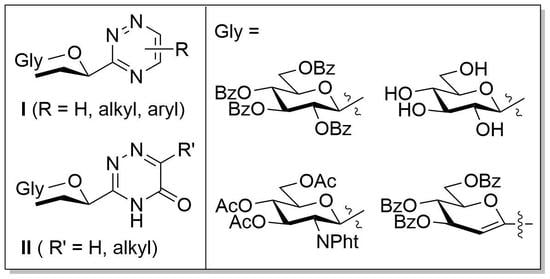
3.6. Synthesis and Characterisation of the New Compounds
3-(2,3,4,6-Tetra-O-benzoyl-β-d-glucopyranosyl)-1,2,4-triazine (2a)
Prepared from amidrazone 1 (0.80 g, 1.25 mmol) and an aq. 40 wt.% solution of glyoxal (144 µL, 1.25 mmol) according to general procedure 1. Reaction time: 3 h. Purified by column chromatography (1:2 EtOAc-hexane) to yield 0.75 g (90%) of pale yellow amorphous solids. Rf = 0.48 (1:1 EtOAc-hexane); [α]D = −40 (c 0.20, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.17, 8.70 (2 × 1H, 2 d, J = 2.1 Hz in each, H-5, H-6), 8.00, 7.95, 7.85, 7.75 (4 × 2H, 4 d, J = 7.92 Hz in each, Ar), 7.53-7.26 (12H, m, Ar), 6.18 (1H, pt, J = 9.5, 9.5 Hz, H-2′ or H-3′ or H-4′), 6.08 (1H, pt, J = 9.7, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.90 (1H, pt, J = 9.5, 9.1 Hz, H-2′ or H-3′ or H-4′), 5.39 (1H, d, J = 9.7 Hz, H-1′), 4.69 (1H, dd, J = 12.1, < 1 Hz, H-6′a), 4.58 (1H, dd, J = 12.1, 5.3 Hz, H-6′b), 4.48-4.44 (1H, m, H-5′); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.8, 165.2, 164.8, 164.3 (4 × C=O, C-3), 149.5, 149.4 (C-5, C-6), 133.4, 133.3, 133.2, 133.0, 129.8-129.6, 129.4, 128.7, 128.6, 128.5, 128.4-128.2 (Ar), 80.1, 76.9, 74.1, 71.6, 69.5 (C-1′ − C-5′), 63.4 (C-6′). ESI-HRMS positive mode (m/z): calcd for C37H30N3O9+ [M+H]+ 660.1977; C37H29N3NaO9+ [M+Na]+ 682.1796. Found: [M+H]+ 660.1972; [M+Na]+ 682.1785.
3-(2,3,4,6-Tetra-O-benzoyl-β-d-glucopyranosyl)-5-methyl-1,2,4-triazine (2b)
Prepared from amidrazone 1 (0.10 g, 0.16 mmol) and methyl glyoxal (24 µL, 0.16 mmol) according to general procedure 1. Reaction time: 3 h. Purified by column chromatography (2:3 EtOAc-hexane) to yield 89 mg (84%) of pale yellow amorphous solids. Rf = 0.38 (1:1 EtOAc-hexane); [α]D = −12 (c 0.20, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.03 (1H, s, H-6), 8.01, 7.94, 7.85, 7.75 (4 × 2H, 4 dd, J = 7.2, 1.2 Hz in each, Ar), 7.54-7.28 (12H, m, Ar), 6.14 (1H, pt, J = 9.5, 9.1 Hz, H-2′ or H-3′ or H-4′), 6.11 (1H, pt, J = 9.7, 9.5 Hz, H-2′ or H-3′ or H-4′), 5.90 (1H, pt, J = 9.6, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.31 (1H, d, J = 9.3 Hz, H-1′), 4.67 (1H, dd, J = 12.3, 3.0 Hz, H-6′a), 4.56 (1H, dd, J = 12.3, 5.2 Hz, H-6′b), 4.42 (1H, ddd, J = 9.7, 5.2, 3.0 Hz, H-5′), 2.56 (3H, s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.8, 165.2, 164.7, 163.2 (4 × C=O, C-3), 160.6 (C-5), 149.9 (C-6), 133.4, 133.2, 133.1, 133.0, 129.8-129.7, 129.5, 128.8, 128.7, 128.6, 128.4-128.2 (Ar), 80.1, 76.9, 74.3, 71.4, 69.5 (C-1′ − C-5′), 63.4 (C-6′), 21.8 (CH3). ESI-HRMS positive mode (m/z): calcd for C38H32N3O9+ [M+H]+ 674.2133; C38H31N3NaO9+ [M+Na]+ 696.1953. Found: [M+H]+ 674.2134; [M+Na]+ 696.1950.
3-(2,3,4,6-Tetra-O-benzoyl-β-d-glucopyranosyl)-5-tert-butyl-1,2,4-triazine (2c)
Prepared from amidrazone 1 (0.30 g, 0.47 mmol) and 3,3-dimethyl-2-oxobutanal (62 mg, 0.47 mmol) according to general procedure 1. Reaction time: 7 h. Purified by column chromatography (3:7 EtOAc-hexane) to yield 0.28 g (83%) of pale yellow amorphous solids. Rf = 0.26 (3:7 EtOAc-hexane); [α]D = −11 (c 0.20, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.23 (1H, s, H-6), 8.02, 7.95, 7.86, 7.73 (4 × 2H, 4 dd, J = 7.1, 1.2 Hz in each, Ar), 7.54-7.24 (12H, m, Ar), 6.25 (1H, pt, J = 9.7, 9.7 Hz, H-2′ or H-3′ or H-4′), 6.13 (1H, pt, J = 9.6, 9.5 Hz, H-2′ or H-3′ or H-4′), 5.90 (1H, pt, J = 9.7, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.35 (1H, d, J = 9.7 Hz, H-1′), 4.69 (1H, dd, J = 12.2, 2.9 Hz, H-6′a), 4.53 (1H, dd, J = 12.2, 5.0 Hz, H-6′b), 4.44 (1H, ddd, J = 9.7, 5.0, 2.9 Hz, H-5′), 1.31 (9H, s, C(CH3)3); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.0, 166.1, 165.9, 165.2, 164.5 (4 × C=O, C-3), 162.5 (C-5), 146.8 (C-6), 133.4, 133.1, 133.1, 133.0, 130.0-129.5, 129.5, 128.9, 128.8, 128.4-128.2 (Ar), 80.1, 76.9, 74.4, 71.0, 69.5 (C-1′ − C-5′), 63.2 (C-6′), 36.8 (C(CH3)3), 28.6 (C(CH3)3). ESI-HRMS positive mode (m/z): calcd for C41H38N3O9+ [M+H]+ 716.2603; C41H37N3NaO9+ [M+Na]+ 738.2422. Found: [M+H]+ 716.2602; [M+Na]+ 738.2419.
3-(2,3,4,6-Tetra-O-benzoyl-β-d-glucopyranosyl)-5-phenyl-1,2,4-triazine (2d).
Method A: Prepared from amidrazone 1 (1.0 g, 1.57 mmol) and phenyl glyoxal monohydrate (0.29 g, 1.88 mmol) according to general procedure 1. Reaction time: 4 h. Purified by column chromatography (1:2 EtOAc-hexane) to yield 0.95 g (83%) of pale yellow amorphous solids.
Method B: To a solution of acetophenone (19 µL, 0.16 mmol, 1 equiv.) in DMSO (2 mL), SeO2 (21 mg, 0.19 mmol, 1.2 equiv.) was added. The mixture was heated at 110 °C until the TLC (1:9 EtOAc-hexane) showed the complete transformation of acetophenone (4 h). Formamidrazine 1 (0.10 g, 0.16 mmol) was then added to the mixture, and the heating was continued. When the TLC (1:1 EtOAc-hexane) showed the completion of the reaction (2 h), the mixture was diluted with EtOAc (50 mL) and extracted with water (20 mL). The separated aqueous phase was washed two times with EtOAc (2 × 50 mL). The combined organic phase was dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue was purified by column chromatography (1:2 EtOAc-hexane). Yield: 57 mg (50%).
