Gas Phase Computational Study of Diclofenac Adsorption on Chitosan Materials


Abstract
1. Introduction
2. Results
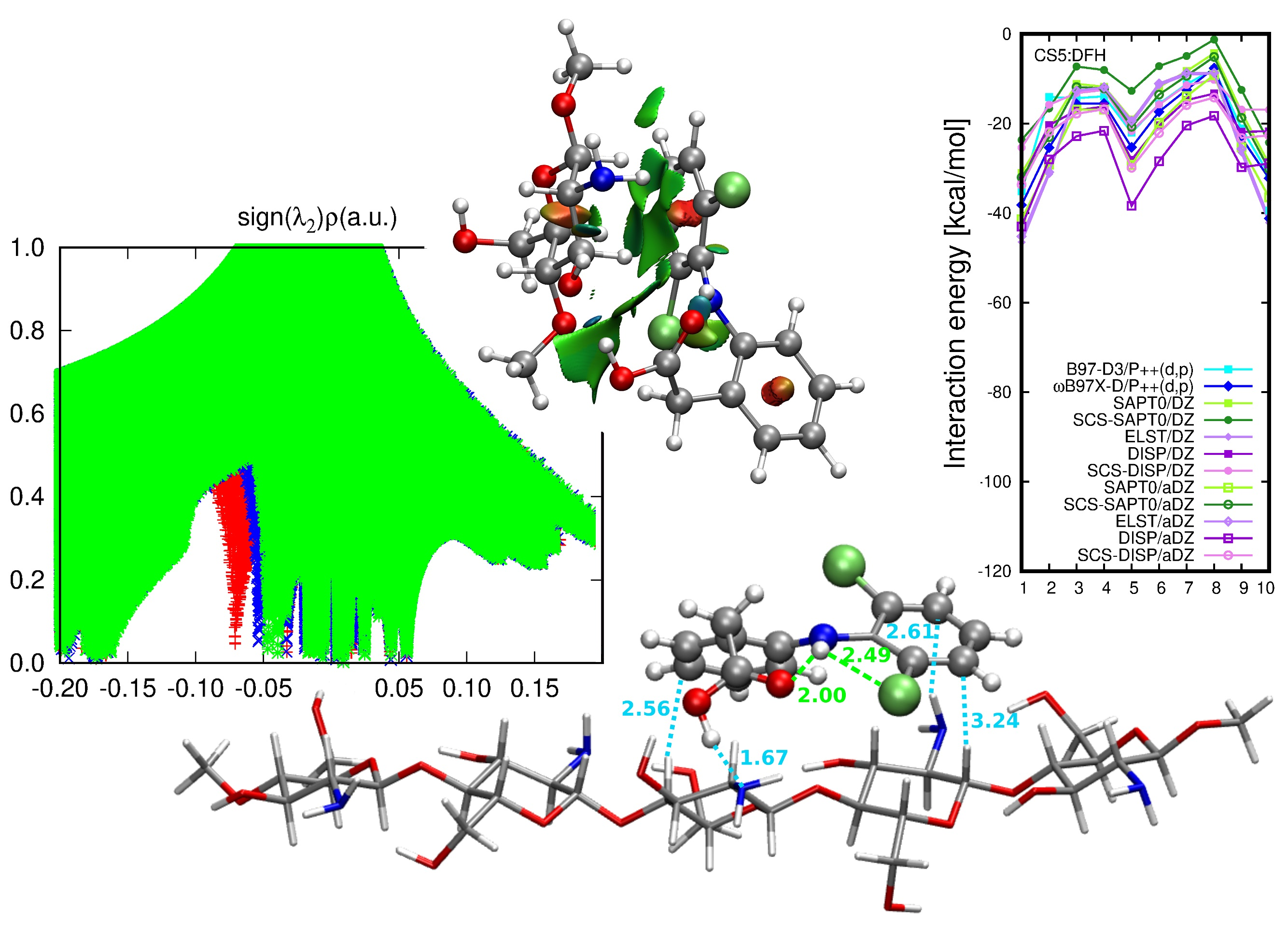
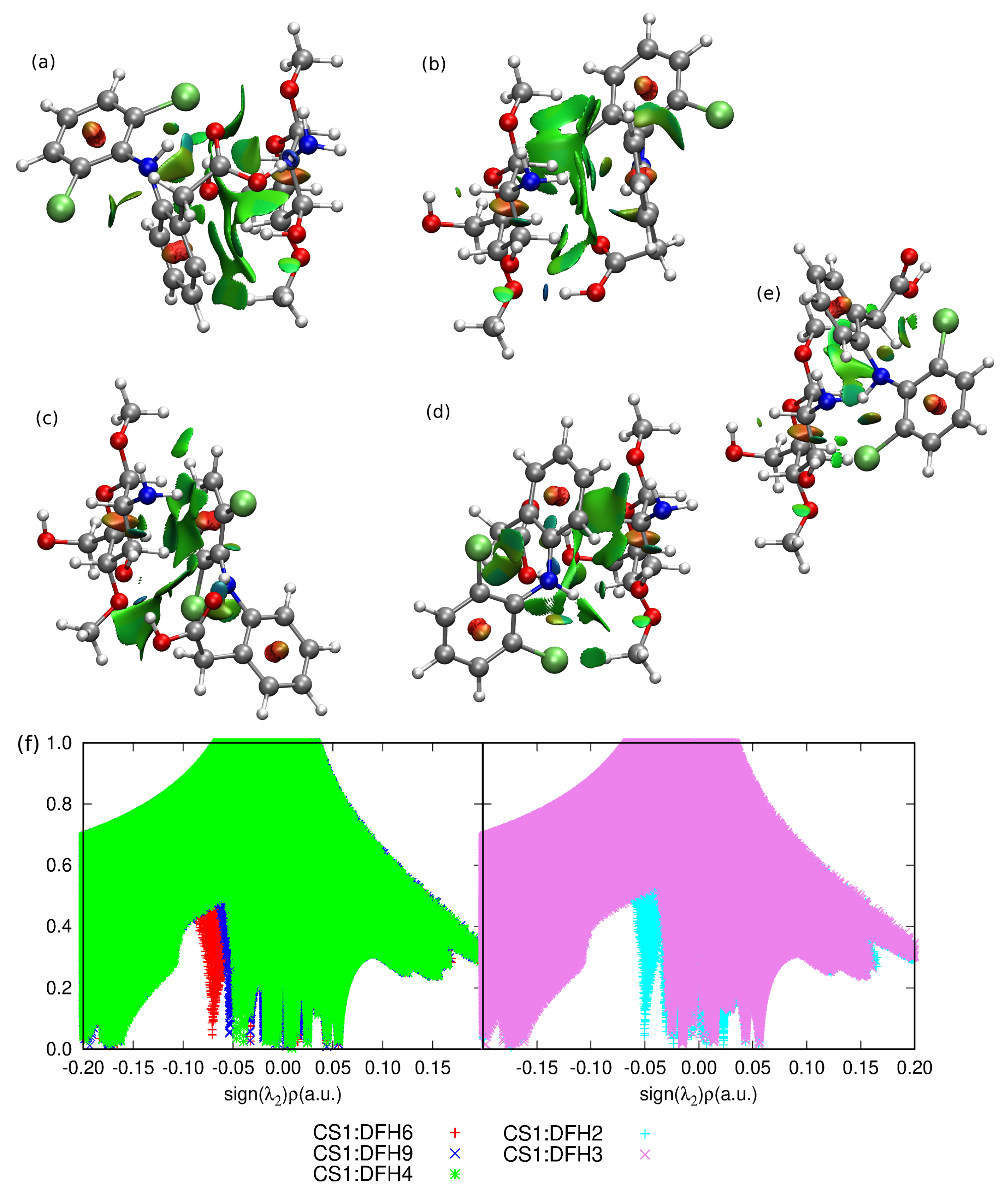
2.1. Diclofenac Interaction with Single Pristine Chitosan Unit
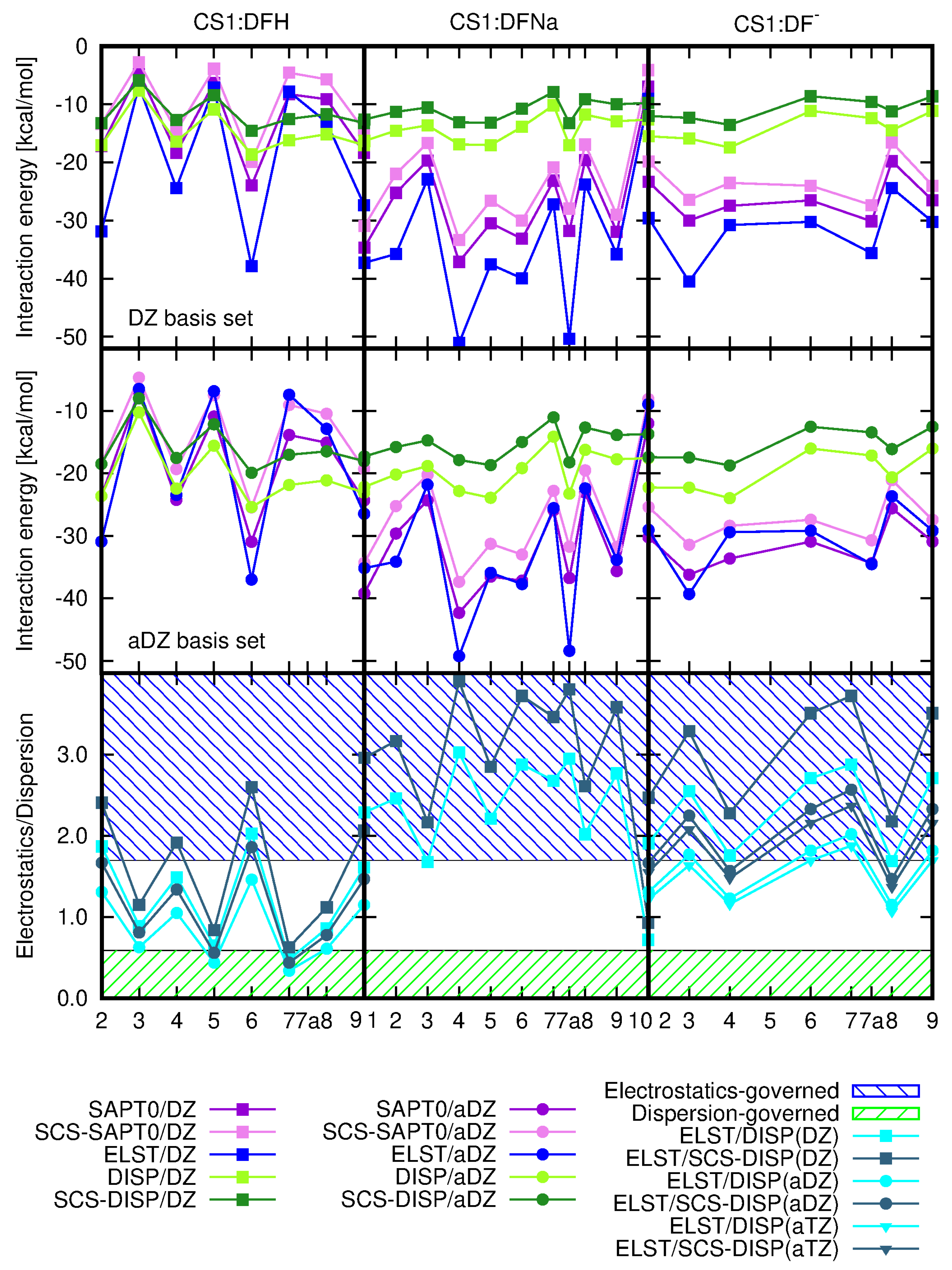
Nature of the CS1: Diclofenac Interaction
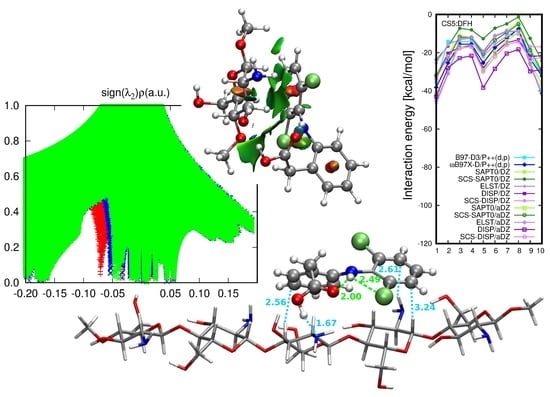
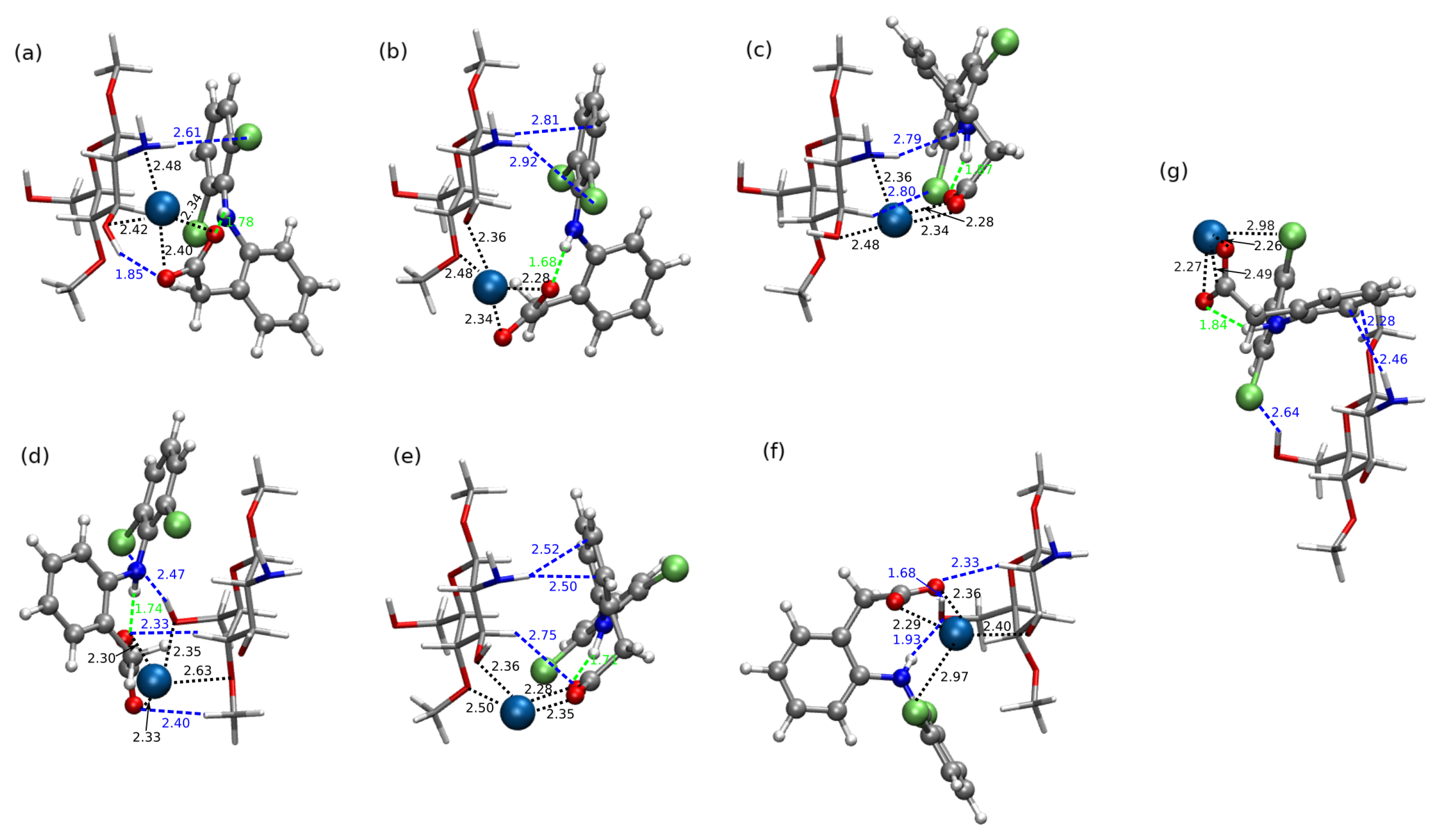
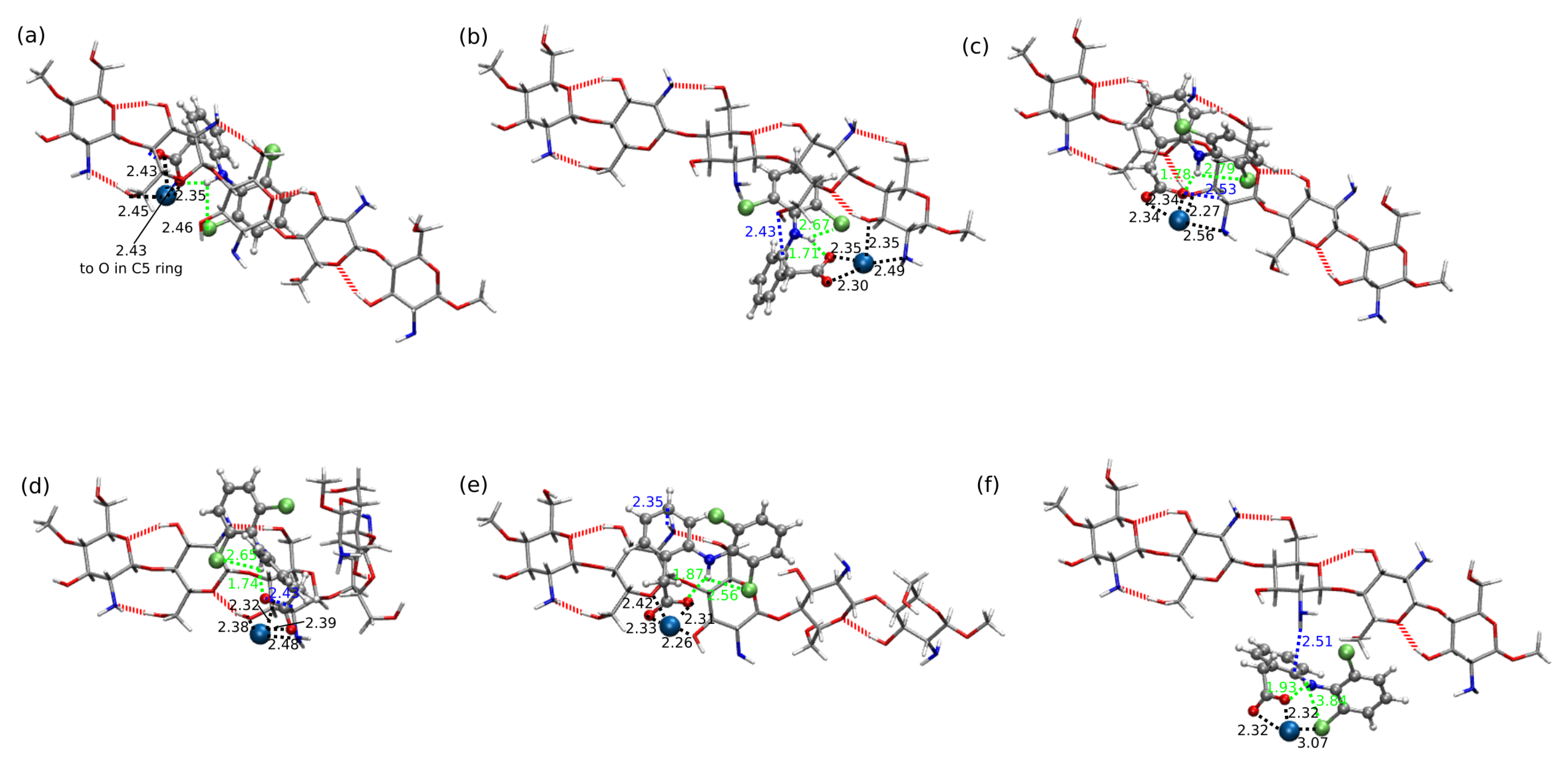
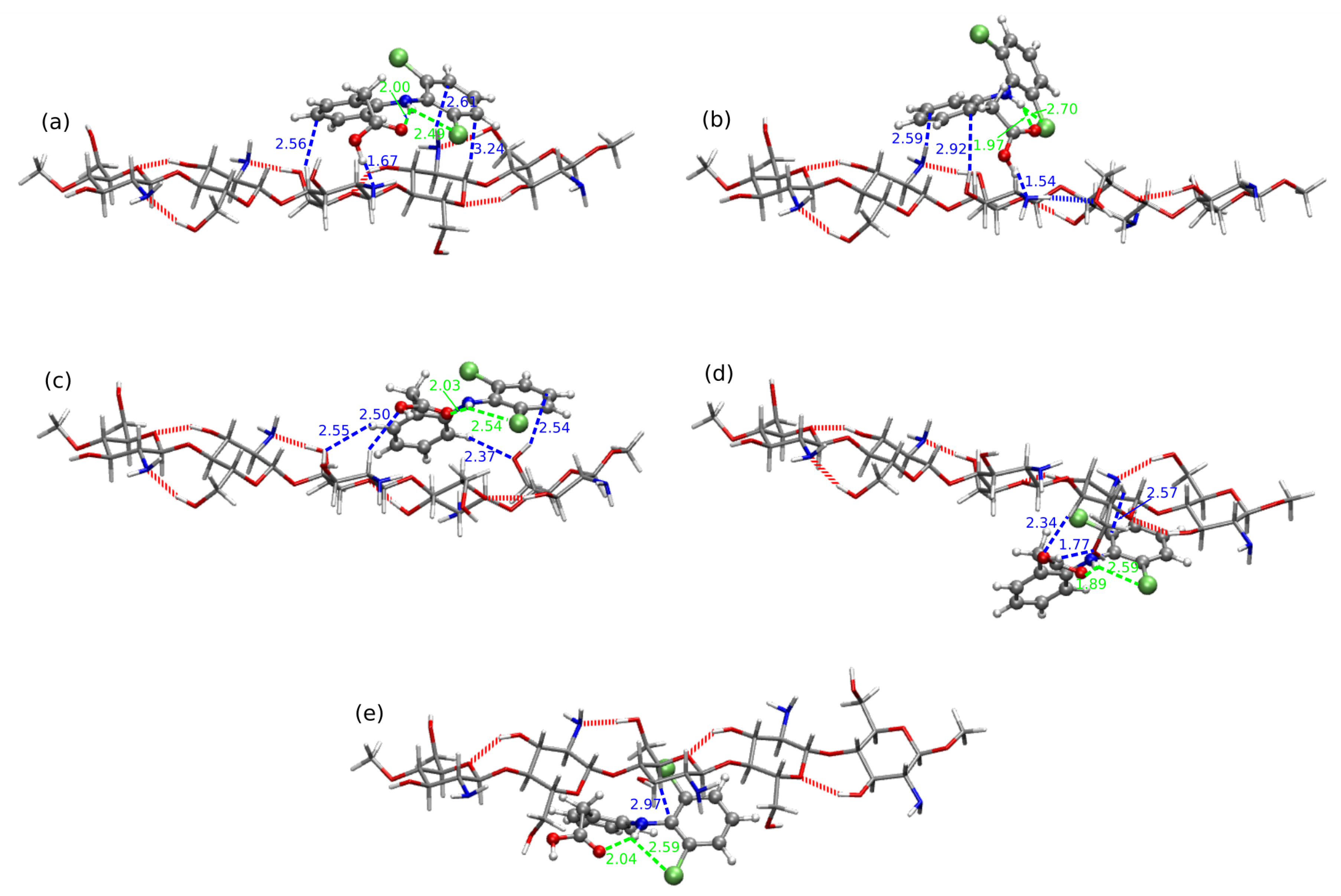
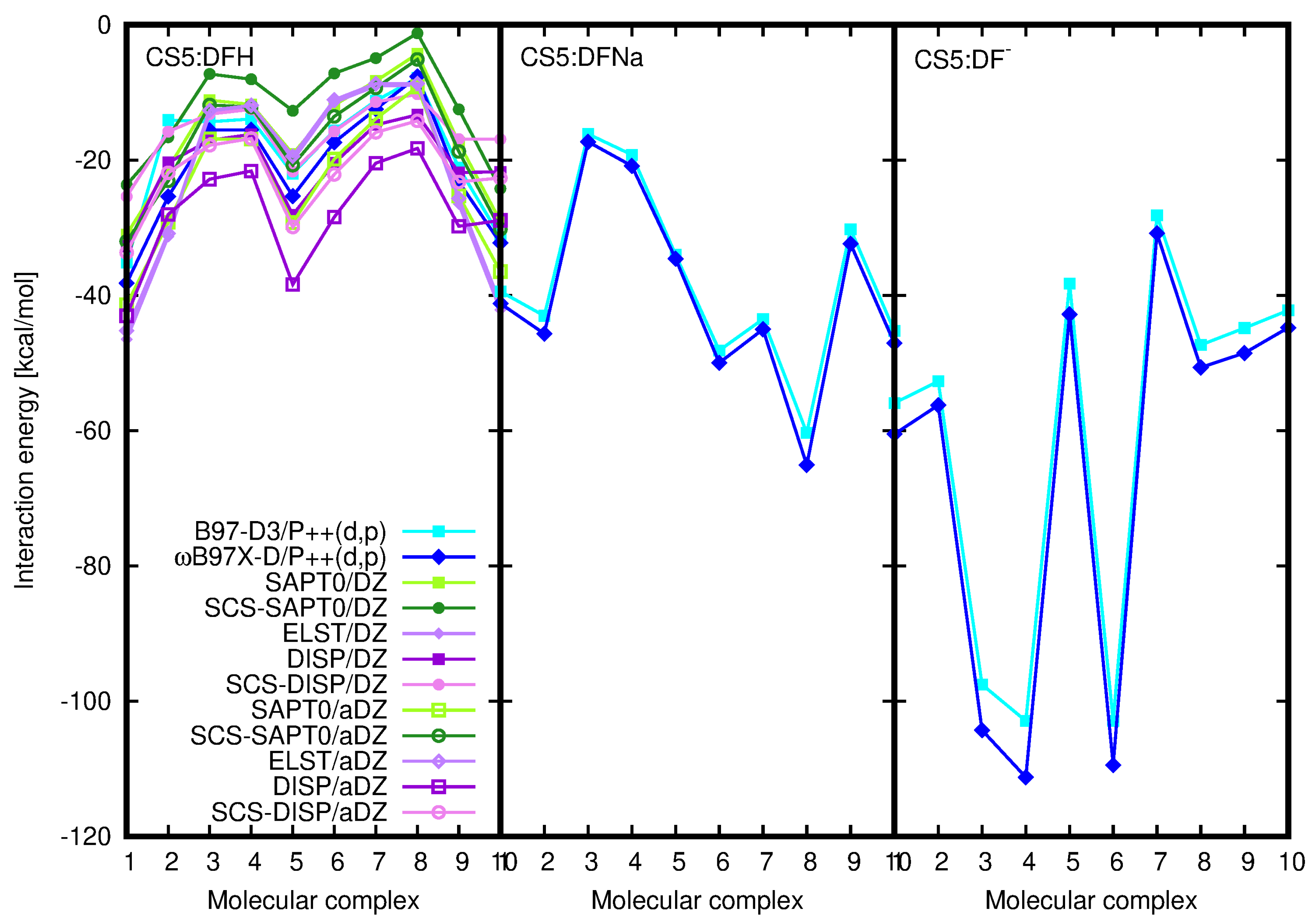
2.2. Diclofenac Interaction with Pristine Five-Unit Chitosan Chain
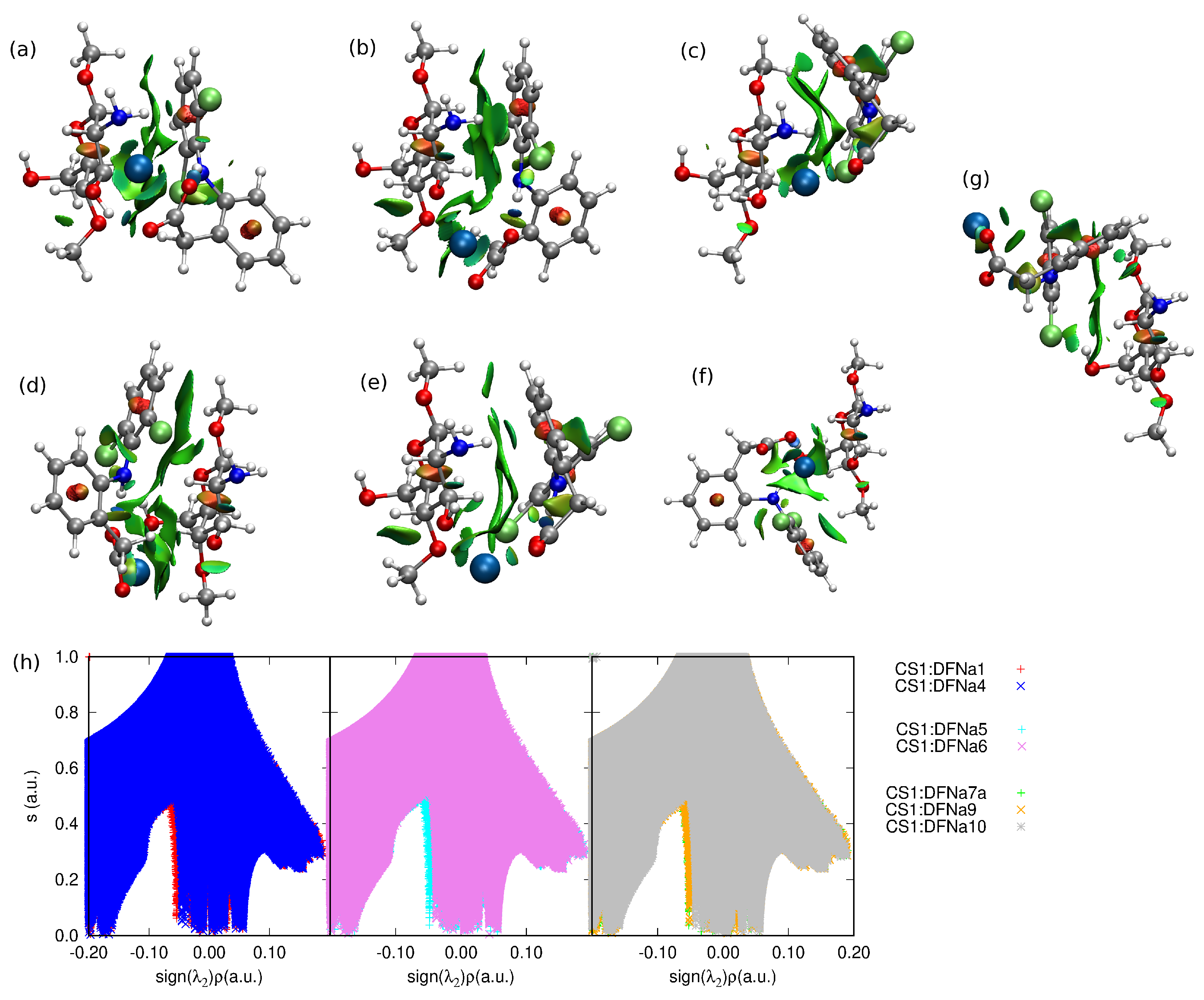
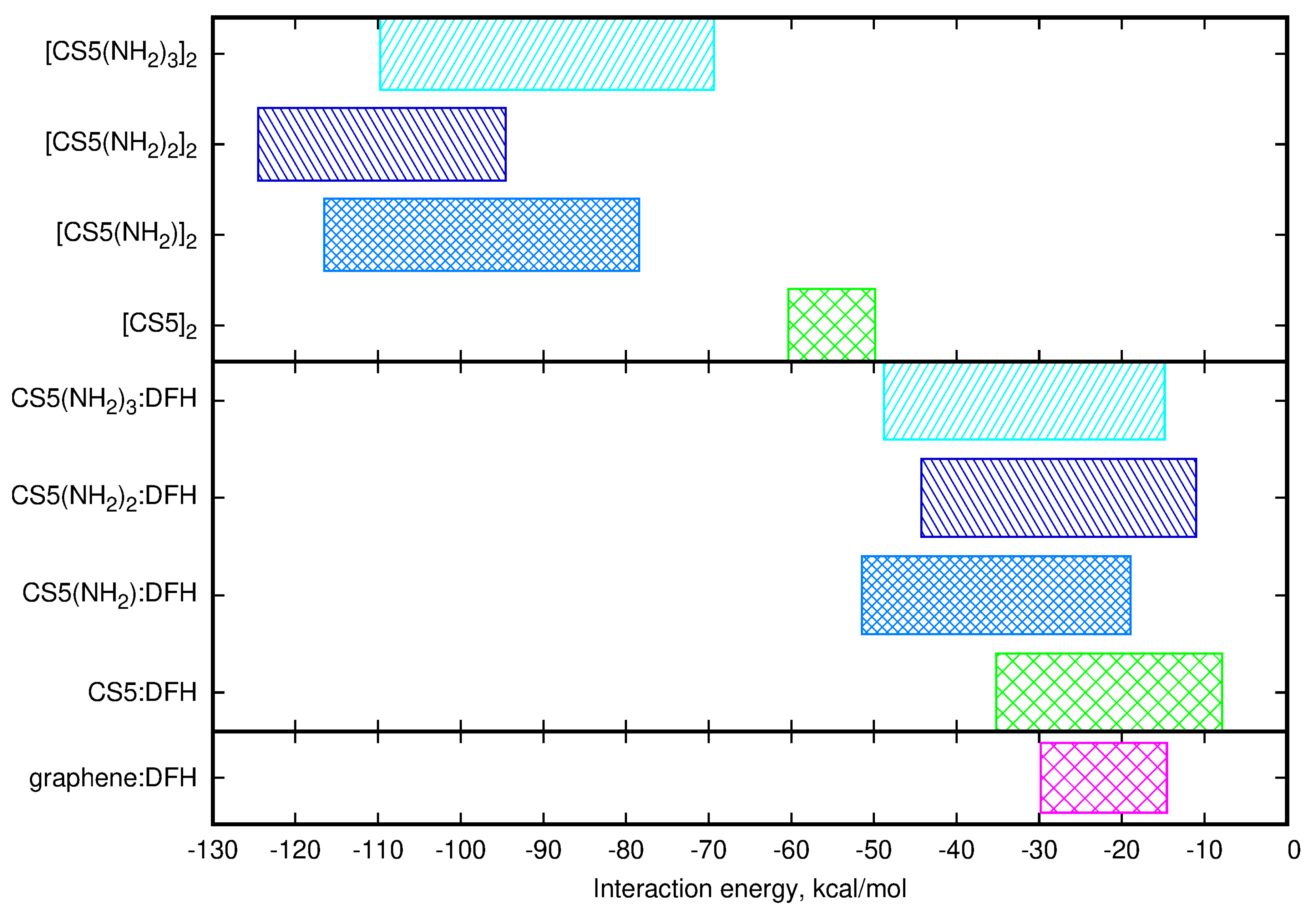
2.3. Diclofenac Interaction with Substituted Chitosan Chain
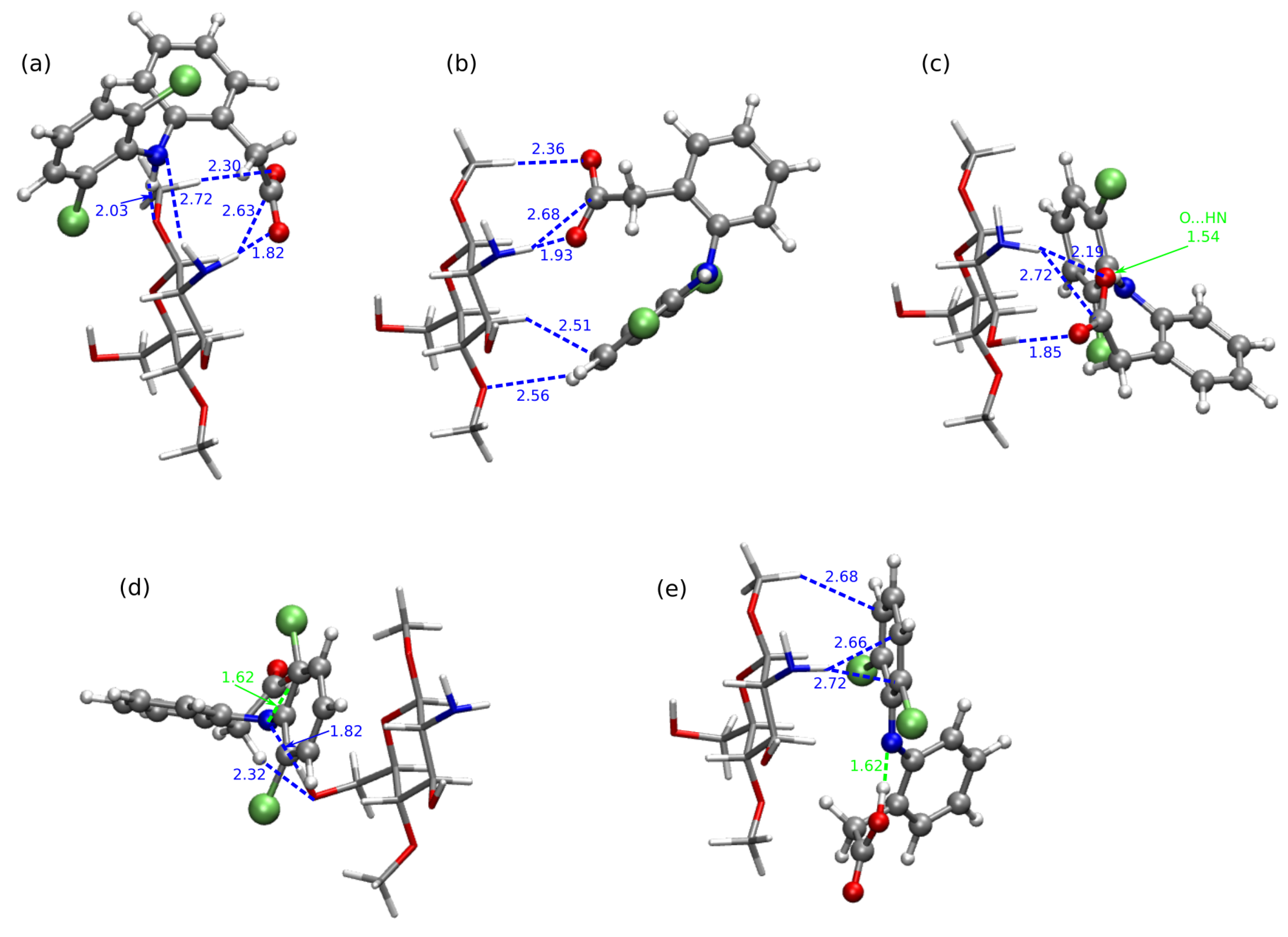
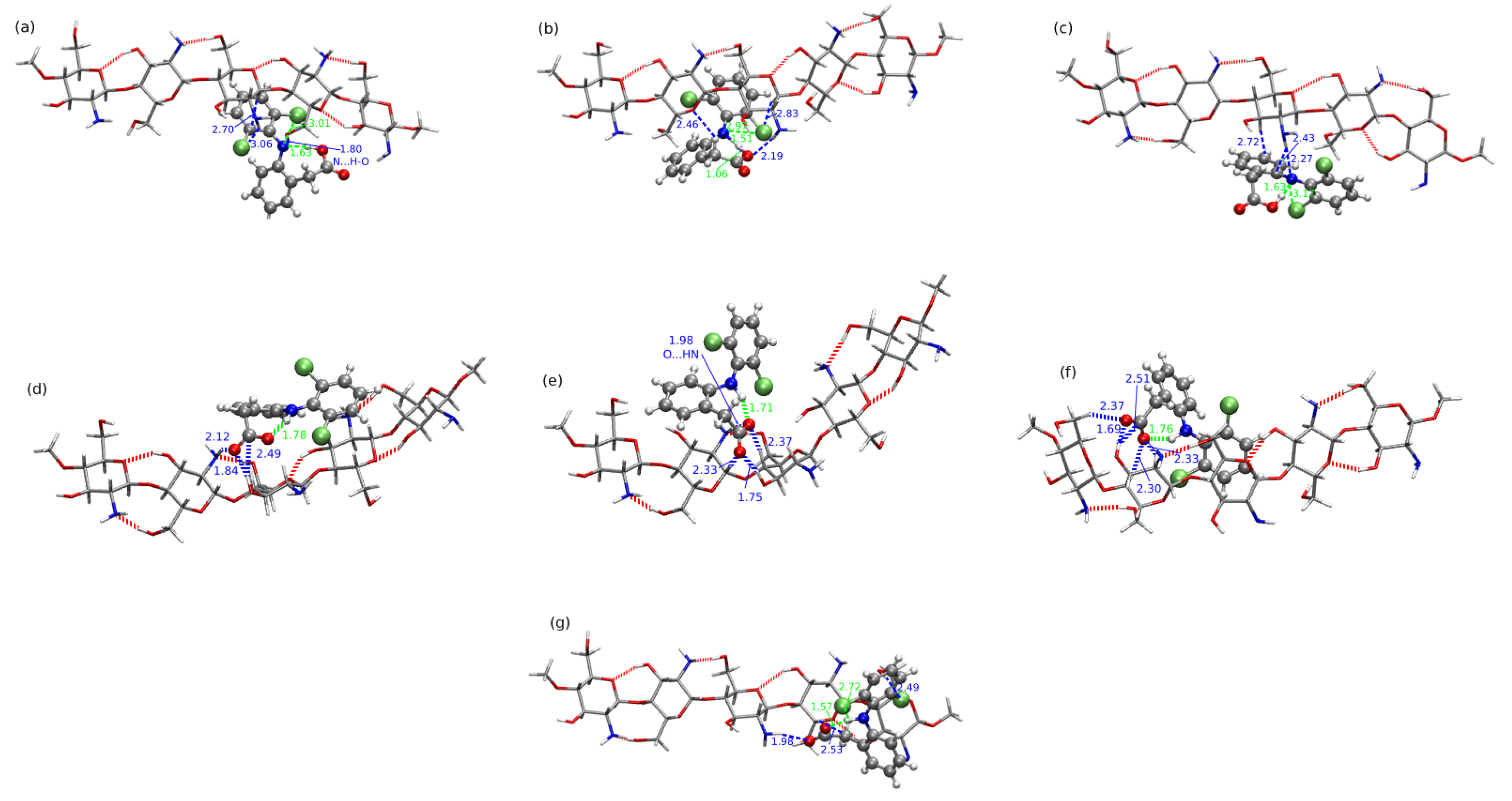
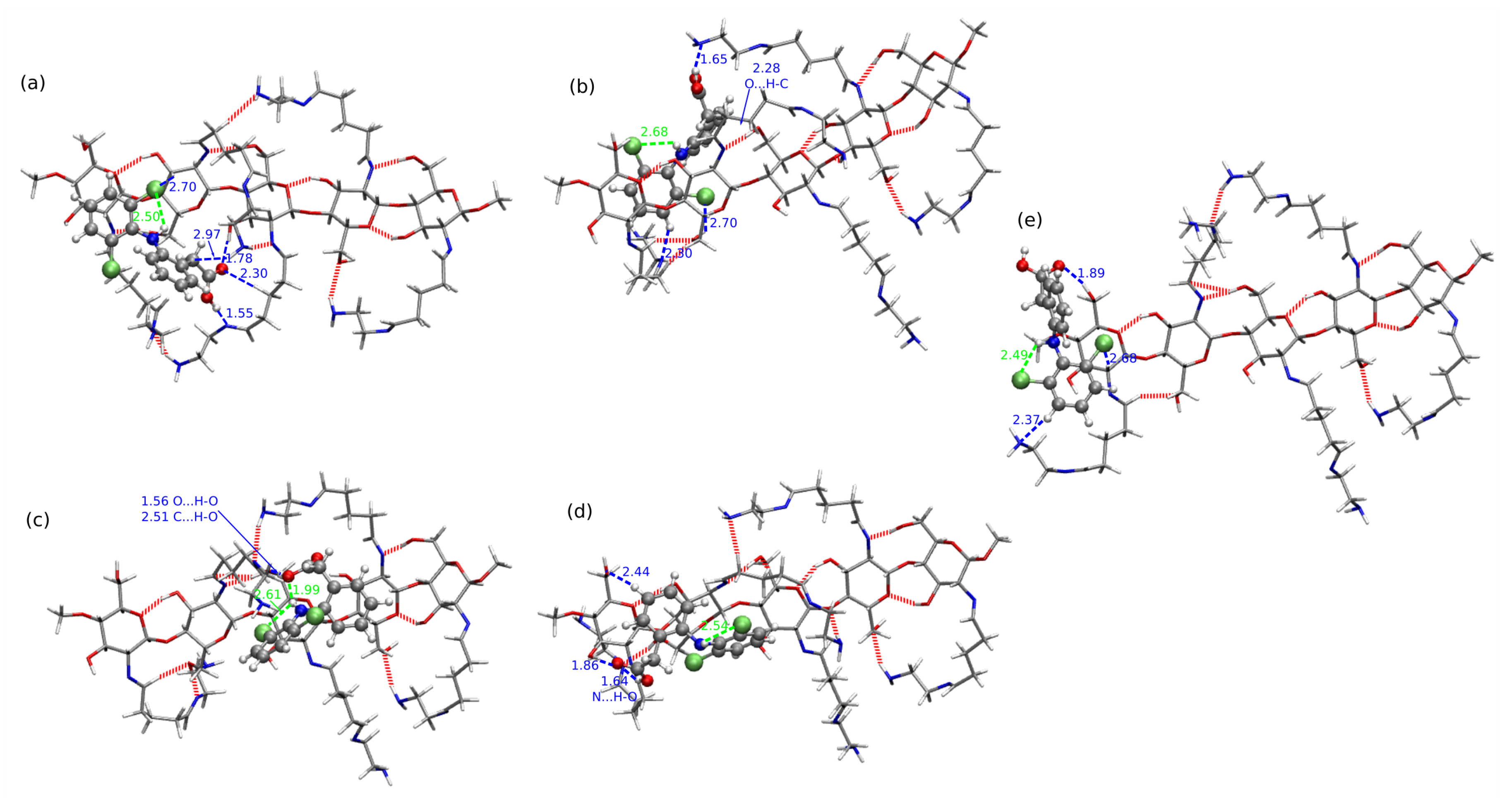
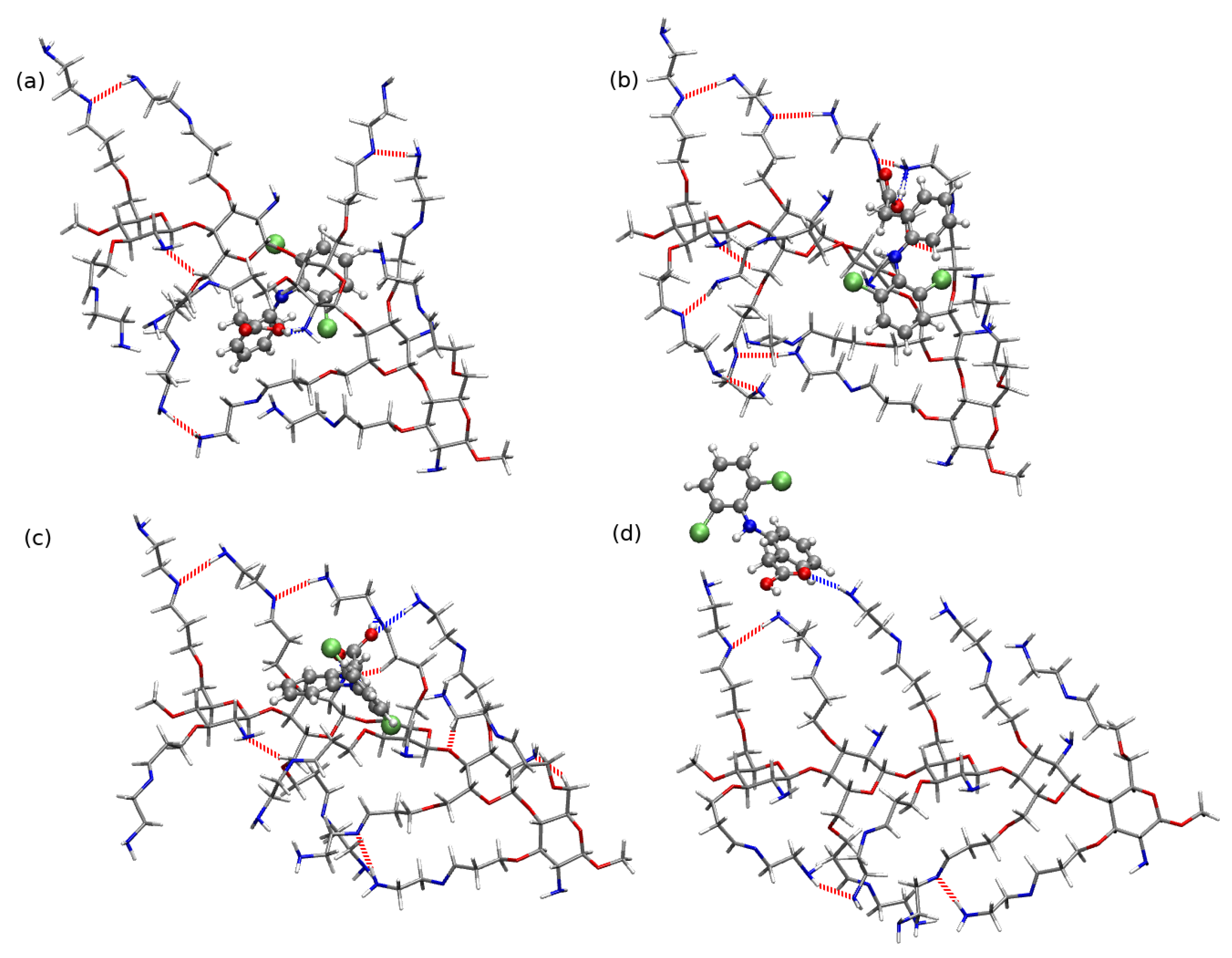
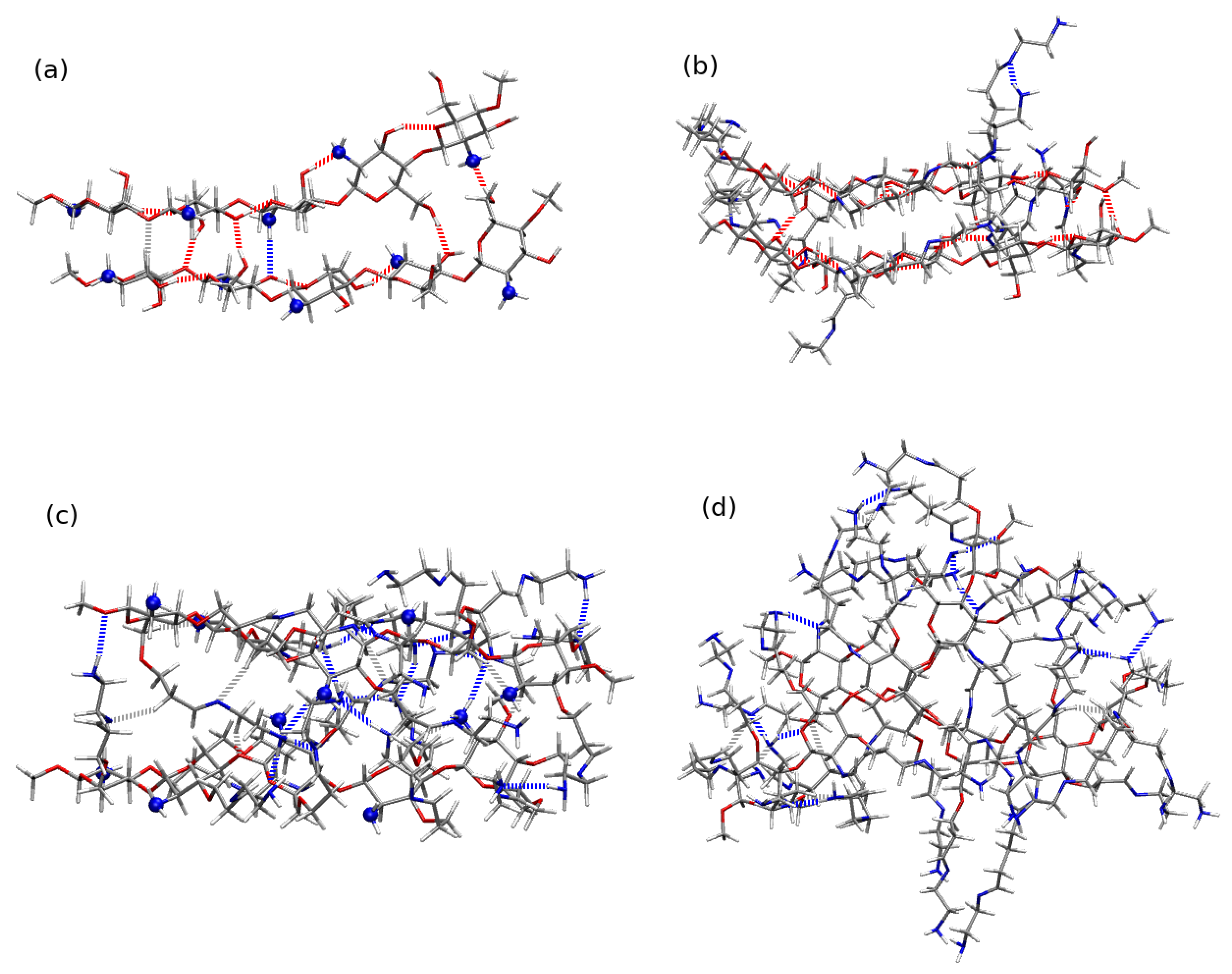
2.4. Chitosan Dimers
3. Discussion
4. Materials and Methods
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| COX | cyclooxygenase |
| NSAID | Non-Steroidal Anti-Inflammatory Drugs |
| CS | Chitosan |
| DFH | Diclofenac |
| DFT | Density Functional Theory |
| SAPT | Symmetry-Adapted Perturbation Theory |
| CPK | Corey–Pauling–Koltun Representation |
| NCI | Non-Covalent Interactions |
| NCIPLOT | Non-Covalent Interaction PLOTting program |
References
- Cunha, R.A.; Soares, T.A.; Rusu, V.H.; Pontes, F.J.S.; Franca, E.F.; Lins, R.D. The Molecular Structure and Conformational Dynamics of Chitosan Polymers: An Integrated Perspective from Experiments and Computational Simulations. In The Complex World of Polysaccharides; Karunaratne, D.N., Ed.; InTech Open: London, UK, 2012. [Google Scholar]
- Franca, E.F.; Lins, R.D.; Freitas, L.C.G.; Straatsma, T.P. Characterization of Chitin and Chitosan Molecular Structure in Aqueous Solution. J. Chem. Theory Comput. 2008, 4, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Chitin and chitosan: Properties and applications. J. Chem. Theory Comput. 2008, 4, 2141–2149. [Google Scholar]
- Kumar, M.N.V.R.; Muzzarelli, R.A.A.; Muzzarelli, C.; Sashiwa, H.; Domb, A.J. Chitosan Chemistry and Pharmaceutical Perspectives. Chem. Rev. 2004, 104, 6017–6084. [Google Scholar] [CrossRef] [PubMed]
- Junior, A.P.D.; Tavares, E.J.M.; Alves, T.V.G.; de Moura, M.R.; da Costa, C.E.F.; Júnior, J.O.C.S.; Costa, R.M.R. Chitosan nanoparticles as a modified diclofenac drug release system. J. Nanopart. Res. 2017, 19, 274. [Google Scholar] [CrossRef]
