
Advances in the Synthesis of Ring-Fused Benzimidazoles and Imidazobenzimidazoles



Abstract
1. Introduction
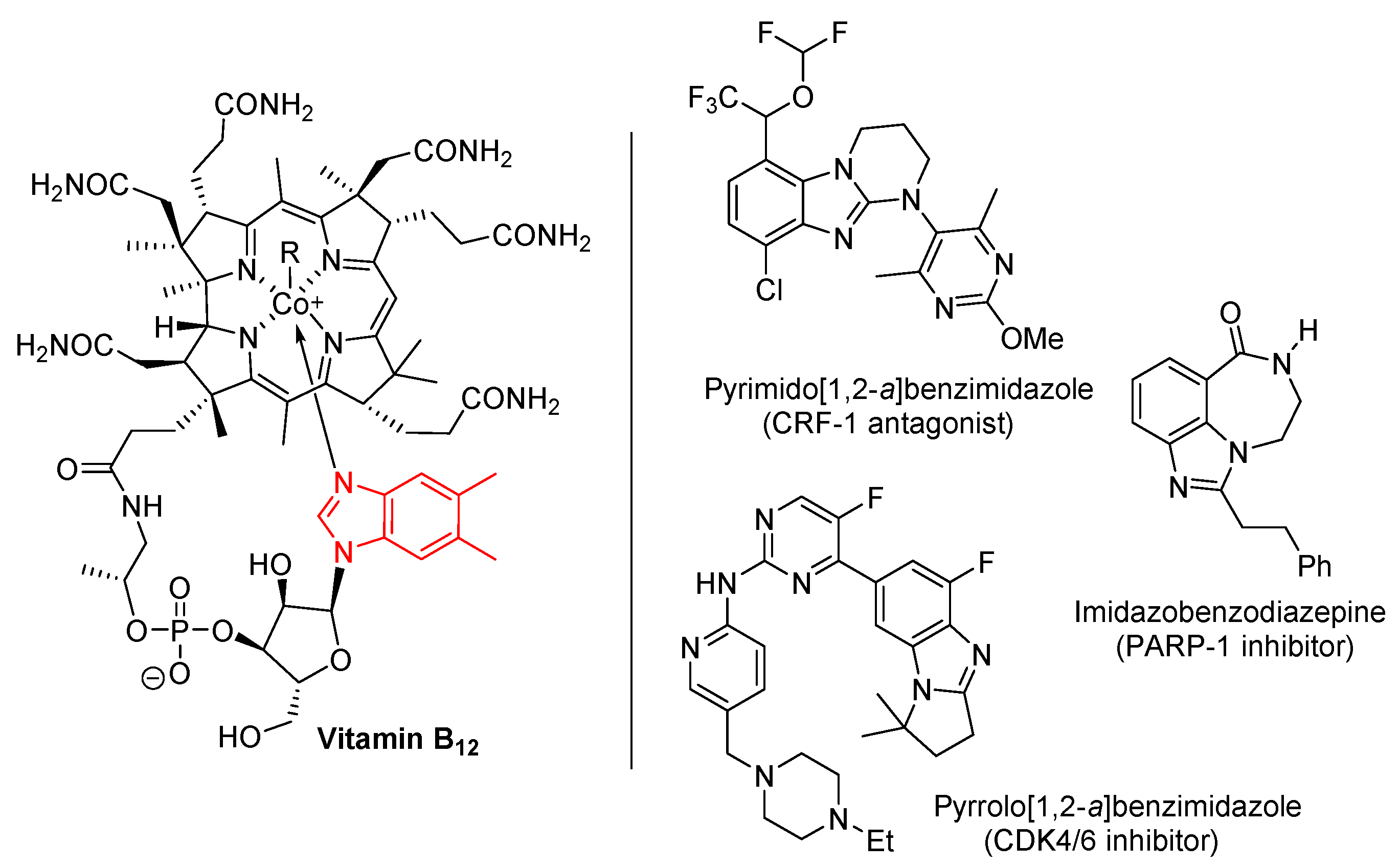
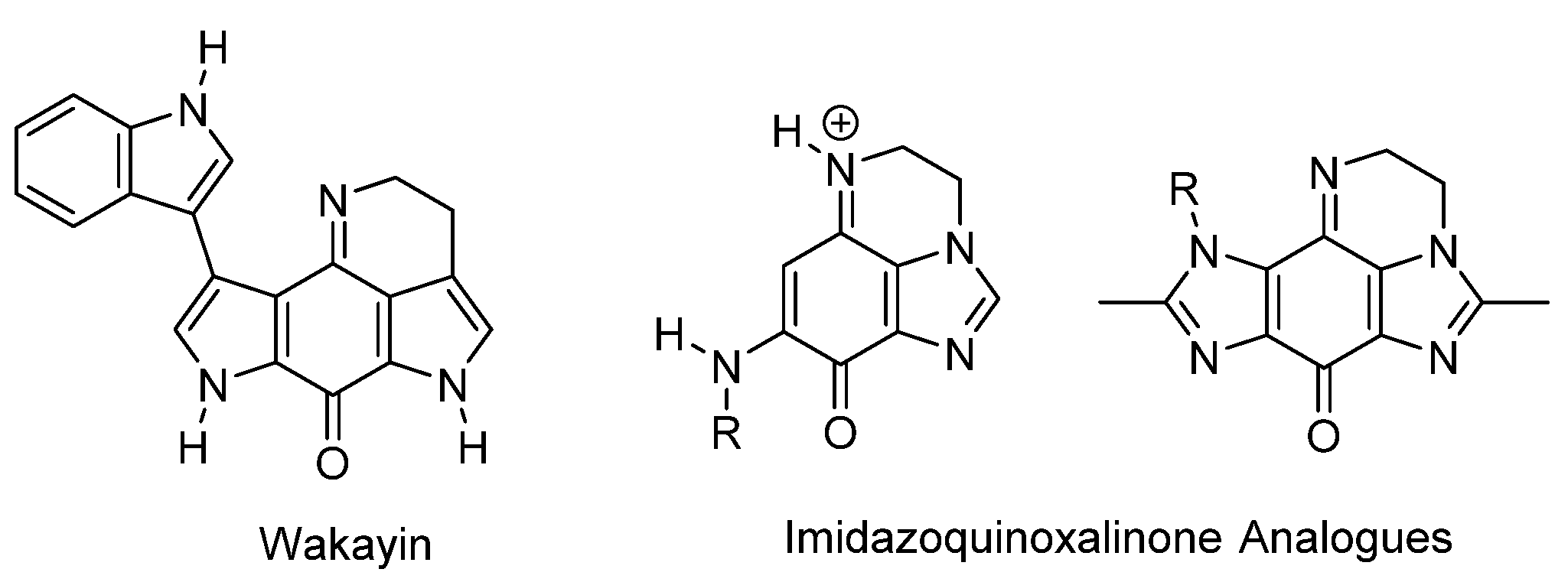
1.1. Significance and Biological Activity
1.2. Available Synthetic Methods
2. Syntheses of Ring-Fused Benzimidazoles and Imidazobenzimidazoles
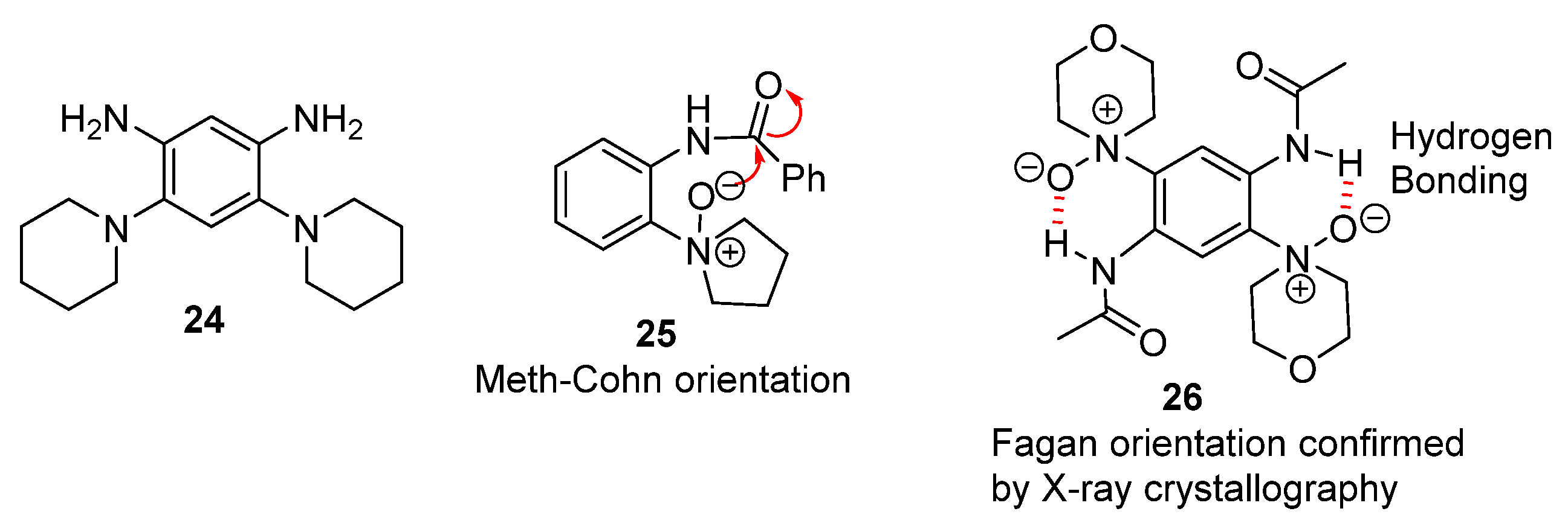
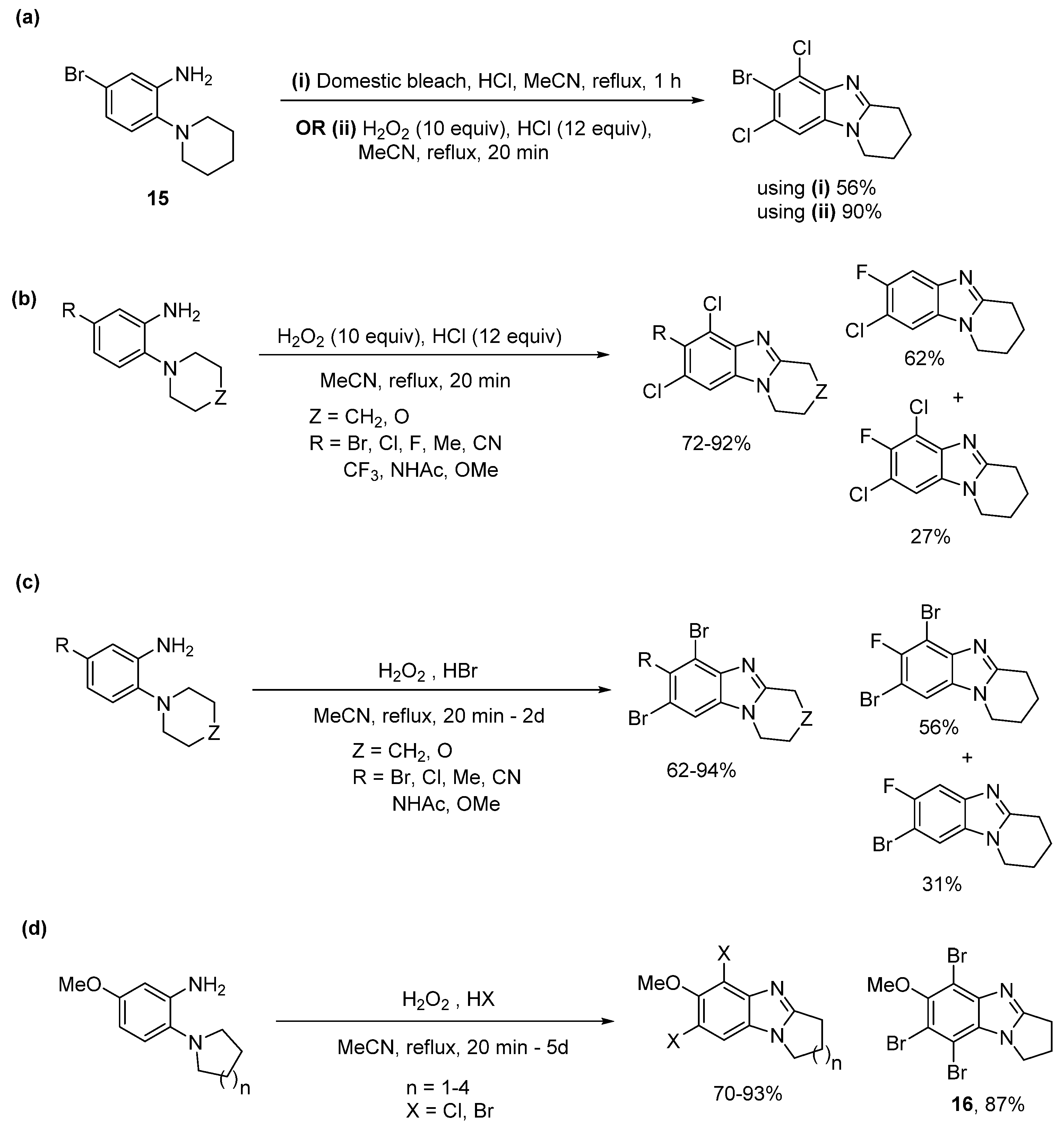
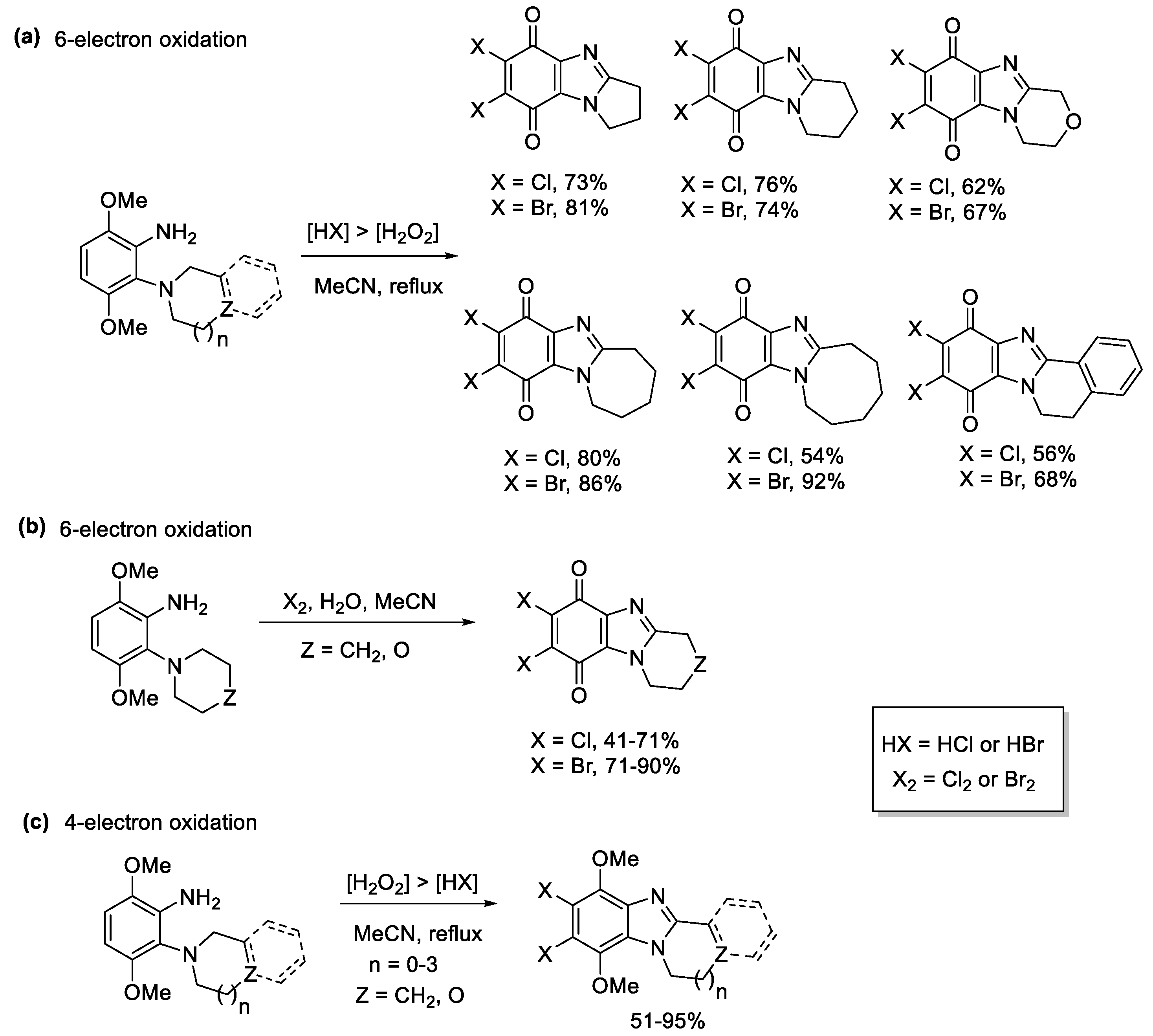
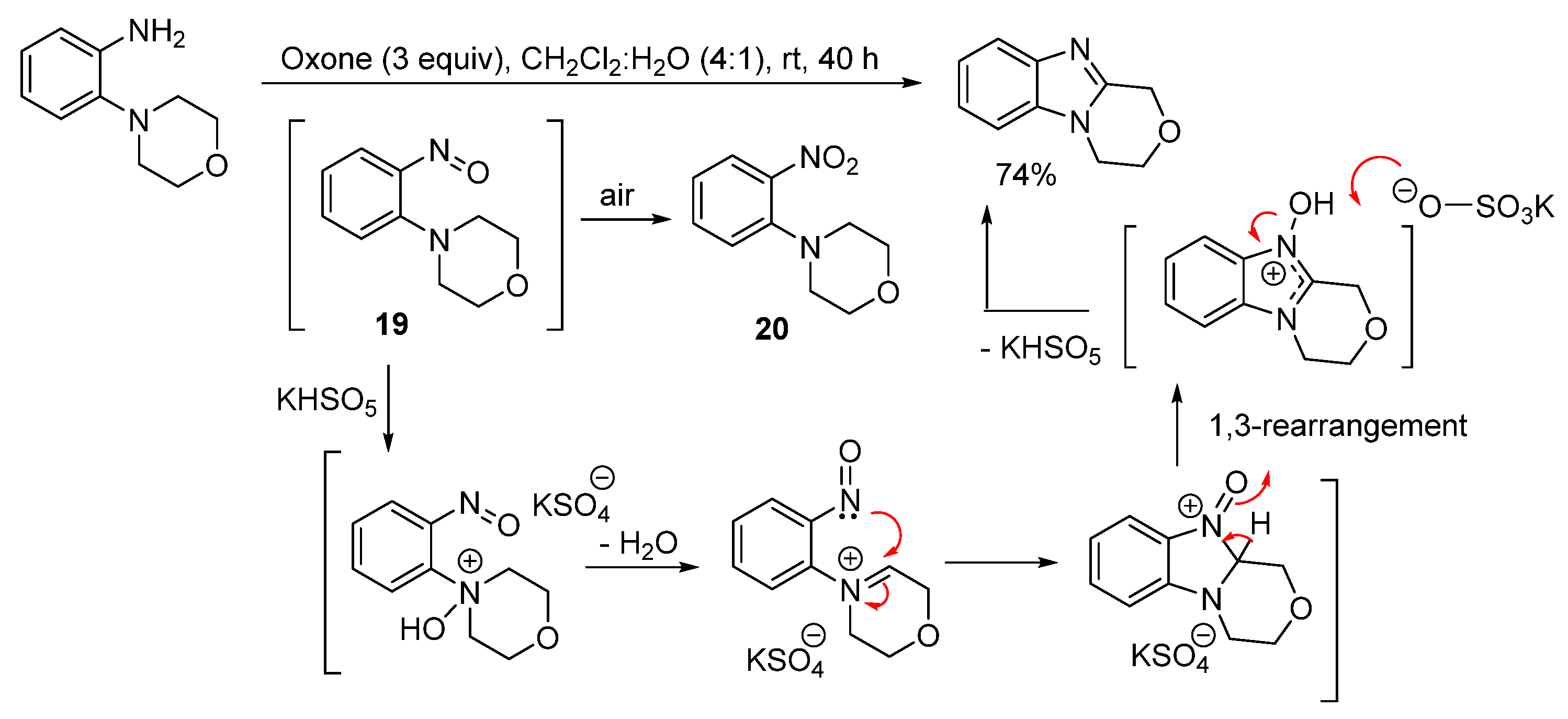
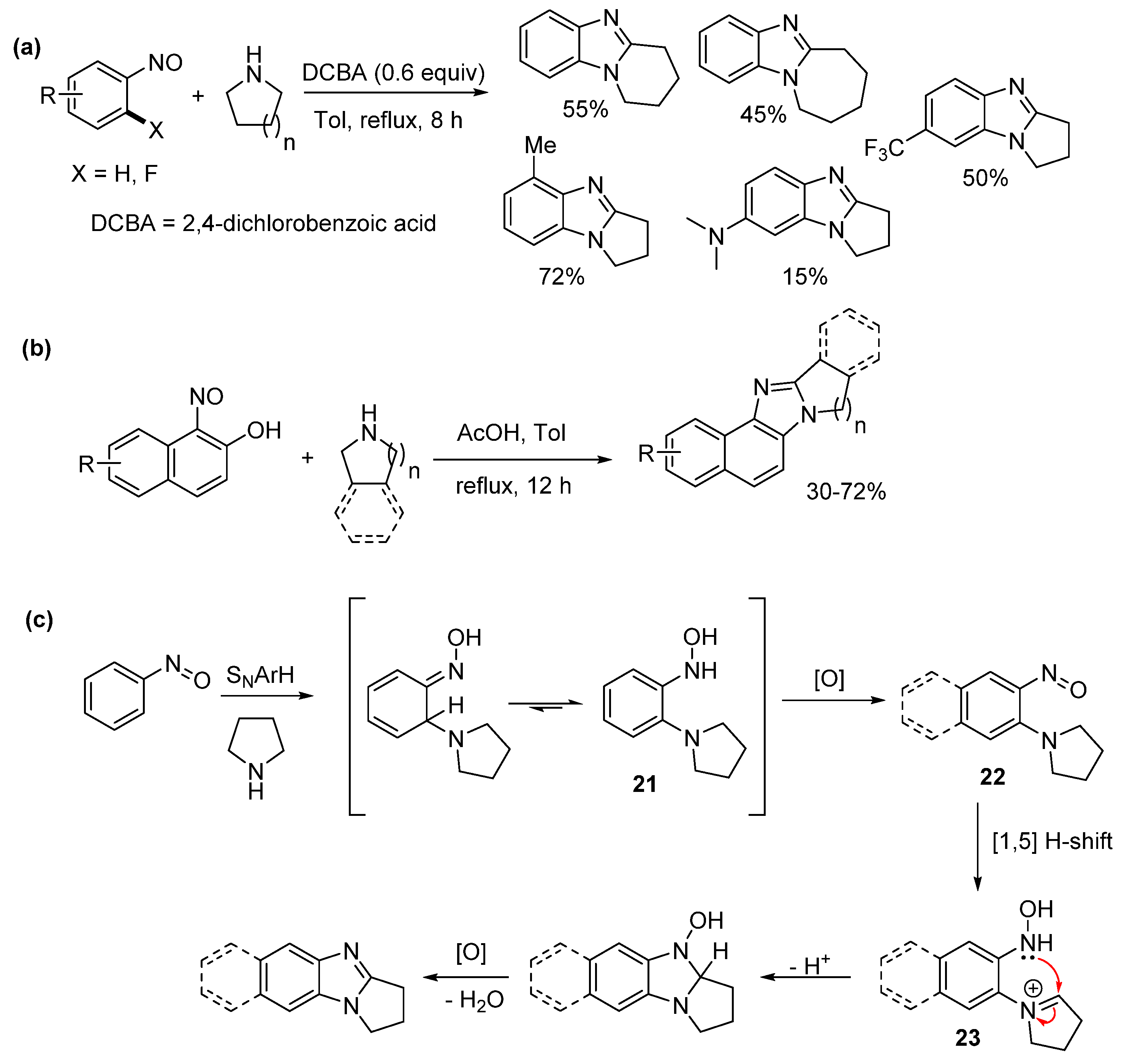
2.1. Oxidations of o-Cycloaminoanilines and Anilide Derivatives (Route A)
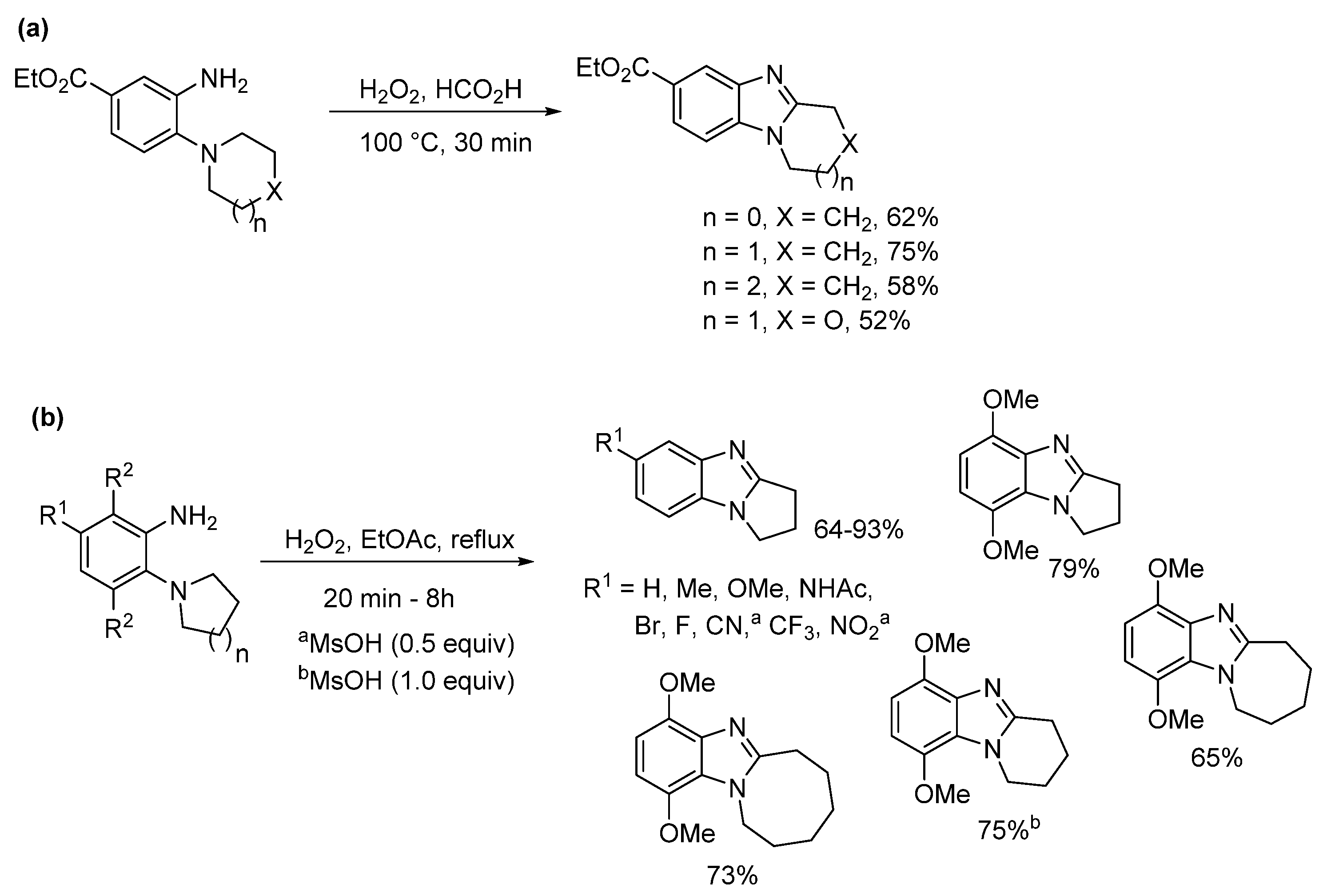
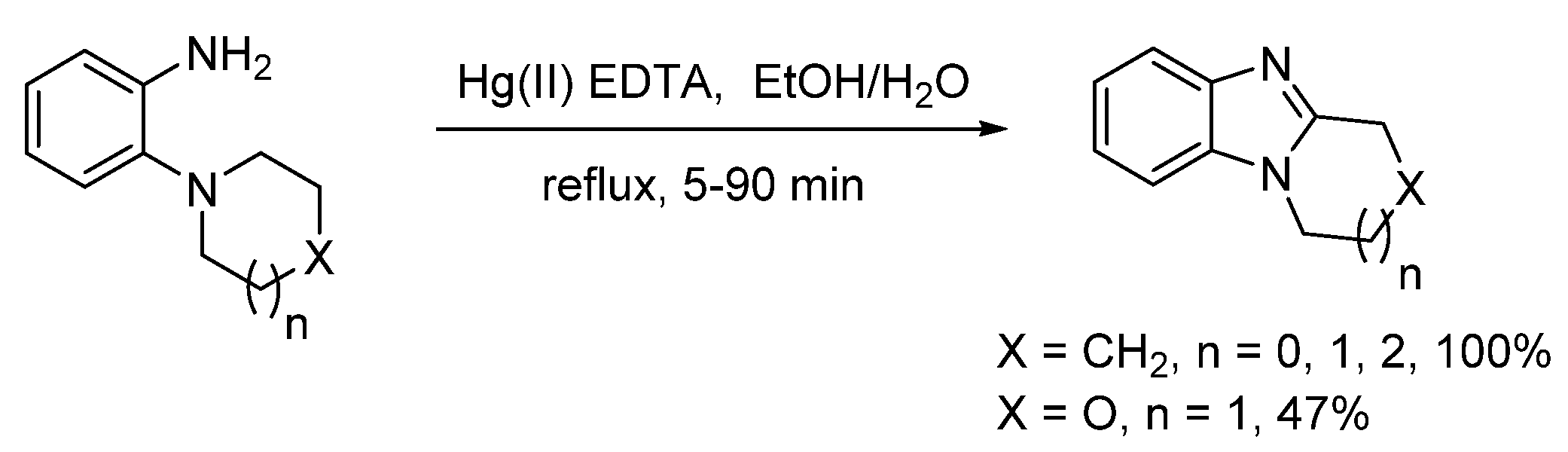
2.1.1. Forming Ring-Fused Benzimidazoles
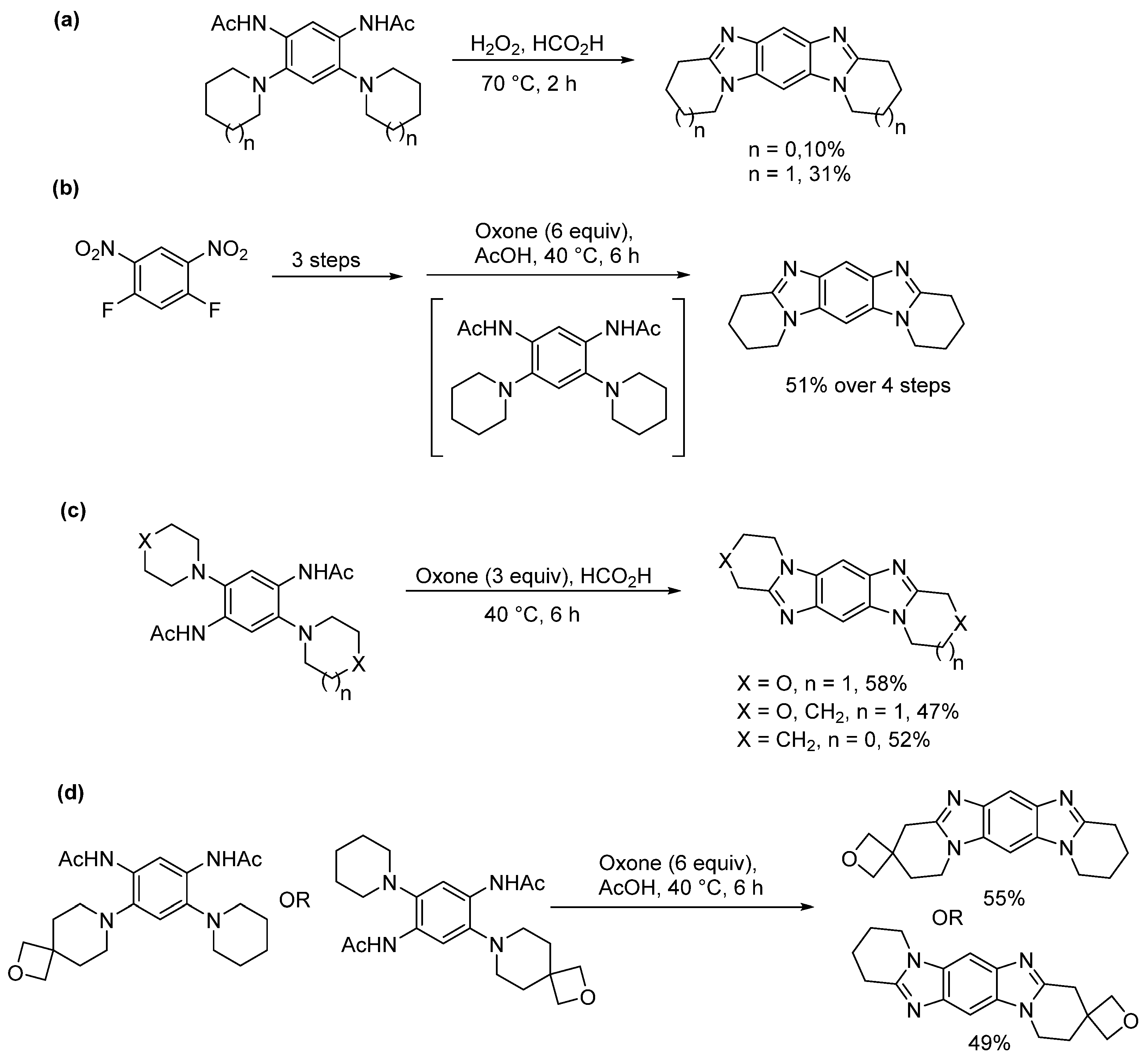
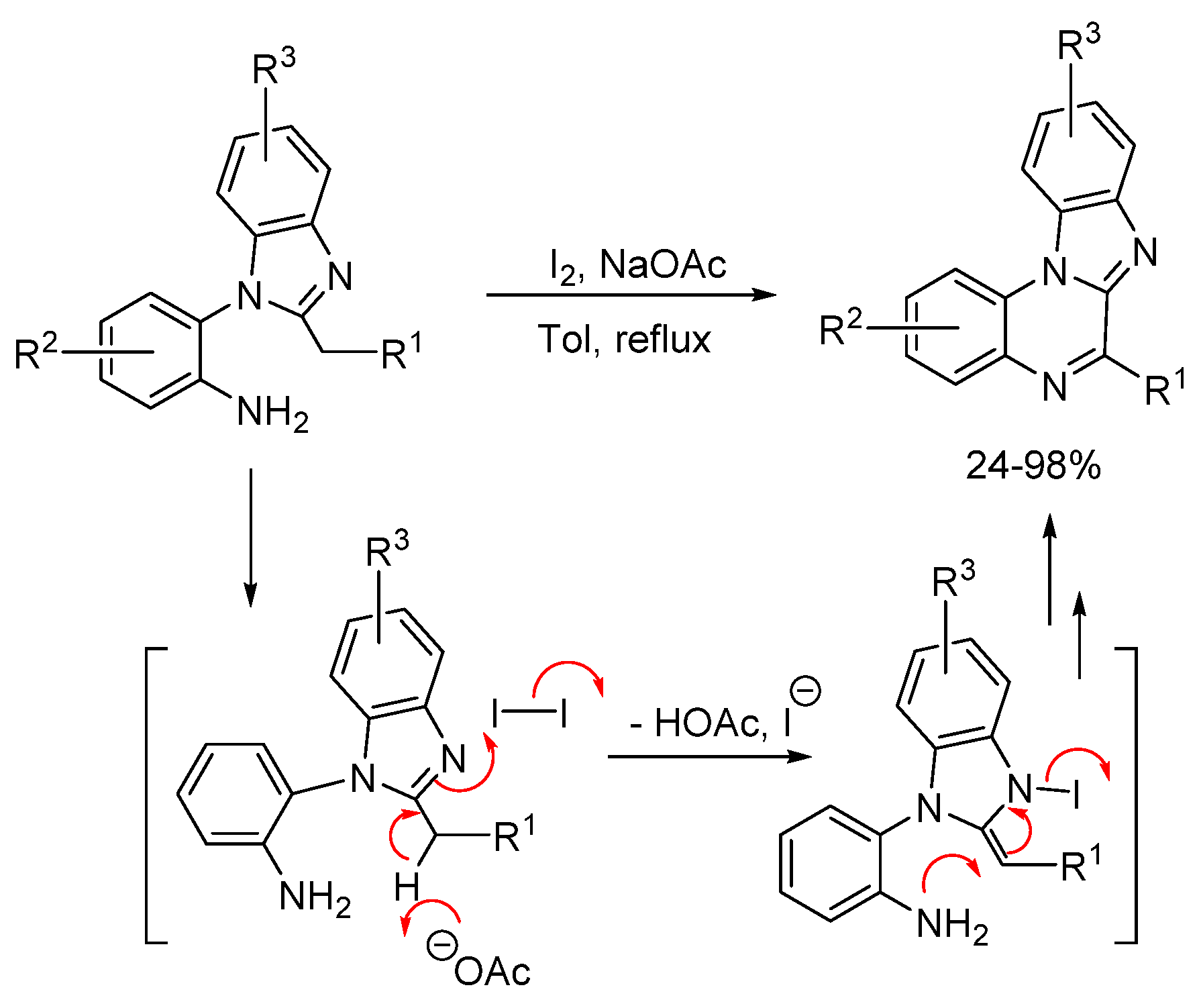
2.1.2. Forming Ring-Fused Imidazobenzimidazoles
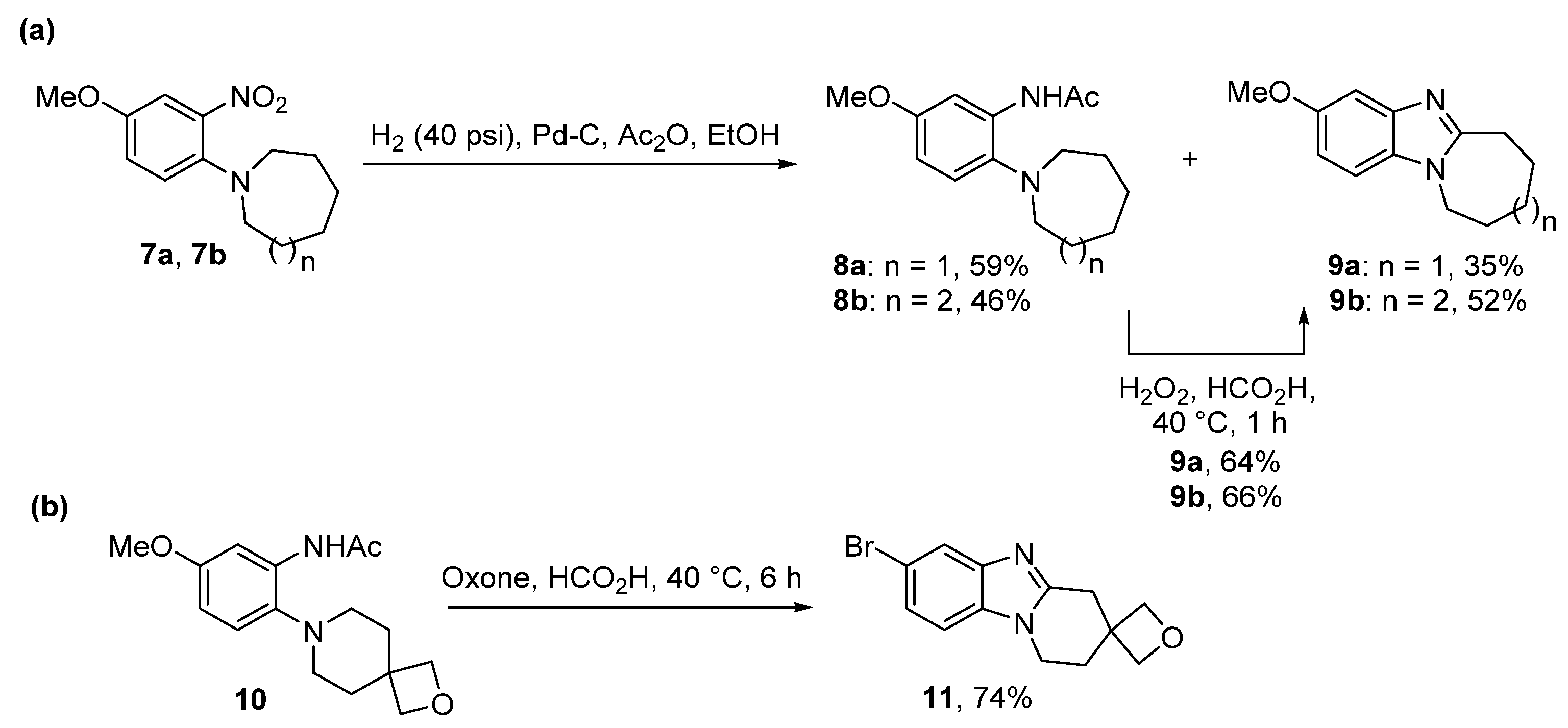
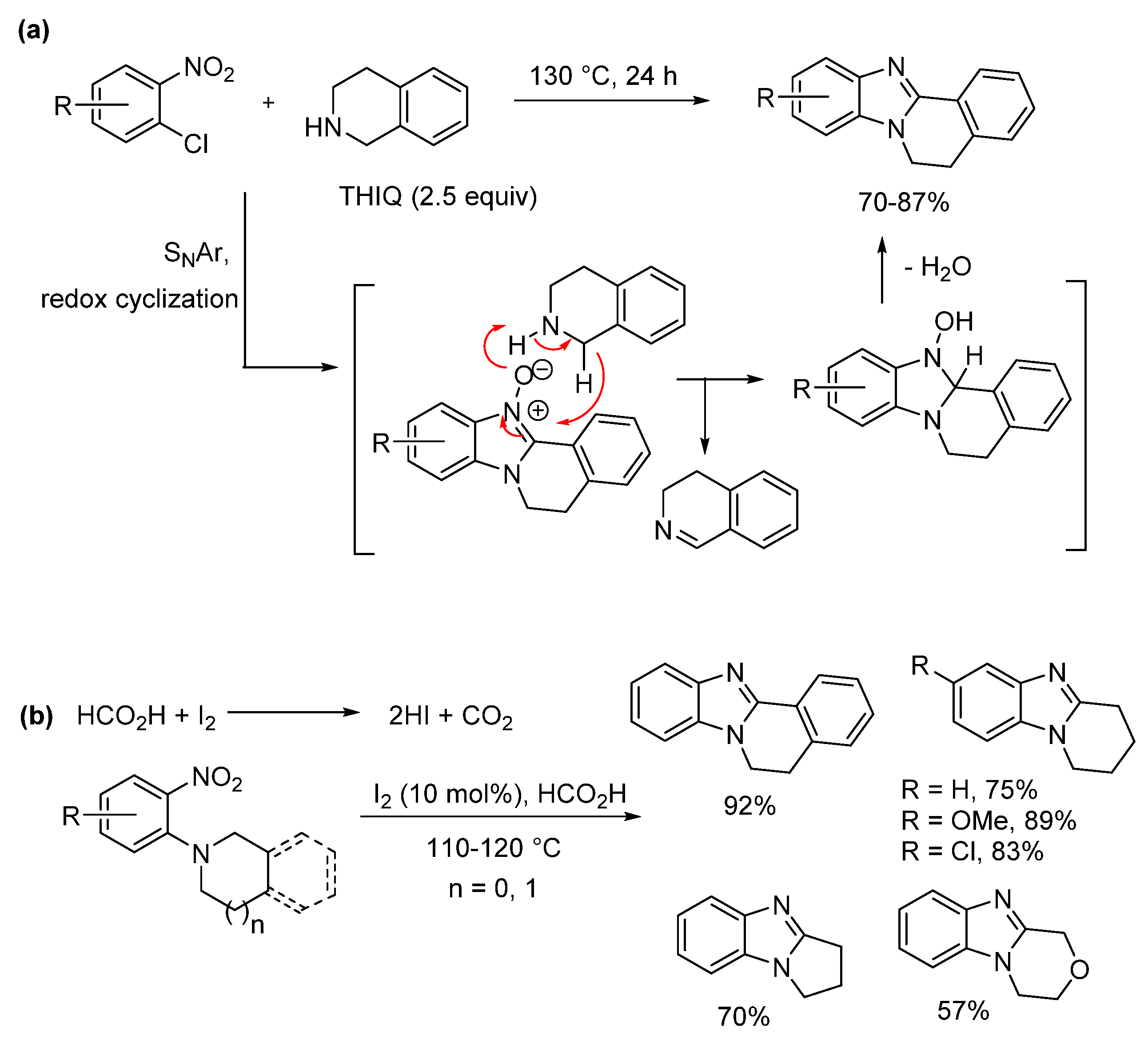
2.2. Reductions of Nitrobenzene-o-Cycloamines (Route B)
2.3. Using Aromatic Amidines, Lactams, and Isothiocyanates (Route C)
2.4. Condensations (Route D)
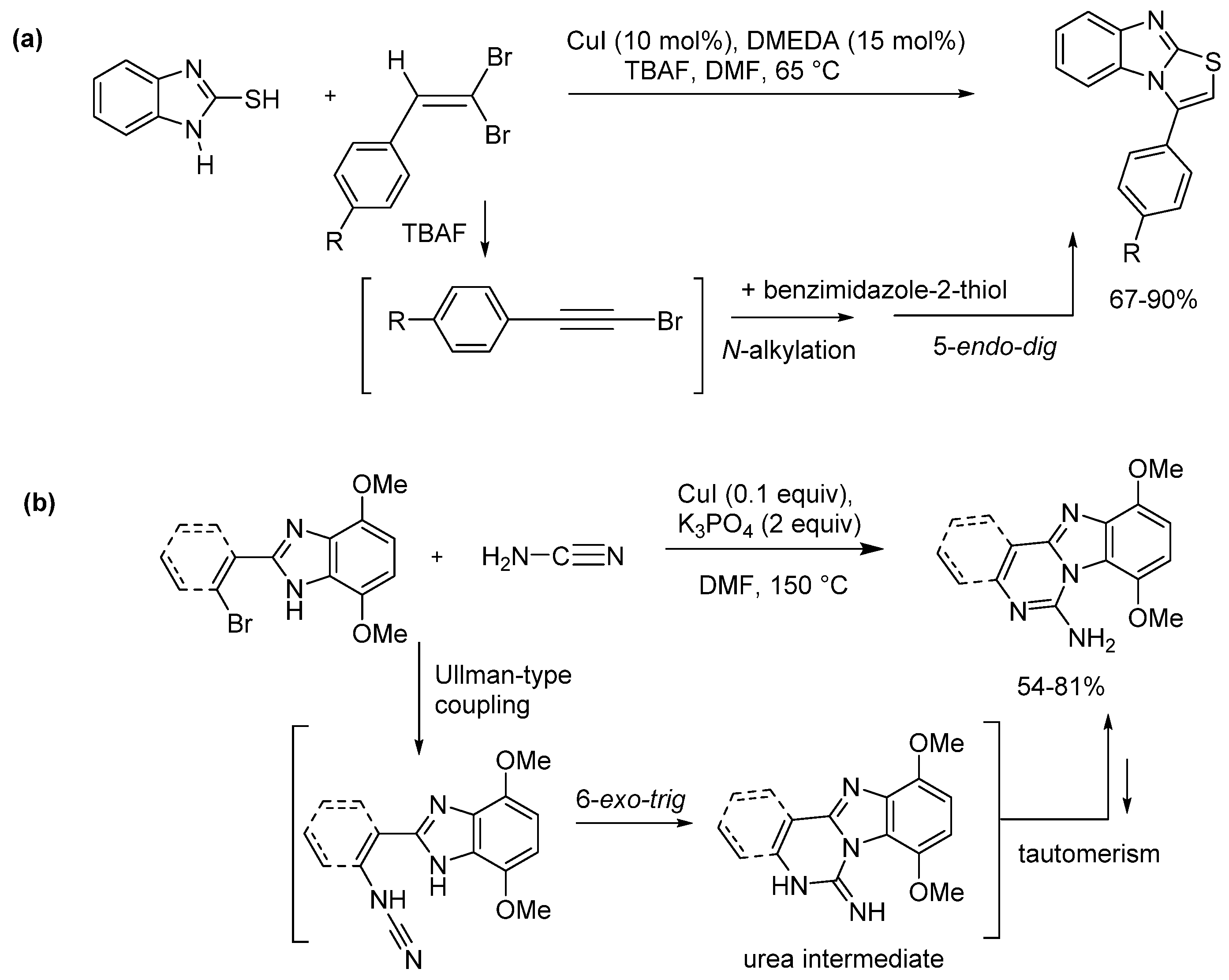
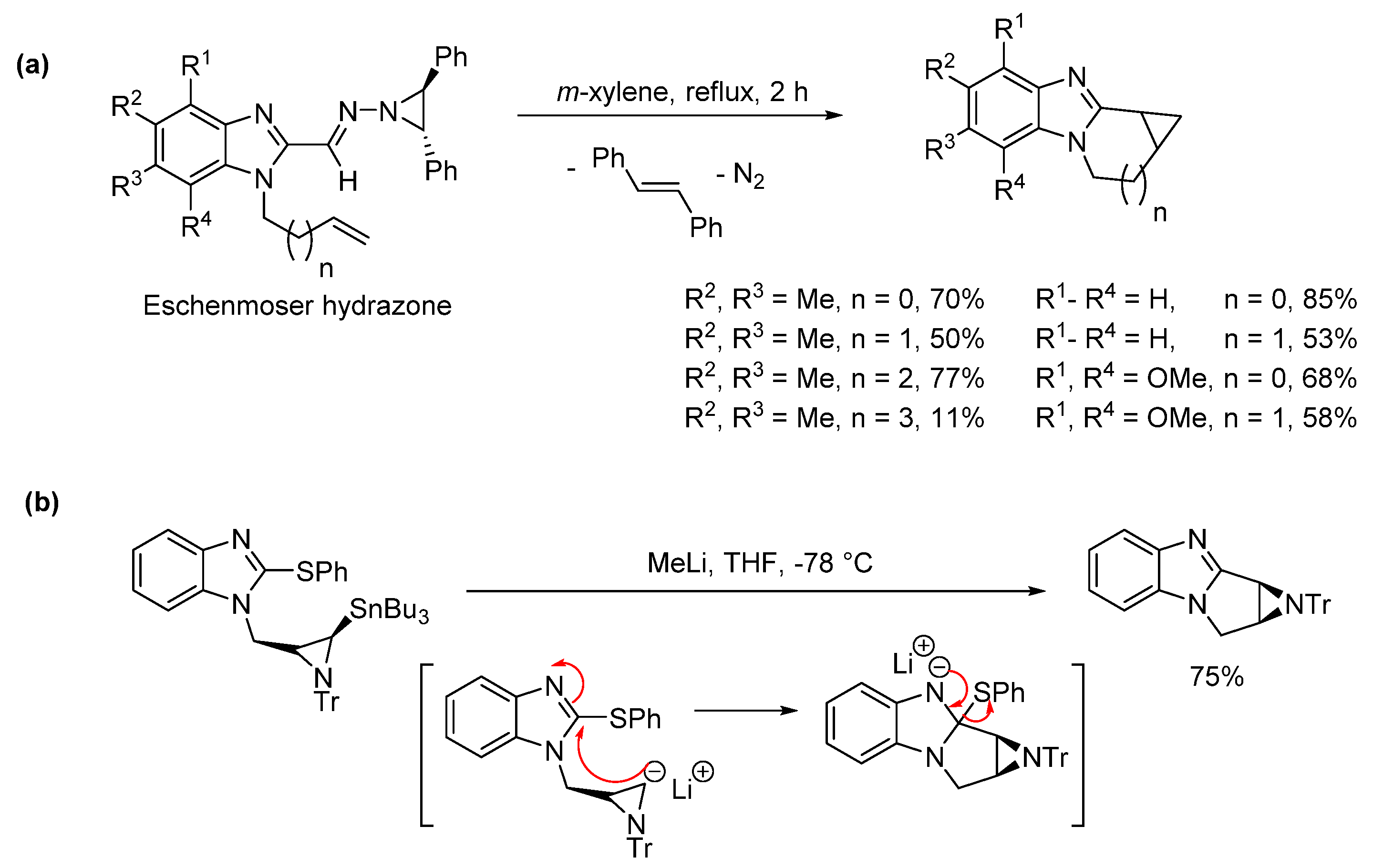
2.5. Annulations onto Benzimidazoles (Route E)
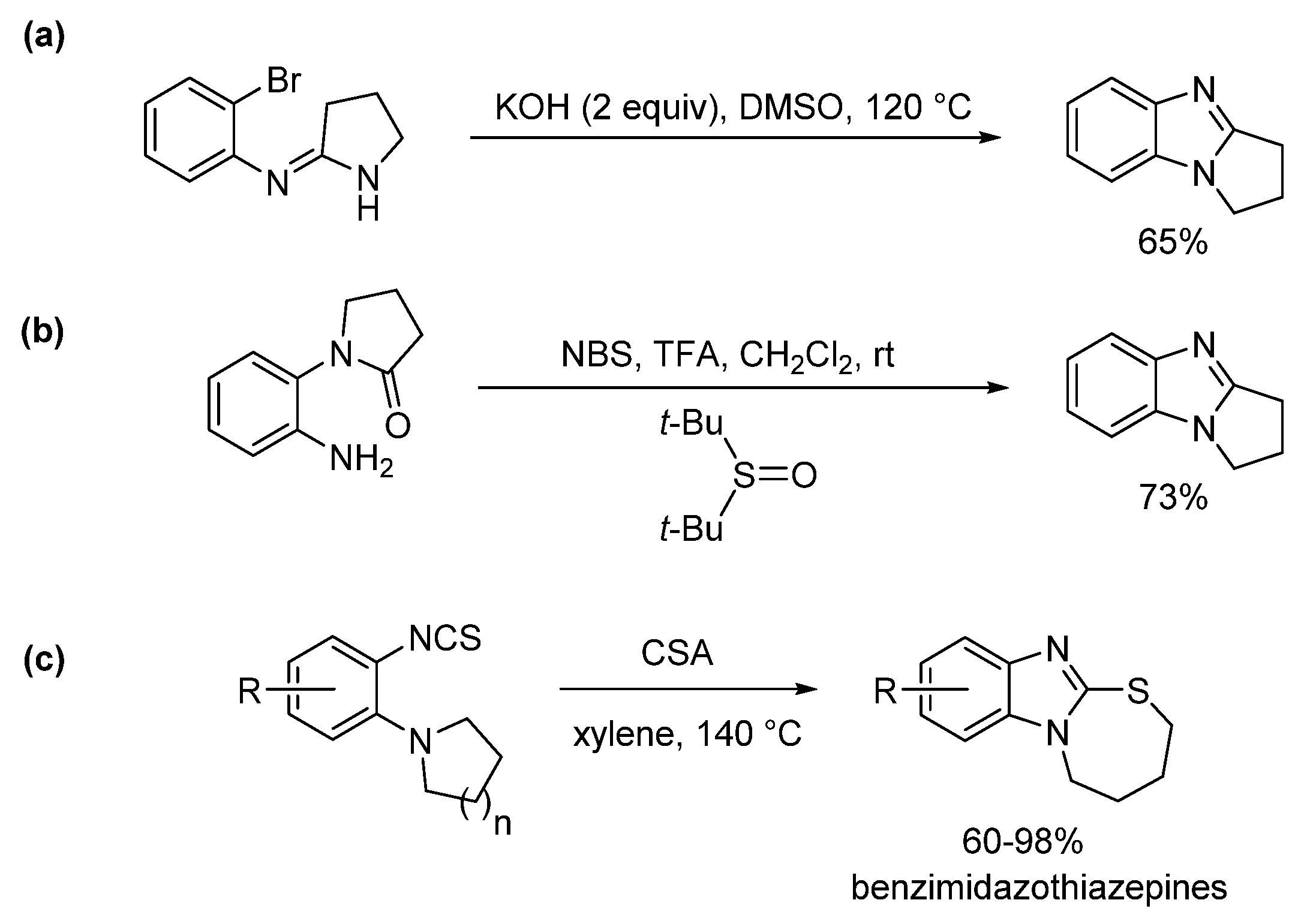
2.5.1. Base-Mediated Methods
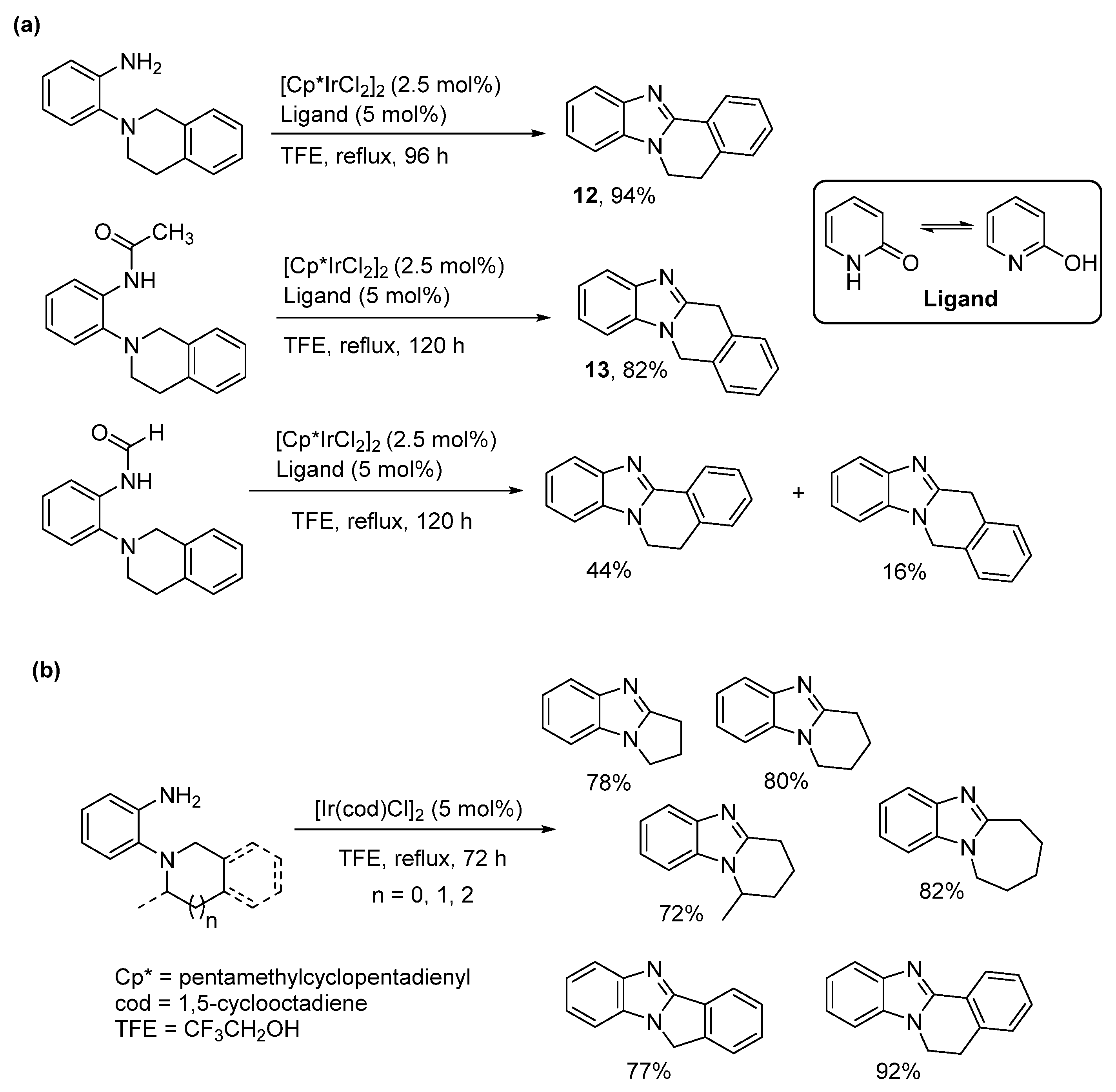
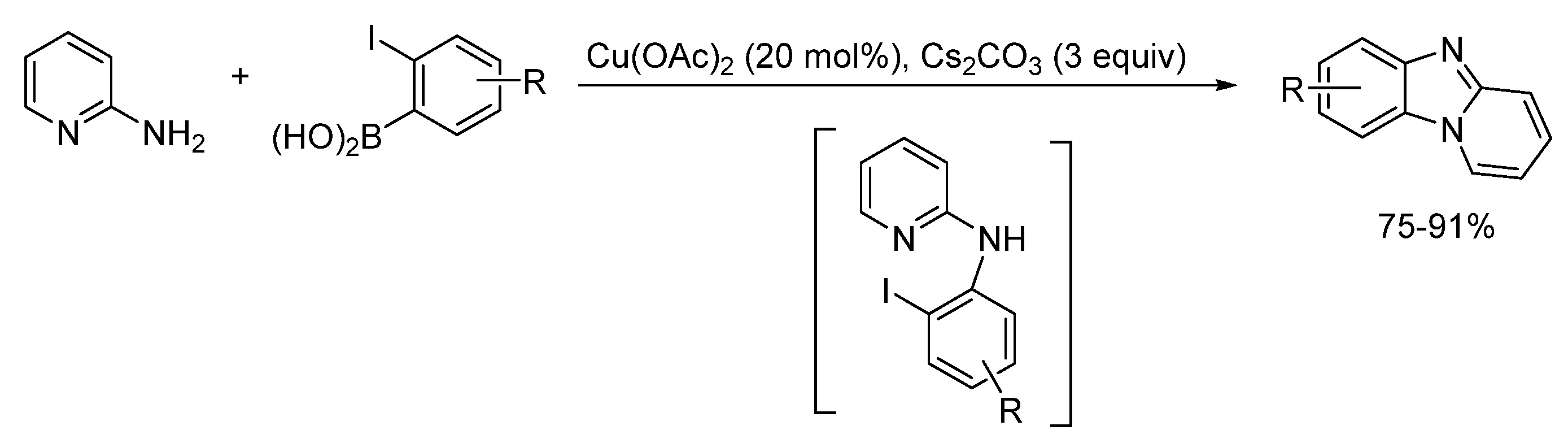
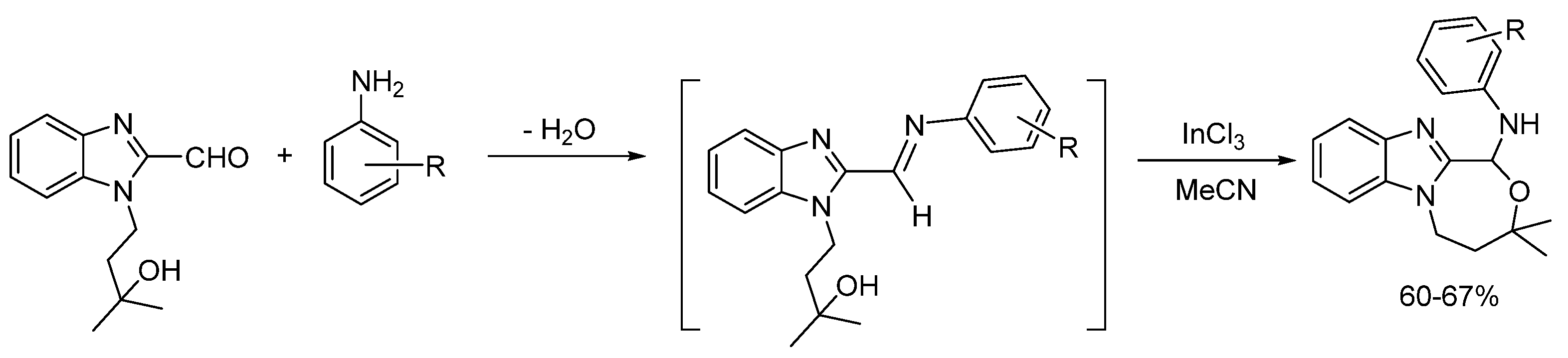
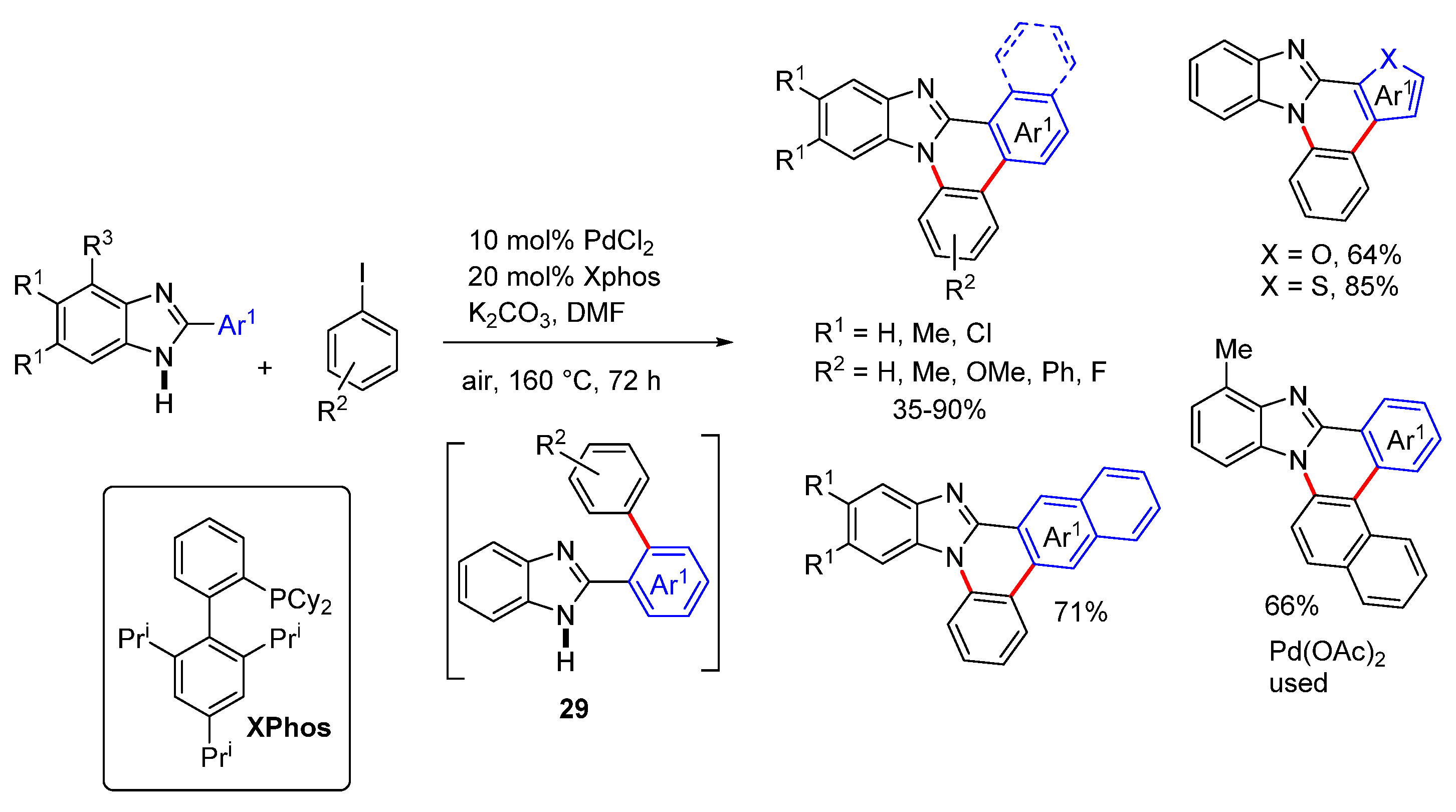
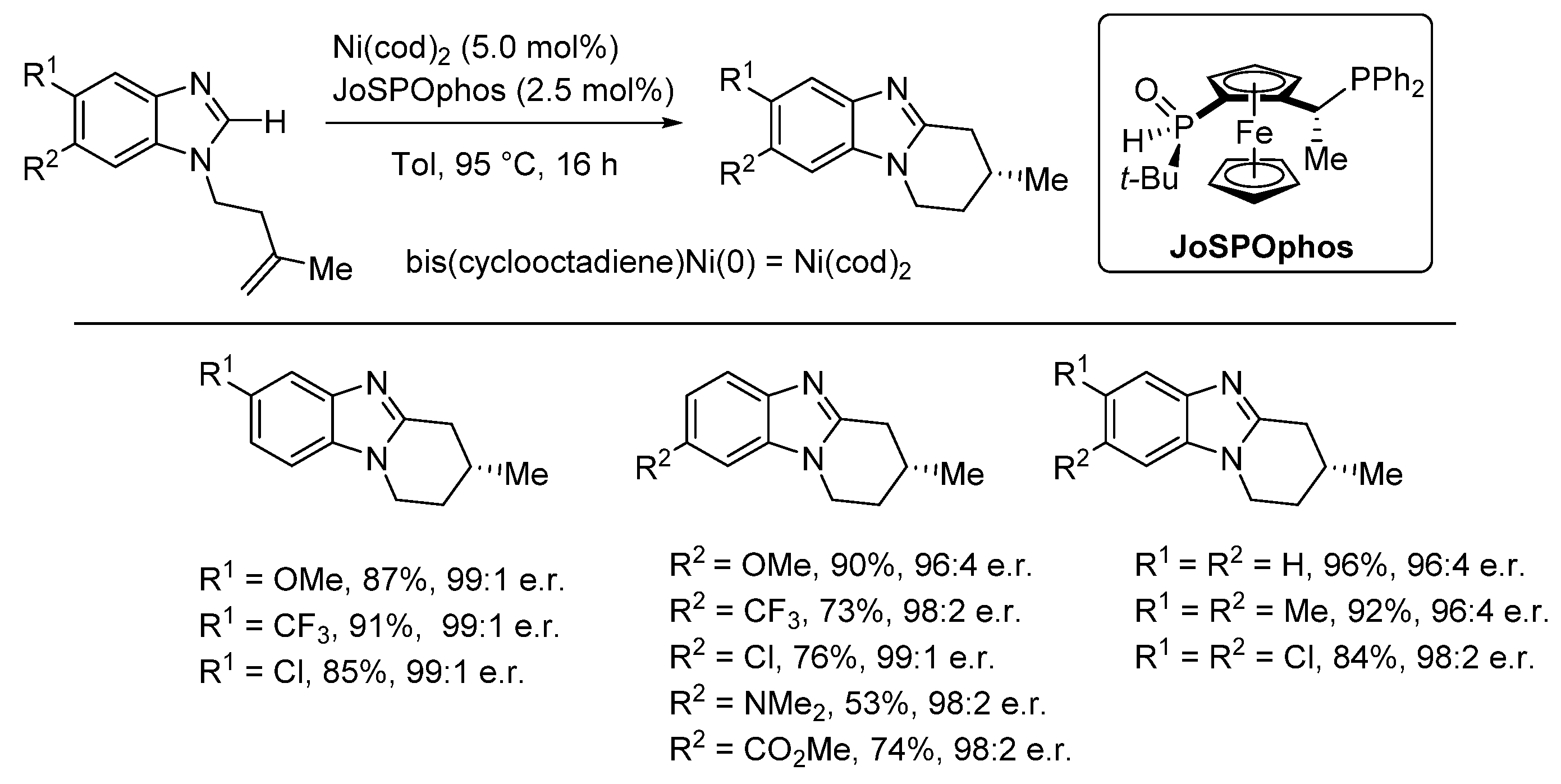
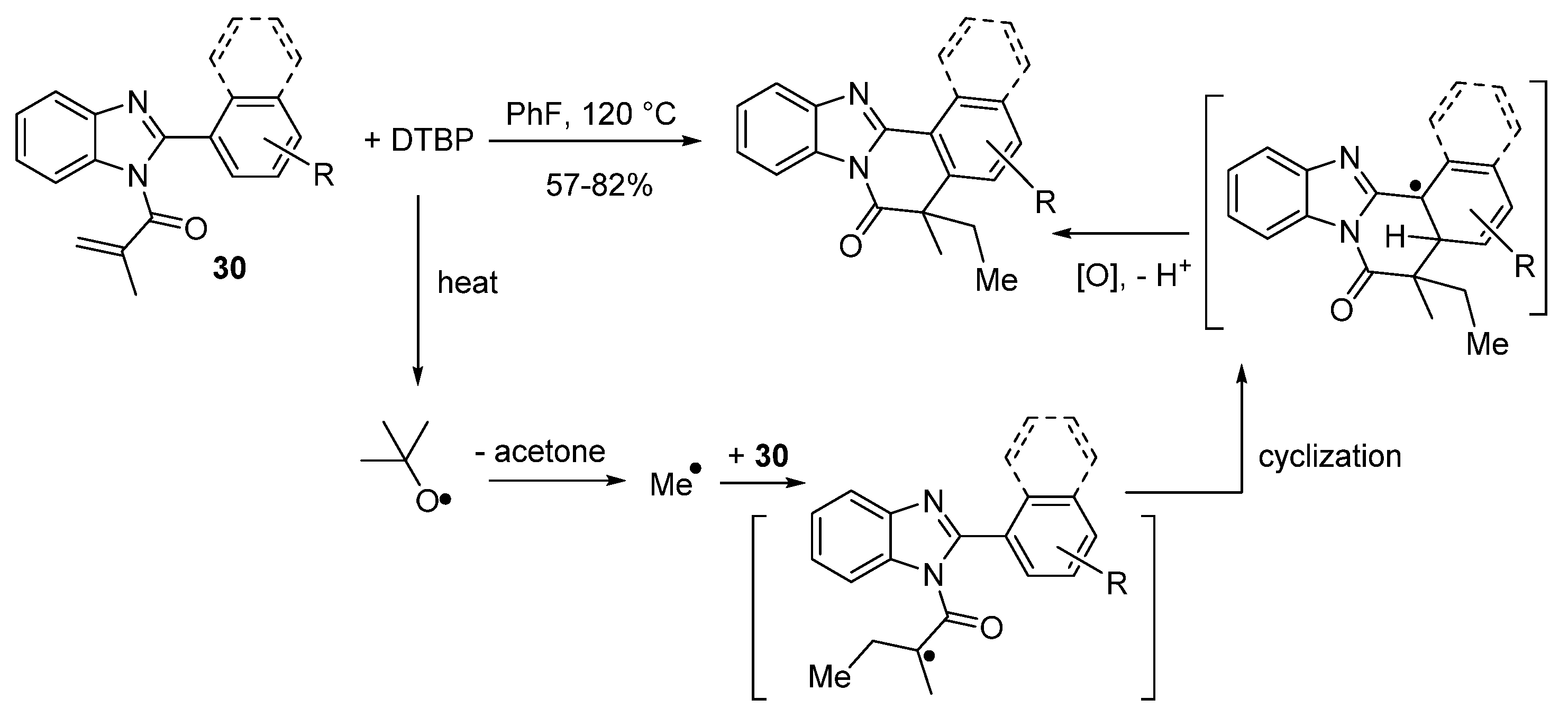
2.5.2. Transition Metal and Lewis Acid Catalyzed Methods
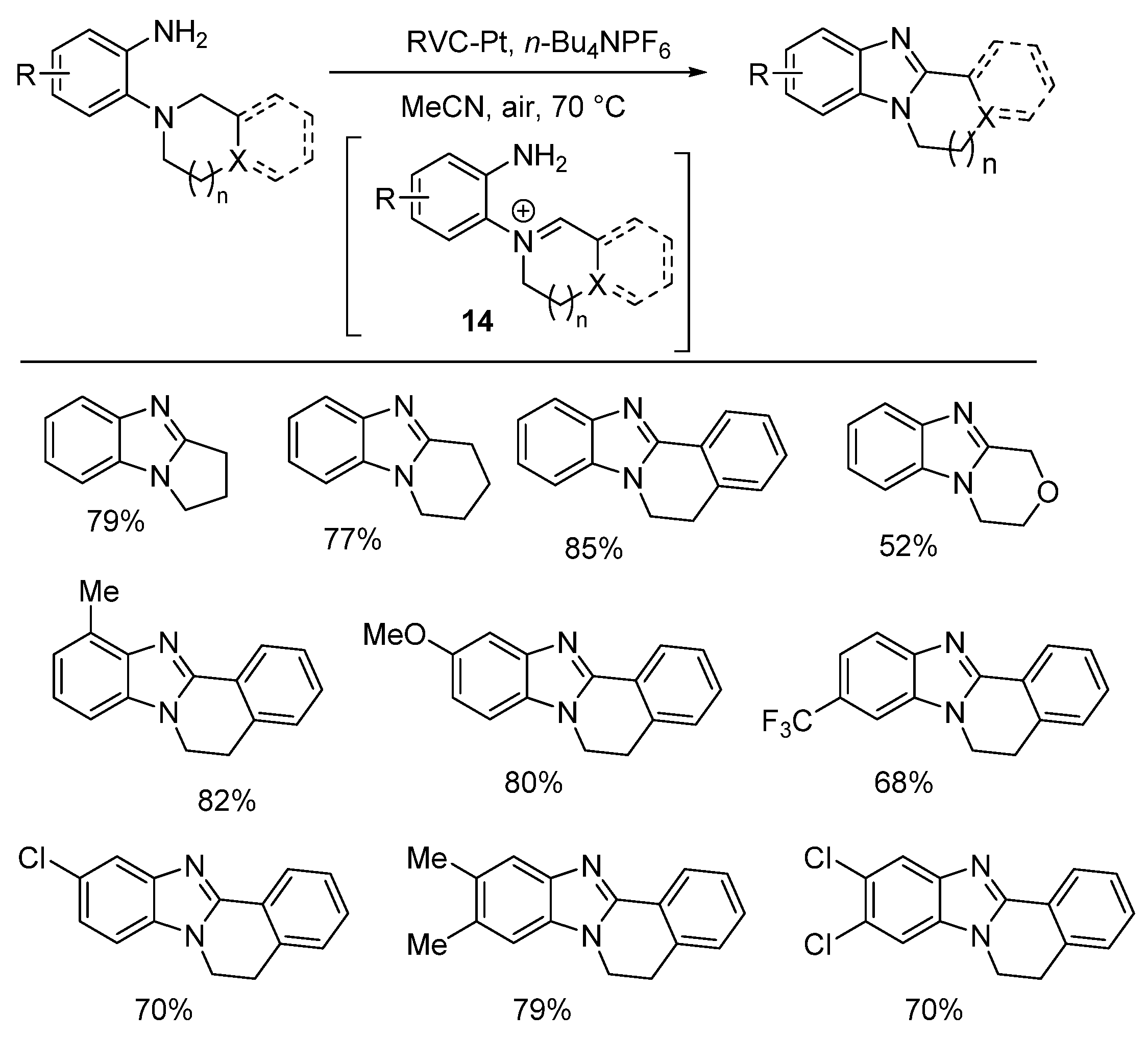
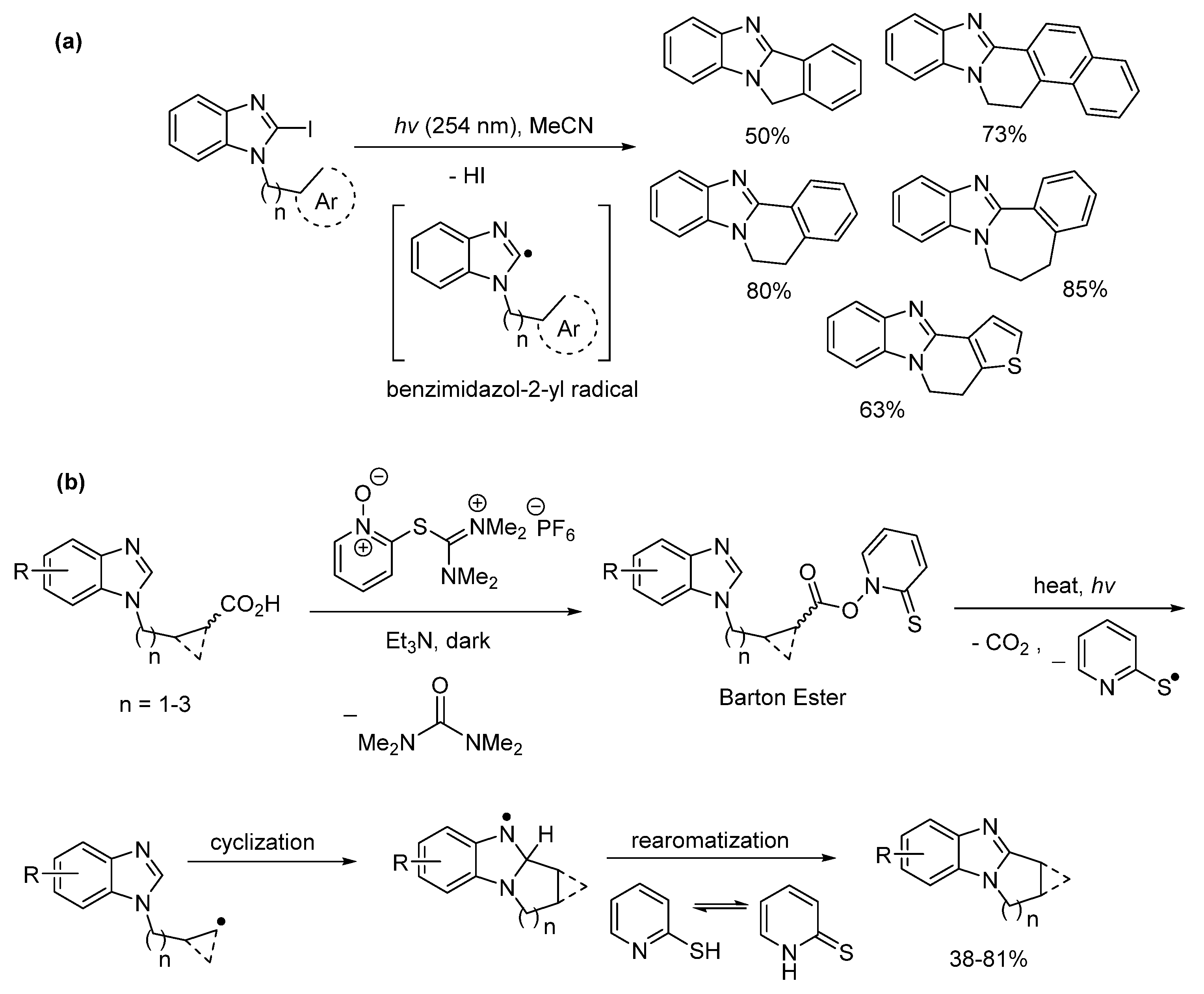
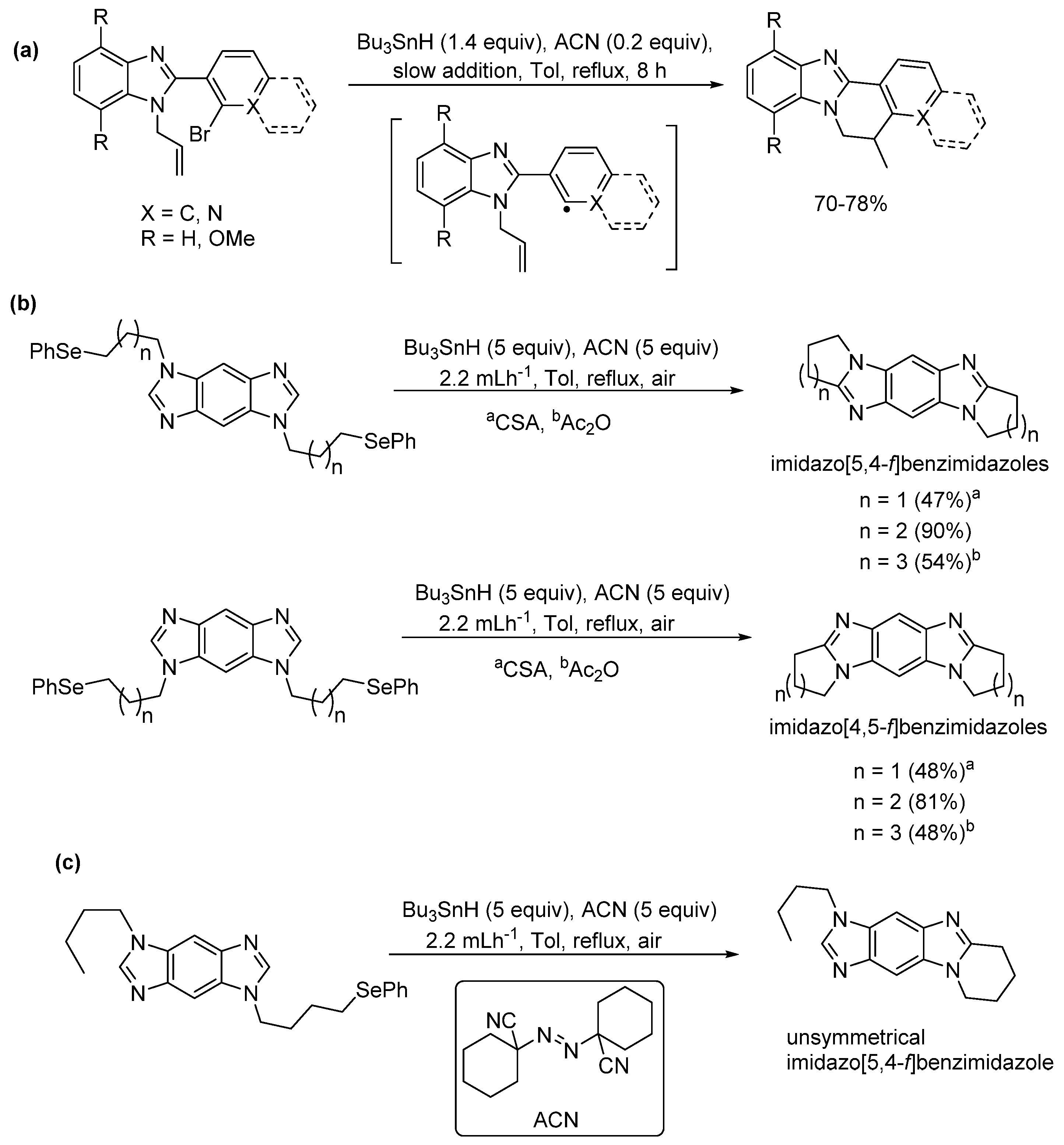
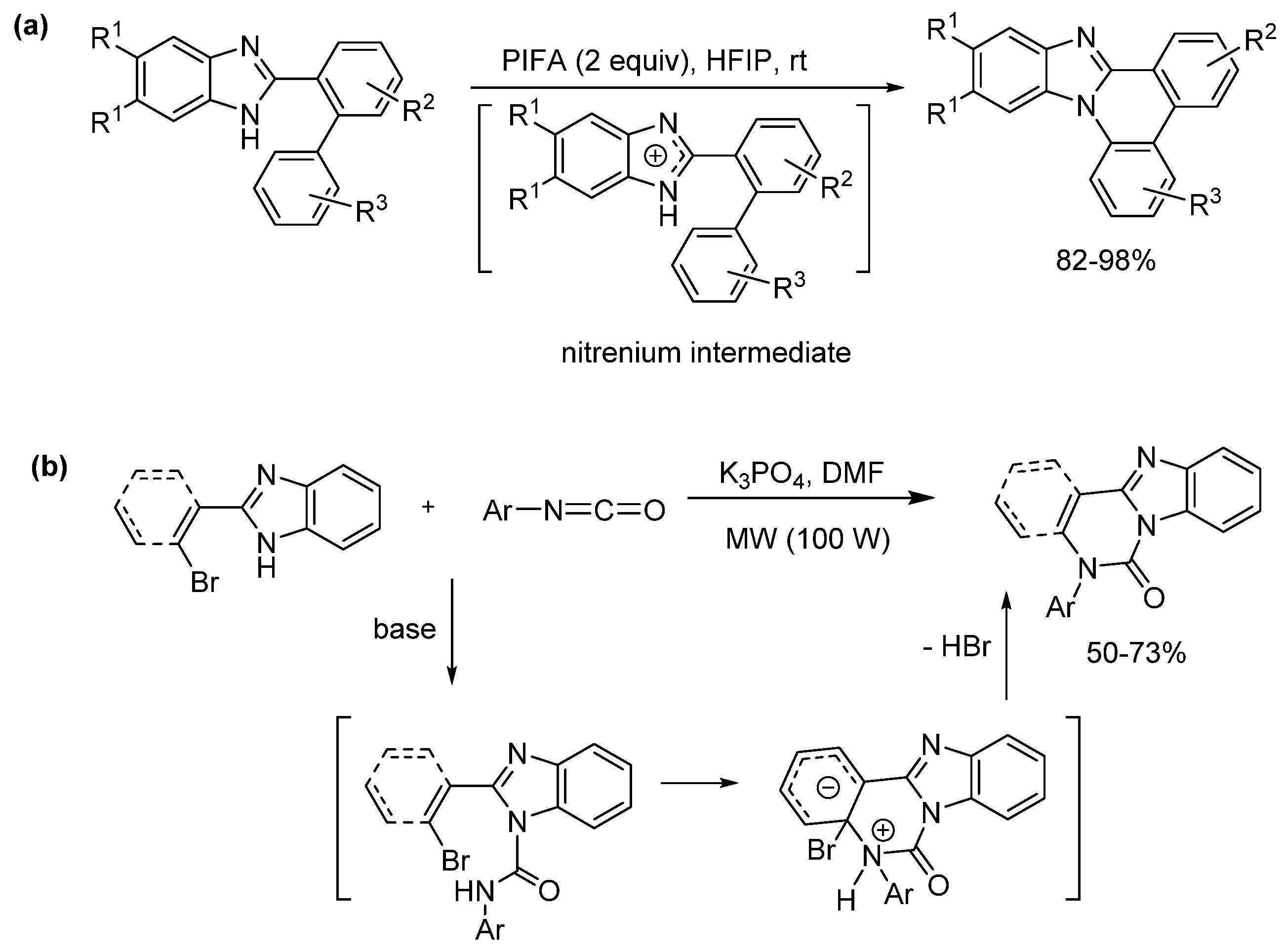
2.5.3. Radical Cyclization Methods
2.5.4. Other Metal-Free Methods
2.5.5. Miscellaneous: Syntheses of Mitomycin Analogues
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brink, N.G.; Folkers, K. Vitamin B12 VI. 5,6-Dimethylbenzimidazole, a. degradation product of vitamin B12. J. Am. Chem. Soc. 1949, 71, 2951. [Google Scholar] [CrossRef]
- Floyd, J.C.; Holliday, E.R.; Petrow, V. Letters to the Editor. J. Pharm. Pharmacol. 1949, 1, 734–735. [Google Scholar] [CrossRef]
- Yadav, G.; Ganguly, S. Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review. Eur. J. Med. Chem. 2015, 97, 419–443. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Hiremathad, A.; Budagumpi, S.; Nagaraja, B.M. Comprehensive review in current developments of benzimidazole-based medicinal chemistry. Chem. Biol. Drug Des. 2015, 86, 19–65. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 97, 419–443. [Google Scholar] [CrossRef] [PubMed]
- Leadbeater, A.J. Plant health management: Fungicides and antibiotics. In Encyclopedia of Agriculture and Food Systems; Van Alfen, N.K., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 408–424. [Google Scholar] [CrossRef]
- Kojima, T.; Mochizuki, M.; Takai, T.; Hoashi, Y.; Morimoto, S.; Seto, M.; Nakamura, M.; Kobayashi, K.; Sako, Y.; Tanaka, M.; et al. Discovery of 1,2,3,4-tetrahydropyrimido[1,2-a]benzimidazoles as novel class of corticotropin releasing factor 1 receptor antagonists. Bioorg. Med. Chem. 2018, 26, 2229–2250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, W.-J.; Yin, L.; Li, H.; Chen, Z.-H.; Zhu, D.-X.; Song, X.-Q.; Cheng, Z.-Z.; Song, P.; Wang, Z.; et al. Design and synthesis of 4-(2,3-dihydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-7-yl)-N-(5-(piperazin-1-ylmethyl)pyridine-2-yl)pyrimidin-2-amine as a highly potent and selective cyclin-dependent kinases 4 and 6 inhibitors and the discovery of structure-activity relationships. Bioorg. Med. Chem. Lett. 2018, 28, 974–978. [Google Scholar] [CrossRef]