Method C: A solution of phenylacetylene (35 µL, 0.32 mmol, 2 equiv.), NIS (43 mg, 0.19 mmol, 1.2 equiv.) and TsOH (3.4 mg, 0.02 mmol, 0.1 equiv.) in DMSO (2 mL) was heated at 110 °C until the TLC (1:9 EtOAc-hexane) indicated the complete conversion of phenylacetylene (4 h). Formamidrazine 1 (0.10 g, 0.16 mmol) was then added to the mixture, and the heating was continued. After completion of the reaction monitored by TLC (2 h), the same steps as described in method B were carried out. Yield: 66 mg (60%). Rf = 0.55 (1:1 EtOAc-hexane); [α]D = −68 (c 0.20, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.58 (1H, s, H-6), 8.17, 8.02, 7.96, 7.87, 7.75 (5 × 2H, 5 d, J = 7.2 Hz in each, Ar), 7.59-7.21 (15H, m, Ar), 6.33, 6.18, 5.94 (3 × 1H, 3 pt, J = 9.7, 9.7 Hz in each, H-2′, H-3′, H-4′), 5.43 (1H, d, J = 9.7 Hz, H-1′), 4.72 (1H, dd, J = 12.2, 2.8 Hz, H-6′a), 4.55 (1H, dd, J = 12.2, 5.1 Hz, H-6′b), 4.49 (1H, ddd, J = 9.7, 5.1, 2.8 Hz, H-5′); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.8, 165.2, 164.7, 163.4 (4 × C=O, C-3), 156.0 (C-5), 145.9 (C-6), 133.4, 133.1, 133.0, 132.8, 132.7, 129.8–129.3, 128.8, 128.7, 128.6, 128.5, 128.4–127.9 (Ar), 80.1, 76.9, 74.4, 71.1, 69.5 (C-1′ − C-5′), 63.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C43H33N3NaO9+ [M+Na]+ 758.2109. Found: 758.2107.
3-(2,3,4,6-Tetra-O-benzoyl-β-d-glucopyranosyl)-5-(p-methoxyphenyl)-1,2,4-triazine (2e)
Prepared from amidrazone 1 (0.10 g, 0.16 mmol) and p-methoxyphenyl glyoxal monohydrate (0.034 g, 0.19 mmol) according to general procedure 1. Reaction time: 4 h. Purification by column chromatography (1:2 EtOAc-hexane), followed by the crystallisation of the resulting syrup from a mixture of Et2O (3 mL) and hexane (2 mL), which gave 116 mg (97%) pale yellow crystals. Rf = 0.45 (1:1 EtOAc-hexane); mp = 150–151 °C; [α]D = −98 (c 0.53, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.50 (1H, s, H-6), 8.17 (2H, d, J = 8.9 Hz, Ar), 8.02, 7.96, 7.87, 7.74 (4 × 2H, 4 dd, J = 7.3, 1.2 Hz in each, Ar), 7.54–7.23 (12H, m, Ar), 7.01 (2H, d, J = 8.9 Hz, Ar), 6.33, 6.15, 5.92 (3 × 1H, 3 pt, J = 9.7, 9.7 Hz in each, H-2′, H-3′, H-4′), 5.37 (1H, d, J = 9.7 Hz, H-1′), 4.70 (1H, dd, J = 12.3, 2.7 Hz, H-6′a), 4.54 (1H, dd, J = 12.3, 5.1 Hz, H-6′b), 4.46 (1H, ddd, J = 9.7, 5.1, 2.7 Hz, H-5′), 3.89 (3H, s, OCH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.9, 165.2, 164.8, 163.6, 163.1 (4 × C=O, C-3, Ar-Cq), 155.5 (C-5), 145.3 (C-6), 133.4, 133.1, 133.0, 129.8–129.6, 128.9, 128.8, 128.8, 128.4–128.2 (Ar), 125.0 (Ar-Cq), 114.8 (Ar-CH), 80.1, 76.9, 74.5, 70.9, 69.5 (C-1′ − C-5′), 63.3 (C-6′), 55.5 (OCH3). ESI-HRMS positive mode (m/z): calcd for C44H36N3O10+ [M+H]+ 766.2395; C44H35N3NaO10+ [M+Na]+ 788.2215. Found: [M+H]+ 766.2390; [M+Na]+ 788.2208.
3-(2,3,4,6-Tetra-O-benzoyl-β-d-glucopyranosyl)-5-(p-chlorophenyl)-1,2,4-triazine (2f)
Prepared from amidrazone 1 (0.10 g, 0.16 mmol) and p-chlorophenyl glyoxal monohydrate (0.035 g, 0.19 mmol) according to general procedure 1. Reaction time: 4 h. Purification by column chromatography (1:2 EtOAc-hexane) gave 109 mg (90%) of pale yellow amorphous solids. Rf = 0.60 (1:1 EtOAc-hexane); [α]D = −82 (c 0.50, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.57 (1H, s, H-6), 8.13 (2H, d, J = 8.4 Hz, Ar), 8.02, 7.96, 7.86, 7.74 (4 × 2H, 4 d, J = 7.5 Hz in each, Ar), 7.56-7.24 (14H, m, Ar), 6.28, 6.16, 5.92 (3 × 1H, 3 pt, J = 9.6, 9.6 Hz in each, H-2′, H-3′, H-4′), 5.39 (1H, d, J = 9.6 Hz, H-1′), 4.72 (1H, dd, J = 12.2, 2.1 Hz, H-6′a), 4.54 (1H, dd, J = 12.2, 4.9 Hz, H-6′b), 4.47 (1H, ddd, J = 9.6, 4.9, 2.1 Hz, H-5′); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.9, 165.2, 164.8, 163.5 (4 × C=O, C-3), 154.9 (C-5), 145.6 (C-6), 139.4 (Ar-Cq), 133.5, 133.3, 133.2, 133.1 (Ar), 131.3 (Ar-Cq), 129.9-128.3 (Ar), 80.1, 77.0, 74.4, 71.1, 69.5 (C-1′ − C-5′), 63.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C43H33ClN3O9+ [M+H]+ 770.1900; C43H32ClN3NaO9+ [M+Na]+ 792.1719. Found: [M+H]+ 770.1900; [M+Na]+ 792.1718.
3-(2,3,4,6-Tetra-O-benzoyl-β-d-glucopyranosyl)-5,6-diphenyl-1,2,4-triazine (2g)
Prepared from amidrazone 1 (1.0 g, 1.57 mmol) and benzil (0.33 g, 1.57 mmol) according to general procedure 1. Reaction time: 7 h. Purified by column chromatography (1:1 EtOAc-hexane) to yield 0.67 g (53%) of pale yellow solids. Rf = 0.71 (3:7 EtOAc-hexane); mp = 175–179 °C; [α]D = −26 (c 0.20, CHCl3); 1H NMR (360 MHz, CDCl3) δ (ppm): 8.02, 7.95, 7.87, 7.76 (4 × 2H, 4 d, J = 7.4 Hz in each, Ar), 7.55–7.24 (22H, m, Ar), 6.36 (1H, pt, J = 9.7, 9.7 Hz, H-2′ or H-3′ or H-4′), 6.15 (1H, pt, J = 9.6, 9.5 Hz, H-2′ or H-3′ or H-4′), 5.91 (1H, pt, J = 9.7, 9.7 Hz, H-2′ or H-3′ or H-4′), 5.45 (1H, d, J = 9.7 Hz, H-1′), 4.69 (1H, dd, J = 12.3, 2.6 Hz, H-6′a), 4.54 (1H, dd, J = 12.3, 5.1 Hz, H-6′b), 4.48 (1H, ddd, J = 9.5, 5.1, 2.6 Hz, H-5′); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.9, 165.2, 164.7 (4 × C=O), 161.2, 157.3, 156.5 (C-3, C-5, C-6), 135.0–128.2 (Ar), 79.9, 77.0, 74.6, 71.1, 69.5 (C-1′ − C-5′), 63.3 (C-6′). ESI-HRMS positive mode (m/z): calcd for C49H38N3O9+ [M+H]+ 812.2603. Found: 812.2602.