- Boonsongrita, Y.; Mitrevej, A.; Mueller, B.W. Chitosan drug binding by ionic interaction. Eur. J. Pharm. Biopharm. 2006, 62, 267–274. [Google Scholar] [CrossRef]
- Sabnis, S.; Rege, P.; Block, L.H. Use of Chitosan in Compressed Tablets of Diclofenac Sodium: Inhibition of Drug Release in an Acidic Environment. Pharm. Dev. Technol. 1997, 2, 243–255. [Google Scholar] [CrossRef]
- Karuna, D.S.; Rathnam, G.; Ubaidulla, U.; Ganesh, M.; Jang, H.T. Chitosan phthalate: A novel polymer for the multiparticulate drug delivery system for diclofenac sodium. Adv. Polym. Technol. 2017, 37, 2013–2020. [Google Scholar] [CrossRef]
- Finšgar, M.; Uzunalić, A.P.; Stergar, J.; Gradišnik, L.; Maver, U. Novel chitosan/diclofenac coatings on medical grade stainless steel for hip replacement applications. Sci. Rep. 2016, 6, 26653. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; Tubertini, O. Chitin and chitosan as chromatographic supports and adsorbents for collection of metal ions from organic and aqueous solutions and sea-water. Talanta 1969, 16, 1571–1577. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Uptake of nitrosyl Ru106 on chitin and chitosan from waste sollutions and polluted sea-water. Water Res. 1970, 4, 451. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; Sipos, L. Chitosan for collection from seawater of naturally occurring zinc, cadmium, lead and copper. Talanta 1971, 18, 853. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Selective collection of trace metal ions by precipitation of chitosan and new derivatives of chitosan. Anal. Chim. Acta 1971, 54, 133. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, Z.; Dai, C.; Zhou, X. Removal of selected pharmaceuticals from aqueous solution using magnetic chitosan: Sorption behavior and mechanism. Environ. Sci. Pollut. Res. 2014, 21, 12780–12789. [Google Scholar] [CrossRef] [PubMed]
- Gerente, C.; Lee, V.K.C.; Cloirec, P.L.; McKay, G. Application of Chitosan for the Removal of Metals From Wastewaters by Adsorption—Mechanisms and Models Review. Crit. Rev. Environ. Sci. Technol. 2007, 37, 41–127. [Google Scholar] [CrossRef]
- Kyzas, G.Z.; Bikiaris, D.N. Recent Modifications of Chitosan for Adsorption Applications: A Critical and Systematic Review. Mar. Drugs 2015, 13, 312–337. [Google Scholar] [CrossRef]
