An Approach to Paracyclophane-Based Tetrathiafulvalenes: Synthesis and Characterization of a Pseudo-Geminal [2.2]Paracyclophane 1,3-Dithia-2-Thione

 ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
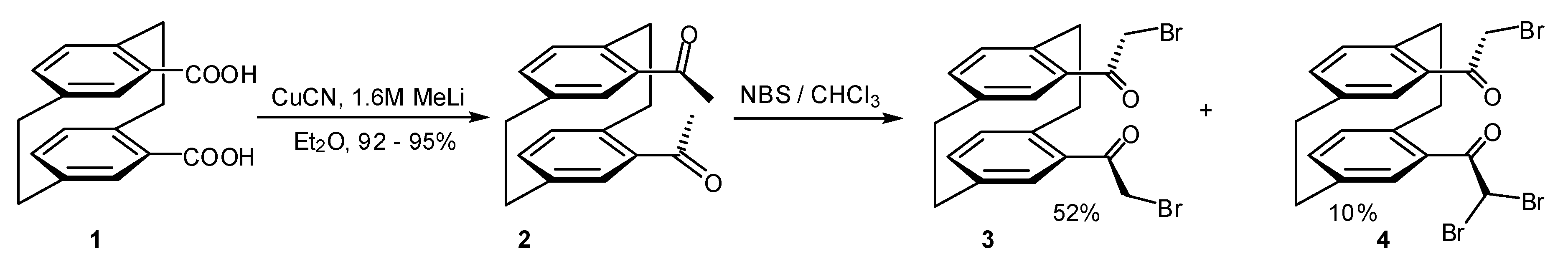
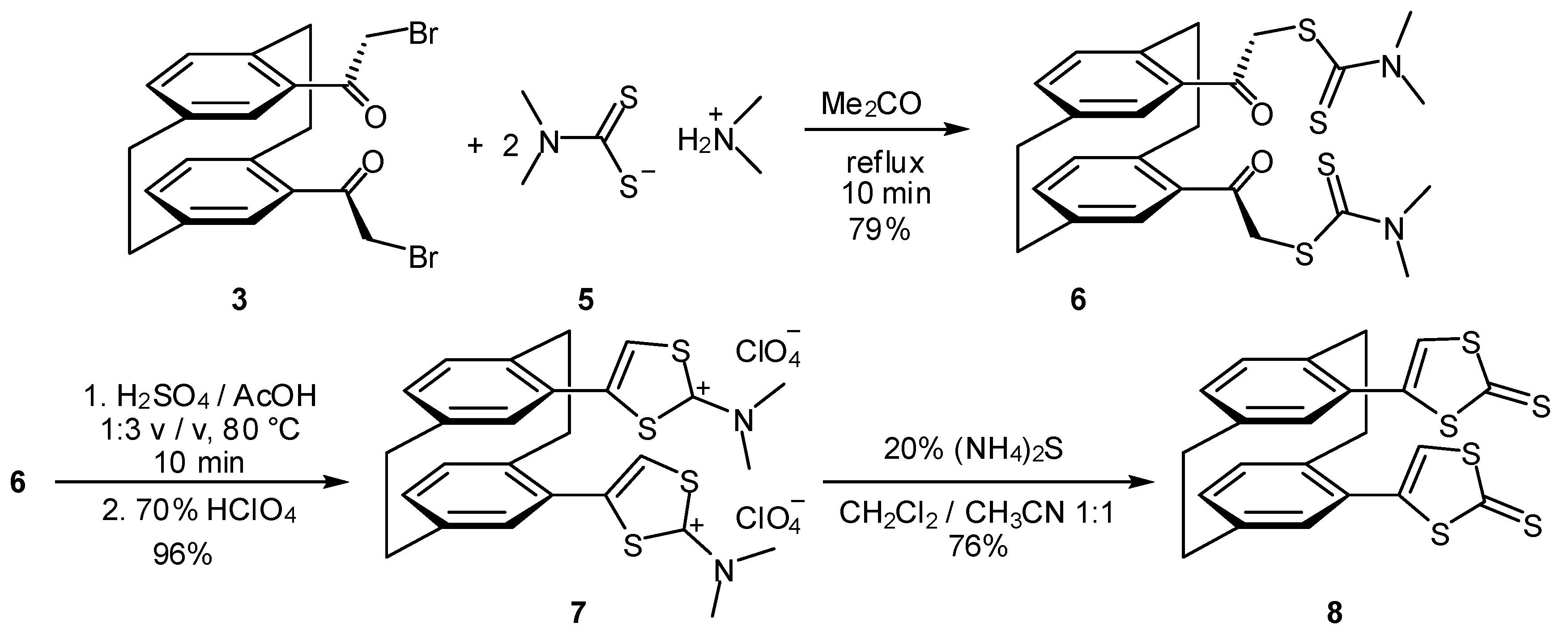
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.1.1. 4,15-Bis(acetyl)[2.2]paracyclophane (2)
3.1.2. Bromination of 4,15-bis(acetyl)[2.2]paracyclophane
3.1.3. 4,15-Bis(bromoacetyl)[2.2]paracyclophane (3)
3.1.4. 4-Bromoacetyl-15-(dibromo)acetyl[2.2]paracyclophane (4)
3.1.5. 4,15-Bis(N,N-dimethyldithiocarbamate) (6)
3.1.6. 4,15-Bis(1,3-dithiol-2-ylium) perchlorate (7)
3.1.7. 4,15-Bis(1,3-dithia-2-thione) (8)
3.2. X-ray Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Brown, C.J.; Farthing, A.C. Preparation and structure of di-p-xylylene. Nature (London) 1949, 164, 915–916. [Google Scholar] [CrossRef]
- Cram, D.J.; Steinberg, H. Macro Rings. I. Preparation and Spectra of the Paracyclophanes. J. Am. Chem. Soc. 1951, 73, 5691–5704. [Google Scholar] [CrossRef]
- Vögtle, F.; Neumann, P. The Synthesis of [2.2] Phanes. Synthesis 1973, 85–103. [Google Scholar] [CrossRef]
- Staab, H.A.; Knaus, G.H.; Henke, H.-E.; Krieger, C. Elektron-Donor-Acceptor-Verbindungen, XXIX. Elektron-Donor-Acceptor-Paracyclophane mit 7,7,8,8-Tetracyanchinodimethan (TCNQ) als Acceptor-Einheit. Chem. Ber. 1983, 116, 2785–2807. [Google Scholar] [CrossRef]
- Boekelheide, V. Syntheses and properties of the [2n]Cyclophanes. Top. Curr. Chem. 1983, 113, 87–143. [Google Scholar] [CrossRef]
- Hopf, H.; Marquard, C. Strain and its Implications in Organic Chemistry; de Meijere, A., Blechert, S., Eds.; Kluwer: Dordrecht, Germany, 1983; pp. 297–332. [Google Scholar] [CrossRef]
- Hopf, H. Step by step--from nonnatural to biological molecular ladders. Angew. Chem. Int. Ed. 2003, 42, 2822–2825. [Google Scholar] [CrossRef] [PubMed]