3-(β-d-Glucopyranosyl)-1,2,4-triazine (3a)
Prepared from triazine 2a (0.25 g, 0.38 mmol) according to general procedure 2. Reaction time: 3 h. Purified by column chromatography (4:1 CHCl3-MeOH) to yield 58 mg (63%) of pale yellow amorphous solids. Rf = 0.32 (7:3 CHCl3-MeOH); [α]D = −138 (c 0.22, DMSO). 1H NMR (360 MHz, CD3OD) δ (ppm): 9.30, 8.84 (2 × 1H, 2 d, J = 2.5 Hz in each, H-5, H-6), 4.61 (1H, d, J = 9.7 Hz, H-1′), 3.84 (1H, pt, J = 9.5, 9.1 Hz, H-2′ or H-3′ or H-4′), 3.83 (1H, dd, J = 12.1, 2.0 Hz, H-6′a), 3.68 (1H, dd, J = 12.1, 5.0 Hz, H-6′b), 3.56 (1H, pt, J = 9.1, 8.9 Hz, H-2′ or H-3′ or H-4′), 3.48-3.46 (2H, m, H-2′ or H-3′ or H-4′, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 167.6 (C-3), 151.7, 150.9 (C-5, C-6), 83.1, 82.8, 79.4, 74.3, 71.4 (C-1′ − C-5′), 62.8 (C-6′). ESI-HRMS positive mode (m/z): calcd for C9H14N3O5+ [M+H]+ 244.0928; C9H13N3NaO5+ [M+Na]+ 266.0747. Found: [M+H]+ 244.0930; [M+Na]+ 266.0746.
3-(β-d-Glucopyranosyl)-5-methyl-1,2,4-triazine (3b)
Prepared from triazine 2b (0.20 g, 0.3 mmol) according to general procedure 2. Reaction time: 5 h. Purified by column chromatography (4:1 CHCl3-MeOH) to yield 44 mg (58%) pale yellow syrup. Rf = 0.30 (4:1 CHCl3-MeOH); [α]D = +57 (c 0.20, MeOH). 1H NMR (360 MHz, CD3OD) δ (ppm): 9.26 (1H, s, H-6), 4.58 (1H, d, J = 9.7 Hz, H-1′), 3.88 (1H, dd, J = 11.9, 2.2 Hz, H-6′a), 3.87 (1H, pt, J = 9.4, 9.2 Hz, H-2′ or H-3′ or H-4′), 3.72 (1H, dd, J = 11.9, 3.3 Hz, H-6′b), 3.61-3.50 (3H, m, H-2′ and/or H-3′ and/or H-4′, H-5′), 2.63 (3H, s, CH3); 13C NMR (90 MHz, CD3OD) δ (ppm): 166.5, 163.0 (C-3, C-5), 151.2 (C-6), 83.0, 82.8, 79.4, 74.3, 71.4 (C-1′ − C-5′), 62.9 (C-6′), 21.9 (CH3). ESI-HRMS positive mode (m/z): calcd for C10H15N3NaO5+ [M+Na]+ 280.0904. Found: 280.0903.
3-(β-d-Glucopyranosyl)-5-tert-butyl-1,2,4-triazine (3c)
Prepared from triazine 2c (0.20 g, 0.28 mmol) according to general procedure 2. Reaction time: 5 h. Purified by column chromatography (9:1 CHCl3-MeOH) to yield 73 mg (88%) of pale yellow solids. Rf = 0.46 (4:1 CHCl3-MeOH); mp = 196–200 °C; [α]D = +17 (c 0.20, MeOH). 1H NMR (360 MHz, CD3OD) δ (ppm): 9.46 (1H, s, H-6), 4.59 (1H, d, J = 9.7 Hz, H-1′), 3.98 (1H, pt, J = 9.4, 9.2 Hz, H-2′ or H-3′ or H-4′), 3.90 (1H, dd, J = 12.0, 1.5 Hz, H-6′a), 3.70 (1H, dd, J = 12.0, 5.0 Hz, H-6′b), 3.59 (1H, pt, J = 9.0, 8.9 Hz, H-2′ or H-3′ or H-4′), 3.54–3.46 (2H, m, H-2′ or H-3′ or H-4′, H-5′), 1.43 (9H, s, C(CH3)3); 13C NMR (90 MHz, CD3OD) δ (ppm): 172.0, 166.2 (C-3, C-5), 148.1 (C-6), 83.4, 82.9, 79.5, 74.2, 71.7 (C-1′ − C-5′), 63.0 (C-6′), 38.0 (C(CH3)3), 29.1 (C(CH3)3). ESI-HRMS positive mode (m/z): calcd for C13H22N3O5+ [M+H]+ 300.1554; C13H21N3NaO5+ [M+Na]+ 322.1373. Found: [M+H]+ 300.1550; [M+Na]+ 322.1369.
3-(β-d-Glucopyranosyl)-5-phenyl-1,2,4-triazine (3d)
Prepared from triazine 2d (0.20 g, 0.27 mmol) according to general procedure 2. Reaction time: 4 h. Purified by column chromatography (9:1 CHCl3-MeOH) to yield 62 mg (72%) of pale yellow solids. Rf = 0.43 (4:1 CHCl3-MeOH); mp = 236–240 °C; [α]D = +44 (c 0.20, DMSO). 1H NMR (360 MHz, DMSO-d6) δ (ppm): 10.06 (1H, s, H-6), 8.36 (2H, d, J = 8.3 Hz, Ar), 7.71–7.62 (3H, m, Ar), 5.11–5.02, 5.57–4.55 (4H, OH), 4.52 (1H, d, J = 9.9 Hz, H-1′), 3.98–3.92, 3.75–3.71, 3.48–3.37, 3.27–3.20 (6H, m, H-2′, H-3′, H-4′, H-5′, H-6′a,b); 13C NMR (90 MHz, DMSO-d6) δ (ppm): 165.2 (C-3), 154.8 (C-5), 146.1 (C-6), 133.0, 132.6, 129.3 (2), 127.8 (2) (Ar), 82.1, 82.0, 77.9, 72.0, 70.4 (C-1′ − C-5′), 61.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C15H18N3O5+ [M+H]+ 320.1241; C15H17N3NaO5+ [M+Na]+ 342.1060. Found: [M+H]+ 320.1239; [M+Na]+ 342.1054.
3-(β-d-Glucopyranosyl)-5-(p-methoxyphenyl)-1,2,4-triazine (3e)
Prepared from triazine 2e (96 mg, 0.13 mmol) according to general procedure 2. Reaction time: 2 h. Purified by column chromatography (8:1 CHCl3-MeOH) to yield 32 mg (73%) of pale yellow amorphous solids. Rf = 0.50 (7:3 CHCl3-MeOH); [α]D = +24 (c 0.49, DMSO). 1H NMR (400 MHz, CD3OD) δ (ppm): 9.75 (1H, s, H-6), 8.33, 7.12 (2 × 2H, 2 d, J = 8.8 Hz in each, Ar), 4.62 (1H, d, J = 9.7 Hz, H-1′), 4.02 (1H, pt, J = 9.3, 9.2 Hz, H-2′ or H-3′ or H-4′), 3.92-3.90 (4H, m, H-6′a, OCH3), 3.74 (1H, dd, J = 11.5, 2.9 Hz, H-6′b), 3.62 (1H, pt, J = 9.0, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.55-3.53 (2H, m, H-2′ or H-3′ or H-4′, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 166.8, 165.4 (C-3, Ar-Cq), 157.4 (C-5), 146.5 (C-6), 131.1 (2), 126.6, 116.0 (2) (Ar), 83.4, 82.9, 79.5, 74.3, 71.6 (C-1′ − C-5′), 62.9 (C-6′), 56.2 (OCH3). ESI-HRMS positive mode (m/z): calcd for C16H20N3O6+ [M+H]+ 350.1347; C16H19N3NaO6+ [M+Na]+ 372.1166. Found: [M+H]+ 350.1340; [M+Na]+ 372.1159.