- Ferraris, D.; Ficco, R.P.; Dain, D.; Ginski, M.; Lautar, S.; Lee-Wisdom, K.; Liang, S.; Lin, Q.; Lu, M.X.-C.; Morgan, L.; et al. Design and synthesis of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. Part 4: Biological evaluation of imidazobenzodiazepines as potent PARP-1 inhibitors for treatment of ischemic injuries. Bioorg. Med. Chem. 2003, 11, 3695–3707. [Google Scholar] [CrossRef]
- Hoang, H.; Huang, X.; Skibo, E.B. Synthesis and in vitro evaluation of imidazole-based wakayin analogues. Org. Biomol. Chem. 2008, 6, 3059–3064. [Google Scholar] [CrossRef]
- Bass, P.D.; Gubler, D.A.; Judd, T.C.; Williams, R.M. Mitomycinoid alkaloids: Mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev. 2013, 113, 6816–6863. [Google Scholar] [CrossRef]
- O’Donovan, L.; Carty, M.P.; Aldabbagh, F. First synthesis of N-[(aziridin-2-yl)methyl]benzimidazolequinone and analysis of toxicity towards normal and Fanconi anemia cells. Chem. Commun. 2008, 43, 5592–5594. [Google Scholar] [CrossRef]
- Fahey, K.; O’Donovan, L.; Carr, M.; Carty, M.P.; Aldabbagh, F. The influence of the aziridinyl substituent of benzimidazoles and benzimidazolequinones on toxicity towards normal and Fanconi anaemia cells. Eur. J. Med. Chem. 2010, 45, 1873–1879. [Google Scholar] [CrossRef] [PubMed]
- Bonham, S.; O’Donovan, L.; Carty, M.P.; Aldabbagh, F. First synthesis of an aziridinyl fused pyrrolo[1,2-a]benzimidazole and toxicity evaluation towards normal and breast cancer cell lines. Org. Biomol. Chem. 2011, 9, 6700–6706. [Google Scholar] [CrossRef]
- Islam, I.; Skibo, E.B.; Dorr, R.T.; Alberta, D.S. Structure-activity studies of antitumor agents based on pyrrolo[l,2-a]benzimidazoles: New reductive alkylating DNA cleaving agents. J. Med. Chem. 1991, 34, 2954–2961. [Google Scholar] [CrossRef] [PubMed]
- Skibo, E.B.; Gordon, S.; Bess, L.; Boruah, R.; Heileman, M.J. Studies of pyrrolo[1,2-a]benzimidazolequinone DT-diaphorase substrate activity, topoisomerase II inhibition activity, and DNA reductive alkylation. J. Med. Chem. 1997, 40, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Craigo, W.A.; LeSueur, B.W.; Skibo, E.B. Design of highly active analogues of the pyrrolo[1,2-a]benzimidazole antitumor agents. J. Med. Chem. 1999, 42, 3324–3333. [Google Scholar] [CrossRef] [PubMed]
- Skibo, E.B.; Jamil, A.; Austin, B.; Hansen, D.; Ghodousi, A. Triple molecular target approach to selective melanoma cytotoxicity. Org. Biomol. Chem. 2010, 8, 1577–1587. [Google Scholar] [CrossRef]
- Sharma, A.; Arambula, J.F.; Koo, S.; Kumar, R.; Singh, H.; Sessler, J.L.; Kim, J.S. Hypoxia-targeted drug delivery. Chem. Soc. Rev. 2019, 48, 771–813. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) as a therapeutic and diagnostic target in cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef]
- Lynch, M.; Hehir, S.; Kavanagh, P.; Leech, D.; O’Shaughnessy, J.; Carty, M.P.; Aldabbagh, F. Synthesis by radical cyclization and cytotoxicity of highly potent bioreductive alicyclic ring fused [1,2-a] benzimidazolequinones. Chem. Eur. J. 2007, 13, 3218–3226. [Google Scholar] [CrossRef]
- Hehir, S.; O’Donovan, L.; Carty, M.P.; Aldabbagh, F. Synthesis of dimethyl substituted benzimidazoles containing cyclopropane fused onto five to eight membered [1,2-a] alicyclic rings and influence of methyl group substituents on cytotoxicity of benzimidazolequinones. Tetrahedron 2008, 64, 4196–4203. [Google Scholar] [CrossRef]
- Moriarty, E.; Carr, M.; Bonham, S.; Carty, M.P.; Aldabbagh, F. Synthesis and toxicity towards normal and cancer cell lines of benzimidazolequinones containing fused aromatic rings and 2-aromatic ring substituents. Eur. J. Med. Chem. 2010, 45, 3762–3769. [Google Scholar] [CrossRef]
