
Small Molecule Inhibitors of KDM5 Histone Demethylases Increase the Radiosensitivity of Breast Cancer Cells Overexpressing JARID1B


 , , and
, , and
Abstract

1. Introduction
2. Results
2.1. RS3195 Is a KDM5 Enzymes Inhibitor That Induces a Strong G2/M Arrest in MCF-7 Cells
2.2. KDM5 Enzymes Inhibition Does Not Significantly Change the Transcriptome of MCF-7 Cells
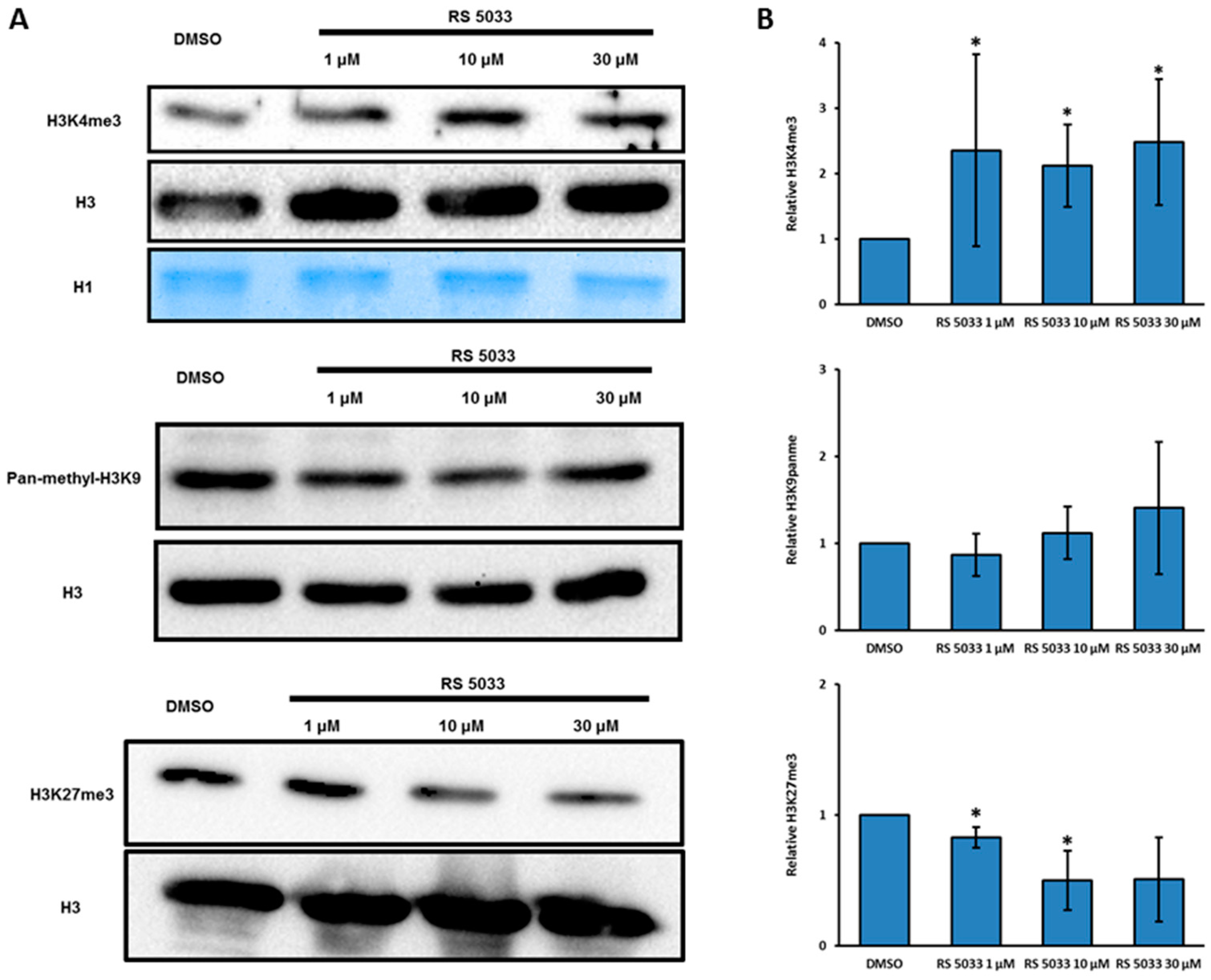
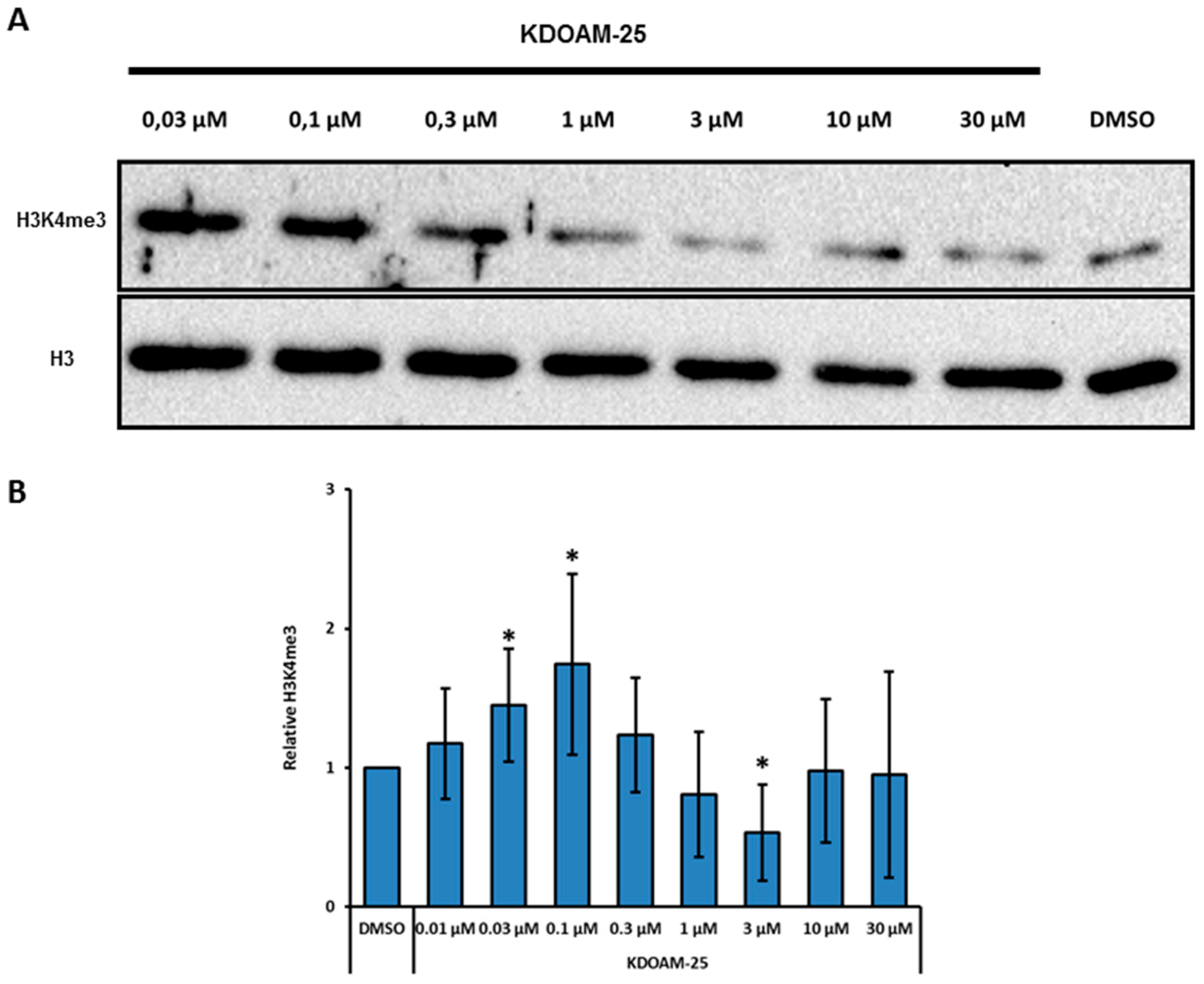
2.3. Designing RS 5033, a Selective KDM5 Enzymes Inhibitor with No Effects on Cell Cycle Dynamics
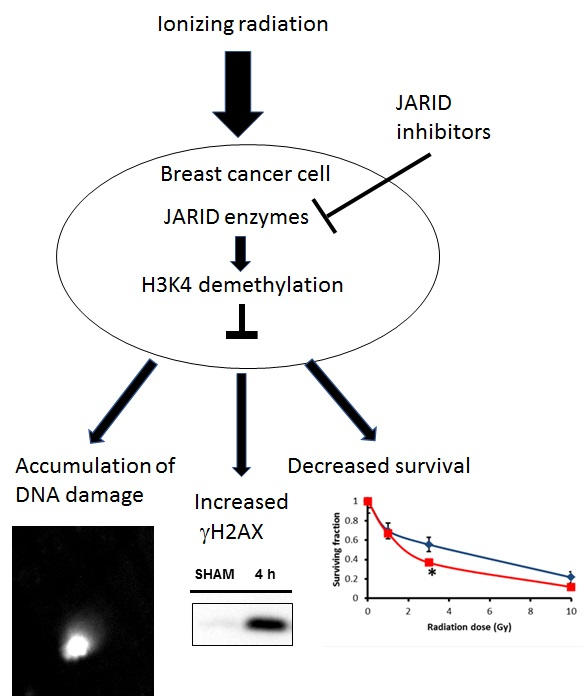
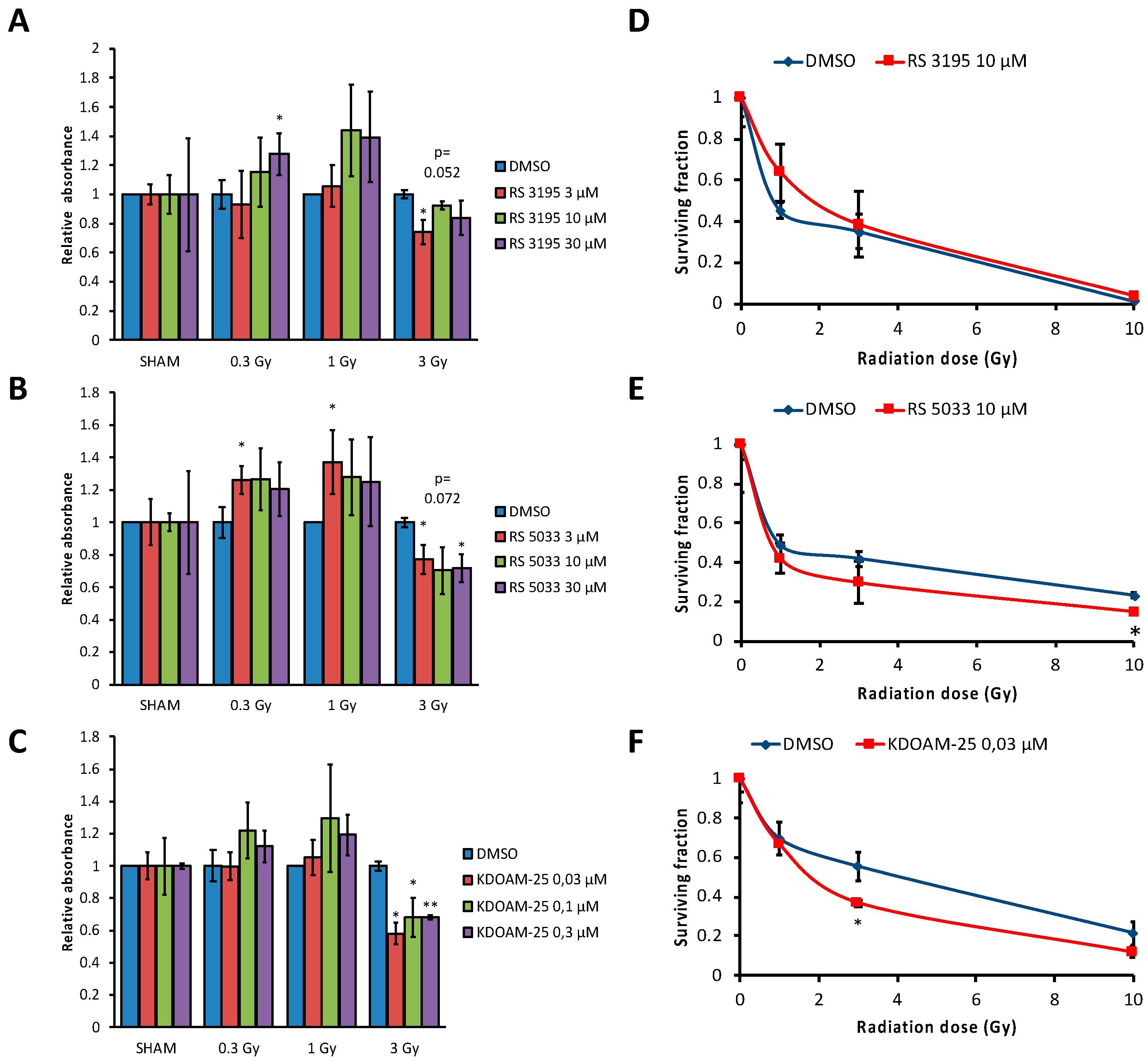
2.4. KDM5 Enzymes Inhibition Increases Breast Cancer Cells Sensitivity to Ionizing Radiation
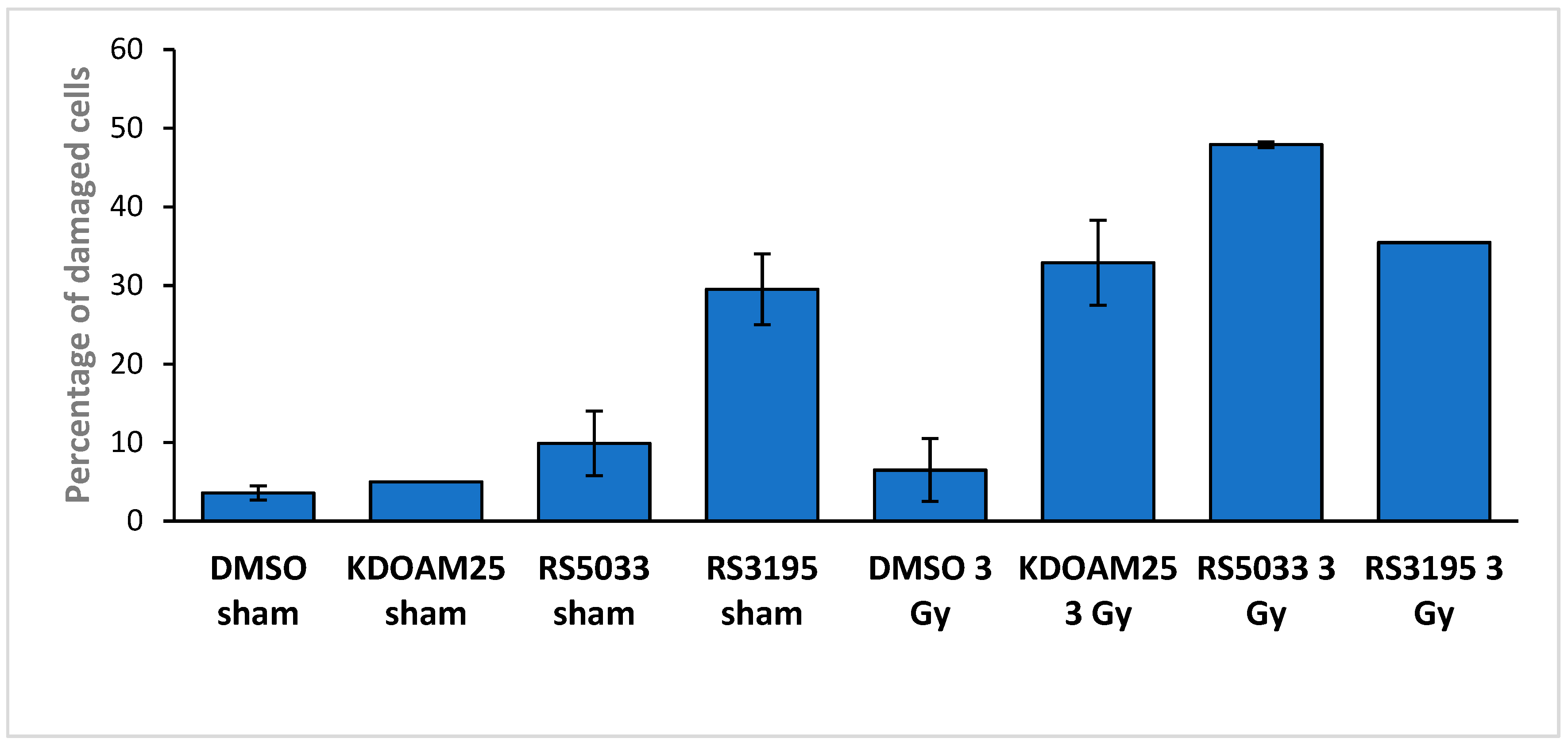