3-(β-d-Glucopyranosyl)-5-(p-chlorophenyl)-1,2,4-triazine (3f)
Prepared from triazine 2f (79 mg, 0.10 mmol) according to general procedure 2. Reaction time: 2 h. Purified by column chromatography (8:1 CHCl3-MeOH) to yield 26 mg (72%) of pale yellow amorphous solids. Rf = 0.47 (7:3 CHCl3-MeOH); [α]D = +30 (c 0.53, DMSO). 1H NMR (400 MHz, CD3OD) δ (ppm): 9.87 (1H, s, H-6), 8.36, 7.62 (2 × 2H, 2 d, J = 8.6 Hz in each, Ar), 4.67 (1H, d, J = 9.7 Hz, H-1′), 4.02 (1H, pt, J = 9.3, 9.3 Hz, H-2′ or H-3′ or H-4′), 3.90 (1H, dd, J = 12.3, < 1 Hz, H-6′a), 3.73 (1H, dd, J = 12.3, 4.9 Hz, H-6′b), 3.62 (1H, pt, J = 9.0, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.54-3.53 (2H, m, H-2′ or H-3′ or H-4′, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 13C NMR (90 MHz, CD3OD) δ (ppm): 167.0 (C-3), 156.8 (C-5), 147.0 (C-6), 140.4, 133.3, 130.8 (4) (Ar), 83.4, 82.9, 79.5, 74.3, 71.6 (C-1′ − C-5′), 62.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C15H17ClN3O5+ [M+H]+ 354.0851; C15H16ClN3NaO5+ [M+Na]+ 376.0671. Found: [M+H]+ 354.0851; [M+Na]+ 376.0668.
3-(β-d-Glucopyranosyl)-5,6-diphenyl-1,2,4-triazine (3g)
Prepared from triazine 2g (0.15 g, 0.18 mmol) according to general procedure 2. Reaction time: 4 h. Purified by column chromatography (9:1 CHCl3-MeOH) to yield 61 mg (84%) pale yellow oil. Rf = 0.32 (9:1 CHCl3-MeOH); [α]D = +42 (c 0.44, MeOH). 1H NMR (360 MHz, CD3OD) δ (ppm): 7.60-7.34 (10H, m, Ar), 4.75 (1H, d, J = 9.7 Hz, H-1′), 4.05 (1H, pt, J = 9.5, 9.1 Hz, H-2′ or H-3′ or H-4′), 3.92 (1H, dd, J = 12.3, < 1 Hz, H-6′a), 3.75 (1H, dd, J = 12.3, 4.5 Hz, H-6′b), 3.65 (1H, pt, J = 9.1, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.57-3.55 (2H, m, H-2′ or H-3′ or H-4′, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 13C NMR (90 MHz, CD3OD) δ (ppm): 165.0 (C-3), 158.8, 158.7 (C-5, C-6), 136.8, 136.7, 132.0, 131.2 (2), 131.0, 130.7 (2), 129.8 (2), 129.6 (2) (Ar), 83.0, 82.9, 79.5, 74.3, 71.5 (C-1′ − C-5′), 62.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C21H22N3O5+ [M+H]+ 396.1554; C21H21N3NaO5+ [M+Na]+ 418.1373. Found: [M+H]+ 396.1555; [M+Na]+ 418.1371.
C-(2-Deoxy-2-phthalimido-3,4,6-tri-O-acetyl-β-d-glucopyranosyl)formamidrazone (5)
Ethyl C-(2-deoxy-2-phthalimido-3,4,6-tri-O-acetyl-β-d-glucopyranosyl)formimidate (4, 1.56 g, 3.17 mmol) was dissolved in anhydrous EtOH (30 mL), and hydrazine monohydrate (154 µL, 3.17 mmol) was added. The reaction mixture was stirred at room temperature, and the transformation of 4 was monitored by TLC (3:2 EtOAc-hexane). After the completion of the reaction (4.5 h), the precipitated product was filtered off and washed with EtOH to give 0.77 g of white solids. The mother liquor was evaporated under reduced pressure to give an oil, which was triturated with diethyl ether to give an additional 0.55 g of white solids. The combined yield of the product: 1.31 g (87%). Rf = 0.32 (3:2 EtOAc-hexane); [α]D = −13 (c 0.49, MeOH). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.86-7.74 (4H, m, Ar), 5.98 (1H, pt, J = 9.8, 9.7 Hz, H-2 or H-3 or H-4), 5.19 (1H, pt, J = 9.7, 9.6 Hz, H-2 or H-3 or H-4), 4.81 (1H, d, J = 9.9 Hz, H-1), 4.58 (2H, br s, NH2), 4.50 (1H, pt, J = 9.9, 9.6 Hz, H-2 or H-3 or H-4), 4.35 (1H, dd, J = 12.3, 4.6 Hz, H-6a), 4.17 (1H, dd, J = 12.5, 2.4 Hz, H-6b), 3.98 (1H, ddd, J = 9.9, 4.6, 2.4 Hz H-5′), 3.61-3.27 (2H, br signal, NH2), 2.11, 2.05, 1.88 (3 × 3H, 3 s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.6, 169.9, 169.5 (CH3-C=O), 167.8, 167.4 (Phth-C=O), 148.2 (C=N), 134.2, 134.0, 131.6, 131.0, 123.7, 123.3 (Ar), 75.6, 74.2, 70.9, 68.8 (C-1, C-3 − C-5), 62.1 (C-6), 52.7 (C-2), 20.7, 20.5, 20.4 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C21H25N4O9+ [M+H]+ 477.1616. Found: 477.1613.
3-(2′-Deoxy-2′-phthalimido-3′,4′,6′-tri-O-acetyl-β-d-glucopyranosyl)-1,2,4-triazine (6a)
Prepared from amidrazone 5 (0.10 g, 0.21 mmol) and an aq. 40 wt.% solution of glyoxal (23.8 µL, 0.21 mmol) according to general procedure 1. Purified by column chromatography (1:1 EtOAc-hexane) to yield 58 mg (58%) of pale yellow syrup. Rf = 0.36 (3:2 EtOAc-hexane); [α]D = −52 (c 0.23, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.16, 8.67 (2 × 1H, 2 d, J = 2.4 Hz in each, H-5, H-6), 7.80-7.69 (4H, m, Ar), 6.09 (1H, dd, J = 10.3, 9.2 Hz, H-2′ or H-3′ or H-4′), 5.96 (1H, d, J = 10.5 Hz, H-1′), 5.36 (1H, dd, J = 9.9, 9.3 Hz, H-2′ or H-3′ or H-4′), 5.00 (1H, pt, J = 10.5, 10.5 Hz, H-2′ or H-3′ or H-4′), 4.38 (1H, dd, J = 12.4, 5.1 Hz, H-6′a), 4.23 (1H, dd, J = 12.5, 2.0 Hz, H-6′b), 4.17 (1H, ddd, J = 10.3, 5.1, 2.0 Hz, H-5′), 2.10, 2.08, 1.88 (3 × 3H, 3 s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.6, 170.1, 169.4 (CH3-C=O), 167.6, 166.9 (Phth-C=O), 164.4 (C-3), 149.4 (2) (C-5, C-6), 134.3, 131.3, 130.8, 123.6 (Ar), 76.8, 76.5, 71.3, 68.8 (C-1′, C-3′ − C-5′), 62.2 (C-6′), 53.2 (C-2′), 20.7, 20.6, 20.4 (3 × CH3).). ESI-HRMS positive mode (m/z): calcd for C23H23N4O9+ [M+H]+ 499.1460; C23H22N4NaO9+ [M+Na]+ 521.1279. Found: [M+H]+ 499.1455; [M+Na]+ 521.1279.