- Schulz, W.G.; Skibo, E.B. Inhibitors of topoisomerase II based on the benzodiimidazole and dipyrroloimidazobenzimidazole ring systems: Controlling DT-diaphorase reductive inactivation with steric bulk. J. Med. Chem. 2000, 43, 629–638. [Google Scholar] [CrossRef]
- Suleman, A.; Skibo, E.B. A comprehensive study of the active site residues of DT-diaphorase: Rational design of benzimidazolediones as DT-diaphorase substrates. J. Med. Chem. 2002, 45, 1211–1220. [Google Scholar] [CrossRef]
- Fagan, V.; Bonham, S.; Carty, M.P.; Aldabbagh, F. One-pot double intramolecular homolytic aromatic substitution routes to dialicyclic ring fused imidazobenzimidazolequinones and preliminary analysis of anticancer activity. Org. Biomol. Chem. 2010, 8, 3149–3156. [Google Scholar] [CrossRef] [PubMed]
- Fagan, V.; Bonham, S.; McArdle, P.; Carty, M.P.; Aldabbagh, F. Synthesis and toxicity of new ring-fused imidazo[5,4-f]benzimidazolequinones and mechanism using amine N-oxide cyclizations. Eur. J. Org. Chem. 2012, 2012, 1967–1975. [Google Scholar] [CrossRef]
- Fagan, V.; Bonham, S.; Carty, M.P.; Saenz-Méndez, P.; Eriksson, L.A.; Aldabbagh, F. COMPARE analysis of the toxicity of an iminoquinone derivative of the imidazo[5,4-f]benzimidazoles with NAD(P)H:quinone oxidoreductase 1 (NQO1) activity and computational docking of quinones as NQO1 substrates. Bioorg. Med. Chem. 2012, 20, 3223–3232. [Google Scholar] [CrossRef]
- Conboy, D.; Aldabbagh, F. 6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1′,2′:1,2]imidazo[4,5-f]benzimidazole-13-one: Synthesis and cytotoxicity evaluation. Molbank 2020, 2020, M118. [Google Scholar] [CrossRef]
- Dawood, K.M.; Abdel-Wahab, B.F. Synthetic routes to benzimidazole-based fused polyheterocycles. ARKIVOC 2010, 333–389. [Google Scholar] [CrossRef]
- Dawood, K.M.; Elwan, N.M.; Abdel-Wahab, B.F. Recent advances on the synthesis of azoles, azines and azepines fused to benzimidazole. ARKIVOC 2011, 111–195. [Google Scholar] [CrossRef]
- Khajuria, R.; Rasheed, S.; Khajuria, C.; Kapoor, K.K.; Das, P. Recent developments in the synthesis of pyrido[1,2-a]benzimidazoles. Synthesis 2018, 50, 2131–2149. [Google Scholar] [CrossRef]
- Manna, S.K.; Das, T.; Samanta, S. Polycyclic benzimidazole: Synthesis and photophysical properties. ChemistrySelect 2019, 4, 8781–8790. [Google Scholar] [CrossRef]
- Spiegel, L.; Kaufmann, H. Reduction of dinitrophenylpiperidine. II. Communication. Ber. Dtsch. Chem. Ges. 1908, 41, 679–685. [Google Scholar] [CrossRef]
- Bamberger, E.; Tschirner, F. Direct transformation of the aniline in phenylhydroxylamine. Ber. Dtsch. Chem. Ges. 1899, 32, 1675–1678. [Google Scholar] [CrossRef]
- Nair, M.D.; Adams, R. Benzimidazole syntheses by oxidative cyclization with peroxytrifluoroacetic acid. J. Am. Chem. Soc. 1961, 83, 3518–3521. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Suschitzky, H. Syntheses of heterocyclic compounds. Part IV. Oxidative cyclisation of aromatic amines and their N-acyl derivatives. J. Chem. Soc. 1963, 4666–4669. [Google Scholar] [CrossRef]
- Meth-Cohn, O. Mechanism of formation of benzimidazoles by oxidation of o-acylamino-N,N-dialkylanilines with peroxy-acids. J. Chem. Soc. C 1971, 1356–1357. [Google Scholar] [CrossRef]
- Fahey, K.; Aldabbagh, F. Synthesis of seven- and eight-membered [1,2-a] alicyclic ring-fused benzimidazoles and 3-aziridinylazepino[1,2-a]benzimidazolequinone as a potential antitumour agent. Tetrahedron Lett. 2008, 49, 5235–5237. [Google Scholar] [CrossRef]
- Gurry, M.; McArdle, P.; Aldabbagh, F. Synthesis of a spirocyclic oxetane-fused benzimidazole. Molecules 2015, 20, 13864–13874. [Google Scholar] [CrossRef]
