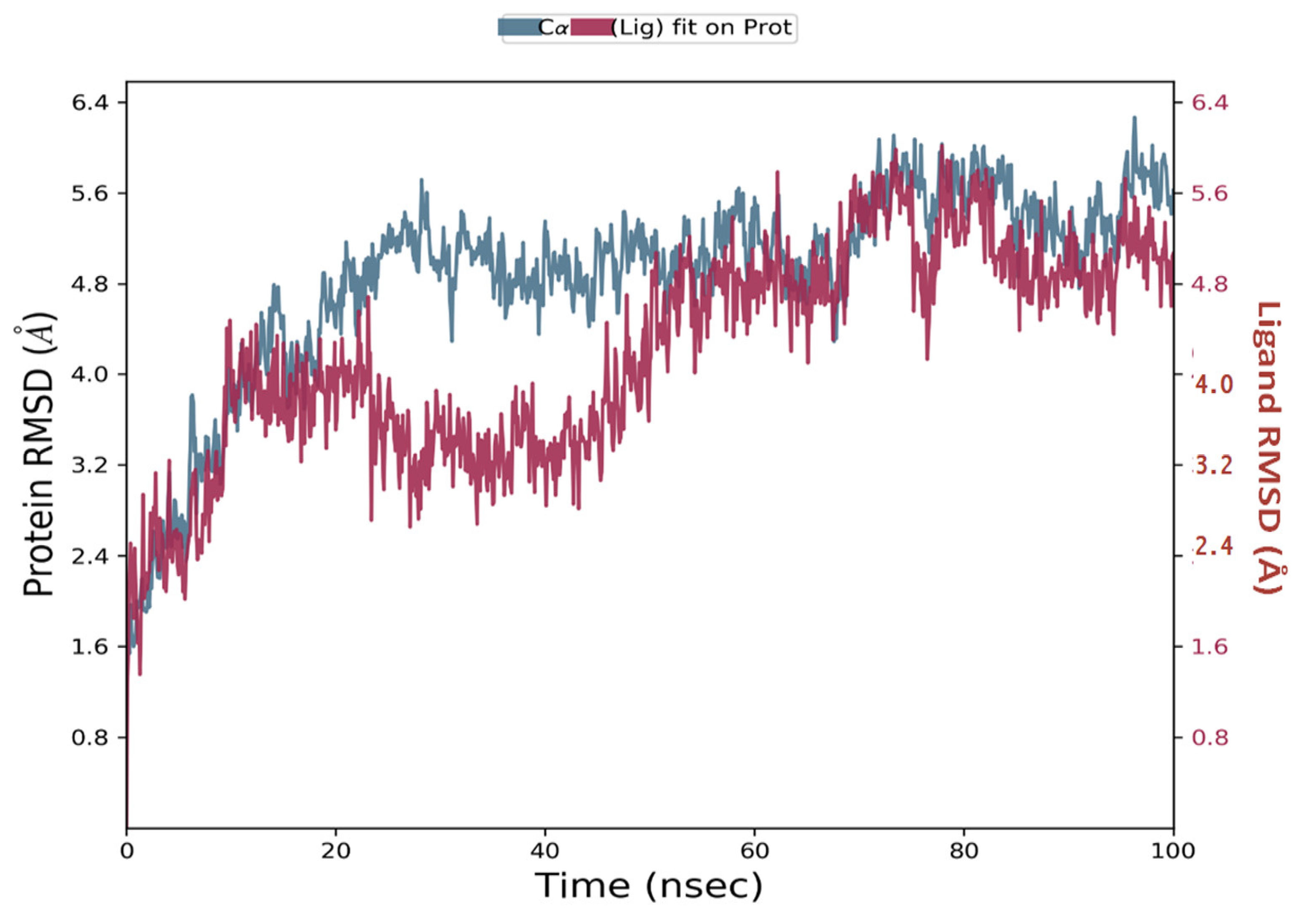
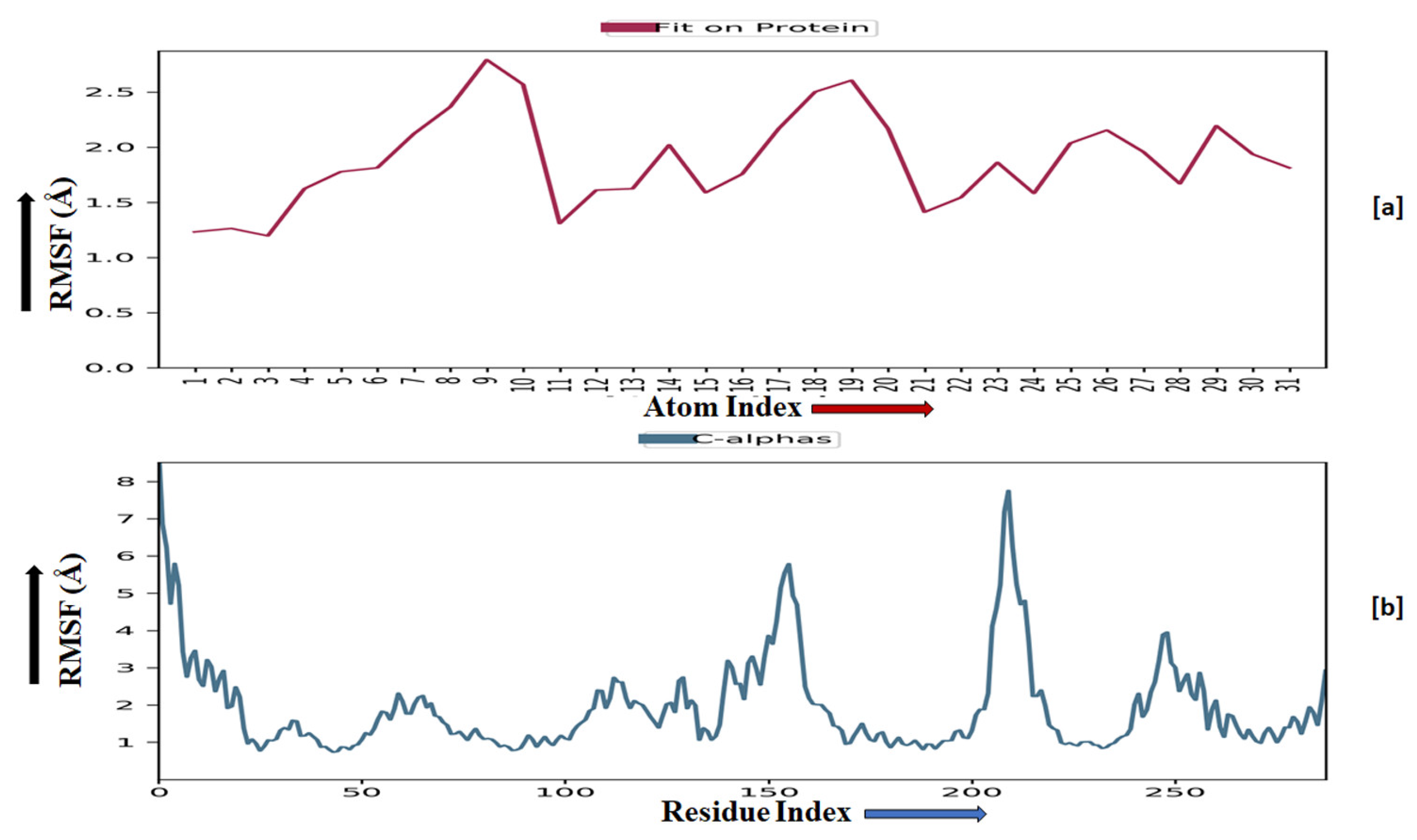
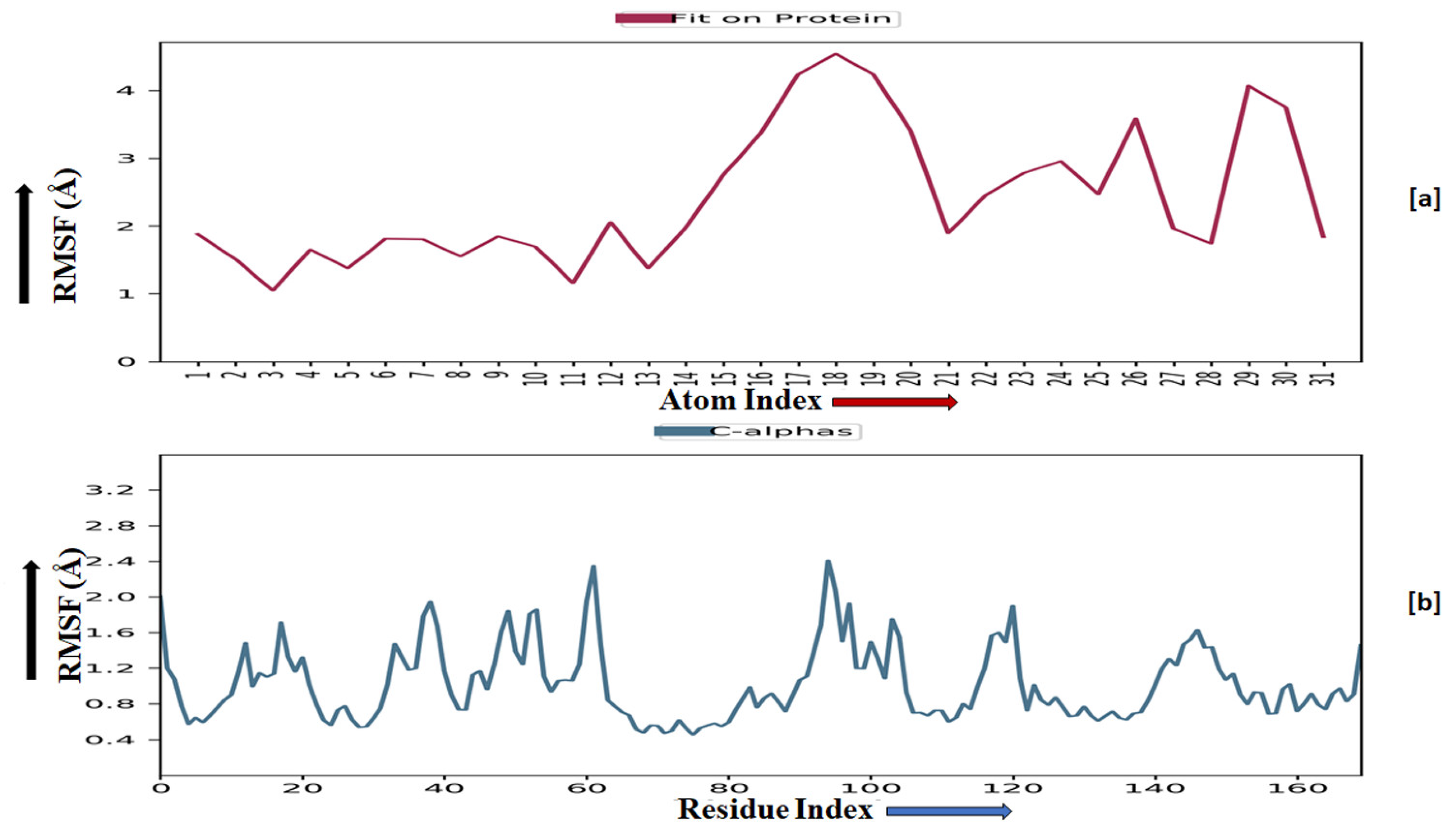
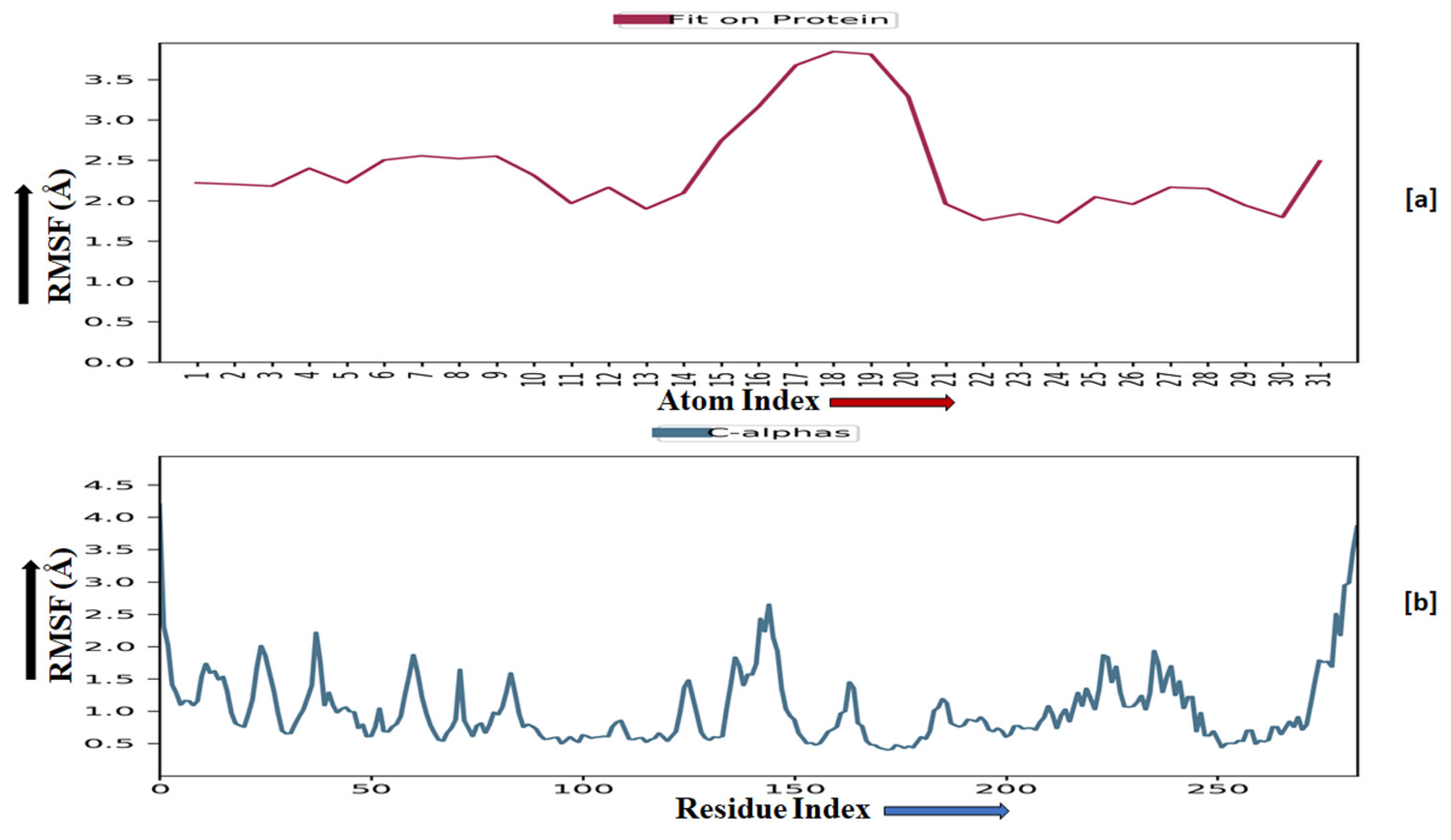
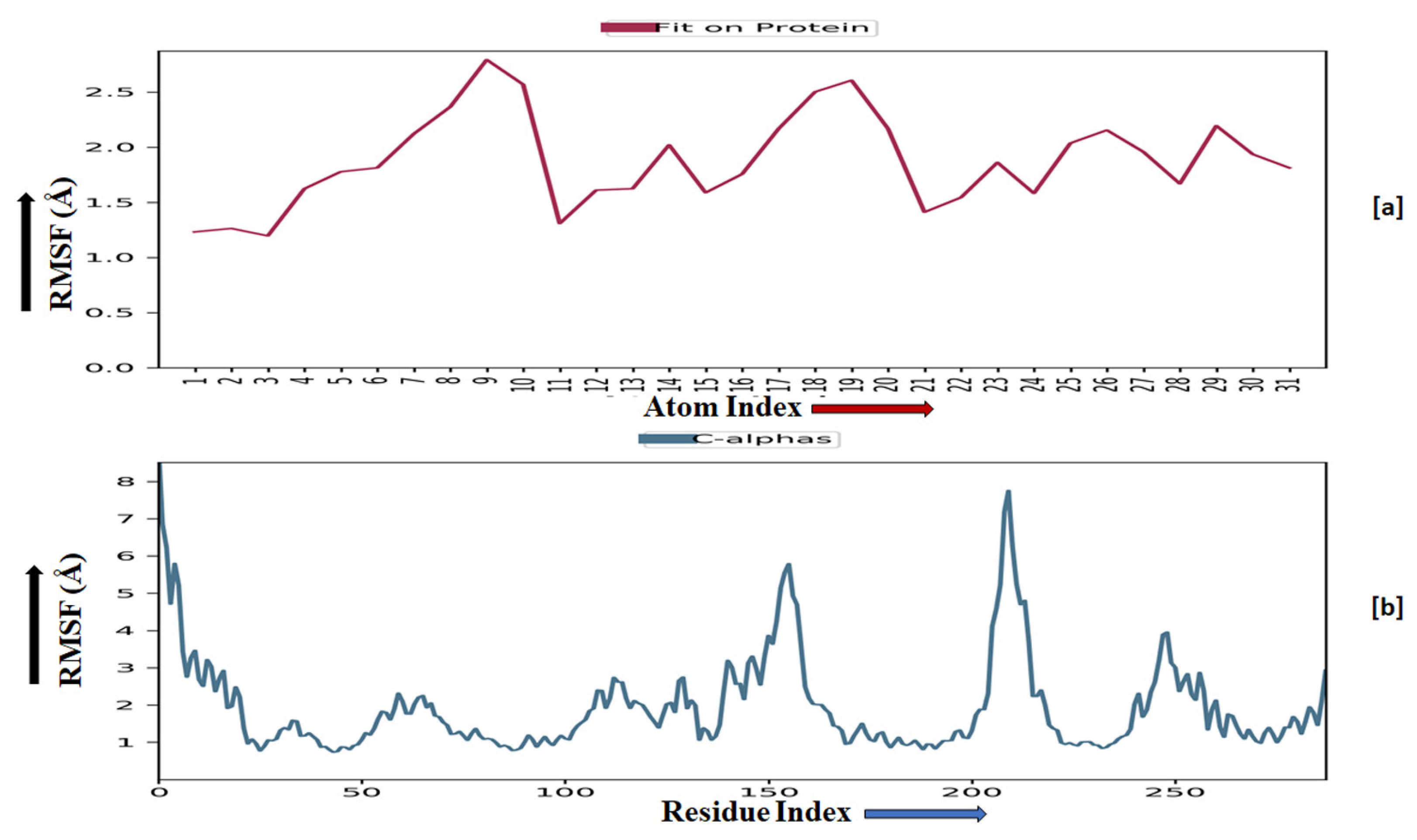
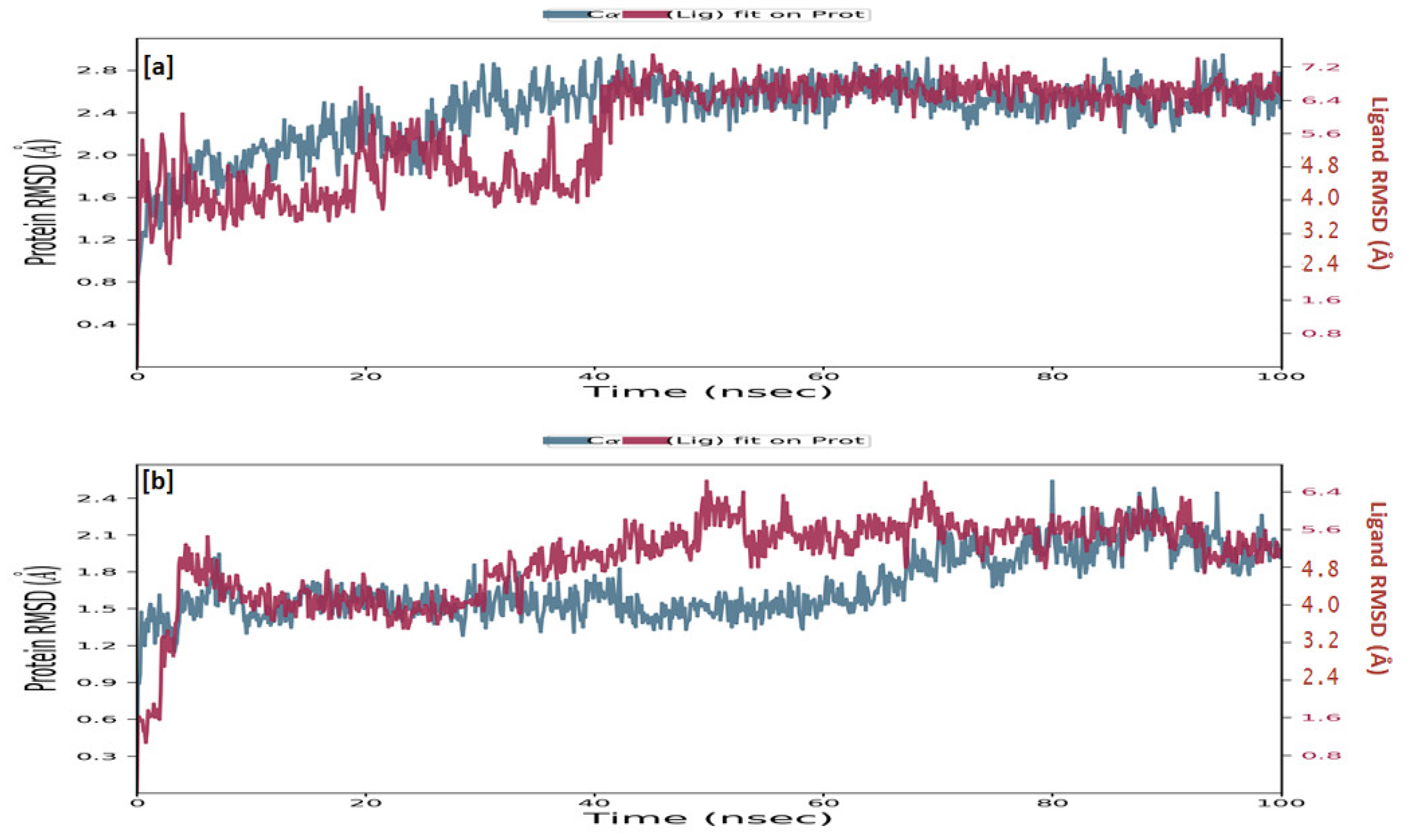
2.3.5. Molecular Dynamics Simulation
2.3.6. Prime MM-GBSA Analysis
3. Discussion
4. Materials and Methods
4.1. Chemical Studies
Synthesis of Betti Bases (MMZs)
- (S,S) and (R,R)-methyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-d-valinate (MMZ-33D)
- (S,S) and (R,R)-methyl ((4-chlorophenyl)(2-hydroxynaphthalen-1-yl)methyl)-d-valinate (MMZ-39AA)
- (S,S) and (R,R)-methyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-l-phenylalaninate (MMZ-45AA)
- (S,R) and (R,S)-methyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-l-phenylalaninate (MMZ-45B)
- (S,S) and (R,R)-methyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-l-prolinate (MMZ-140C)
- (S,S) and (R,R)-dimethyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-l-aspartate (MMZ-147B)
- (S,R) and (R,S)-dimethyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-l-aspartate (MMZ-147C)
- (S,S) and (R,R)-dimethyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-l-glutamate (MMZ-148B)
- (S,R) and (R,S)-dimethyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-l-glutamate (MMZ-148C)
- (S,S) and (R,R)-methyl ((2-hydroxynaphthalen-1-yl)(phenyl)methyl)-l-leucinate (MMZ-167C)
4.2. Biological Studies
4.2.1. Chemicals
4.2.2. Cell Culture
4.2.3. MTT Assay
4.2.4. Dual Acridine Orange/Ethidium Bromide (AO/EB) Fluorescent Staining
4.2.5. Annexin V-FITC and Propidium Iodide Flow Cytometry Analysis
4.3. Computational Analysis
4.3.1. Drug-Likeness and ADMET
4.3.2. DFT Calculations
4.3.3. Molecular Target Prediction
4.3.4. Molecular Docking
4.3.5. Molecular Dynamics
4.3.6. Prime MM-GBSA Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| ADMET | absorption, distribution, metabolism, excretion, and toxicity |
| ADORA1 | adenosine A1 receptor |
| AKT | protein kinase B |
| AO | acridine orange |
| ARE | antioxidant response element |
| ATAD5 | ATPase family AAA domain-containing protein 5 |
| BBB | blood–brain barrier |
| CADD | computer-assisted drug design |
| CDK2 | cyclin-dependent kinase 2 |
| CYP | cytochrome P |
| DFT | density functional theory |
| DMSO | dimethyl sulfoxide |
| EB | ethidium bromide |
| EMM | molecular mechanic energies |
| ERK | extracellular signal-regulated kinase |
| FBS | fetal bovine serum |
| FITC | fluorescein isothiocyanate |
| GB/SA | generalized Born/Surface |
| GI | gastrointestinal |
| GNP | nonpolar solvation |
| GPF | grid parameter file |
| GSGB | SGB polar solvation model |
| GSK-3β | glycogen synthase kinase-3 beta |
| Herg | ether-a-go-go-related gene |
| HLG | HOMO-LUMO energy gap |
| HOMO | highest occupied molecular orbital |
| HSE | heat shock response |
| JNK | c-Jun N-terminal kinase |
| LQTS | long QT syndrome |
| LUMO | lowest occupied molecular orbital |