3-(2′-Deoxy-2′-phthalimido-3′,4′,6′-tri-O-acetyl-β-d-glucopyranosyl)-5-methyl-1,2,4-triazine (6b)
Prepared from amidrazone 5 (0.10 g, 0.21 mmol) and an aq. 40 wt.% solution of methylglyoxal (32 µL, 0.21 mmol) according to general procedure 1. Purified by column chromatography (2:1 EtOAc-hexane) to yield 67 mg (62%) of pale yellow syrup. Rf = 0.41 (2:1 EtOAc-hexane); [α]D = −16 (c 0.25, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.00 (1H, s, H-6), 7.86-7.69 (4H, m, Ar), 6.07 (1H, dd, J = 10.3, 9.3 Hz, H-2′ or H-3′ or H-4′), 5.88 (1H, d, J = 10.5 Hz, H-1′), 5.37 (1H, pt, J = 9.9, 9.4 Hz, H-2′ or H-3′ or H-4′), 5.05 (1H, pt, J = 10.5, 10.5 Hz, H-2′ or H-3′ or H-4′), 4.38 (1H, dd, J = 12.5, 5.0 Hz, H-6′a), 4.23 (1H, dd, J = 12.5, 2.1 Hz, H-6′b), 4.15 (1H, ddd, J = 10.2, 5.0, 2.1 Hz, H-5′), 2.52 (3H, s, CH3), 2.10, 2.08, 1.88 (3 × 3H, 3 s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.7, 170.2, 169.5 (CH3-C=O), 167.8, 166.4 (Phth-C=O), 163.2. 160.5 (C-3, C-5), 149.9 (C-6), 134.2, 131.4, 131.0, 123.6 (Ar), 76.8, 76.5, 71.4, 68.8 (C-1′, C-3′ − C-5′), 62.2 (C-6′), 53.0 (C-2′), 21.7, 20.7, 20.6, 20.4 (4 × CH3). ESI-HRMS positive mode (m/z): calcd for C24H25N4O9+ [M+H]+ 513.1616; C24H24N4NaO9+ [M+Na]+ 535.1435. Found: [M+H]+ 513.1612; [M+Na]+ 535.1429.
3-(2′-Deoxy-2′-phthalimido-3′,4′,6′-tri-O-acetyl-β-d-glucopyranosyl)-5-tert-butyl-1,2,4-triazine (6c)
Prepared from amidrazone 5 (0.10 g, 0.21 mmol) and 3,3-dimethyl-2-oxobutanal (0.028 g, 0.21 mmol) according to general procedure 1. Purified by column chromatography (4:5 EtOAc-hexane) to yield 78 mg (67%) of white syrup. Rf = 0.51 (4:5 EtOAc-hexane); [α]D = +14 (c 0.28, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.21 (1H, s, H-6), 7.81-7.68 (4H, m, Ar), 6.05 (1H, pt, J = 10.3, 9.3 Hz, H-2′ or H-3′ or H-4′), 5.93 (1H, d, J = 10.5 Hz, H-1′), 5.37 (1H, pt, J = 10.0, 9.3 Hz, H-2′ or H-3′ or H-4′), 5.11 (1H, pt, J = 10.5, 10.5 Hz, H-2′ or H-3′ or H-4′), 4.35 (1H, dd, J = 12.4, 4.9 Hz, H-6′a), 4.25 (1H, dd, J = 12.4, 1.8 Hz, H-6′b), 4.15 (1H, ddd, J = 10.0, 4.9, 1.8 Hz, H-5′), 2.10, 2.08, 1.88 (3 × 3H, 3 s, CH3), 1.28 (9H, s, C(CH3)3); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.6, 170.1, 169.7, 169.5 (3 × CH3-C=O, C-5), 167.5, 166.5 (2 × Phth-C=O), 162.6 (C-3), 146.8 (C-6), 134.2, 131.4, 130.9, 123.4 (Ar), 76.9, 76.5, 71.5, 68.9 (C-1′, C-3′ − C-5′), 62.2 (C-6′), 52.9 (C-2′), 36.7 (C(CH3)3), 28.5 (C(CH3)3), 20.7, 20.6, 20.4 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C27H31N4O9+ [M+H]+ 555.2086; C27H30N4NaO9+ [M+Na]+ 577.1905. Found: [M+H]+ 555.2088; [M+Na]+ 577.1904.
3-(2′-Deoxy-2′-phthalimido-3′,4′,6′-tri-O-acetyl-β-d-glucopyranosyl)-5-phenyl-1,2,4-triazine (6d)
Prepared from amidrazone 5 (0.10 g, 0.21 mmol) and phenylglyoxal monohydrate (0.032 g, 0.21 mmol) according to general procedure 1. Purified by column chromatography (1:1 EtOAc-hexane) to yield 88 mg (73%) of yellow syrup. Rf = 0.41 (3:2 EtOAc-hexane); [α]D = −86 (c 0.22, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.57 (1H, s, H-6), 8.17–7.52 (9H, m, Ar), 6.10 (1H, pt, J = 9.8, 9.7 Hz, H-2′ or H-3′ or H-4′), 6.01 (1H, d, J = 10.5 Hz, H-1′), 5.40 (1H, pt, J = 9.7, 9.5 Hz, H-2′ or H-3′ or H-4′), 5.24 (1H, pt, J = 10.5, 10.5 Hz, H-2′ or H-3′ or H-4′), 4.38 (1H, dd, J = 12.3, 4.8 Hz, H-6′a), 4.26-4.17 (2H, m, H-5′, H-6′b), 2.09, 1.90 (9H, 2 s, CH3). 13C NMR (90 MHz, CDCl3) δ (ppm): 170.7, 170.2, 169.5 (CH3-C=O), 167.8, 167.7 (Phth-C=O), 163.4, 155.8 (C-3, C-5), 145.9 (C-6), 134.2, 132.8, 132.6, 129.3, 127.8, 123.5 (Ar), 76.9, 76.6, 71.6, 68.8 (C-1′, C-3′ − C-5′), 62.2 (C-6′), 52.8 (C-2′), 20.7, 20.6, 20.4 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C29H27N4O9+ [M+H]+ 575.1773; C29H26N4NaO9+ [M+Na]+ 597.1592. Found: [M+H]+ 575.1777; [M+Na]+ 597.1593.
3-(2′-Deoxy-2′-phthalimido-3′,4′,6′-tri-O-acetyl-β-d-glucopyranosyl)-5,6-diphenyl-1,2,4-triazine (6e)
Prepared from amidrazone 5 (0.10 g, 0.21 mmol) and benzil (0.042 g, 0.21 mmol) according to general procedure 1. Purified by column chromatography (4:5 EtOAc-hexane) to yield 84 mg (65%) of pale yellow syrup. Rf = 0.51 (4:5 EtOAc-hexane); [α]D = −60 (c 0.20, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.81-7.27 (14H, m, Ar), 6.10 (1H, dd, J = 10.3, 9.3 Hz, H-2′ or H-3′ or H-4′), 6.05 (1H, d, J = 10.6 Hz, H-1′), 5.40 (1H, pt, J = 10.0, 9.3 Hz, H-2′ or H-3′ or H-4′), 5.30 (1H, pt, J = 10.5, 10.5 Hz, H-2′ or H-3′ or H-4′), 4.39 (1H, dd, J = 12.6, 5.1 Hz, H-6′a), 4.25 (1H, dd, J = 12.6, < 1 Hz, H-6′a), 4.21-4.19 (1H, m, H-5′), 2.08, 1.90 (9H, 2 s, CH3). 13C NMR (90 MHz, CDCl3) δ (ppm): 170.6, 170.1, 169.4 (CH3-C=O), 167.8, 166.8 (Phth-C=O), 161.1, 157.1, 156.2 (C-3, C-5, C-6), 134.8-123.4 (Ar), 76.6, 76.5, 71.6, 68.8 (C-1′, C-3′ − C-5′), 62.2 (C-6′), 52.7 (C-2′), 20.7, 20.6, 20.4 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C35H31N4O9+ [M+H]+ 651.2086; C35H30N4NaO9+ [M+Na]+ 673.1905. Found: [M+H]+ 651.2085; [M+Na]+ 673.1902.