- Hussain, H.; Green, I.R.; Ahmed, I. Journey describing applications of Oxone in synthetic chemistry. Chem. Rev. 2013, 113, 3329–3371. [Google Scholar] [CrossRef]
- Alkhader, M.A.; Perera, R.C.; Sinha, R.P.; Smalley, R.K. Synthesis of polynuclear heterocycles. Part 4.l imidazo[4,5-g] [3,1]-benzoxazinones, imidazo[4,5-g]quinazolinones, imidazo[4,5-g]quinazolinediones, and imidazo[4,5-f]indazolinones. J. Chem. Soc. Perkin 1 1979, 1056–1062. [Google Scholar] [CrossRef]
- Sweeney, M.; Gurry, M.; Keane, L.-A.J.; Aldabbagh, F. Greener synthesis using hydrogen peroxide in ethyl acetate of alicyclic ring-fused benzimidazoles and anti-tumour benzimidazolequinones. Tetrahedron Lett. 2017, 58, 3565–3567. [Google Scholar] [CrossRef]
- Gernon, M.D.; Wu, M.; Buszta, T.; Janney, P. Environmental benefits of methanesulfonic acid. Comparative properties and advantages. Green Chem. 1999, 1, 127–140. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Suschitzky, H.; Sutton, M.E. Oxidative cyclisations of o-substituted anilines and benzoic acids with manganese dioxide. J. Chem. Soc. C 1968, 1722–1726. [Google Scholar] [CrossRef]
- Möhrle, H. Gerloff, Tricyclische benzimidazolderivate. J. Arch. Pharm. 1978, 311, 381–393. [Google Scholar] [CrossRef]
- Sun, X.; Hu, Y.; Nie, S.-Z.; Yan, Y.-Y.; Zhang, X.-J.; Yan, M. Efficient construction of C=N double bonds via acceptorless dehydrogenative coupling. Adv. Synth. Catal. 2013, 355, 2179–2184. [Google Scholar] [CrossRef]
- Sun, X.; Lv, X.-H.; Ye, L.-M.; Hu, Y.; Chen, Y.-Y.; Zhang, X.-J.; Yan, M. Synthesis of benzimidazoles via iridium-catalyzed acceptorless dehydrogenative coupling. Org. Biomol. Chem. 2015, 13, 7381–7383. [Google Scholar] [CrossRef]
- Xue, D.; Long, Y.-Q. Metal-free TEMPO-promoted C(sp3)−H amination to afford multisubstituted benzimidazoles. J. Org. Chem. 2014, 79, 4727–4734. [Google Scholar] [CrossRef]
- Thapa, P.; Palacios, P.M.; Tran, T.; Pierce, B.S.; Foss, F.W., Jr. 1,2-Disubstituted benzimidazoles by the iron catalyzed cross-dehydrogenative coupling of isomeric o-phenylenediamine substrates. J. Org. Chem. 2020, 85, 1991–2009. [Google Scholar] [CrossRef]
- Li, Q.-Y.; Cheng, S.-Y.; Tang, H.-T.; Pan, Y.-M. Synthesis of rutaecarpine alkaloids via an electrochemical cross dehydrogenation coupling reaction. Green Chem. 2019, 21, 5517–5520. [Google Scholar] [CrossRef]
- Begunov, R.S.; Sakulina, V.O.; Syroeshkin, M.A.; Saverina, E.A.; Sokolov, A.A.; Minyaev, M.E. Electroreductive heterocyclization of ortho-piperidino substituted nitro(het)arenes. Mendeleev Commun. 2020, 30, 633–635. [Google Scholar] [CrossRef]
- Martin, J.; Meth-Cohn, O.; Suschitzky, H. A simple route to polychlorobenzimidazoles and related systems. Tetrahedron Lett. 1973, 14, 4495–4498. [Google Scholar] [CrossRef]
- Gurry, M.; Sweeney, M.; McArdle, P.; Aldabbagh, F. One-pot hydrogen peroxide and hydrohalic acid induced ring closure and selective aromatic halogenation to give new ring-fused benzimidazoles. Org. Lett. 2015, 17, 2856–2859. [Google Scholar] [CrossRef]
- Sweeney, M.; Keane, L.-A.J.; Gurry, M.; McArdle, P.; Aldabbagh, F. One-pot synthesis of dihalogenated ring-fused benzimidazolequinones from 3,6-dimethoxy-2-(cycloamino)anilines using hydrogen peroxide and hydrohalic acid. Org. Lett. 2018, 20, 6970–6974. [Google Scholar] [CrossRef]
- Conboy, D.; Mirallai, S.I.; Craig, A.; McArdle, P.; Al-Kinani, A.A.; Barton, S.; Aldabbagh, F. Incorporating morpholine and oxetane into benzimidazolequinone antitumor agents: The discovery of 1,4,6,9-tetramethoxyphenazine from hydrogen peroxide and hydroiodic acid-mediated oxidative cyclizations. J. Org. Chem. 2019, 84, 9811–9818. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.v.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Suschitzky, H. Heterocycles by ring closure of ortho-substituted t-anilines (The t-amino effect). Adv. Heterocycl. Chem. 1972, 14, 211–278. [Google Scholar] [CrossRef]