| MCR | multicomponent reactions |
| MDR | multidrug resistance |
| MESP | molecular electrostatic potential |
| MMP | mitochondrial membrane potential |
| NF-KB1 | nuclear factor-kappa B1 |
| OPLS-AA | optimized potential liquid solvation-all atom |
| PBS | buffered saline |
| PDB | protein data bank |
| P-gp | P-glycoprotein |
| PI | propidium iodide |
| PI3K | phosphatidylinositol 3-kinase |
| PLK1 | polo-like kinase 1 |
| RMSD | root-mean-square deviation |
| RMSF | root-mean-square fluctuation |
| SLC6A14 | sodium- and chloride-dependent neutral and basic amino acid transporter B(0+) |
| TP53 | cellular tumor antigen p53 |
| TRIM24 | transcription intermediary factor 1-alpha |
References
- Olgen, S. Overview on Anticancer Drug Design and Development. Curr. Med. Chem. 2018, 25, 1704–1719. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Stewart, A.A.; Wheatley-Price, P.; Batist, G.; Kantarjian, H.M.; Schiller, J.; Clemons, M.; Bradford, J.-P.; Gillespie, L.; Kurzrock, R. The Importance of Greater Speed in Drug Development for Advanced Malignancies. Cancer Med. 2018, 7, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Connors, T. Anticancer Drug Development: The Way Forward. Oncologist 1996, 1, 180–181. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Mirza, Z.; Ashraf, G.M.D.; Kamal, M.A.; Ansari, S.A.; Damanhouri, G.A.; Abuzenadah, A.M.; Chaudhary, A.G.; Sheikh, I.A. New Anticancer Agents: Recent Developments in Tumor Therapy. Anticancer Res. 2012, 32, 2999–3005. [Google Scholar]
- Geromichalos, G.D. Importance of Molecular Computer Modeling in Anticancer Drug Development. J. BUON Off. J. Balk. Union Oncol. 2007, 12 (Suppl. S1), S101–S118. [Google Scholar]
- Garofalo, M.; Grazioso, G.; Cavalli, A.; Sgrignani, J. How Computational Chemistry and Drug Delivery Techniques Can Support the Development of New Anticancer Drugs. Molecules 2020, 25, 1756. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Rabbani, G.; Danishuddin, M.; Choi, I. Computer Aided Drug Design and Its Application to the Development of Potential Drugs for Neurodegenerative Disorders. Curr. Neuropharmacol. 2018, 16, 740–748. [Google Scholar] [CrossRef]
- Geromichalos, G.D.; Alifieris, C.E.; Geromichalou, E.G.; Trafalis, D.T. Overview on the Current Status on Virtual High-Throughput Screening and Combinatorial Chemistry Approaches in Multi-Target Anticancer Drug Discovery; Part II. J. BUON Off. J. Balk. Union Oncol. 2016, 21, 1337–1358. [Google Scholar]
- Cui, W.; Aouidate, A.; Wang, S.; Yu, Q.; Li, Y.; Yuan, S. Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 2020, 11, 733. [Google Scholar] [CrossRef]
- Mohanram, I.; Meshram, J. Synthesis and Biological Activities of 4-Aminoantipyrine Derivatives Derived from Betti-Type Reaction. ISRN Org. Chem. 2014, 2014, 639392. [Google Scholar] [CrossRef]
- Cardellicchio, C.; Capozzi, M.A.M.; Naso, F. The Betti Base: The Awakening of a Sleeping Beauty. Tetrahedron Asymmetry 2010, 21, 507–517. [Google Scholar] [CrossRef]
