

Identification of Dihydrolipoamide Dehydrogenase as Potential Target of Vemurafenib-Resistant Melanoma Cells

 , , , , and
, , , , and
Abstract
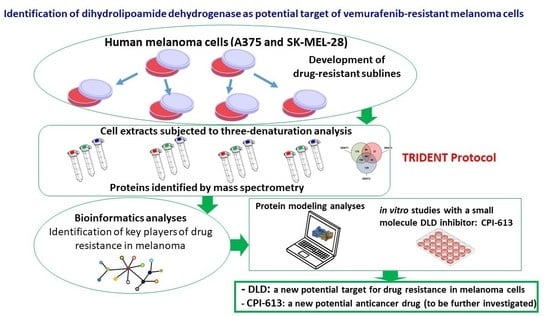
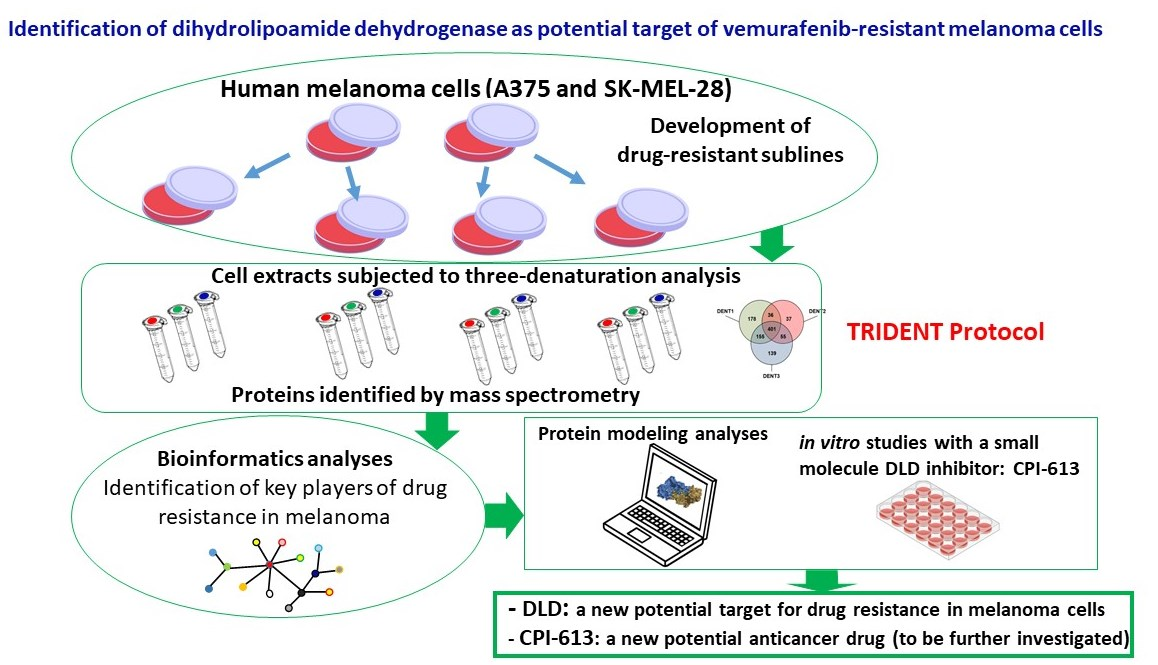{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
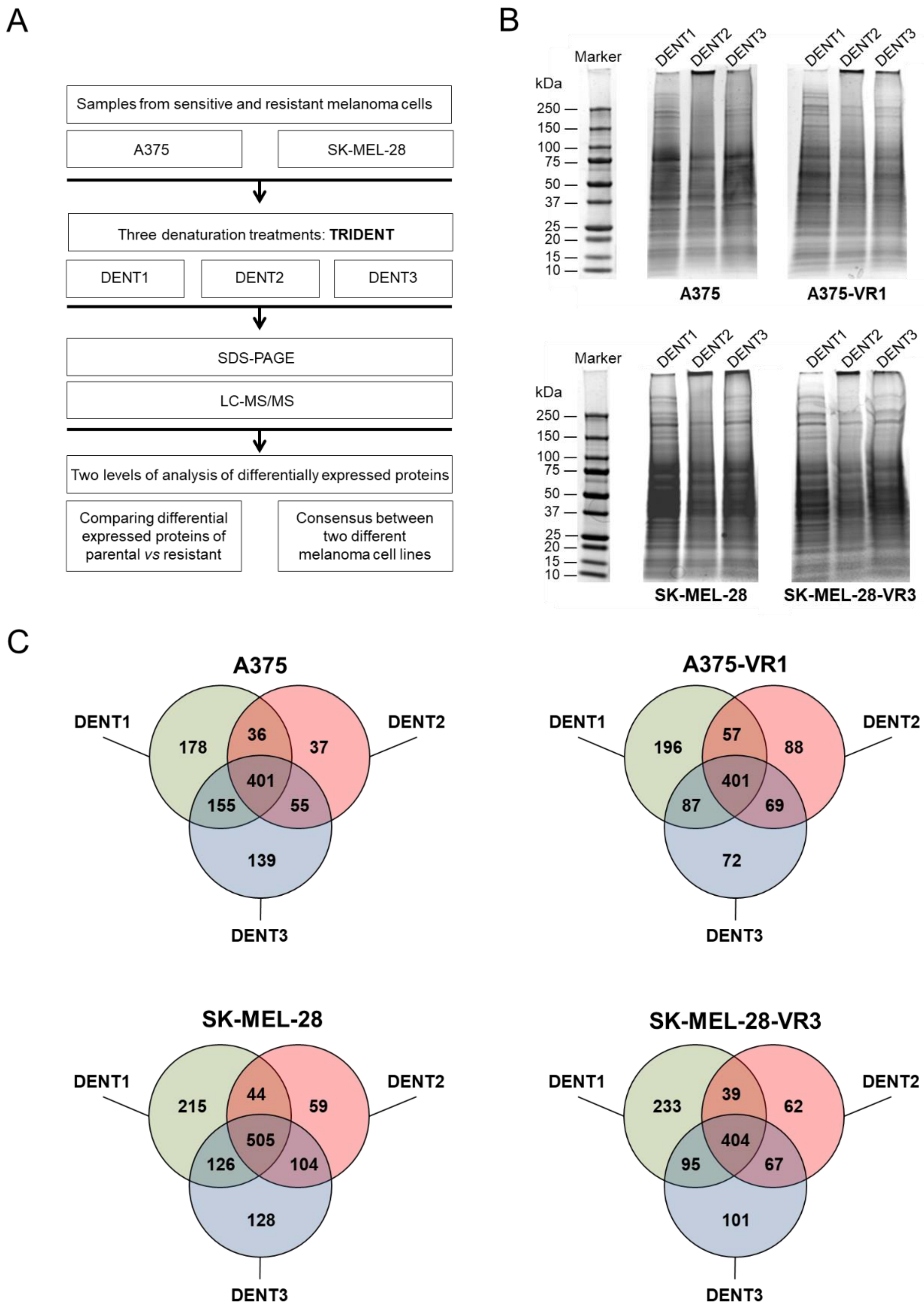
2.1. Comparative Evaluation of Proteomes Expressed by Vemurafenib-Sensitive and -Resistant Cells
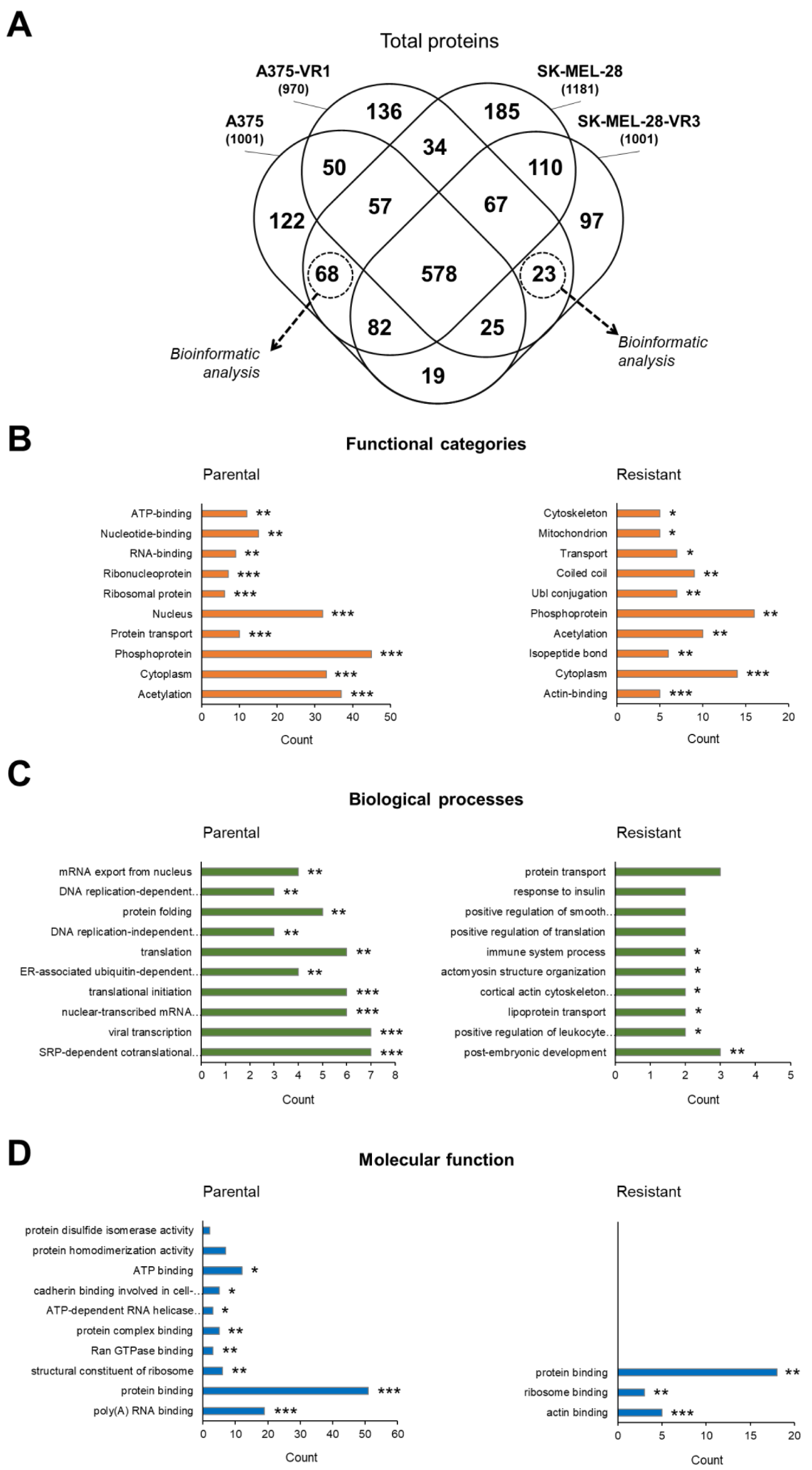
2.2. Bioinformatic Analysis of Total Differentially Expressed Proteins
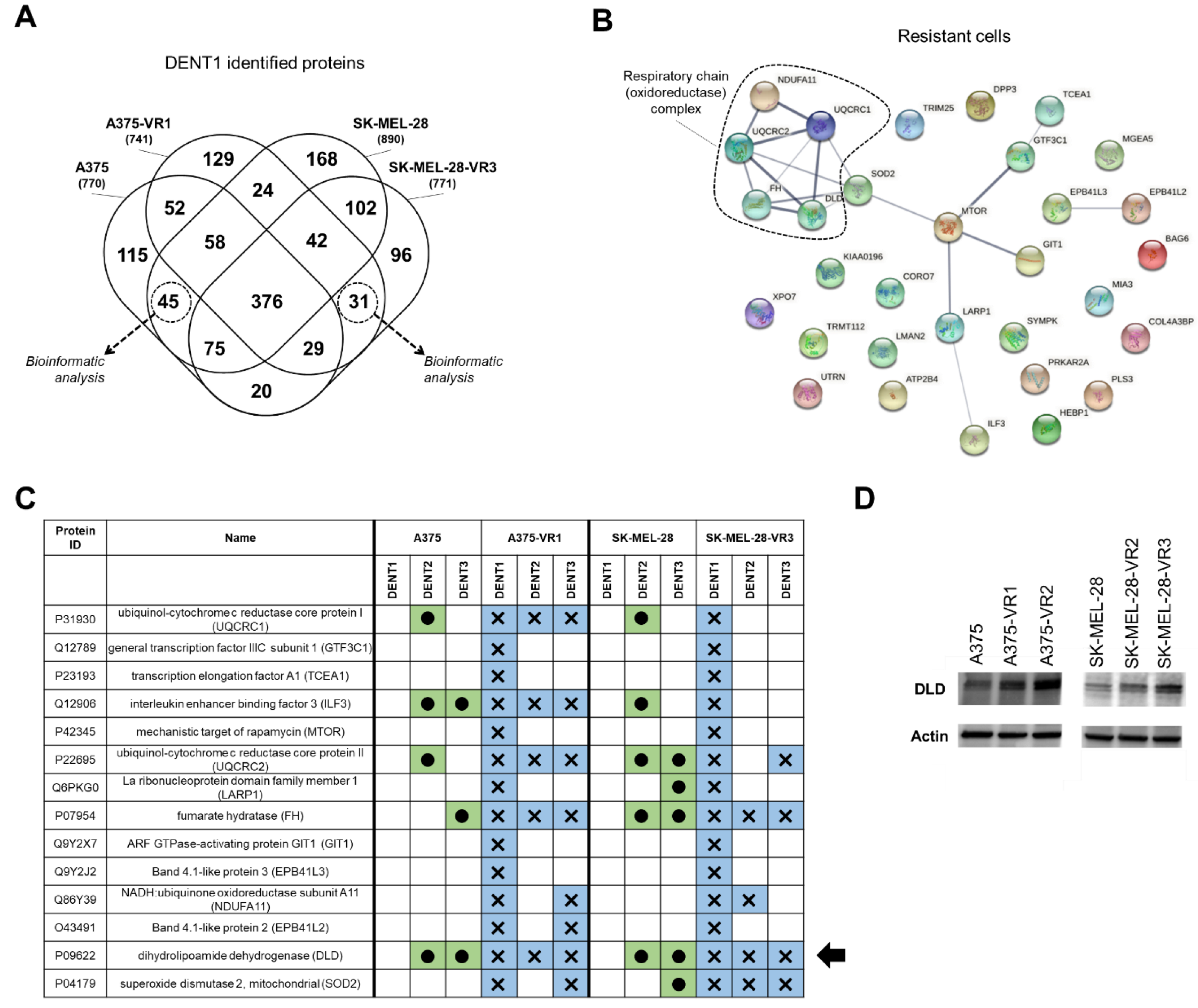
2.3. Bioinformatic Analysis of DENT1 Differentially Expressed Proteins
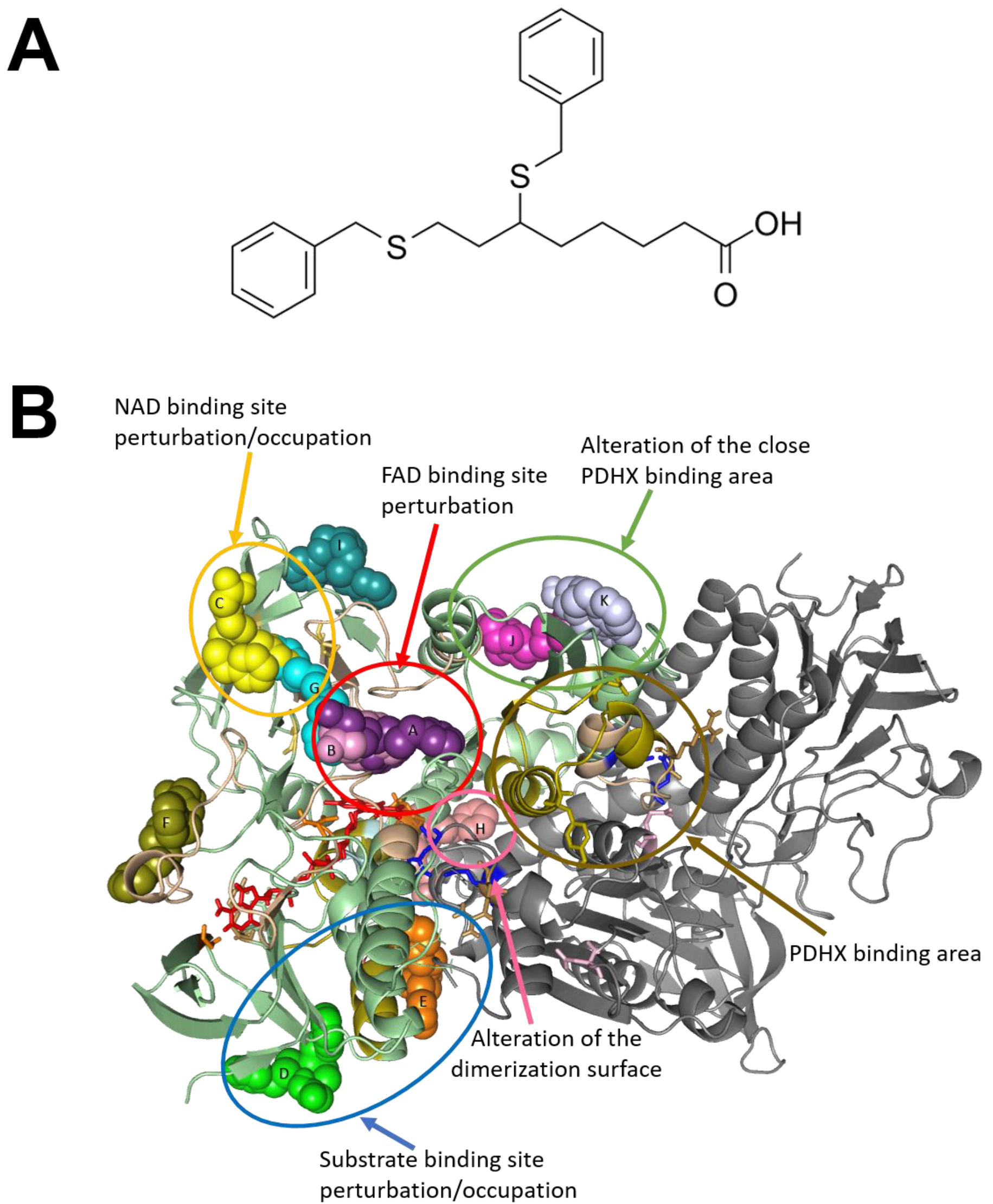
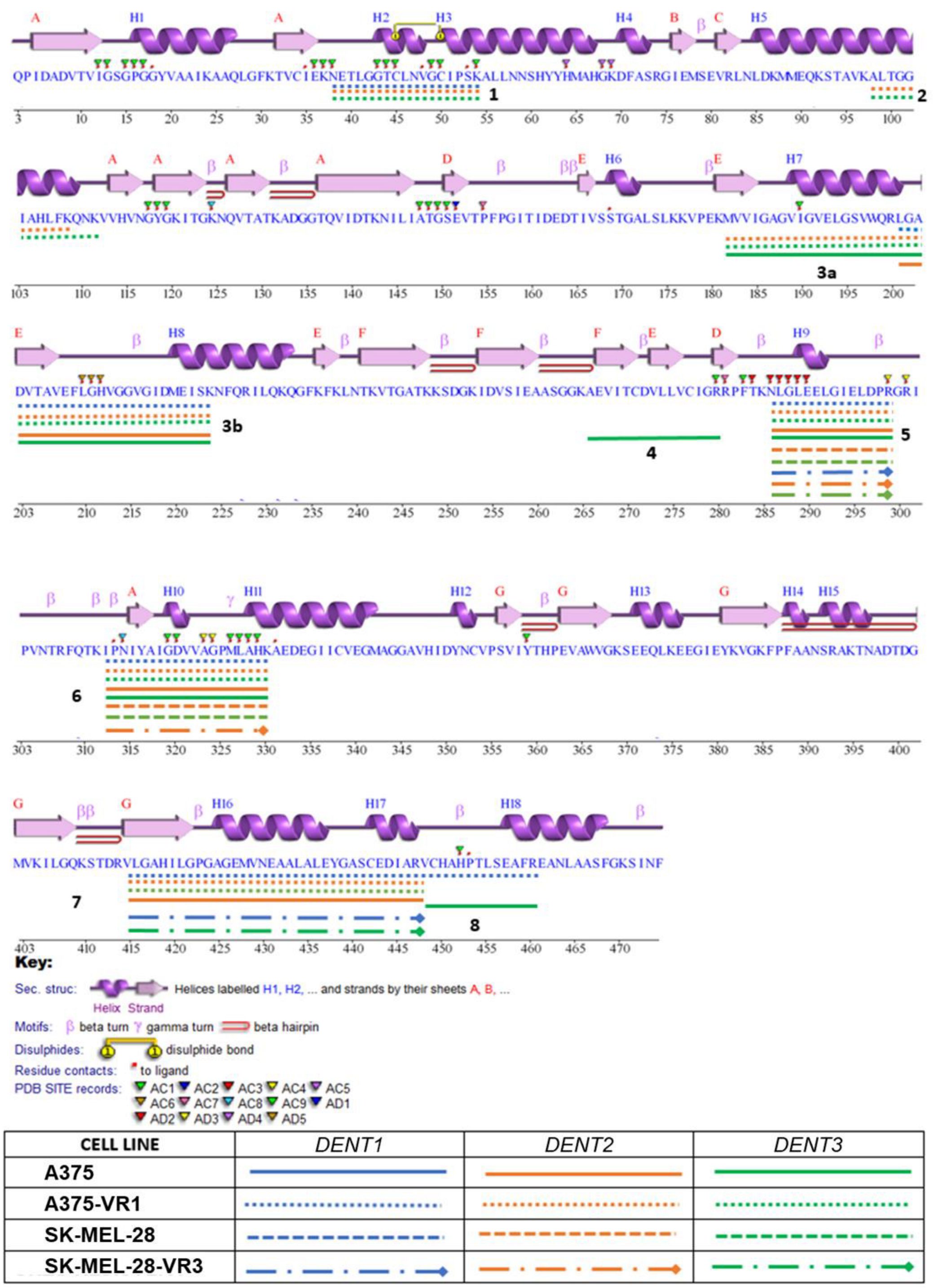
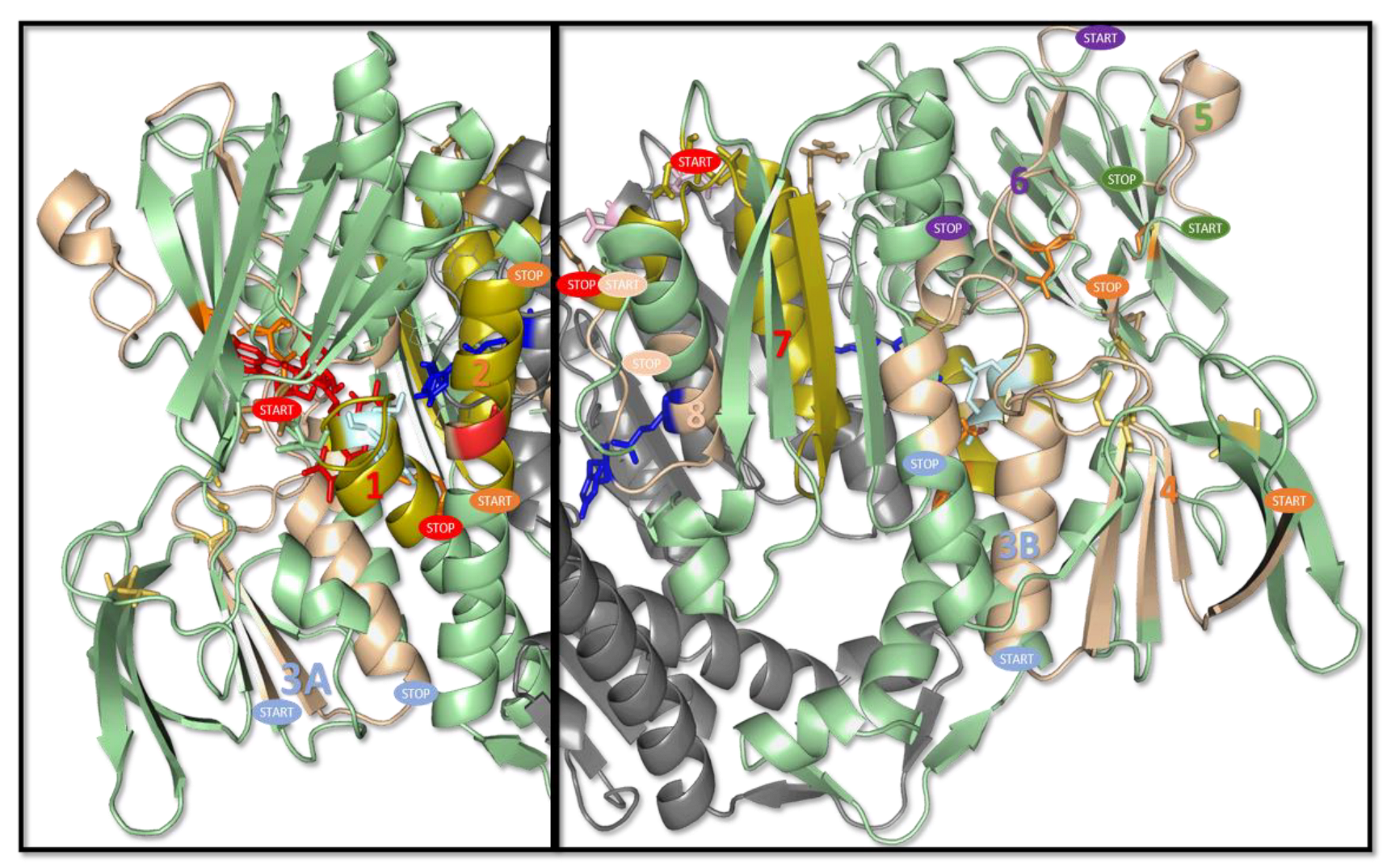
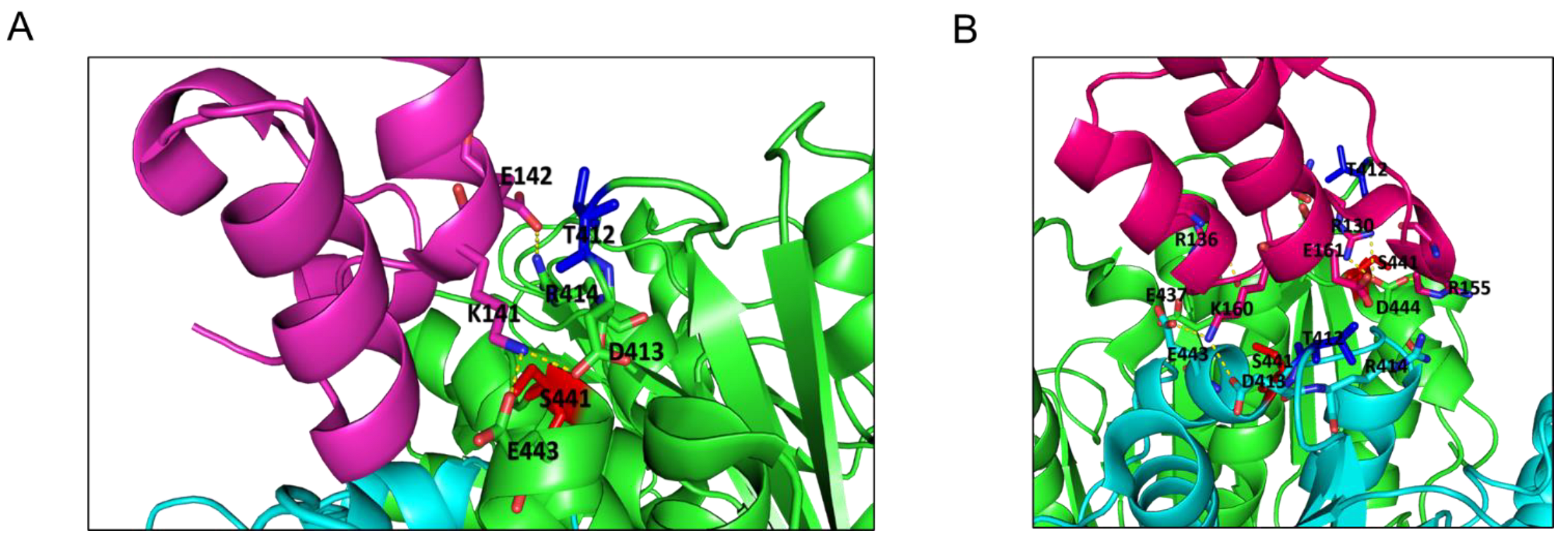
2.4. Structural Profiling of Dihydrolipoamide Dehydrogenase
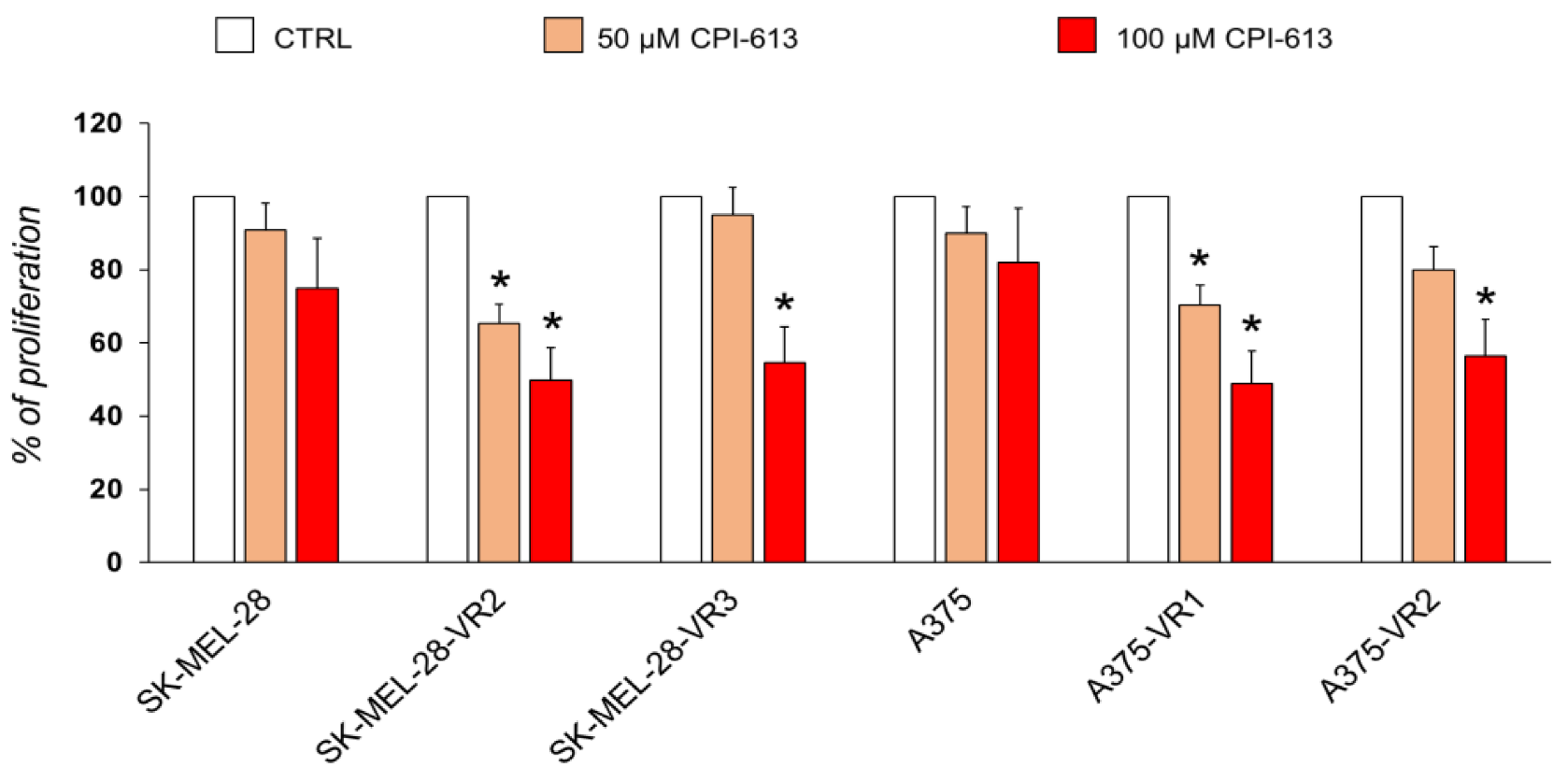
2.5. Targeting Vemurafenib-Resistant Cells with DLD Inhibitor
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture and Generation of Vemurafenib-Resistant Sublines
4.3. Mass Spectrometry Analysis and Protein Identification
4.4. Bioinformatics Analyses
4.5. Immunoblotting
4.6. In Silico 3D Structure Analyses
4.7. Cell Proliferation and Cell Cycle Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Azoury, S.C.; Lange, J.R. Epidemiology, risk factors, prevention, and early detection of melanoma. Surg. Clin. North. Am. 2014, 94, 945–962. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, R.R.; Postow, M.A. Combinatorial Approaches to the Treatment of Advanced Melanoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 145–158. [Google Scholar] [CrossRef]
- Sood, S.; Jayachandiran, R.; Pandey, S. Current Advancements and Novel Strategies in the Treatment of Metastatic Melanoma. Integr. Cancer Ther. 2021, 20, 1534735421990078. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [PubMed]