- Kwast, A.; Stachowska, K.; Trawczyński, A.; Wróbel, Z. N-Aryl-2-nitrosoanilines as intermediates in the synthesis of substituted phenazines from nitroarenes. Tetrahedron Lett. 2011, 52, 6484–6488. [Google Scholar] [CrossRef]
- Guttenberger, N.; Blankenfeldt, W.; Breinbauer, R. Recent developments in the isolation, biological function, biosynthesis, and synthesis of phenazine natural products. Bioorg. Med. Chem. 2017, 25, 6149–6166. [Google Scholar] [CrossRef] [PubMed]
- Conboy, D.; Aldabbagh, F. The reactivity of Oxone towards 4,6-di(cycloamino)-1,3-phenylenediamines: Synthesis of spirocyclic oxetane ring-fused imidazobenzimidazoles. ARKIVOC 2020, 180–191. [Google Scholar] [CrossRef]
- Purkait, A.; Roy, S.K.; Srivastava, H.K.; Jana, C.K. Metal-free sequential C(sp2)−H/OH and C(sp3)−H aminations of nitrosoarenes and N-heterocycles to ring-fused imidazoles. Org. Lett. 2017, 19, 2540–2543. [Google Scholar] [CrossRef]
- Saunders, K.H. Syntheses in the pyrido- and piperido-(1′:2′-1:2)benzimidazole series. J. Chem. Soc. 1955, 3275–3287. [Google Scholar] [CrossRef]
- Grantham, R.K.; Meth-Cohn, O. The formation of benzimidazolones and quinoxalines from o-nitrophenyldialkylanilines: A re-investigation. J. Chem. Soc. C 1969, 70–74. [Google Scholar] [CrossRef]
- Skibo, E.B.; Islam, I.; Schulz, W.G.; Zhou, R.; Bess, L.; Boruah, R. The organic chemistry of the pyrrolo[1,2-a]benzimidazole antitumor agents. An example of rational drug design. Synlett 1996, 297–309. [Google Scholar] [CrossRef]
- Suschitzky, H.; Sutton, M.E. Reductive cyclization of aromatic nitro compounds to benzimidazoles with titanous chloride. Tetrahedron 1968, 24, 4581–4587. [Google Scholar] [CrossRef]
- Alonso, J.; Halland, N.; Nazaré, M.; R’kyek, O.; Urmann, M.; Lindenschmidt, A. A direct, regioselective palladium-catalyzed synthesis of N-substituted benzimidazoles and imidazopyridines. Eur. J. Org. Chem. 2011, 234–237. [Google Scholar] [CrossRef]
- Hubbard, J.W.; Piegols, A.M.; Söderberg, B.C.G. Palladium-catalyzed N-heteroannulation of N-allyl- or N-benzyl-2-nitrobenzenamines: Synthesis of 2-substituted benzimidazoles. Tetrahedron 2007, 63, 7077–7085. [Google Scholar] [CrossRef]
- Joardar, S.; Bhattacharyya, A.; Das, S. A palladium on carbon catalyzed one-pot synthesis of substituted benzimidazoles. Synthesis 2014, 46, 3121–3132. [Google Scholar] [CrossRef]
- Lu, C.; Su, Z.; Jing, D.; Jin, S.; Xie, L.; Li, L.; Zheng, K. Intramolecular reductive cyclization of o-nitroarenes via biradical recombination. Org. Lett. 2019, 21, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Ermolenko, L.; Al-Mourabit, A. Redox condensation of o-halonitrobenzene with 1,2,3,4-tetrahydroisoquinoline: Involvement of an unexpected auto-catalyzed redox cascade. Chem. Commun. 2016, 52, 4914–4917. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Ermolenko, L.; Al-Mourabit, A. Formic acid as a sustainable and complementary reductant: An approach to fused benzimidazoles by molecular iodine-catalyzed reductive redox cyclization of o-nitro-t-anilines. Green Chem. 2016, 18, 2966–2970. [Google Scholar] [CrossRef]
- Deng, X.; McAllister, H.; Mani, N.S. CuI-catalyzed amination of arylhalides with guanidines or amidines: A facile synthesis of 1H-2-substituted benzimidazoles. J. Org. Chem. 2009, 74, 5742–5745. [Google Scholar] [CrossRef]
- Liubchak, K.; Nazarenko, K.; Tolmachev, A. Synthesis of annulated benzimidazoles via amidine cyclization. Tetrahedron 2012, 68, 2993–3000. [Google Scholar] [CrossRef]