- Cardellicchio, C.; Ciccarella, G.; Naso, F.; Schingaro, E.; Scordari, F. The Betti Base: Absolute Configuration and Routes to a Family of Related Chiral Nonracemic Bases. Tetrahedron Asymmetry 1998, 9, 3667–3675. [Google Scholar] [CrossRef]
- Adrián, P.; Alexis, R.G.; Roderick, A.; Kaylie, D.; Miguel, X.F.; Giovanna, B.; José, M.P. Naphthol-Derived Betti Bases as Potential SLC6A14 Blockers. J. Mol. Clin. Med. 2019, 2, 35–40. [Google Scholar] [CrossRef]
- Capozzi, M.A.M.; Capitelli, F.; Bottoni, A.; Calvaresi, M.; Cardellicchio, C. Stacked Naphthyls and Weak Hydrogen-Bond Interactions Govern the Conformational Behavior of P-Resolved Cyclic Phosphonamides: A Combined Experimental and Computational Study. J. Org. Chem. 2014, 79, 11101–11109. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.A.M.; Cardellicchio, C.; Magaletti, A.; Bevilacqua, A.; Perricone, M.; Corbo, M.R. Bioactivity of a Family of Chiral Nonracemic Aminobenzylnaphthols towards Candida Albicans. Molecules 2014, 19, 5219–5230. [Google Scholar] [CrossRef]
- Yellapurkar, I.; Shaikh, S.; Pavale, G.; Bhabal, S.; Ramana, M.M.V. Kaolin-Catalysed One-Pot Synthesis of Thiophene Containing Aminonaphthols under Solvent-Free Condition and Their in Vitro Anticancer and Antioxidant Activity. Res. Chem. Intermed. 2021, 47, 4067–4082. [Google Scholar] [CrossRef]
- Gyémánt, N.; Engi, H.; Schelz, Z.; Szatmári, I.; Tóth, D.; Fülöp, F.; Molnár, J.; de Witte, P.A.M. In Vitro and in Vivo Multidrug Resistance Reversal Activity by a Betti-Base Derivative of Tylosin. Br. J. Cancer 2010, 103, 178–185. [Google Scholar] [CrossRef]
- Nagaraju, B.; Kovvuri, J.; Kumar, C.G.; Routhu, S.R.; Shareef, M.A.; Kadagathur, M.; Adiyala, P.R.; Alavala, S.; Nagesh, N.; Kamal, A. Synthesis and Biological Evaluation of Pyrazole Linked Benzothiazole-β-Naphthol Derivatives as Topoisomerase I Inhibitors with DNA Binding Ability. Bioorg. Med. Chem. 2019, 27, 708–720. [Google Scholar] [CrossRef]
- Mallamaci, R.; Capozzi, M.A.M.; Cardellicchio, C. Antiproliferative Activity of Aminobenzylnaphthols Deriving from the Betti Reaction. Appl. Sci. 2022, 12, 7779. [Google Scholar] [CrossRef]
- Lee, D.-Y.; Lin, H.-Y.; Ramasamy, M.; Kuo, S.-C.; Lee, P.-C.; Hsieh, M.-T. Synthesis and Characterization of the Ethylene-Carbonate-Linked L-Valine Derivatives of 4,4-Dimethylcurcumin with Potential Anticancer Activities. Molecules 2021, 26, 7050. [Google Scholar] [CrossRef]
- Viswanathan, A.; Sebastianelli, G.; Brown, K.; Raunio, J.; Sipilä, V.; Yli-Harja, O.; Candeias, N.R.; Kandhavelu, M. In Vitro Anti-Glioblastoma Activity of L-Valine Derived Boroxazolidones. Eur. J. Pharmacol. 2019, 854, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Cimarelli, C.; Mazzanti, A.; Palmieri, G.; Volpini, E. Solvent-Free Asymmetric Aminoalkylation of Electron-Rich Aromatic Compounds: Stereoselective Synthesis of Aminoalkylnaphthols by Crystallization-Induced Asymmetric Transformation. J. Org. Chem. 2001, 66, 4759–4765. [Google Scholar] [CrossRef]