2.5. Effects of KDM5 Inhibitors on DNA Damage Accumulation
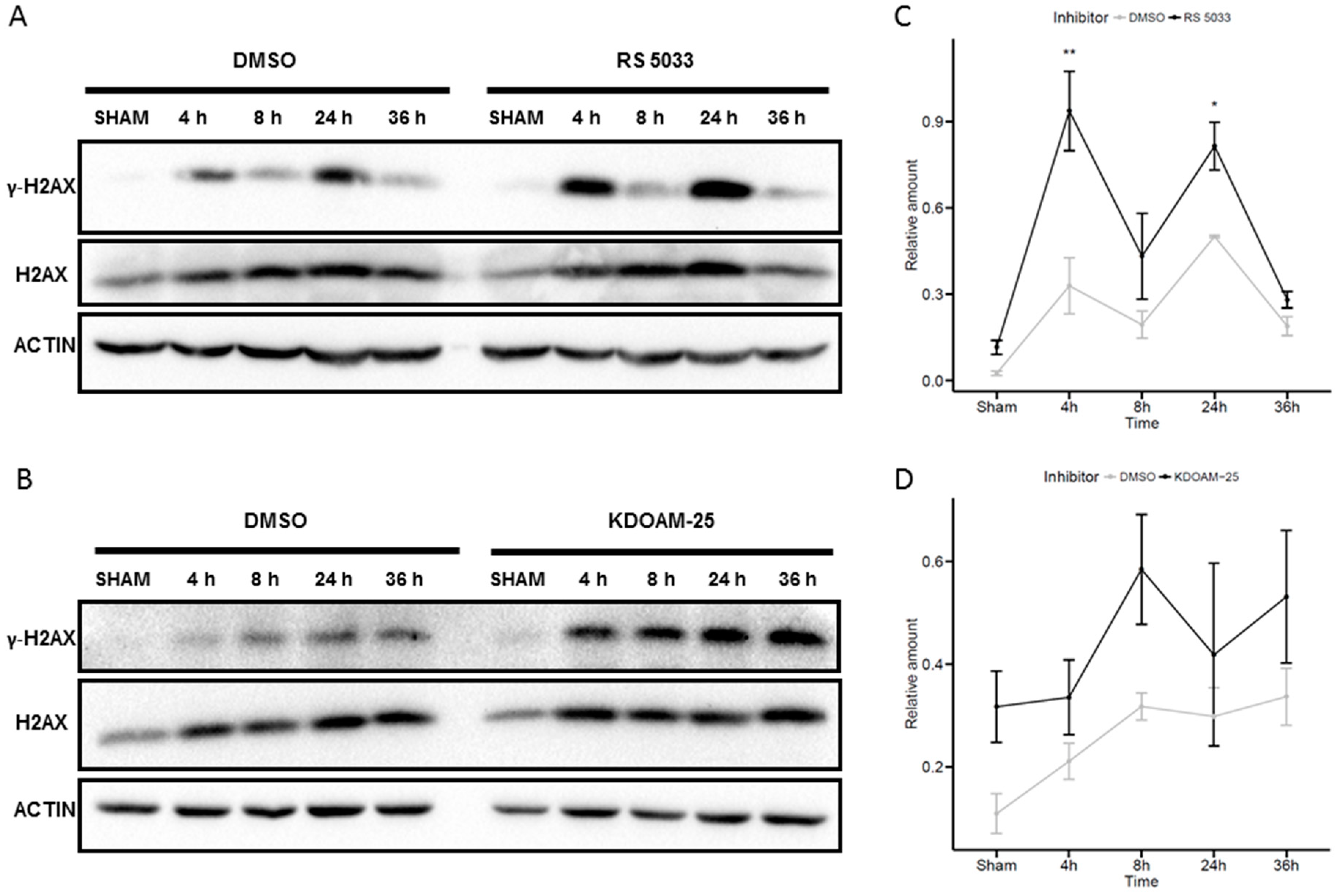
2.6. Effects of KDM5 Inhibitors on H2AX Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. RNA-Sequencing
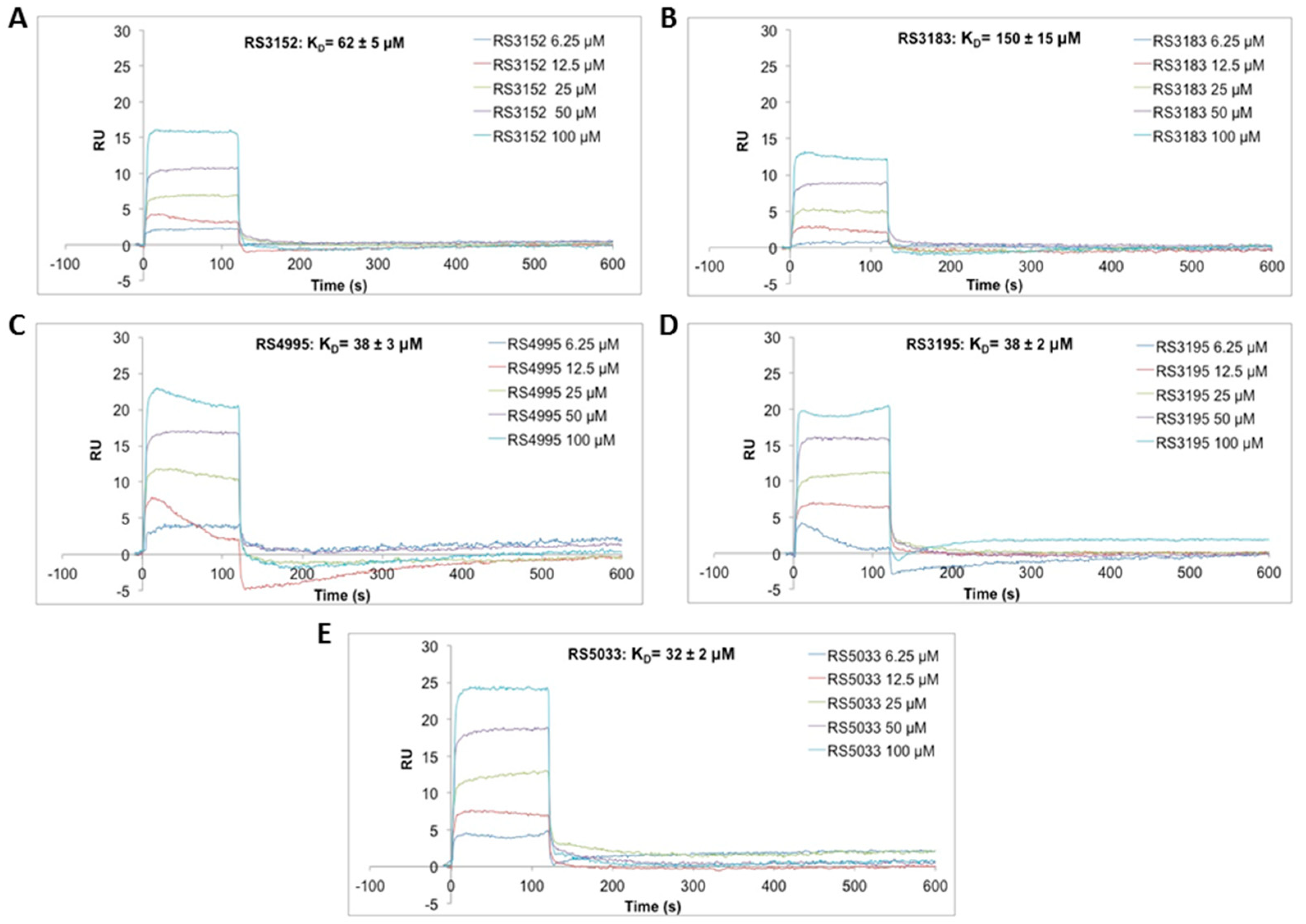
4.3. Surface Plasmon Resonance (SPR)
4.4. Chemical Compounds
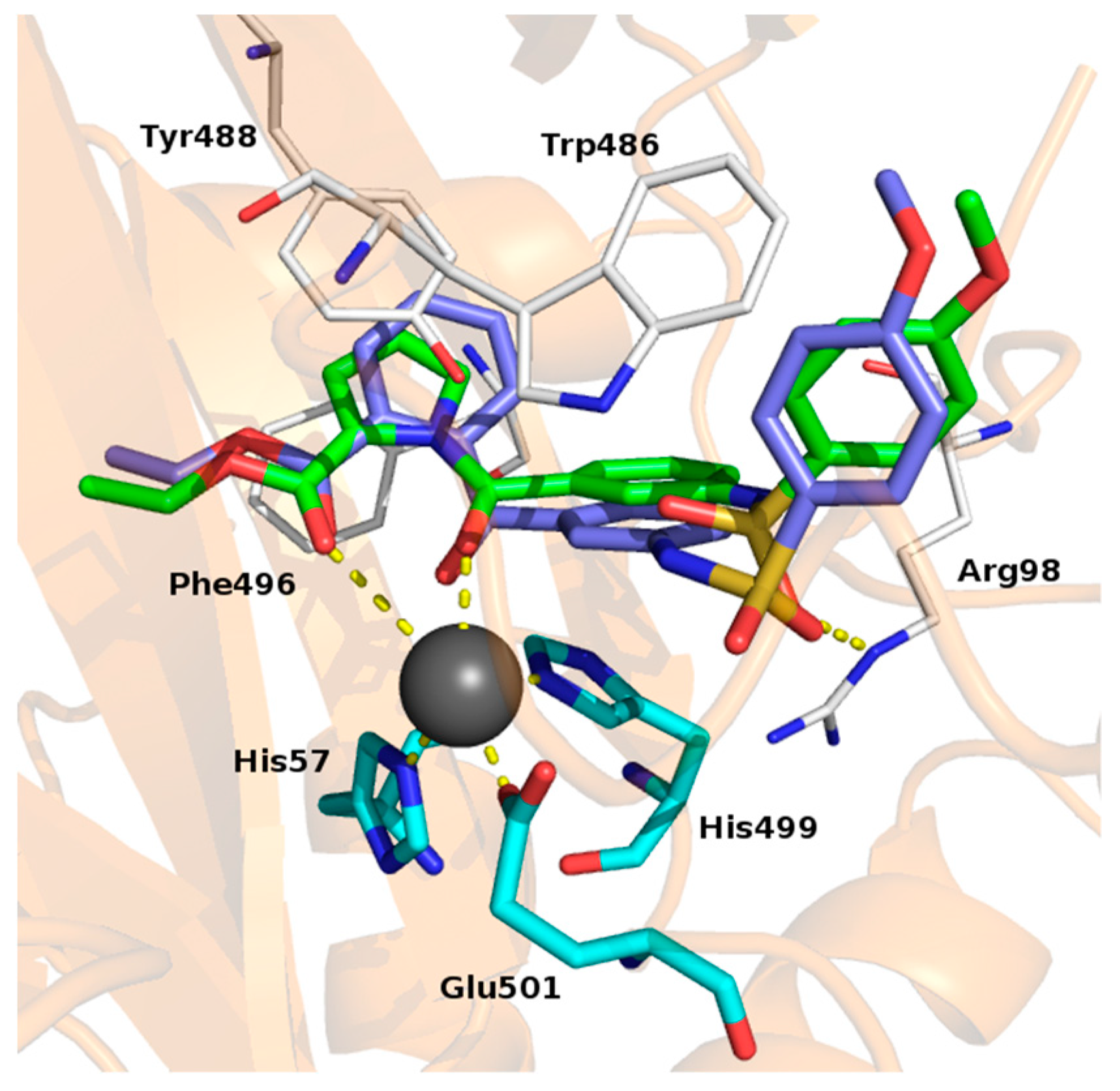
Molecular Modelling
4.5. Enzymatic Inhibition Assay
4.6. Protein Analysis
4.6.1. Preparation of Whole Cellular Lysate
4.6.2. Western Blotting Analysis
4.7. Gene Expression Analysis
4.7.1. RNA Extraction and Reverse Transcription
4.7.2. Quantitative RT-PCR
- -3′ CYP1A1 mRNA:
- - Fw: 5′- CCCCACAGCACAACAAGAGA-3′
- CYP1A1 mRNA:
- - Rv: 5′- CAGGGGTGAGAAACCGTTCA-3′
- AHRR mRNA:
- - Fw: 5′- CAATTACTCAGCAGGAAGGAGC-3′
- AHRR mRNA:
- - Rv: 5′- CTTGGGGTCAAGGACAAGGTC-3′-3′
- GAPDH mRNA:
- - Fw: 5′- TCCTCTGACTTCAACAGCGAC-3′
- GAPDH mRNA:
- - Rv: 5′- CGTTGTCATACCAGGAAAT-3′
4.8. Clonogenic Assay
4.9. Citotoxicity Assay
4.10. Flow-Cytometry
4.11. COMET Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NuRD | Nucleosome Remodeling and Deacetylase |
| HDAC | Histone DeACetylase |
| PARP | Poly (ADP-ribose) polymerase |
| KDM | histone lysine demethylase |
| LSD | Lysine-Specific histone Demethylase |
| JHDMS | Jumonji Histone Demethylases |
| SPR | Surface Plasmon Resonance |
| QRT-PCR | Quantitative Reverse Transcription-PCR |
| IC50 | half maximal inhibitory concentration |
References
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell. 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Forneris, E.; Binda, C.; Vanoni, M.A.; Mattevi, A.; Battaglioli, E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005, 579, 2203–2207. [Google Scholar] [CrossRef]
- Karytinos, A.; Forneris, F.; Profumo, A.; Clossani, G.; Battaglioli, E.; Binda, C.; Mattevi, A. A novel mammalian flavin-dependent histone demethylase. J. Biol. Chem. 2009, 284, 17775–17782. [Google Scholar] [CrossRef]
- Klose, J.R.; Kallin, E.M.; Khang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Gen. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Lohse, B.; Kristensen, J.L.; Kristensen, L.H.; Agger, K.; Helin, K.; Gajhede, M.; Clausen, R.P. Inhibitors of histone demethylases. Bioorg. Med. Chem. 2011, 19, 3625–3636. [Google Scholar] [CrossRef]