N1-Ethoxycarbonylmethylidene-C-(2′,3′,4′,6′-tetra-O-benzoyl-β-d-glucopyranosyl)formamidrazone (7a)
A solution of amidrazone 1 (0.10 g, 0.16 mmol) and ethyl glyoxalate (31 µL, 0.16 mmol, an 50% solution in toluene) was boiled in dry EtOH (3 mL) until the TLC (1:1 and 7:3 EtOAc-hexane) showed the complete conversion of 1. After the completion of the reaction (1 h), the solvent was removed under reduced pressure. After column chromatographic purification (2:3 EtOAc-hexane), 44 mg (39%) of the title compound was obtained as a pale yellow syrup. Rf = 0.53 (1:1 EtOAc-hexane); [α]D = +23 (c 0.27, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.06-7.83, 7.59-7.26, 7.07 (21H, m, Ar, CH=N), 6.02 (1H, pt, J = 9.6, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.76 (1H, pt, J = 9.8, 9.7 Hz, H-2′ or H-3′ or H-4′), 5.67 (1H, pt, J = 9.7, 9.7 Hz, H-2′ or H-3′ or H-4′), 4.69 (1H, dd, J = 12.4, 2.7 Hz, H-6′a), 4.54 (1H, dd, J = 12.4, 5.2 Hz, H-6′b), 4.51 (1H, d, J = 9.7 Hz, H-1′), 4.35-4.25 (1H, m, H-5′), 4.23 (2H, q, J = 7.1 Hz, CH2), 1.28 (3H, t, J = 7.1 Hz, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.2, 165.6, 165.3, 165.2, 163.7, 160.9 (5 × C=O, C=N), 146.1 (CH=N), 133.6, 133.3, 133.2, 129.9-128.3 (Ar), 76.4, 76.3, 73.4, 70.5, 69.1 (C-1′ − C-5′), 62.8, 61.2 (C-6′, CH2), 14.1 (CH3).
N1-Methoxycarbonylethylidene-C-(2′,3′,4′,6′-tetra-O-benzoyl-β-d-glucopyranosyl)formamidrazone (7b)
A solution of amidrazone 1 (0.10 g, 0.16 mmol) and methyl pyruvate (14 µL, 0.16 mmol) was boiled in dry EtOH (3 mL) until the TLC (1:1 and 7:3 EtOAc-hexane) showed the complete conversion of 1. After the completion of the reaction (1.5 h), the solvent was removed under reduced pressure. After column chromatographic purification (2:3 EtOAc-hexane), 67 mg (59%) of the title compound was obtained as a pale yellow syrup. Rf = 0.53 (1:1 EtOAc-hexane). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.05-7.83, 7.58-7.25, 7.07 (20H, m, Ar), 6.03 (1H, pt, J = 9.6, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.94-5.83 (2H, broad signal, NH), 5.75 (1H, pt, J = 9.8, 9.7 Hz, H-2′ or H-3′ or H-4′), 5.73 (1H, pt, J = 9.6, 9.6 Hz, H-2′ or H-3′ or H-4′), 4.68 (1H, dd, J = 12.3, < 1 Hz, H-6′a), 4.55 (1H, dd, J = 12.4, 5.4 Hz, H-6′b), 4.55 (1H, d, J = 9.7 Hz, H-1′), 4.27 (1H, ddd, J = 9.7, 5.4, 2.3 Hz, H-5′), 3.75 (3H, s, OCH3), 1.60 (3H, s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.7, 165.6, 165.2, 165.0 (5 × C=O), 158.3, 153.7 (2 × C=N), 133.6, 133.5, 133.4, 133.2, 129.8-128.3 (Ar), 76.8, 76.4, 73.6, 70.5, 69.2 (C-1′ − C-5′), 62.9 (C-6′), 52.3 (OCH3), 13.0 (CH3).
3-(3′,4′,6′-Tri-O-benzoyl-2′-deoxy-d-arabino-hex-1′-enopyranosyl)-1,2,4-triazin-5(4H)-one (8a)
Prepared from amidrazone 1 (0.10 g, 0.16 mmol) and ethyl glyoxalate (31 µL, 0.16 mmol, an 50% solution in toluene) according to general procedure 3. Reaction time: 3 d. Purified by column chromatography (7:3 EtOAc-hexane) to yield 50 mg (57%) of pale yellow amorphous solids. Rf = 0.24 (7:3 EtOAc-hexane); [α]D = −44 (c 0.26, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 14.05 (1H, br s, NH), 7.97-7.93, 7.67-7.64, 7.53-7.49 (16H, m, Ar, H-6), 6.60 (1H, d, J = 3.5 Hz, H-2′), 6.00 (1H, dd, J = 5.6, 3.5 Hz, H-3′), 5.87 (1H, pt, J = 7.6, 5.6 Hz, H-4′), 5.15 (1H, ddd, J = 7.6, 4.6, 3.3 Hz, H-5′), 4.82 (1H, dd, J = 12.4, 4.6 Hz, H-6′a), 4.74 (1H, dd, J = 12.4, 3.3 Hz, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 165.3, 164.9, 164.2 (3 × C=O), 161.8, 152.6, 144.6 (C-3, C-5, C-6), 145.4 (C-1′), 133.8, 133.6, 133.4, 129.3-128.6 (Ar), 104.0 (C-2′), 74.8, 67.8, 66.8 (C-3′ − C-5′), 61.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C30H23N3NaO8+ [M+Na]+ 576.1377. Found: 576.1377.
3-(3′,4′,6′-Tri-O-benzoyl-2′-deoxy-d-arabino-hex-1′-enopyranosyl)-6-methyl-1,2,4-triazin-5(4H)-one (8b)
Prepared from amidrazone 1 (0.10 g, 0.16 mmol) and methyl pyruvate (14 µL, 0.16 mmol) according to general procedure 3. Reaction time: 6 h. Purified by column chromatography (7:3 EtOAc-hexane) to yield 67 mg (75%) of pale yellow amorphous solids. Rf = 0.42 (7:3 EtOAc-hexane); [α]D = −29 (c 0.23, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 13.82 (1H, br s, NH), 7.96-7.95, 7.68-7.64, 7.53-7.49 (15H, m, Ar), 6.37 (1H, broad signal, H-2′), 5.98 (1H, broad signal, H-3′), 5.87 (1H, pt, J = 7.0, 6.0 Hz, H-4′), 5.14 (1H, broad signal, H-5′), 4.82 (1H, dd, J = 12.3, 4.6 Hz, H-6′a), 4.73 (1H, dd, J = 12.3, 2.5 Hz, H-6′b), 2.18 (3H, s, CH3); 13C NMR (90 MHz, DMSO-d6) δ (ppm): 165.2, 164.9, 164.4 (3 × C=O), 162.3, 153.2, 152.4 (C-3, C-5, C-6), 145.4 (C-1′), 133.7, 133.6, 133.3, 129.3-128.6 (Ar), 103.5 (C-2′), 74.7, 67.7, 66.8 (C-3′ − C-5′), 61.2 (C-6′), 17.1 (CH3). ESI-HRMS positive mode (m/z): calcd for C31H26N3O8+ [M+H]+ 568.1714; C31H25N3NaO8+ [M+Na]+ 590.1534. Found: [M+H]+ 568.1711; [M+Na]+ 590.1531.
3-(2′-Deoxy-2′-phthalimido-3′,4′,6′-tri-O-acetyl-β-d-glucopyranosyl)-1,2,4-triazin-5(4H)-one (9a)
Prepared from amidrazone 5 (0.10 g, 0.21 mmol) and ethyl glyoxylate (43 µL, 0.21 mmol) according to general procedure 3. Reaction time: 2 d. Purified by column chromatography (3:2 EtOAc-hexane) to yield 55 mg (51%) of pale yellow syrup. Rf = 0.25 (5:2 EtOAc-hexane); [α]D = +17 (c 0.21, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 11.73 (1H, s, NH), 7.90–7.72 (5H, m, Ar, H-6), 6.07 (1H, pt, J = 9.8, 9.8 Hz, H-2′ or H-3′ or H-4′), 5.35 (1H, d, J = 9.4 Hz, H-1′), 5.23 (1H, pt, J = 9.8, 9.6 Hz, H-2′ or H-3′ or H-4′), 4.54 (1H, pt, J = 10.5, 10.5 Hz, H-2′ or H-3′ or H-4′), 4.40 (1H, dd, J = 12.6, 5.3 Hz, H-6′a), 4.26 (1H, dd, J = 12.6, 1.7 Hz, H-6′b), 4.12 (1H, ddd, J = 10.0, 5.3, 1.7 Hz, H-5′), 2.13, 2.09, 1.90 (3 × 3H, 3 s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 171.1, 169.8, 169.6 (CH3-C=O), 167.8 (2 × Phth-C=O), 161.2, 159.2 (C-3, C-5), 144.3 (C-6), 134.5, 131.3, 123.9 (Ar), 76.2, 71.4, 70.5, 68.5 (C-1′, C-3′ − C-5′), 62.1 (C-6′), 52.5 (C-2′), 20.8, 20.6, 20.4 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C23H23N4O10+ [M+H]+ 515.1409; C23H22N4NaO10+ [M+Na]+ 537.1228. Found: [M+H]+ 515.1409; [M+Na]+ 537.1226.