- Gleiter, R.; Hopf, H. (Eds.) Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Gibson, S.E.; Knight, J.D. [2.2]Paracyclophane derivatives in asymmetric catalysis. Org. Biomol. Chem. 2003, 1, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Spuling, E.; Knoll, D.M.; Lahann, J.; Bräse, S. Planar chiral [2.2]Paracyclophanes: From synthetic curiosity to applications in asymmetric synthesis and materials. Chem. Soc. Rev. 2018, 47, 6947–6963. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Spuling, E.; Knoll, D.M.; Lahann, J.; Bräse, S. Regioselective Functionalization of [2.2]Paracyclophanes: Recent Synthetic Progress and Perspectives. Angew. Chem. Int. Ed. 2020, 59, 2156–2170. [Google Scholar] [CrossRef]
- Bartholomew, G.P.; Bazan, G.C. Bichromophoric Paracyclophanes: Models for Interchromophore Delocalization. Acc. Chem. Res. 2001, 34, 30–39. [Google Scholar] [CrossRef]
- Hopf, H. [2.2]Paracyclophanes in Polymer Chemistry and Materials Science. Angew. Chem. Int. Ed. 2008, 47, 9808–9812. [Google Scholar] [CrossRef]
- Vögtle, F. Cyclophane Chemistry, Synthesis, Structure and Reactions; Wiley: Chichester, UK, 1993; pp. 71–111. [Google Scholar] [CrossRef]
- Yamada, J. TTF Chemistry Fundamentals and Applications of Tetrathiafulvalene; Sugimoto, T., Ed.; Kodansha: Tokyo, Japan; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar] [CrossRef]
- Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, Oligoacenenes, and Their Buckminsterfullerene Derivatives: The Brick and Mortar of Organic Electronics. Chem. Rev. 2004, 104, 4891–4946. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, L.G.; Bahrin, L.G.; Jones, P.G.; Birsa, M.L.; Hopf, H. [2.2]Paracyclophane derivatives containing tetrathiafulvalene moieties. Beilstein J. Org. Chem. 2015, 11, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Zitt, H.; Dix, I.; Hopf, H.; Jones, P.G. 4,15-Diamino[2.2]Paracyclophane, a Reusable Template for Topochemical Reaction Control in Solution. Eur. J. Org. Chem. 2002, 2298–2307. [Google Scholar] [CrossRef]
- CCDC-1545015 contains the supplementary crystallographic data for compound 2. The data were deposited as a database communication: Jones, P.G.; Bahrin, L.G.; Birsa, M.L.; Hopf, H. CSD Communications, 2017, doi:10.5517/ccdc.csd.cc1nvq6f. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
- CCDC-2036116 and CCDC-2036117 contain the supplementary crystallographic data for compounds 3 and 4 respectively. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