- Bruce, W.J.; Koljonen, J.L.; Romanelli, M.R.; Khan, A.U.; Neumeister, M.W. Adjuvant and Neoadjuvant Therapeutics for the Treatment of Cutaneous Melanoma. Clin. Plast. Surg. 2021, 48, 651–658. [Google Scholar] [CrossRef]
- Bruno, W.; Martinuzzi, C.; Andreotti, V.; Pastorino, L.; Spagnolo, F.; Dalmasso, B.; Cabiddu, F.; Gualco, M.; Ballestrero, A.; Bianchi-Scarrà, G.; et al. Italian Melanoma Intergroup (IMI). Heterogeneity and frequency of BRAF mutations in primary melanoma: Comparison between molecular methods and immunohistochemistry. Oncotarget 2017, 8, 8069–8082. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Greaves, W.O.; Verma, S.; Patel, K.P.; Davies, M.A.; Barkoh, B.A.; Galbincea, J.M.; Yao, H.; Lazar, A.J.; Aldape, K.D.; Medeiros, L.J.; et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J. Mol. Diagn. 2013, 15, 220–226. [Google Scholar] [CrossRef]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco. Targets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Eckburg, A.; Gantiwala, S.; Hart, Z.; Dein, J.; Lam, K.; Puri, N. Resistance to Molecularly Targeted Therapies in Melanoma. Cancers 2021, 13, 1115. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Roberto, M.; Panebianco, M.; Botticelli, A.; Mazzuca, F.; Marchetti, P. Drug resistance of BRAF-mutant melanoma: Review of up-to-date mechanisms of action and promising targeted agents. Eur. J. Pharmacol. 2019, 862, 172621. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, C.; Jiménez-Cervantes, C.; Sánchez-Laorden, B.; García-Borrón, J.C. Functional interplay between secreted ligands and receptors in melanoma. Semin. Cell Dev. Biol. 2018, 78, 73–84. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, C.; Cordella, M.; Mariotti, S.; Rossi, S.; Senatore, C.; Lintas, C.; Levati, L.; D’Arcangelo, D.; Facchiano, A.; D’Atri, S.; et al. Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals. Biomedicines 2021, 9, 79. [Google Scholar] [CrossRef]