- Schichl, D.A.; Enthaler, S.; Holla, W.; Riermeier, T.; Kragl, U.; Beller, M. Dynamic Kinetic Resolution of α-Amino Acid Esters in the Presence of Aldehydes. Eur. J. Org. Chem. 2008, 2008, 3506–3512. [Google Scholar] [CrossRef]
- Cardellicchio, C.; Capozzi, M.A.M.; Alvarez-Larena, A.; Piniella, J.F.; Capitelli, F. Investigation on the Weak Interactions Assembling the Crystal Structures of Betti Bases. CrystEngComm 2012, 14, 3972–3981. [Google Scholar] [CrossRef]
- Capozzi, M.A.M.; Cardellicchio, C. (S,S)-1-(Phenyl((1′-4-Nitrophenyl-Ethyl)Amino)Methyl)-2-Naphthol. Molbank 2022, 2022, M1522. [Google Scholar] [CrossRef]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-Fluorouracil and Other Fluoropyrimidines in Colorectal Cancer: Past, Present and Future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef] [PubMed]
- Isacoff, W.H.; Reber, H.A.; Bedford, R.; Hoos, W.; Rahib, L.; Upfill-Brown, A.; Donahue, T.; Hines, O.J. Low-Dose Continuous 5-Fluorouracil Combined with Leucovorin, Nab-Paclitaxel, Oxaliplatin, and Bevacizumab for Patients with Advanced Pancreatic Cancer: A Retrospective Analysis. Target. Oncol. 2018, 13, 461–468. [Google Scholar] [CrossRef]
- Wang, W.-B.; Yang, Y.; Zhao, Y.-P.; Zhang, T.-P.; Liao, Q.; Shu, H. Recent Studies of 5-Fluorouracil Resistance in Pancreatic Cancer. World J. Gastroenterol. WJG 2014, 20, 15682–15690. [Google Scholar] [CrossRef]
- Kasibhatla, S.; Amarante-Mendes, G.P.; Finucane, D.; Brunner, T.; Bossy-Wetzel, E.; Green, D.R. Acridine Orange/Ethidium Bromide (AO/EB) Staining to Detect Apoptosis. CSH Protoc. 2006, 2006, 4493. [Google Scholar] [CrossRef]
- Wlodkowic, D.; Skommer, J.; Darzynkiewicz, Z. Flow Cytometry-Based Apoptosis Detection. In Apoptosis Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 559. [Google Scholar] [CrossRef]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-Score—A Comprehensive Scoring Function for Evaluation of Chemical Drug-Likeness. MedChemComm 2018, 10, 148–157. [Google Scholar] [CrossRef]
- Agoni, C.; Olotu, F.A.; Ramharack, P.; Soliman, M.E. Druggability and Drug-Likeness Concepts in Drug Design: Are Biomodelling and Predictive Tools Having Their Say? J. Mol. Model. 2020, 26, 120. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, S.Z.; Jevrić, L.R.; Podunavac Kuzmanović, S.O.; Lončar, E.S. Prediction of In-Silico ADME Properties of 1,2-O-Isopropylidene Aldohexose Derivatives. Iran. J. Pharm. Res. IJPR 2014, 13, 899–907. [Google Scholar]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Bernacki, J.; Dobrowolska, A.; Nierwińska, K.; Małecki, A. Physiology and Pharmacological Role of the Blood-Brain Barrier. Pharmacol. Rep. PR 2008, 60, 600–622. [Google Scholar]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood-Brain Barrier (BBB) Score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, H.; Yang, K.; Xu, W.; Tang, W.; Li, X. The Structure and Functions of P-Glycoprotein. Curr. Med. Chem. 2010, 17, 786–800. [Google Scholar] [CrossRef]