- Sarbu, L.G.; Shova, S.; Peptanariu, D.; Sandu, I.A.; Birsa, L.M.; Bahrin, L.G. The Cytotoxic Properties of Some Tricyclic 1,3-Dithiolium Flavonoids. Molecules 2019, 24, 2459. [Google Scholar] [CrossRef]
- Bahrin, L.G.; Apostu, M.O.; Birsa, L.M.; Stefan, M. The antibacterial properties of sulfur containing flavonoids. Bioorg. Med. Chem. Lett. 2014, 24, 2315–2318. [Google Scholar] [CrossRef]
- Bahrin, L.G.; Hopf, H.; Jones, P.G.; Sarbu, L.G.; Babii, C.; Mihai, A.C.; Stefan, M.; Birsa, L.M. Antibacterial structure–activity relationship studies of several tricyclic sulfur-containing flavonoids. Beilstein J. Org. Chem. 2016, 12, 1065–1071. [Google Scholar] [CrossRef]
- Bahrin, L.G.; Jones, P.G.; Hopf, H. Tricyclic flavonoids with 1,3-dithiolium substructure. Beilstein J. Org. Chem. 2012, 8, 1999–2003. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. They can be prepared according with the reported experimental procedures. |





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahrin, L.G.; Hopf, H.; Jones, P.G.; Birsa, M.L.; Sarbu, L.G. An Approach to Paracyclophane-Based Tetrathiafulvalenes: Synthesis and Characterization of a Pseudo-Geminal [2.2]Paracyclophane 1,3-Dithia-2-Thione. Molecules 2020, 25, 5262. https://doi.org/10.3390/molecules25225262
Bahrin LG, Hopf H, Jones PG, Birsa ML, Sarbu LG. An Approach to Paracyclophane-Based Tetrathiafulvalenes: Synthesis and Characterization of a Pseudo-Geminal [2.2]Paracyclophane 1,3-Dithia-2-Thione. Molecules. 2020; 25(22):5262. https://doi.org/10.3390/molecules25225262
Chicago/Turabian StyleBahrin, Lucian G., Henning Hopf, Peter G. Jones, Mihail L. Birsa, and Laura G. Sarbu. 2020. "An Approach to Paracyclophane-Based Tetrathiafulvalenes: Synthesis and Characterization of a Pseudo-Geminal [2.2]Paracyclophane 1,3-Dithia-2-Thione" Molecules 25, no. 22: 5262. https://doi.org/10.3390/molecules25225262
APA StyleBahrin, L. G., Hopf, H., Jones, P. G., Birsa, M. L., & Sarbu, L. G. (2020). An Approach to Paracyclophane-Based Tetrathiafulvalenes: Synthesis and Characterization of a Pseudo-Geminal [2.2]Paracyclophane 1,3-Dithia-2-Thione. Molecules, 25(22), 5262. https://doi.org/10.3390/molecules25225262