- Liang, X.X.; Omer, A.M.; Hu, Z.H.; Wang, Y.; Yu, D.; Ouyang, X.K. Efficient adsorption of diclofenac sodium from aqueous solutions using magnetic amine-functionalized chitosan. Chemosphere 2019, 217, 270–278. [Google Scholar] [CrossRef]
- Ziegler-Borowska, M.; Chełminiak, D.; Kaczmarek, H.; Kaczmarek-Kędziera, A. Effect of side substituents on thermal stability of the modified chitosan and its nanocomposites with magnetite. J. Therm. Anal. Calorim. 2016, 124, 1267–1280. [Google Scholar] [CrossRef]
- Mourya, V.K.; Inamdar, N.N. Chitosan-modifications and applications: Opportunities galore. React. Funct. Polym. 2008, 68, 1013–1051. [Google Scholar] [CrossRef]
- Zhang, J.; Xia, W.; Liu, P.; Cheng, Q.; Tahi, T.; Gu, W.; Li, B. Chitosan Modification and Pharmaceutical/Biomedical Applications. Mar. Drugs 2010, 8, 1962–1987. [Google Scholar] [CrossRef]
- Ji, J.; Wang, L.; Yu, H.; Chen, Y.; Zhao, Y.; Zhang, H.; Amer, W.A.; Sun, Y.; Huang, L.; Saleem, M. Chemical Modifications of Chitosan and Its Applications. Polym. Plast. Technol. Eng. 2014, 53, 1494–1505. [Google Scholar] [CrossRef]
- Ruiz, G.A.M.; Corrales, H.F.Z. Chitosan, Chitosan Derivatives and their Biomedical Applications. In Biological Activities and Application of Marine Polysaccharides; Shalaby, E.A., Ed.; InTech Open: London, UK, 2017. [Google Scholar]
- Prashanth, K.V.H.; Tharanathan, R.N. Chitin/chitosan: modifications and their unlimited application potential—An overview. Trends Food Sci. Technol. 2007, 18, 117–131. [Google Scholar] [CrossRef]
- Brasselet, C.; Pierre, G.; Dubessay, P.; Dols-Lafargue, M.; Coulon, J.; Maupeu, J.; Vallet-Courbin, A.; de Baynast, H.; Doco, T.; Michaud, P.; et al. Modification of Chitosan for the Generation of Functional Derivatives. Appl. Sci. 2019, 9, 1321. [Google Scholar] [CrossRef]
- Kador, K.E.; Subramanian, A. Selective Modification of Chitosan to Enable the Formation of Chitosan-DNA Condensates by Electron Donator Stabilization. Int. J. Carbohydr. Chem. 2011, 2011, 146419. [Google Scholar] [CrossRef]
- Thomas, D.; Thomas, S. Chemical Modification of Chitosan and Its Biomedical Application. In Biopolymer Nanocomposites: Processing, Properties, and Applications; Dufresne, A., Thomas, S., Pothen, L.A., Eds.; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Ziegler-Borowska, M.; Mylkie, K.; Kozlowska, M.; Nowak, P.; Chelminiak-Dudkiewicz, D.; Kozakiewicz, A.; Ilnicka, A.; Kaczmarek-Kedziera, A. Effect of Geometrical Structure, Drying, and Synthetic Method on Aminated Chitosan-Coated Magnetic Nanoparticles Utility for HSA Effective Immobilization. Molecules 2019, 24, 1925. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Zhang, J.; Dai, C.M.; Zhou, X.F.; Liu, S.G. Sorption of carbamazepine from water by magnetic molecularly imprinted polymers based on chitosan-Fe3O4. Carbohydr. Polym. 2013, 97, 809–816. [Google Scholar] [CrossRef]
- Ziegler-Borowska, M.; Chełminiak, D.; Siódmiak, T.; Sikora, A.; Marszałł, M.P.; Kaczmarek, H. Synthesis of new chitosan coated magnetic nanoparticles with surfacemodified with long-distanced amino groups as a supportfor bioligands binding. Mater. Lett. 2014, 132, 63–65. [Google Scholar] [CrossRef]
- Sallmann, A.R. The history of diclofenac. Am. J. Med. 1986, 80, 29–33. [Google Scholar] [CrossRef]
- Dreve, S.; Kacso, I.; Popa, A.; Raita, O.; Dragan, F.; Bende, A.; Borodi, G.; Bratu, I. Structural investigation of chitosan-based microspheres with some anti-inflammatory drugs. J. Mol. Struct. 2011, 997, 78–86. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Orozco, M.; Luque, F.J. Theoretical Methods for the Description of the Solvent Effect in Biomolecular Systems. Chem. Rev. 2000, 100, 4187–4225. [Google Scholar] [CrossRef]
- Chen, A.; Pu, X.; He, S.; Guo, Y.; Wen, Z.; Li, M.; Wong, N.B.; Tian, A. Solvent effects on isolated formamide and its monohydrated complex: observations from PCM study. New J. Chem. 2009, 33, 1709–1719. [Google Scholar] [CrossRef]
- Aquino, A.J.A.; Tunega, D.; Haberhauer, G.; Gerzabek, M.G.; Lischka, H. Solvent Effects on Hydrogen Bonds—A Theoretical Study. J. Phys. Chem. A 2002, 106, 1862–1871. [Google Scholar] [CrossRef]
- Grabowski, S.J. (Ed.) Hydrogen Bonding—New Insights; Challenges and Advanced in Computational Chemistry and Physics; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Rospenk, M.; Sobczyk, L.; Schah-Mohammedi, P.; Limbach, H.H.; Golubev, N.S.; Melikova, S.M. Dimerization and solvent-assisted proton dislocation in the low-barrier hydrogen bond of a Mannich base: a low-temperature NMR study. Magn. Res. Chem. 2001, 39, S81–S90. [Google Scholar] [CrossRef]
- Pawelka, Z.; Sobczyk, L. Effect of Solvents on Dielectric, Spectroscopic and Thermodynamic Manifestations of Intermolecular Interactions. Iodine Complexes of Pyridine and Picoline. J. Solut. Chem. 1983, 12, 355–367. [Google Scholar] [CrossRef]