3-(2′-Deoxy-2′-phthalimido-3′,4′,6′-tri-O-acetyl-β-d-glucopyranosyl)-6-methyl-1,2,4-triazin-5(4H)-one (9b)
Prepared from amidrazone 5 (0.10 g, 0.21 mmol) and methyl pyruvate (23 µL, 0.21 mmol) according to general procedure 3. Reaction time: 2 d. Purified by column chromatography (3:2 EtOAc-hexane) to yield 56 mg (50%) of pale yellow syrup. Rf = 0.25 (5:2 EtOAc-hexane); [α]D = +45 (c 0.21, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 11.39 (1H, s, NH), 7.86–7.72 (4H, m, Ar), 6.06 (1H, pt, J = 9.8, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.31 (1H, d, J = 10.5 Hz, H-1′), 5.23 (1H, pt, J = 9.8, 9.5 Hz, H-2′ or H-3′ or H-4′), 4.54 (1H, pt, J = 10.5, 9.8 Hz, H-2′ or H-3′ or H-4′), 4.39 (1H, dd, J = 12.1, 5.0 Hz, H-6′a), 4.27 (1H, dd, J = 12.3, < 1 Hz, H-6′b), 4.16-4-07 (1H, m, H-5′), 2.20 (3H, s, CH3), 2.14, 2.09, 1.89 (3 × 3H, 3 s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 171.0, 169.9, 169.6 (CH3-C=O), 167.9, 167.7 (2 × Phth-C=O), 161.9, 158.8, 153.8 (C-3, C-5, C-6), 134.4 (2), 131.4, 131.3, 123.8 (2) (Ar), 76.1, 71.3, 70.5, 68.5 (C-1′, C-3′ − C-5′), 62.1 (C-6′), 52.6 (C-2′), 20.8, 20.6, 20.4 (3 × CH3), 17.4 (CH3). ESI-HRMS positive mode (m/z): calcd for C24H25N4O10+ [M+H]+ 529.1565; C24H24N4NaO10+ [M+Na]+ 551.1385. Found: [M+H]+ 529.1569; [M+Na]+ 551.1386.
((6aS,7R,7aR)-1-(2′,3′,4′,6′-Tetra-O-benzoyl-β-d-glucopyranosyl)-6,6a,7,7a,8,9-hexahydro-5H-cyclopropa[5,6]cyclookta[1,2-c]pyridin-7-yl)methanol and ((6aR,7S,7aS)-1-(2′,3′,4′,6′-tetra-O-benzoyl-β-d-glucopyranosyl)-6,6a,7,7a,8,9-hexahydro-5H-cyclopropa[5,6]cyclookta[1,2-c]pyridin-7-yl)methanol (11a)
Prepared from triazine 2a (0.10 g, 0.15 mmol) and ((1R,8S,9r)-bicyclo[6.1.0]non-4-yn-9-yl)methanol (10, 46 mg, 0.30 mmol) in CH2Cl2 according to general procedure 4. Reaction time: 2 d. Purified by column chromatography (1:1 EtOAc-hexane) to give 115 mg (97%) of colourless syrup. Diastereomeric ratio: 5:4. Rf = 0.15 (1:1 EtOAc-hexane). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.29, 8.26 (2 × 1H, 2 d, J = 4.8 Hz in each, 2 × H-3), 8.02–7.21 (2 × 20H, m, Ar), 6.94, 6.92 (2 × 1H, 2 d, J = 4.8 Hz in each, 2 × H-4), 6.44 (2H, pt, J = 9.6, 9.5 Hz, 2 × (H-2′ or H-3′ or H-4′)), 6.11 (2H, pt, J = 9.5, 9.5 Hz, 2 × (H-2′ or H-3′ or H-4′)), 5.88, 5.87 (2 × 1H, 2 pt, J = 9.7, 9.6 Hz in each, 2 × (H-2′ or H-3′ or H-4′)), 5.27, 5.25 (2 × 1H, 2 d, J = 9.7 Hz in each, 2 × H-1′), 4.81, 4.71 (2 × 1H, 2 dd, J = 12.2, 2.1 Hz in each, 2 × H-6′a), 4.51, 4.46 (2 × 1H, 2 dd, J = 12.2, 5.1 Hz in each, 2 × H-6′b), 4.44–4.37 (2 × 1H, m, 2 × H-5′), 3.36–3.31, 3.18–2.87, 2.75–2.45, 2.38–2.33, 1.47–1.26, 0.66–0.52, 0.36–0.20 (2 × 14H, m, aliphatics, OH); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.2, 166.1, 166.0, 165.9, 165.2, 165.2, 164.5, 164.4 (2 × 4 × C=O), 152.4, 152.3, 151.2, 151.1 (2 × C-1, 2 × C-4a), 146.4 (2 × C-3), 137.6, 137.4 (2 × C-9a), 133.3-132.7, 130.0-128.0 (Ar), 125.9, 125.8 (2 × C-4), 79.0, 78.3, 76.8, 76.8, 75.1, 75.1, 71.3, 71.2, 69.9, 69.8 (2 × (C-1′ − C-5′)), 66.1, 66.1 (2 × C-6′), 63.5, 63.2 (2 × CH2OH), 33.3, 33.2, 30.0, 29.7, 29.2, 28.5, 28.3, 28.0, 25.8, 25.4, 21.4, 20.8, 20.4, 19.9 (2 × (C-5, C-6, C-6a, C-7, C-7a, C-8, C-9)). ESI-HRMS positive mode (m/z): calcd for C47H44NO10+ [M+H]+: 782.2960; C47H43NNaO10+ [M+Na]+: 804.2779. Found: [M+H]+: 782.2959; [M+Na]+: 804.2779.
((6aS,7R,7aR)-3-Methyl-1-(2′,3′,4′,6′-tetra-O-benzoyl-β-d-glucopyranosyl)-6,6a,7,7a,8,9-hexahydro-5H-cyclopropa[5,6]cyclookta[1,2-c]pyridin-7-yl)methanol and ((6aR,7S,7aS)-3-methyl-1-(2′,3′,4′,6′-tetra-O-benzoyl-β-d-glucopyranosyl)-6,6a,7,7a,8,9-hexahydro-5H-cyclopropa[5,6]cyclookta[1,2-c]pyridin-7-yl)methanol (11b)
Prepared from triazine 2a (0.05 g, 0.074 mmol) and ((1R,8S,9r)-bicyclo[6.1.0]non-4-yn-9-yl)methanol (10, 22 mg, 0.148 mmol) in CH2Cl2 according to general procedure 4. Reaction time: 6 d. Purified by column chromatography (4:5 EtOAc-hexane) to give 24 mg (41%) of colourless amorphous solids. Diastereomeric ratio: 5:4. Rf = 0.28 (1:1 EtOAc-hexane). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.05-7.23 (2 × 20H, m, Ar), 6.77, 6.76 (2 × 1H, 2 s, 2 × H-4), 6.47, 6.43 (2 × 1H, 2 pt, J = 9.7, 9.6 Hz in each, 2 × (H-2′ or H-3′ or H-4′)), 6.07 (2H, pt, J = 9.6, 9.6 Hz, 2 × (H-2′ or H-3′ or H-4′)), 5.86, 5.84 (2 × 1H, 2 pt, J = 9.7, 9.6 Hz in each, 2 × (H-2′ or H-3′ or H-4′)), 5.17, 5.16 (2 × 1H, 2 d, J = 9.7 Hz in each, 2 × H-1′), 4.88, 4.70 (2 × 1H, 2 dd, J = 12.1, 2.8 Hz in each, 2 × H-6′a), 4.49 (1H, dd, J = 12.1, 5.3 Hz, H-6′b), 4.42-4.33 (3H, m, H-6′b, 2 × H-5′), 3.50-2.32 (m, aliphatics), 2.30, 2.26 (2 × 3H, 2 s, 2 × CH3), 1.40-0.31 (m, aliphatics, OH); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.3, 166.2, 166.1, 166.0, 165.3, 165.3, 164.7, 164.6 (2 × 4 × C=O), 154.8, 154.6, 152.4, 152.3, 150.4, 150.3 (2 × C-1, 2 × C-4a, 2 × C-3), 134.4, 134.3 (2 × C-9a), 133.3-132.6, 130.1-128.0 (Ar), 125.4, 125.3 (2 × C-4), 79.6, 78.8, 76.9, 76.9, 75.3, 75.2, 71.1, 70.8, 70.0, 69.8 (2 × (C-1′ − C-5′)), 66.3, 65.7 (2 × C-6′), 63.6, 63.0 (2 × CH2OH), 33.1, 33.0, 30.0, 29.9, 28.8, 28.2, 28.1, 26.8, 25.5, 25.3, 23.5, 23.4, 21.6, 20.8, 20.6, 20.4 (2 × (C-5, C-6, C-6a, C-7, C-7a, C-8, C-9), 2 × CH3). ESI-HRMS positive mode (m/z): calcd for C48H45NNaO10+ [M+Na]+: 818.2936. Found: 818.2935.