- Indini, A.; Grossi, F.; Mandalà, M.; Taverna, D.; Audrito, V. Metabolic Interplay between the Immune System and Melanoma Cells: Therapeutic Implications. Biomedicines 2021, 9, 607. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Invest. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef]
- Kumar, P.R.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A.; Moncrieff, M.D. Mitochondrial oxidative phosphorylation in cutaneous melanoma. Br. J. Cancer. 2021, 124, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Murtas, D.; Tomei, S. Cutaneous Melanoma Classification: The Importance of High-Throughput Genomic Technologies. Front. Oncol. 2021, 11, 635488. [Google Scholar] [CrossRef] [PubMed]
- Paulitschke, V.; Haudek-Prinz, V.; Griss, J.; Berger, W.; Mohr, T.; Pehamberger, H.; Kunstfeld, R.; Gerner, C. Functional Classification of Cellular Proteome Profiles Support the Identification of Drug Resistance Signatures in Melanoma Cells. J. Proteome Res. 2013, 12, 3264–3276. [Google Scholar] [CrossRef] [PubMed]
- Paulitschke, V.; Eichhoff, O.; Gerner, C.; Paulitschke, P.; Bileck, A.; Mohr, T.; Cheng, P.F.; Leitner, A.; Guenova, E.; Saulite, I.; et al. Proteomic identification of a marker signature for MAPKi resistance in melanoma. EMBO J. 2019, 38, e95874. [Google Scholar] [CrossRef]
- Song, H.; Liu, D.; Dong, S.; Zeng, L.; Wu, Z.; Zhao, P.; Zhang, L.; Chen, Z.S.; Zou, C. Epitranscriptomics and epiproteomics in cancer drug resistance: Therapeutic implications. Signal Transduct. Target Ther. 2020, 5, 193. [Google Scholar] [CrossRef]
- Tikhonov, D.; Kulikova, L.; Kopylov, A.T.; Rudnev, V.; Stepanov, A.; Malsagova, K.; Izotov, A.; Kulikov, D.; Zulkarnaev, A.; Enikeev, D.; et al. Proteomic and molecular dynamic investigations of PTM-induced structural fluctuations in breast and ovarian cancer. Sci. Rep. 2021, 11, 19318. [Google Scholar] [CrossRef]