- Vijay, U.; Gupta, S.; Mathur, P.; Suravajhala, P.; Bhatnagar, P. Microbial Mutagenicity Assay: Ames Test. Bio-Protocol 2018, 8, e2763. [Google Scholar] [CrossRef]
- Lee, H.-M.; Yu, M.-S.; Kazmi, S.R.; Oh, S.Y.; Rhee, K.-H.; Bae, M.-A.; Lee, B.H.; Shin, D.-S.; Oh, K.-S.; Ceong, H.; et al. Computational Determination of hERG-Related Cardiotoxicity of Drug Candidates. BMC Bioinform. 2019, 20, 250. [Google Scholar] [CrossRef]
- Vernetti, L.A.; Vogt, A.; Gough, A.; Taylor, D.L. Evolution of Experimental Models of the Liver to Predict Human Drug Hepatotoxicity and Efficacy. Clin. Liver Dis. 2017, 21, 197–214. [Google Scholar] [CrossRef]
- Ballet, F. Hepatotoxicity in Drug Development: Detection, Significance and Solutions. J. Hepatol. 1997, 26 (Suppl. 2), 26–36. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A Web Server for the In Silico Prediction of Rodent Oral Toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef]
- Descotes, J. Immunotoxicology: Role in the Safety Assessment of Drugs. Drug Saf. 2005, 28, 127–136. [Google Scholar] [CrossRef]
- Amati, M.; Stoia, S.; Baerends, E.J. The Electron Affinity as the Highest Occupied Anion Orbital Energy with a Sufficiently Accurate Approximation of the Exact Kohn-Sham Potential. J. Chem. Theory Comput. 2020, 16, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Curpăn, R.; Avram, S.; Vianello, R.; Bologa, C. Exploring the Biological Promiscuity of High-Throughput Screening Hits through DFT Calculations. Bioorg. Med. Chem. 2014, 22, 2461–2468. [Google Scholar] [CrossRef]
- Ye, N.; Yang, Z.; Liu, Y. Applications of Density Functional Theory in COVID-19 Drug Modeling. Drug Discov. Today 2022, 27, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A Web Server for Target Prediction of Bioactive Small Molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.R.; Yadav, P.; Bedekar, A.V. Application of Optically Active Aminonaphthols as NMR Solvating Agents for Chiral Discrimination of Mandelic Acid. Tetrahedron Asymmetry 2014, 25, 767–774. [Google Scholar] [CrossRef]
- Capozzi, M.A.M.; Cardellicchio, C. Stereoselection in the Betti Reaction of Valine Methyl Esters. Tetrahedron Asymmetry 2017, 28, 1792–1796. [Google Scholar] [CrossRef]
- Da Violante, G.; Zerrouk, N.; Richard, I.; Provot, G.; Chaumeil, J.C.; Arnaud, P. Evaluation of the Cytotoxicity Effect of Dimethyl Sulfoxide (DMSO) on Caco2/TC7 Colon Tumor Cell Cultures. Biol. Pharm. Bull. 2002, 25, 1600–1603. [Google Scholar] [CrossRef]
- Gallo, K.; Goede, A.; Preissner, R.; Gohlke, B.-O. SuperPred 3.0: Drug Classification and Target Prediction-a Machine Learning Approach. Nucleic Acids Res. 2022, 50, W726–W731. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Weight | Hydrogen Bond Acceptors | Hydrogen Bond Donors | Consensus Log p Value | Drug-Likeness |
|---|---|---|---|---|---|
| MMZ-33 | 363.45 | 4 | 2 | 4.32 | Yes; 0 violation |