- Baars, H.; Beyer, A.; Kohlhepp, S.V.; Bolm, C. Transition-metal-free synthesis of benzimidazoles mediated by KOH/DMSO. Org. Lett. 2014, 16, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, F.; Sun, Z. Synthesis of 1,2-disubstituted benzimidazoles using an aza-Wittig-equivalent process. RSC Adv. 2017, 7, 44421–44425. [Google Scholar] [CrossRef]
- Geng, X.; Liu, S.; Wang, W.; Qu, J.; Wang, B. tert-Amino effect-promoted rearrangement of aryl isothiocyanate: A versatile approach to benzimidazothiazepines and benzimidazothioethers. J. Org. Chem. 2020, 85, 12635–12643. [Google Scholar] [CrossRef]
- Huang, J.; He, Y.; Wang, Y.; Zhu, Q. Synthesis of benzimidazoles by PIDA-promoted direct C(sp2)-H imidation of N-arylamidines. Chem. Eur. J. 2012, 18, 13964–13967. [Google Scholar] [CrossRef] [PubMed]
- Kutsumura, N.; Kunimatsu, S.; Kagawa, K.; Otani, T.; Saito, T. Synthesis of benzimidazole-fused heterocycles by intramolecular oxidative C-N bond formation using hypervalent iodine reagents. Synthesis 2011, 3235–3240. [Google Scholar] [CrossRef]
- Rasheed, S.; Rao, D.N.; Das, P. Copper-catalyzed inter- and intramolecular C−N bond formation: Synthesis of benzimidazole-fused heterocycles. J. Org. Chem. 2015, 80, 9321–9327. [Google Scholar] [CrossRef]
- Elder, M.S.; Melson, G.A.; Busch, D.I. Reactions of coordinated ligands. XII. The synthesis of o-benzylene-2,1-benzimidazole in the presence of nickel(II) ions, and a study of some of its metal complexes. Inorg. Chem. 1966, 5, 74–77. [Google Scholar] [CrossRef]
- Chen, J.; Qu, J.; Zhang, Y.; Chen, Y.; Liu, N.; Chen, B. Metal-free construction of tricyclic or tetracyclic compounds, acid-promoted synthesis of benzo[4,5]imidazo[2,1-a]isoindole and 1,2-dialkyl-2,3-dihydrobenzimidazoles. Tetrahedron 2013, 69, 316–319. [Google Scholar] [CrossRef]
- Anastasiou, D.; Campi, E.M.; Chaouk, H.; Jackson, W.R. Synthesis of benzimidazoles containing a fused alicyclic ring by rhodium-catalysed hydroformylation of N-alkenyl-1,2-diaminobenzenes. Tetrahedron 1992, 48, 7467–7478. [Google Scholar] [CrossRef]
- De Selms, R.C. Benzimidazoles. II. Synthesis of N-heterocyclic ring systems containing 1,2-fused benzimidazole moieties. J. Org. Chem. 1962, 27, 2165–2167. [Google Scholar] [CrossRef]
- Haque, M.R.; Rasmussen, M. Ambident heterocyclic reactivity: Intramolecular alkylations of 2,4-disubstituted benzimidazoles. Tetrahedron 1997, 53, 6937–6958. [Google Scholar] [CrossRef]
- Qin, H.; Miao, Y.; Xu, J.; Bi, Q.; Qu, W.; Liu, W.; Feng, F.; Sun, H. A facile and efficient [4 + 2] annulation reaction of sulfur ylides: Access to N-fused benzimidazoles. Org. Chem. Front. 2019, 6, 205–208. [Google Scholar] [CrossRef]
- Almansour, A.I.; Arumugam, N.; Kumar, R.S.; Soliman, S.M.; Altaf, M.; Ghabbour, H.A. Synthesis, spectroscopic, X-ray diffraction and DFT studies of novel benzimidazole fused-1,4-oxazepines. Molecules 2016, 23, 724. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Chen, C.; Yue, Y.; Yu, Y.; Peng, J. Palladium(II)-catalyzed sequential C−H arylation/aerobic oxidative C−H amination: One-pot synthesis of benzimidazole-fused phenanthridines from 2-arylbenzimidazoles and aryl halides. J. Org. Chem. 2015, 80, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.L.; Vasudevan, A.; Bergman, R.G.; Ellman, J.A.; Souers, A.J. Microwave-assisted C-H bond activation: A rapid entry into functionalized heterocycles. Org. Lett. 2003, 5, 2131–2134. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Qi, S.-L.; Luan, Y.-X.; Han, X.-W.; Wang, S.; Chen, H.; Ye, M. Enantioselective Ni−Al bimetallic catalyzed exo-selective C−H cyclization of imidazoles with alkenes. J. Am. Chem. Soc. 2018, 140, 5360–5364. [Google Scholar] [CrossRef]