- Verdoliva, V.; Senatore, C.; Polci, M.L.; Rossi, S.; Cordella, M.; Carlucci, G.; Marchetti, P.; Antonini-Cappellini, G.; Facchiano, A.; D’Arcangelo, D.; et al. Differential denaturation of serum proteome reveals a significant amount of hidden information in complex mixtures of proteins. PLoS ONE 2013, 8, e57104. [Google Scholar] [CrossRef] [PubMed]
- Fleminger, G.; Dayan, A. The moonlighting activities of dihydrolipoamide dehydrogenase: Biotechnological and biomedical applications. J. Mol. Recognit. 2021, 34, e2924. [Google Scholar] [CrossRef]
- Rossi, S.; Cordella, M.; Tabolacci, C.; Nassa, G.; D’Arcangelo, D.; Senatore, C.; Pagnotto, P.; Magliozzi, R.; Salvati, A.; Weisz, A.; et al. TNF-alpha and metalloproteases as key players in melanoma cells aggressiveness. J. Exp. Clin. Cancer Res. 2018, 37, 326. [Google Scholar] [CrossRef]
- Atefi, M.; von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS ONE 2011, 6, e28973. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Zhang, G.; Kwong, L.; Lu, H.; Tan, J.; Sadek, N.; Xiao, M.; Zhang, J.; Labrie, M.; et al. Targeting mTOR signaling overcomes acquired resistance to combined BRAF and MEK inhibition in BRAF-mutant melanoma. Oncogene 2021, 40, 5590–5599. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Szabo, E.; Wilk, P.; Nagy, B.; Zambo, Z.; Bui, D.; Weichsel, A.; Arjunan, P.; Torocsik, B.; Hubert, A.; Furey, W.; et al. Underlying molecular alterations in human dihydrolipoamide dehydrogenase deficiency revealed by structural analyses of disease-causing enzyme variants. Hum. Mol. Genet. 2019, 28, 3339–3354. [Google Scholar] [CrossRef] [PubMed]
- Cordella, M.; Tabolacci, C.; Senatore, C.; Rossi, S.; Mueller, S.; Lintas, C.; Eramo, A.; D’Arcangelo, D.; Valitutti, S.; Facchiano, A.; et al. Theophylline induces differentiation and modulates cytoskeleton dynamics and cytokines secretion in human melanoma-initiating cells. Life Sci. 2019, 230, 121–131. [Google Scholar] [CrossRef]