| MMZ-39 | 397.89 | 4 | 2 | 4.86 | Yes; 0 violation |
| MMZ-45 | 411.49 | 4 | 2 | 4.86 | Yes; 1 violation: MLOGP > 4.15 |
| MMZ-140 | 361.43 | 4 | 1 | 3.79 | Yes; 0 violation |
| MMZ-147 | 393.43 | 6 | 2 | 3.37 | Yes; 0 violation |
| MMZ-167 | 377.48 | 4 | 2 | 4.62 | Yes; 0 violation |
| Compound/Property | Gastrointestinal (GI) Absorption (SwissADME) | CYP2D6 Inhibitor (SwissADME/pkCSM) | CYP3A4 Inhibitor (SwissADME/pkCSM) | Blood–Brain Barrier (BBB) Permeability (SwissADME) | P-glycoprotein Substrate (SwissADME) | Ames Toxicity (pkCSM/PreADMET) | Cardiotoxicity (hERG Inhibition) (PreADMET) | Hepatotoxicity (pkCSM) |
|---|---|---|---|---|---|---|---|---|
| MMZ-33 | High | Yes/No | No/No | Yes | Yes | No/No | Ambiguous | Yes |
| MMZ-39 | High | Yes/No | Yes/No | Yes | Yes | Yes/No | Medium risk | Yes |
| MMZ-45 | High | Yes | Yes | Yes | Yes | Yes/Yes | Ambiguous | Yes |
| MMZ-140 | High | Yes | No/Yes | Yes | No | Yes/Yes | Low risk | Yes |
| MMZ-147 | High | Yes/No | Yes | No | No | Yes/No | Ambiguous | Yes |
| MMZ-167 | High | Yes | Yes | Yes | Yes | Yes/No | Ambiguous | Yes |
| Compound /Property | Hepatotoxicity | Immunotoxicity | Mutagenicity | ATAD5 | HSE | MMP | nrf2/ARE | TP53 |
|---|---|---|---|---|---|---|---|---|
| MMZ-33 | Inactive (p = 0.55) | Inactive (p = 0.98) | Inactive (p = 0.56) | Inactive (p = 0.85) | Inactive (p = 0.92) | Active (p = 0.59) | Inactive (p = 0.92) | Inactive (p = 0.8) |
| MMZ-39 | Active (p = 0.5) | Inactive (p = 0.93) | Inactive (p = 0.64) | Inactive (p = 0.88) | Inactive (p = 0.86) | Active (p = 0.6) | Inactive (p = 0.86) | Inactive (p = 0.72) |
| MMZ-45 | Inactive (p = 0.64) | Inactive (p = 0.99) | Inactive (p = 0.54) | Inactive (p = 0.88) | Inactive (p = 0.93) | Inactive (p = 0.61) | Inactive (p = 0.93) | Inactive (p = 0.85) |
| MMZ-140 | Inactive (p = 0.82) | Inactive (p = 0.86) | Inactive (p = 0.51) | Inactive (p = 0.95) | Inactive (p = 0.95) | Inactive (p = 0.69) | Inactive (p = 0.95) | Inactive (p = 0.92) |
| MMZ-147 | Inactive (p = 0.65) | Inactive (p = 0.99) | Inactive (p = 0.5) | Inactive (p = 0.86) | Inactive (p = 0.94) | Inactive (p = 0.56) | Inactive (p = 0.94) | Inactive (p = 0.74) |
| MMZ-167 | Inactive (p = 0.62) | Inactive (p = 0.86) | Inactive (p = 0.6) | Inactive (p = 0.86) | Inactive (p = 0.63) | Inactive (p = 0.61) | Inactive (p = 0.93) | Inactive (p = 0.83) |
| Compound ID | HOMO (eV) | LUMO (eV) | HLG (eV) |
|---|---|---|---|
| MMZ-33 | −0.213 | −0.058 | 0.15 |
| MMZ-39 | −0.214 | −0.058 | 0.15 |
| MMZ-45 | −0.201 | −0.046 | 0.15 |
| MMZ-140 | −0.201 | −0.046 | 0.15 |
| MMZ-147 | −0.201 | −0.045 | 0.15 |
| MMZ-148 | −0.201 | −0.046 | 0.15 |
| MMZ-167 | −0.203 | −0.047 | 0.15 |
| Protein | PDB id | ΔGcoulomb a | ΔGvdw b | ΔGcovalent c | ΔGsolv d | ΔGsolvlipo e | ΔGbind f |
|---|---|---|---|---|---|---|---|
| ADORA1 | 6D9H | −12.86 | −52.55 | 17.9 | 35.45 | −35.2 | −51.57 |
| CDK1 | 6GU6 | −7.7 | −47.87 | 15.01 | 38.36 | −24.03 | −28.53 |