- Loup, J.; Müller, V.; Ghorai, D.; Ackermann, L. Enantioselective aluminum-free alkene hydroarylations through C-H activation by a chiral nickel/JoSPOphos manifold. Angew. Chem. Int. Ed. 2019, 58, 1749–1753. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Y.; Wang, H.; Guo, D.; Ye, D.; Xu, Y.; Jiang, H.; Liu, H. Silver-catalyzed intramolecular hydroamination of alkynes in aqueous media: Efficient and regioselective synthesis for fused benzimidazoles. Green Chem. 2011, 13, 397–405. [Google Scholar] [CrossRef]
- Dhole, S.; Sun, C.-M. Direct access to dihydrobenzoimidazo[2,1-a]isoquinolines through ruthenium-catalyzed formal [4+2] annulation. Adv. Synth. Catal. 2019, 361, 535–541. [Google Scholar] [CrossRef]
- Pereira, K.C.; Porter, A.L.; DeBoef, B. Intramolecular arylation of benzimidazoles via Pd(II)/Cu(I) catalyzed cross-dehydrogenative coupling. Tetrahedron Lett. 2014, 55, 1729–1732. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, X.; Hu, J.; Zhang, M.; Wang, L. Palladium-catalyzed C(sp2)-H activation for the formation of C-N bonds: Rapid access to benzimidazoquinazolines. Asian J. Org. Chem. 2019, 8, 417–421. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, Y.; Huang, J.; Chen, W. Copper-catalyzed synthesis of N-fused heterocycles through regioselective 1,2-aminothiolation of 1,1-dibromoalkenes. Org. Lett. 2010, 12, 3704–3707. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.P.; Dao, P.D.Q.; Cho, C.S. Synthesis of 2-aminoquinazoline- and 2-aminopyrimidine-fused hybrid scaffolds by copper-catalyzed C(sp2)–N coupling and cyclization followed by oxidation. Eur. J. Org. Chem. 2020, 2020, 3468–3474. [Google Scholar] [CrossRef]
- Diep, T.D.; Dao, P.D.Q.; Ho, S.L.; Cho, C.S. Copper-catalyzed synthesis of trinuclear N-fused hybrid scaffolds by double C(sp2)–N bond formation between 2-(2-bromoaryl)indoles and 2-aminoazoles. Eur. J. Org. Chem. 2020, 2020, 2807–2812. [Google Scholar] [CrossRef]
- Lopes, A.B.; Wagner, P.; Gulea, M. Synthesis of benzimidazole-fused medium-sized N,S-heterocycles via palladium-catalyzed cyclizations. Eur. J. Org. Chem. 2019, 1361–1370. [Google Scholar] [CrossRef]
- Joyce, E.; McArdle, P.; Aldabbagh, F. Acetic anhydride generated imidazolium ylide in ring closures onto carboxylic acids; part of the synthesis of new potential bioreductive antitumor agents. Synlett 2011, 1097–1100. [Google Scholar] [CrossRef]
- Joyce, E.; Kavanagh, P.; Leech, D.; Karpinska, J.; McArdle, P.; Aldabbagh, F. Acetic anhydride mediated condensation of aromatic o-diacid dichlorides with benzimidazoles to provide electro-reducible p-dione adducts. Tetrahedron Lett. 2012, 53, 3788–3791. [Google Scholar] [CrossRef]
- O’Connell, J.M.; Moriarty, E.; Aldabbagh, F. Access to aromatic ring-fused benzimidazoles using photochemical substitutions of the benzimidazol-2-yl radical. Synthesis 2012, 44, 3371–3377. [Google Scholar] [CrossRef]
- Clyne, M.A.; Aldabbagh, F. Photochemical intramolecular aromatic substitutions of the imidazol-2-yl radical are superior to those mediated by Bu3SnH. Org. Biomol. Chem. 2006, 4, 268–277. [Google Scholar] [CrossRef]
- Coyle, R.; Fahey, K.; Aldabbagh, F. Barton esters for initiator-free radical cyclisation with heteroaromatic substitution. Org. Biomol. Chem. 2013, 11, 1672–1682. [Google Scholar] [CrossRef]
- Moriarty, E.; Aldabbagh, F. Synthesis of aryl ring-fused benzimidazolequinones using 6-exo-trig radical cyclizations. Tetrahedron Lett. 2009, 50, 5251–5253. [Google Scholar] [CrossRef]