- Pawelka, Z.; Rospenk, M.; Sobczyk, L. Solvent Effect Upon Dipole Moment and Proton Transfer Equilibrium in Ortho Mannich Bases. Bulletin des Sociétés Chimiques Belges 1987, 96, 415–421. [Google Scholar] [CrossRef]
- Grech, E.; Nowicka-Scheibe, J.; Olejnik, Z.; Lis, T.; Pawelka, Z.; Malarski, Z.; Sobczyk, L. An IR, NMR, dipole moment and X-ray study on intramolecular O-H...N hydrogen bonding in 8-hydroxy-N,N-dimethyl-1-naphthylamine. J. Chem. Soc. Perkin Trans. 1996, 2, 343–348. [Google Scholar] [CrossRef]
- Schreiber, V.M.; Rospenk, M.; Kulbida, I.; Sobczyk, L. Shaping of broad IR absorption in proton transfer equilibrating OH...N hydrogen bonded systems. Spectrochim. Acta A 1997, 53, 2067–2078. [Google Scholar] [CrossRef]
- Koeppe, B.; Pylaeva, S.; Allolio, C.; Sebastiani, D.; Nibbering, E.T.J.; Denisov, G.S.; Limbach, H.H.; Tolstoy, P.M. Polar solvent fluctuations drive proton transfer in hydrogen bonded complexes of carboxylic acid with pyridines: NMR, IR and ab initio MD study. Phys. Chem. Chem. Phys. 2017, 19, 1010–1028. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Denisov, G.S.; Borissova, A.O.; Goloveshkin, A.S.; Greindl, J.; Limbach, H.H.; Shenderovich, I.G. NMR Study of Solvation Effect on the Geometry of Proton-Bound Homodimers of Increasing Size. J. Phys. Chem. A 2017, 121, 8697–8705. [Google Scholar] [CrossRef]
- Mennucci, J.T.B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar]
- Pollice, R.; Bot, M.; Kobylianskii, I.J.; Shenderovich, I.; Chen, P. Attenuation of London Dispersion in Dichloromethane Solutions. J. Am. Chem. Soc. 2017, 139, 13126–13140. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Guerard, J.J.; Arey, J.S. Critical Evaluation of Implicit Solvent Models for Predicting Aqueous Oxidation Potentials of Neutral Organic Compounds. J. Chem. Theory Comput. 2013, 9, 5046–5058. [Google Scholar] [CrossRef] [PubMed]
- Kromann, J.C.; Steinmann, C.; Jensen, J.H. Improving solvation energy predictions using the SMD solvation method and semiempirical electronic structure methods. J. Chem. Phys. 2018, 149, 104102. [Google Scholar] [CrossRef]