- Keil, B. Specificity of Proteolysis; Springer-Verlag Berlin and Heidelberg GmbH & Co. KG: Berlin, Germany, 1992; p. 335. [Google Scholar]
- Ferrucci, P.F.; Lens, M.; Cocorocchio, E. Combined BRAF-Targeted Therapy with Immunotherapy in BRAF-Mutated Advanced Melanoma Patients. Curr. Oncol. Rep. 2021, 23, 138. [Google Scholar] [CrossRef]
- Smalley, K.S.; Eroglu, Z.; Sondak, V.K. Combination Therapies for Melanoma: A New Standard of Care? Am. J. Clin. Dermatol. 2016, 17, 99–105. [Google Scholar] [CrossRef]
- Dumaz, N.; Lebbé, C. New perspectives on targeting RAF, MEK and ERK in melanoma. Curr. Opin. Oncol. 2021, 33, 120–126. [Google Scholar] [CrossRef]
- Okamoto, H.; Yoshimatsu, Y.; Tomizawa, T.; Kunita, A.; Takayama, R.; Morikawa, T.; Komura, D.; Takahashi, K.; Oshima, T.; Sato, M.; et al. Interleukin-13 receptor α2 is a novel marker and potential therapeutic target for human melanoma. Sci. Rep. 2019, 9, 1281. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Jóźwicki, W.; Roszkowski, K.; Filipiak, J.; Slominski, A.T. Melanin content in melanoma metastases affects the outcome of radiotherapy. Oncotarget 2016, 7, 17844–17853. [Google Scholar] [CrossRef]
- Sarna, M.; Krzykawska-Serda, M.; Jakubowska, M.; Zadlo, A.; Urbanska, K. Melanin presence inhibits melanoma cell spread in mice in a unique mechanical fashion. Sci. Rep. 2019, 9, 9280. [Google Scholar] [CrossRef]
- Slominski, R.M.; Sarna, T.; Płonka, P.M.; Raman, C.; Brożyna, A.A.; Slominski, A.T. Melanoma, Melanin, and Melanogenesis: The Yin and Yang Relationship. Front. Oncol. 2022, 12, 842496. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.A.; Annis, M.G.; Frederick, D.T.; Biondini, M.; Dong, Z.; Kwong, L.; Chin, L.; Keler, T.; Hawthorne, T.; Watson, I.R.; et al. MAPK Pathway Inhibitors Sensitize BRAF-Mutant Melanoma to an Antibody-Drug Conjugate Targeting GPNMB. Clin. Cancer Res. 2016, 22, 6088–6098. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Delgado-Goni, T.; Casals Galobart, T.; Wantuch, S.; Jamin, Y.; Leach, M.O.; Robinson, S.P.; Bamber, J.; Beloueche-Babari, M. Detecting human melanoma cell re-differentiation following BRAF or heat shock protein 90 inhibition using photoacoustic and magnetic resonance imaging. Sci. Rep. 2017, 7, 8215. [Google Scholar] [CrossRef] [PubMed]
- Yumnam, S.; Kang, M.C.; Oh, S.H.; Kwon, H.C.; Kim, J.C.; Jung, E.S.; Lee, C.H.; Lee, A.Y.; Hwang, J.I.; Kim, S.Y. Downregulation of dihydrolipoyl dehydrogenase by UVA suppresses melanoma progression via triggering oxidative stress and altering energy metabolism. Free Radic. Biol. Med. 2021, 162, 77–87. [Google Scholar] [CrossRef]
- Alkaraki, A.; McArthur, G.A.; Sheppard, K.E.; Smith, L.K. Metabolic Plasticity in Melanoma Progression and Response to Oncogene Targeted Therapies. Cancers 2021, 13, 5810. [Google Scholar] [CrossRef]
- Ghoneum, A.; Abdulfattah, A.Y.; Warren, B.O.; Shu, J.; Said, N. Redox Homeostasis and Metabolism in Cancer: A Complex Mechanism and Potential Targeted Therapeutics. Int. J. Mol. Sci. 2020, 21, 3100. [Google Scholar] [CrossRef]
- Neitzel, C.; Demuth, P.; Wittmann, S.; Fahrer, J. Targeting Altered Energy Metabolism in Colorectal Cancer: Oncogenic Reprogramming, the Central Role of the TCA Cycle and Therapeutic Opportunities. Cancers 2020, 12, 1731. [Google Scholar] [CrossRef]
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2014, 20, 5255–5264. [Google Scholar] [CrossRef]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef]
- Pardee, T.S.; Luther, S.; Buyse, M.; Powell, B.L.; Cortes, J. Devimistat in combination with high dose cytarabine and mitoxantrone compared with high dose cytarabine and mitoxantrone in older patients with relapsed/refractory acute myeloid leukemia: ARMADA 2000 Phase III study. Future Oncol. 2019, 15, 3197–3208. [Google Scholar] [CrossRef]