| CDK2 | 2FVD | −10.49 | −49.28 | 12.96 | 28.05 | −46.92 | −66.73 |
| CK | 6TLS | −7.57 | −31.17 | 3.54 | 21.06 | −26.48 | −41.89 |
| NFKB1 | 1SVC | −16.51 | −35.67 | 2.8 | 37.3 | −10.43 | −23.71 |
| PLK1 | 3FC2 | −18.94 | −34.95 | 10.71 | 26.68 | −22.78 | −42.18 |
| TRIM24 | 4YBM | −18.94 | −34.95 | 10.71 | 26.68 | −22.78 | −42.18 |
| Drug Target | PDB Code | x-D | y-D | z-D | Spacing (Ả) | x Center | y Center | z Center |
|---|---|---|---|---|---|---|---|---|
| AKT1 | 6CCY | 40 | 50 | 48 | 0.425 | −9.801 | 15.312 | −31.398 |
| AURKB | 4AF3 | 40 | 50 | 40 | 0.408 | 21.226 | −21.921 | −10.221 |
| CDK1 | 6GU6 | 40 | 40 | 40 | 0.469 | 23.159 | 21.848 | −2.268 |
| CDK2 | 2FVD | 40 | 40 | 40 | 0.397 | 1.231 | 28.133 | 8.792 |
| EGFR | 7VRA | 40 | 40 | 40 | 0.397 | 50.153 | 1.467 | −19.629 |
| FGFR | 5AM6 | 40 | 40 | 48 | 0.392 | 217.536 | −7.806 | 24.459 |
| FLT3 | 4RT7 | 40 | 50 | 48 | 0.425 | −38.825 | 11.685 | −15.423 |
| JAK2 | 2B7A | 40 | 40 | 40 | 0.419 | 114.221 | 64.945 | 10.271 |
| JAK3 | 3PJC | 40 | 50 | 40 | 0.403 | 8.857 | −5.345 | 10.707 |
| PLK1 | 3FC2 | 40 | 50 | 40 | 0.431 | 47.588 | −5.939 | 9.028 |
| PRKCQ | 4Q9Z | 40 | 50 | 40 | 0.408 | 21.315 | −8.295 | −8.006 |
| RIPK2 | 5W5O | 40 | 50 | 48 | 0.431 | −1.027 | 16.243 | 94.25 |
| ADORA1 | 6D9H | 40 | 50 | 48 | 0.419 | 92.16 | 120.297 | 92.496 |
| BACE1 | 4IVT | 40 | 50 | 40 | 0.414 | 22.376 | 22.871 | 0.873 |
| BACE2 | 2EWY | 40 | 40 | 40 | 0.442 | 106.825 | 24.801 | 2.867 |
| LTA4H | 5N3W | 40 | 50 | 48 | 0.431 | 11.962 | −1.41 | 0.812 |
| CAPN1 | 2NQG | 40 | 40 | 40 | 0.419 | 13.64 | 8.089 | −21.839 |
| MMP16 | 1RM8 | 40 | 40 | 40 | 0.431 | 0.381 | 3.249 | 48.361 |
| PDE2A | 5U7D | 40 | 50 | 48 | 0.408 | 14.119 | 6.752 | 19.46 |
| PDE5 | 2H42 | 40 | 40 | 40 | 0.408 | 30.79 | 119.342 | 11.038 |
| PDE4B | 4KP6 | 40 | 50 | 40 | 0.386 | −41.761 | 91.222 | 114.399 |
| PDE7A | 4PM0 | 40 | 50 | 40 | 0.403 | −45.444 | 25.125 | 1.399 |
| PDE10A | 3WI2 | 40 | 50 | 40 | 0.414 | 20.523 | 3.048 | 58.144 |
| HSD17B2 | 3HB4 | 40 | 42 | 40 | 0.408 | 11.394 | 6.482 | −10.657 |
| IDH1 | 5LGE | 40 | 42 | 40 | 0.392 | −29.141 | −99.881 | 25.215 |
| LDHA | 6MV8 | 40 | 40 | 40 | 0.414 | 40.266 | 14.748 | 27.266 |
| CYP11B1 | 6M7X | 40 | 40 | 40 | 0.397 | 51.53 | −45.46 | −6.116 |
| CYP11B2 | 4ZGX | 40 | 42 | 40 | 0.419 | 60.088 | −54.484 | 115.987 |
| STAT3 | 6NUQ | 44 | 60 | 44 | 0.403 | 13.619 | 54.024 | −0.083 |
| SMARCA2 | 5DKH | 40 | 42 | 40 | 0.386 | −12.258 | 38.92 | 7.45 |
| UQCRB | 5NMI | 40 | 40 | 40 | 0.403 | 39.838 | 15.607 | 1.039 |
| MCL1 | 5FDR | 40 | 42 | 40 | 0.403 | 37.551 | 1.085 | 19.468 |
| APE1 | 6BOW | 66 | 66 | 66 | 0.731 | 9.292 | −30.663 | −0.237 |
| BCHE | 5LKR | 40 | 42 | 40 | 0.408 | −41.505 | 50.849 | −12.665 |
| C5AR | 6C1R | 40 | 40 | 40 | 0.403 | 12.846 | 1.777 | −45.271 |
| CAPN1 | 1ZCM | 40 | 40 | 40 | 0.386 | −21.825 | 5.536 | 34.882 |
| CHRM4 | 5DSG | 40 | 42 | 40 | 0.392 | 50.41 | 8.435 | 63.576 |
| CHRM5 | 6OL9 | 40 | 40 | 40 | 0.431 | 35.173 | 23.793 | −40.677 |