- Mirzaei, S.; Ivanov, M.V.; Timerghazin, Q.K. Improving Performance of the SMD Solvation Model: Bondi Radii Improve Predicted Aqueous Solvation Free Energies of Ions and pKa Values of Thiols. J. Phys. Chem. A 2019, 123, 44. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Simplified calculation approaches designed to reproduce the geometry of hydrogen bonds in molecular complexes in aprotic solvents. J. Chem. Phys. 2018, 148, 124313. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. What is more important: A macroscopic reaction field or solute-solvent interactions? J. Chem. Phys. 2019, 150, 204505. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. Adduct under Field—A Qualitative Approach to Account for Solvent Effect on Hydrogen Bonding. Molecules 2020, 25, 436. [Google Scholar] [CrossRef]
- Kozlowska, M.; Rodziewicz, P.; Kaczmarek-Kedziera, A. Structural stability of diclofenac vs. inhibition activity from ab initio molecular dynamics simulations. Comparative study with ibuprofen and ketoprofen. Struct. Chem. 2017, 28, 999–1008. [Google Scholar] [CrossRef]
- Kozlowska, M.; Rodziewicz, P.; Kaczmarek-Kedziera, A. Solvation of diclofenac in water from atomistic molecular dynamics simulations - interplay between solute-solute and solute-solvent interactions. Phys. Chem. Chem. Phys. 2019, 20, 8629–8639. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Jaeger, H.M.; Carrell, E.J.; Tschumper, G.S.; Sherrill, C.D. Accurate Interaction Energies for Problematic Dispersion-Bound Complexes: Homogeneous Dimers of NCCN, P2, and PCCP. J. Chem. Theory Comput. 2011, 7, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Parrish, R.M.; Sherrill, C.D.; Turney, J.M.; Schaefer, H.F. Large-scale symmetry-adapted perturbation theory computations via density fitting and Laplace transformation techniques: Investigating the fundamental forces of DNA-intercalator interactions. J. Chem. Phys. 2011, 135, 174107. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting and Cholesky decomposition approximations in symmetry-adapted perturbation theory: Implementation and application to probe the nature of π-π interactions in linear acenes. J. Chem. Phys. 2010, 132, 184111. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Wavefunction methods for noncovalent interactions. WIREs Comput. Mol. Sci. 2012, 2, 304–326. [Google Scholar] [CrossRef]
- Rezač, J.; Riley, K.E.; Hobza, P. S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef]
- Kaczmarek-Kędziera, A. Influence of photodegradation and surface modification on the graphene-diclofenac physisorption process. Int. J. Quant. Chem. 2019, 119, e26030. [Google Scholar] [CrossRef]