- McLoughlin, P.T.F.; Clyne, M.; Aldabbagh, F. Intermolecular “oxidative” aromatic substitution reactions of the imidazol-5-yl radical mediated by the “reductant” Bu3SnH. Tetrahedron 2004, 60, 8065–8071. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Bowry, V.W.; Bowman, W.R.; Mann, E.; Parr, J.; Storey, J.M.D. The mechanism of Bu3SnH-mediated homolytic aromatic substitution. Angew. Chem. Int. Ed. 2004, 43, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Gurry, M.; Aldabbagh, F. A new era for homolytic aromatic substitution: Replacing Bu3SnH with efficient light-induced chain reactions. Org. Biomol. Chem. 2016, 11, 3849–3862. [Google Scholar] [CrossRef]
- Coyle, R.; McArdle, P.; Aldabbagh, F. Tandem reactions via Barton esters with intermolecular addition and vinyl radical substitution onto Indole. J. Org. Chem. 2014, 79, 5903–5907. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pan, C.; Yuan, C.; Yu, J.-T. Peroxide-mediated synthesis of benzimidazo[2,1-a]isoquinoline-6(5H)-ones via cascade methylation/ethylation and intramolecular cyclization. Org. Biomol. Chem. 2021, 19, 619–626. [Google Scholar] [CrossRef]
- Sun, K.; Li, S.-J.; Chen, X.L.; Liu, Y.; Huang, X.-Q.; Wei, D.-H.; Qu, L.-B.; Zhao, Y.-F.; Yu, B. Silver-catalyzed decarboxylative radical cascade cyclization toward benzimidazo[2,1-a]isoquinolin-6(5H)-ones. Chem. Commun. 2019, 55, 2861–2864. [Google Scholar] [CrossRef]
- Bera, S.K.; Alam, M.T.; Mal, P. C−N coupling via antiaromatic endocyclic nitrenium ions. J. Org. Chem. 2019, 84, 12009–12020. [Google Scholar] [CrossRef] [PubMed]
- Dao, P.D.Q.; Cho, C.S. Construction of binuclear benzimidazole-fused quinazolinones and pyrimidinones using aryl isocyanates as building blocks by transition-metal-free C(sp2)−N coupling. J. Org. Chem. 2020, 85, 13354–13362. [Google Scholar] [CrossRef]
- Chen, W.; Du, Y.; Wang, M.; Fang, Y.; Yu, W.; Chang, J. Synthesis of benzo[4,5]imidazo[1,2-a]quinoxalines by I2-mediated sp3 C–H amination. Org. Chem. Front. 2020, 7, 3705–3708. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Cunningham, D.; Kavanagh, P.; Leech, D.; McArdle, P.; Aldabbagh, F. Synthesis of benzimidazolequinone analogue of cyclopropamitosene antitumor agents. Synlett 2004, 2382–2384. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Aldabbagh, F. Synthesis of pyrrolo- and pyrido[1,2-a]benzimidazolequinone anti-tumor agents containing a fused cyclopropane ring. Synthesis 2005, 1069–1076. [Google Scholar] [CrossRef]
- Aldabbagh, F.; Bowman, W.R. Radical cyclisation onto imidazoles and benzimidazoles. Tetrahedron 1999, 55, 4109–4122. [Google Scholar] [CrossRef]
- Conboy, D.; Kielty, P.; Bear, J.C.; Cockcroft, J.K.; Farras, P.; McArdle, P.; Singer, R.J.; Smith, D.A.; Aldabbagh, F. Ring-fused dimethoxybenzimidazole-benzimidazolequinone (DMBBQ): Tunable halogenation and quinone formation using NaX/Oxone. Org. Biomol. Chem. 2021, 19, 2716–2724. [Google Scholar] [CrossRef] [PubMed]












































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
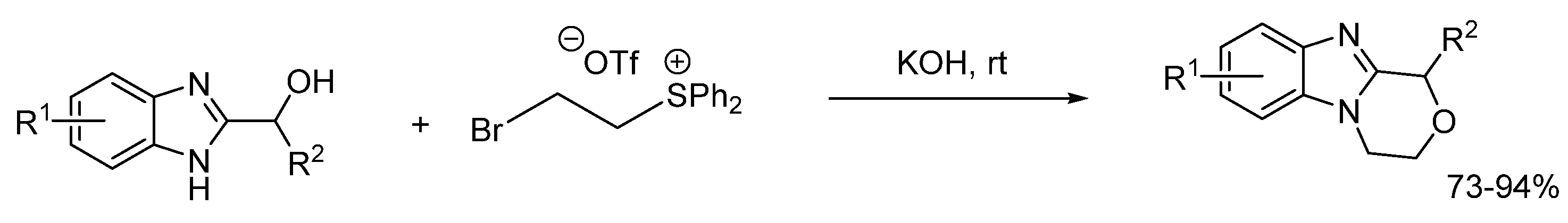{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sweeney, M.; Conboy, D.; Mirallai, S.I.; Aldabbagh, F. Advances in the Synthesis of Ring-Fused Benzimidazoles and Imidazobenzimidazoles. Molecules 2021, 26, 2684. https://doi.org/10.3390/molecules26092684
Sweeney M, Conboy D, Mirallai SI, Aldabbagh F. Advances in the Synthesis of Ring-Fused Benzimidazoles and Imidazobenzimidazoles. Molecules. 2021; 26(9):2684. https://doi.org/10.3390/molecules26092684
Chicago/Turabian StyleSweeney, Martin, Darren Conboy, Styliana I. Mirallai, and Fawaz Aldabbagh. 2021. "Advances in the Synthesis of Ring-Fused Benzimidazoles and Imidazobenzimidazoles" Molecules 26, no. 9: 2684. https://doi.org/10.3390/molecules26092684
APA StyleSweeney, M., Conboy, D., Mirallai, S. I., & Aldabbagh, F. (2021). Advances in the Synthesis of Ring-Fused Benzimidazoles and Imidazobenzimidazoles. Molecules, 26(9), 2684. https://doi.org/10.3390/molecules26092684