| CHRNA4 | 6UR8 | 40 | 40 | 40 | 0.403 | 124.113 | 145.382 | 189.741 |
| CK | 6TLS | 44 | 44 | 44 | 0.403 | 77.273 | 7.948 | 21.258 |
| CLK4 | 6FYV | 40 | 40 | 40 | 0.414 | −28.205 | 22.947 | −18.2 |
| COX2 | 5F19 | 40 | 42 | 40 | 0.436 | 27.852 | 30.441 | 63.17 |
| CPAR | 6C1R | 40 | 40 | 40 | 0.403 | 12.846 | 1.777 | −45.271 |
| CPSD | 4OD9 | 44 | 44 | 44 | 0.408 | −3.204 | 12.697 | −34.841 |
| CYP3A4 | 5VCC | 40 | 40 | 40 | 0.425 | −23.77 | −27.5 | −11.384 |
| DUSP3 | 3F81 | 40 | 42 | 40 | 0.397 | −0.908 | 0.681 | −6.463 |
| FPR2 | 6OMM | 40 | 40 | 40 | 0.486 | 116.151 | 131.184 | 111.263 |
| GLUT1 | 6THA | 40 | 40 | 40 | 0.392 | 18.054 | 59.12 | 11.134 |
| GNAI1 | 6N4B | 40 | 40 | 40 | 0.397 | 92.223 | 131.93 | 120.478 |
| HADH2 | 2O23 | 40 | 42 | 40 | 0.414 | 19.88 | 13.935 | −12.26 |
| HERG | 5VA1 | 100 | 82 | 108 | 1.000 | 93.575 | 57.237 | 60.469 |
| MDM4 | 6Q9Y | 40 | 40 | 40 | 0.392 | −5.108 | 10.302 | −14.609 |
| NFKB1 | 1SVC | 48 | 58 | 58 | 0.394 | 28.358 | 17.443 | 43.771 |
| NR1A1 | 3ILZ | 40 | 42 | 40 | 0.425 | 28.128 | 38.913 | 32.411 |
| NR1I2 | 6TFI | 40 | 40 | 40 | 0.403 | 51.713 | 36.805 | 15.99 |
| PRCP | 3N2Z | 80 | 84 | 96 | 0.686 | 51.603 | 32.164 | 72.575 |
| PSMA2 | 6KWY | 40 | 40 | 40 | 0.436 | 238.36 | 189.562 | 104.143 |
| SLC1A3 | 5LM4 | 40 | 42 | 40 | 0.397 | −476.806 | 301.433 | 12.118 |
| TDP1 | 6N0D | 40 | 40 | 40 | 0.392 | 8.387 | −14.555 | −34.733 |
| TRIM24 | 4YBM | 44 | 44 | 44 | 0.347 | 36.436 | −18.263 | −32.015 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kciuk, M.; Malinowska, M.; Gielecińska, A.; Sundaraj, R.; Mujwar, S.; Zawisza, A.; Kontek, R. Synthesis, Computational, and Anticancer In Vitro Investigations of Aminobenzylnaphthols Derived from 2-Naphtol, Benzaldehydes, and α-Aminoacids via the Betti Reaction. Molecules 2023, 28, 7230. https://doi.org/10.3390/molecules28207230
Kciuk M, Malinowska M, Gielecińska A, Sundaraj R, Mujwar S, Zawisza A, Kontek R. Synthesis, Computational, and Anticancer In Vitro Investigations of Aminobenzylnaphthols Derived from 2-Naphtol, Benzaldehydes, and α-Aminoacids via the Betti Reaction. Molecules. 2023; 28(20):7230. https://doi.org/10.3390/molecules28207230
Chicago/Turabian StyleKciuk, Mateusz, Martyna Malinowska, Adrianna Gielecińska, Rajamanikandan Sundaraj, Somdutt Mujwar, Anna Zawisza, and Renata Kontek. 2023. "Synthesis, Computational, and Anticancer In Vitro Investigations of Aminobenzylnaphthols Derived from 2-Naphtol, Benzaldehydes, and α-Aminoacids via the Betti Reaction" Molecules 28, no. 20: 7230. https://doi.org/10.3390/molecules28207230
APA StyleKciuk, M., Malinowska, M., Gielecińska, A., Sundaraj, R., Mujwar, S., Zawisza, A., & Kontek, R. (2023). Synthesis, Computational, and Anticancer In Vitro Investigations of Aminobenzylnaphthols Derived from 2-Naphtol, Benzaldehydes, and α-Aminoacids via the Betti Reaction. Molecules, 28(20), 7230. https://doi.org/10.3390/molecules28207230