- Jansen, H.B.; Ros, P. Non-empirical molecular orbital calculations on the protonation of carbon monoxide. Chem. Phys. Lett. 1969, 3, 140–143. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. Calculation of small molecular interactions by differences of separate total energies—Some procedures with reduced errors. Mol. Phys. 1970, 19, 553. [Google Scholar] [CrossRef]
- Liu, B.; McLean, A.D. Accurate calculation of the attractive interaction of two ground state helium atoms. J. Chem. Phys. 1973, 59, 4557. [Google Scholar] [CrossRef]
- van Duijneveldt, F.B.; van Duijneveldt-van de Rijdt, J.G.C.M.; van Lenthe, J.H. State of the Art in Counterpoise Theory. Chem. Rev. 1994, 94, 1873–1885. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Hobza, P.; Selzle, H.L.; Schlag, E.W. Potential Energy Surface for the Benzene Dimer. Results of ab Initio CCSD(T) Calculations Show Two Nearly Isoenergetic Structures: T-Shaped and Parallel-Displaced. J. Phys. Chem. 1996, 100, 18790–18794. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Uchimaru, T.; Matsumura, K.; Mikami, M.; Tanabe, K. Effects of the higher electron correlation correction on the calculated intermolecular interaction energies of benzene and naphthalene dimers: comparison between MP2 and CCSD(T) calculations. Chem. Phys. Lett. 2000, 319, 547–554. [Google Scholar] [CrossRef]
- Hobza, P.; Selzle, H.L.; Schlag, E.W. Structure and Properties of Benzene-Containing Molecular Clusters: Nonempirical ab Initio Calculations and Experiments. Chem. Rev. 1994, 94, 1767–1785. [Google Scholar] [CrossRef]
- Tkatchenko, A.; DiStasio, R.A., Jr.; Head-Gordon, M.; Scheffler, M. Dispersion-corrected Møller–Plesset second-order perturbation theory. J. Chem. Phys. 2009, 131, 094106. [Google Scholar] [CrossRef] [PubMed]
- Hesselmann, A. Improved supermolecular second order Moller–Plesset intermolecular interaction energies using time-dependent density functional response theory. J. Chem. Phys. 2008, 128, 144112. [Google Scholar] [CrossRef]
- Pitonak, M.; Hesselmann, A. Accurate Intermolecular Interaction Energies from a Combination of MP2 and TDDFT Response Theory. J. Chem. Theory Comput. 2010, 6, 168. [Google Scholar] [CrossRef]
- Hesselmann, A.; Korona, T. Intermolecular symmetry-adapted perturbation theory study of large organic complexes. J. Chem. Phys. 2014, 141, 094107. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. MOLPRO, Version 2018.1, a Package of ab Initio Programs, 2089. Available online: https://www.molpro.net (accessed on 29 May 2020).