((6aS,7R,7aR)-3-Phenyl-1-(2′,3′,4′,6′-tetra-O-benzoyl-β-d-glucopyranosyl)-6,6a,7,7a,8,9-hexahydro-5H-cyclopropa[5,6]cyclookta[1,2-c]pyridin-7-yl)methanol and ((6aR,7S,7aS)-3-phenyl-1-(2′,3′,4′,6′-tetra-O-benzoyl-β-d-glucopyranosyl)-6,6a,7,7a,8,9-hexahydro-5H-cyclopropa[5,6]cyclookta[1,2-c]pyridin-7-yl)methanol (11d)
Prepared from triazine 2d (0.05 g, 0.068 mmol) and ((1R,8S,9r)-bicyclo[6.1.0]non-4-yn-9-yl)methanol (10, 20 mg, 0.136 mmol) in CH2Cl2 according to general procedure 4. Reaction time: 6 d. Purified by column chromatography (1:1 EtOAc-hexane) to give 43 mg (73%) of colourless amorphous solids. Diastereomeric ratio: 5:4. Rf = 0.28 (1:1 EtOAc-hexane). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.04-7.15 (2 × 26H, m, Ar, 2 × H-4), 6.69, 6.65 (2 × 1H, 2 pt, J = 9.7, 9.7 Hz in each, 2 × (H-2′ or H-3′ or H-4′)), 6.12 (2H, pt, J = 9.6, 9.5 Hz, 2 × (H-2′ or H-3′ or H-4′)), 5.89, 5.86 (2 × 1H, 2 pt, J = 9.8, 9.6 Hz in each, 2 × (H-2′ or H-3′ or H-4′)), 5.31, 5.29 (2 × 1H, 2 d, J = 9.8 Hz in each, 2 × H-1′), 4.90, 4.67 (2 × 1H, 2 dd, J = 12.1, 2.9 Hz in each, 2 × H-6′a), 4.50 (1H, dd, J = 12.1, 5.3 Hz, H-6′b), 4.42–4.36 (3 × 1H, m, H-6′b, 2 × H-5′), 3.43–3.38, 3.20–2.52, 2.46–2.34, 1.52–1.36, 0.74–0.34 (2 × 13H, m, aliphatics); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.3, 166.3, 166.2, 166.1, 165.3, 165.2, 164.8, 164.6 (2 × 4 × C=O), 154.0, 153.6, 153.0, 153.0, 150.9, 150.8 (2 × C-1, 2 × C-3, 2 × C-4a), 138.9, 138.7, 136.4, 136.1 (2 × C-9a, 2 × Ar-Cq), 133.4-132.5, 129.9-126.8 (Ar), 122.6, 122.3 (2 × C-4), 79.0, 78.4, 76.9, 76.9, 75.6, 75.4, 70.8, 70.5, 69.9, 69.6 (2 × (C-1′ − C-5′)), 66.2, 66.2 (2 × C-6′), 63.7, 62.9 (2 × CH2OH), 33.5, 30.0, 29.9, 29.1, 28.8, 28.2, 28.0, 25.6, 25.4, 20.5, 20.5, 20.4, 20.3 (2 × (C-5, C-6, C-6a, C-7, C-7a, C-8, C-9)). ESI-HRMS positive mode (m/z): calcd for C53H48NO10+ [M+H]+: 858.3273; C53H47NNaO10+ [M+Na]+: 880.3092. Found: [M+H]+: 858.3272; [M+Na]+: 880.3093.
((6aS,7R,7aR)-1-(β-d-glucopyranosyl)-3-phenyl-6,6a,7,7a,8,9-hexahydro-5H-cyclopropa[5,6]cyclookta[1,2-c]pyridin-7-yl)methanol and ((6aR,7S,7aS)-1-(β-d-glucopyranosyl)-3-phenyl-6,6a,7,7a,8,9-hexahydro-5H-cyclopropa[5,6]cyclookta[1,2-c]pyridin-7-yl)methanol (12d)
Prepared from triazine
3d (20 mg, 0.063 mmol) and ((1
R,8
S,9
r)-bicyclo[6.1.0]non-4-yn-9-yl)methanol (
10, 19 mg, 0.126 mmol) in MeOH according to general procedure 4. Reaction time: 5 d. Purified by column chromatography (8:1 CHCl
3-MeOH) to give 26 mg (94%) of pale yellow amorphous solids. Diastereomeric ratio: 1:1. R
f = 0.21 (8:1 CHCl
3-MeOH).
1H NMR (360 MHz, CD
3OD) δ (ppm): 8.02–8.01 (2 × 2H, Ph), 7.58 (2H, s, 2 × H-4), 7.47–7.36 (2 × 3H, m, Ph), 4.65, 4.63 (2 × 1H, 2 d,
J = 9.3 Hz in each, H-1′), 4.34, 4.33 (2 × 1H, 2 pt,
J = 9.1, 9.1 Hz in each, 2 × (H-2′ or H-3′ or H-4′)), 3.87–3.51 (2 × 4H, m, 2 × (H-2′ and/or H-3′ and/or H-4′, H-6′a, H6′b), 4.46–3.30 (2H, m, 2 × H-5′), 3.30–3.22, 3.14–3.05, 2.99–2.89, 2.65–2.44, 1.51–1.39, 0.72–0.61 (2 × 13H, m, aliphatics);
13C NMR (90 MHz, CD
3OD) δ (ppm): 155.5, 155.3, 155.3, 155.2, 155.2, 155.1 (2 × C-1, 2 × C-3, 2 × C-4a), 140.8, 140.7, 138.5, 138.5 (2 × C-9a, 2 × Ph-C
q), 129.8, 129.7, 129.6, 129.6, 128.0 (Ph), 123.3, 123.2 (2 × C-4), 82.4, 82.4, 79.7, 79.7, 79.6, 73.8, 73.7, 71.6, 71.5 (2 × (C-1′ − C-5′)), 66.6, 66.5 (2 × C-6′), 62.9, 62.8 (2 × CH
2OH), 35.0, 34.8, 31.1, 31.0, 30.3, 30.1, 29.9, 29.8, 26.6, 26.5, 23.0, 22.9, 22.6, 22.5 (2 × (C-5, C-6, C-6a, C-7, C-7a, C-8, C-9)). ESI-HRMS positive mode (m/z): calcd for C
25H
31NNaO
6+ [M+Na]
+: 464.2044. Found: 464.2042 (
Supplementary Materials).



 ,
, 


{kind=link}
{kind=link}