- Anderson, R.; Miller, L.D.; Isom, S.; Chou, J.W.; Pladna, K.M.; Schramm, N.J.; Ellis, L.R.; Howard, D.S.; Bhave, R.R.; Manuel, M.; et al. Phase II trial of cytarabine and mitoxantrone with devimistat in acute myeloid leukemia. Nat. Commun. 2022, 13, 1673. [Google Scholar] [CrossRef] [PubMed]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R., Jr.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Pal, H.C.; Sharma, S.; Strickland, L.R.; Katiyar, S.K.; Ballestas, M.E.; Athar, M.; Elmets, C.A.; Afaq, F. Fisetin inhibits human melanoma cell invasion through promotion of mesenchymal to epithelial transition and by targeting MAPK and NFκB signaling pathways. PLoS ONE 2014, 9, e86338. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Jin, L.; Jiang, C.C.; Long, G.V.; Scolyer, R.A.; Wu, Q.; Zhang, X.D.; Mei, Y.; Wu, M. AEBP1 upregulation confers acquired resistance to BRAF (V600E) inhibition in melanoma. Cell Death Dis. 2013, 4, e914. [Google Scholar] [CrossRef]
- Houghton, P.; Fang, R.; Techatanawat, I.; Steventon, G.; Hylands, P.J.; Lee, C.C. The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity. Methods 2007, 42, 377–387. [Google Scholar] [CrossRef]
- Graziani, G.; Artuso, S.; De Luca, A.; Muzi, A.; Rotili, D.; Scimeca, M.; Atzori, M.G.; Ceci, C.; Mai, A.; Leonetti, C.; et al. A new water soluble MAPK activator exerts antitumor activity in melanoma cells resistant to the BRAF inhibitor vemurafenib. Biochem. Pharmacol. 2015, 95, 16–27. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioin-formatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, C.A.; Wynn, R.M.; Chuang, J.L.; Naik, M.T.; Young, B.B.; Huang, T.H.; Chuang, D.T. Structural and thermodynamic basis for weak interactions between dihydrolipoamide dehydrogenase and subunit-binding domain of the branched-chain alpha-ketoacid dehydrogenase complex. J. Biol. Chem. 2011, 286, 23476–23488. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, C.A.; Wynn, R.M.; Chuang, J.L.; Machius, M.; Tomchick, D.R.; Chuang, D.T. Structural insight into interactions between dihydrolipoamide dehydrogenase (E3) and E3 binding protein of human pyruvate dehydrogenase complex. Structure 2006, 14, 611–621. [Google Scholar] [CrossRef]
- Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.M.; Journe, F. Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers 2020, 12, 1323. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabolacci, C.; Giordano, D.; Rossi, S.; Cordella, M.; D’Arcangelo, D.; Moschella, F.; D’Atri, S.; Biffoni, M.; Facchiano, A.; Facchiano, F. Identification of Dihydrolipoamide Dehydrogenase as Potential Target of Vemurafenib-Resistant Melanoma Cells. Molecules 2022, 27, 7800. https://doi.org/10.3390/molecules27227800
Tabolacci C, Giordano D, Rossi S, Cordella M, D’Arcangelo D, Moschella F, D’Atri S, Biffoni M, Facchiano A, Facchiano F. Identification of Dihydrolipoamide Dehydrogenase as Potential Target of Vemurafenib-Resistant Melanoma Cells. Molecules. 2022; 27(22):7800. https://doi.org/10.3390/molecules27227800
Chicago/Turabian StyleTabolacci, Claudio, Deborah Giordano, Stefania Rossi, Martina Cordella, Daniela D’Arcangelo, Federica Moschella, Stefania D’Atri, Mauro Biffoni, Angelo Facchiano, and Francesco Facchiano. 2022. "Identification of Dihydrolipoamide Dehydrogenase as Potential Target of Vemurafenib-Resistant Melanoma Cells" Molecules 27, no. 22: 7800. https://doi.org/10.3390/molecules27227800
APA StyleTabolacci, C., Giordano, D., Rossi, S., Cordella, M., D’Arcangelo, D., Moschella, F., D’Atri, S., Biffoni, M., Facchiano, A., & Facchiano, F. (2022). Identification of Dihydrolipoamide Dehydrogenase as Potential Target of Vemurafenib-Resistant Melanoma Cells. Molecules, 27(22), 7800. https://doi.org/10.3390/molecules27227800