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: a general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Remigio, R.D.; Richard, R.M.; Gonthier, J.F.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An open-source ab initio electronic structure program. WIREs Comput. Mol. Sci. 2012, 2, 556. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | B97-D3 | B97X-D | MP2 | MP2 | MP2C | SAPT0 | SAPT0 |
|---|---|---|---|---|---|---|---|
| Basis Set | P++(d,p) | P++(d,p) | P++(d,p) | aDZ | aDZ | DZ | aDZ |
| CS1:DFNa | |||||||
| 1 | −36.10 | −38.68 | −31.08 | −33.34 | −34.66 | −39.20 | |
| 2 | −25.54 | −27.82 | −21.40 | −23.66 | −25.24 | −29.62 | |
| 3 | −23.94 | −25.72 | −17.51 | −19.97 | −19.72 | −24.33 | |
| 4 | −38.46 | −41.20 | −32.89 | −35.76 | −37.12 | −42.32 | |
| 5 | −33.23 | −36.00 | −27.63 | −30.74 | −30.46 | −36.51 | |
| 6 | −34.84 | −36.72 | −29.59 | −31.64 | −33.12 | −37.19 | |
| 7 | −24.45 | −25.96 | −19.64 | −20.89 | −23.13 | −25.90 | |
| 7a | −32.29 | −34.92 | −26.38 | −29.21 | −31.76 | −36.76 | |
| 8 | −21.79 | −22.61 | −17.05 | −18.93 | −19.60 | −23.10 | |
| 9 | −34.19 | −35.50 | −28.65 | −30.40 | −31.93 | −35.66 | |
| 10 | −9.74 | −10.32 | −7.87 | −10.31 | −6.98 | −11.99 | |
| CS1:DF | |||||||
| 2 | −24.11 | −25.60 | −21.56 | −23.32 | −30.24 | ||
| 3 | −28.99 | −32.46 | −27.28 | −30.01 | −36.24 | ||
| 4 | −27.56 | −30.00 | −24.86 | −27.46 | −33.64 | ||
| 6 | −25.72 | −27.79 | −23.87 | −26.54 | −30.92 | ||
| 7a | −27.92 | −30.91 | −26.28 | −30.11 | −34.40 | ||
| 8 | −21.47 | −22.60 | −19.13 | −19.80 | −25.60 | ||
| CS1:DFH | |||||||
| 2 | −19.04 | −20.71 | −15.35 | −18.80 | −16.99 | −17.20 | −23.63 |
| 3 | −5.99 | −6.73 | −4.50 | −5.76 | −5.26 | −4.47 | −6.93 |
| 4 | −20.06 | −22.02 | −16.73 | −19.77 | −17.75 | −18.32 | −24.24 |
| 5 | −8.78 | −9.31 | −10.80 | −9.38 | −7.74 | −6.35 | −10.91 |
| 6 | −26.48 | −27.52 | −22.21 | −25.59 | −23.58 | −23.99 | −30.94 |
| 7 | −11.74 | −13.34 | −9.00 | −11.84 | −9.49 | −8.24 | −13.85 |
| 8 | −11.33 | −13.23 | −9.36 | −12.18 | −9.95 | −9.13 | −15.09 |
| 9 | −20.77 | −22.54 | −16.86 | −19.82 | −17.86 | −18.33 | −24.32 |
| System | SAPT0 | SCS-SAPT0 | ELST | EXCH | IND | DISP | SCS-DISP | ELST/DISP | ELST/SCS-DISP |
|---|---|---|---|---|---|---|---|---|---|
| CS1:DFNa | |||||||||
| 1 | −39.20 | −34.33 | −35.16 | 26.85 | −8.74 | −22.16 | −17.29 | 1.59 | 2.03 |
| 2 | −29.62 | −25.23 | −34.15 | 33.82 | −9.13 | −20.17 | −15.78 | 1.69 | 2.16 |
| 3 | −24.33 | −20.22 | −21.81 | 25.23 | −8.93 | −18.83 | −14.71 | 1.16 | 1.48 |
| 4 | −42.32 | −37.36 | −49.26 | 41.77 | −12.01 | −22.82 | −17.86 | 2.16 | 2.76 |
| 5 | −36.51 | −31.27 | −35.93 | 32.66 | −9.32 | −23.91 | −18.67 | 1.50 | 1.93 |
| 6 | −37.19 | −33.01 | −37.73 | 28.35 | −8.67 | −19.14 | −14.95 | 1.97 | 2.52 |
| 7 | −25.90 | −22.79 | −25.57 | 20.08 | −6.27 | −14.13 | −11.02 | 1.81 | 2.32 |
| 7a | −36.76 | −31.74 | −48.42 | 51.43 | −16.52 | −23.25 | −18.23 | 2.08 | 2.66 |
| 8 | −23.10 | −19.51 | −22.40 | 20.67 | −5.13 | −16.24 | −12.65 | 1.38 | 1.77 |
| 9 | −35.66 | −31.79 | −33.83 | 25.08 | −9.18 | −17.73 | −13.86 | 1.91 | 2.44 |
| 10 | −11.99 | −8.11 | −8.91 | 17.32 | −2.84 | −17.56 | −13.69 | 0.51 | 0.65 |
| CS1:DF | |||||||||
| 2 | −30.25 | −25.39 | −29.07 | 37.47 | −16.39 | −22.26 | −17.41 | 1.31 | 1.67 |
| 3 | −36.24 | −31.42 | −39.32 | 40.69 | −15.34 | −22.27 | −17.45 | 1.77 | 2.25 |
| 4 | −33.64 | −28.41 | −29.41 | 31.60 | −11.88 | −23.96 | −18.73 | 1.23 | 1.57 |
| 6 | −30.92 | −27.43 | −29.17 | 26.50 | −12.24 | −16.01 | −12.52 | 1.82 | 2.33 |
| 7a | −34.40 | −30.71 | −34.54 | 32.32 | −15.04 | −17.13 | −13.44 | 2.02 | 2.57 |
| 8 | −25.60 | −21.07 | −23.67 | 28.47 | −9.77 | −20.63 | −16.10 | 1.15 | 1.47 |
| 9 | −30.91 | −27.42 | −29.17 | 26.50 | −12.23 | −16.00 | −12.52 | 1.82 | 2.33 |
| CS1:DFH | |||||||||
| 2 | −23.63 | −18.49 | −30.92 | 42.40 | −11.47 | −23.63 | −18.49 | 1.31 | 1.67 |
| 3 | −6.93 | −4.67 | −6.47 | 11.59 | −1.80 | −10.25 | −7.99 | 0.63 | 0.81 |
| 4 | −24.24 | −19.31 | −23.45 | 30.65 | −9.00 | −22.44 | −17.51 | 1.05 | 1.34 |
| 5 | −10.91 | −7.45 | −6.84 | 13.46 | −1.96 | −15.58 | −12.12 | 0.44 | 0.56 |
| 6 | −30.94 | −25.46 | −37.00 | 51.19 | −19.74 | −25.40 | −19.91 | 1.46 | 1.86 |
| 7 | −13.85 | −9.02 | −7.41 | 18.23 | −2.83 | −21.85 | −17.02 | 0.34 | 0.44 |
| 8 | −15.09 | −10.45 | −12.85 | 22.01 | −3.13 | −21.12 | −16.47 | 0.61 | 0.78 |
| 9 | −24.32 | −19.28 | −26.47 | 36.91 | −11.67 | −23.09 | −18.05 | 1.15 | 1.47 |
| System | CS5(NH):DFH | CS5(NH):DFH | CS5(NH):DFH |
|---|---|---|---|
| 1 | −38.93 | −35.66 | −22.95 |
| 2 | −18.93 | −31.55 | −34.41 |
| 3 | −41.04 | −19.49 | −27.27 |
| 4 | −29.20 | −14.79 | −22.31 |
| 5 | −51.45 | −10.91 | −34.86 |
| 6 | −30.42 | −34.08 | −32.82 |
| 7 | −40.08 | −26.81 | −18.93 |
| 8 | −35.45 | −40.81 | −38.93 |
| 9 | −32.36 | −37.22 | −37.04 |
| 10 | −37.09 | −40.86 | −48.76 |
| 11 | −35.79 | −28.40 | −45.51 |
| 12 | −36.24 | −19.64 | −46.27 |
| 13 | −21.03 | −36.81 | −103.76 |
| 14 | −31.09 | −44.26 | −14.80 |
| 15 | −29.30 | −30.52 | −28.83 |
| 16 | −35.72 | −29.02 | −25.21 |
| 17 | −35.42 | −30.82 | −38.17 |
| 18 | −31.42 | −22.55 | −46.66 |
| 19 | −36.18 | −31.67 | −39.11 |
| 20 | −27.00 | −31.48 | −47.92 |
| Material | Dimer 1 | Dimer 2 | Dimer 3 | Dimer 4 | Dimer 5 | Diclofenac |
|---|---|---|---|---|---|---|
| [CS5] | −60.32 | −59.64 | −51.61 | −61.25 | −49.84 | −7.86 to −35.19 |
| [CS5(NH)] | −116.50 | −102.48 | −81.53 | −78.43 | −107.35 | −18.93 to −51.45 |
| [CS5(NH)] | −124.50 | −94.55 | −103.94 | −10.91 to −44.26 | ||
| [CS5(NH)] | −109.76 | −69.34 | −99.19 | −14.80 to −48.76 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczmarek-Kędziera, A. Gas Phase Computational Study of Diclofenac Adsorption on Chitosan Materials. Molecules 2020, 25, 2549. https://doi.org/10.3390/molecules25112549
Kaczmarek-Kędziera A. Gas Phase Computational Study of Diclofenac Adsorption on Chitosan Materials. Molecules. 2020; 25(11):2549. https://doi.org/10.3390/molecules25112549
Chicago/Turabian StyleKaczmarek-Kędziera, Anna. 2020. "Gas Phase Computational Study of Diclofenac Adsorption on Chitosan Materials" Molecules 25, no. 11: 2549. https://doi.org/10.3390/molecules25112549
APA StyleKaczmarek-Kędziera, A. (2020). Gas Phase Computational Study of Diclofenac Adsorption on Chitosan Materials. Molecules, 25(11), 2549. https://doi.org/10.3390/molecules25112549