- Eissenberg, J.C.; Shilatifard, A. Histone H3 Lysine 4 (H3K4) Methylation in Development and Differentiation. Dev. Biol. 2010, 339, 240–249. [Google Scholar] [CrossRef]
- Lauberth, S.M.; Nakayama, T.; Wu, X.; Ferris, A.L.; Tang, Z.; Hughes, S.H.; Roeder, R.G. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 2013, 152, 1021–1036. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Berstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Outchkourov, N.S.; Muiño, J.M.; Kaufmann, K.; van Ijcken, W.F.; Groot Koerkamp, M.J.; van Leenen, D.; de Graaf, P.; Holstege, F.C.; Grosveld, F.G.; Timmers, H.T. Balancing of histone H3K4 methylation states by the Kdm5c/SMCX histone demethylase modulates promoter and enhancer function. Cell. Rep. 2013, 3, 1071–1079. [Google Scholar] [CrossRef]
- Blair, L.P.; Cao, J.; Zou, M.R.; Sayegh, J.; Yan, Q. Epigenetic Regulation by Lysine Demethylase 5 (KDM5) Enzymes in Cancer. Cancers 2011, 3, 1383–1404. [Google Scholar] [CrossRef]
- Hayami, S.; Yoshimatsu, M.; Veerakumarasivam, A.; Unoki, M.; Iwai, Y.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Yamaue, H.; et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: Involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol. Cancer 2010, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Sundquist, K.; Baeckstrom, D.; Poulsom, R.; Hanby, A.; Meier-Ewert, S.; Jones, T.; Mitchell, M.; Pitha-Rowe, P.; Freemont, P.; et al. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J. Biol. Chem. 1999, 274, 15633–15645. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhu, Z.; Han, G.; Ye, X.; Xu, B.; Peng, Z.; Ma, Y.; Yu, Y.; Lin, H.; Chen, A.P.; et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 19226–19231. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Ge, Z.; Wang, L.; Li, Q.; Wang, N.; Bjorkholm, M.; Jia, J.; Xu, D. The histone demethylase RBP2 Is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology 2010, 138, 981–992. [Google Scholar] [CrossRef]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell. 2007, 25, 801–812. [Google Scholar] [CrossRef]
- Defeo-Jones, D.; Huang, P.S.; Jones, R.E.; Haskell, K.M.; Vuocolo, G.A.; Hanobik, M.G.; Huber, H.E.; Oliff, A. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature 1991, 352, 251–254. [Google Scholar] [CrossRef]
- Benevolenskaya, E.V.; Murray, H.; Branton, P.; Young, R.A.; Kaelin, W.G., Jr. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol. Cell. 2005, 18, 623–635. [Google Scholar] [CrossRef]
- Hidalgo, A.; Baudis, M.; Petersen, I.; Arreola, H.; Piña, P.; Vázquez-Ortiz, G.; Hernández, D.; González, J.; Lazos, M.; López, R.; et al. Microarray comparative genomic hybridization detection of chromosomal imbalances in uterine cervix carcinoma. BMC Cancer 2005, 5, 77. [Google Scholar] [CrossRef]
- Wang, Q.; Wei, J.; Su, P.; Gao, P. Histone demethylase JARID1C promotes breast cancer metastasis cells via down regulating BRMS1 expression. Biochem. Biophys. Res. Commun. 2015, 464, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.J.; Piao, L.; Xi, Y.; Rincon-Arano, H.; Rothbart, S.B.; Peng, D.; Wen, H.; Larson, C.; Zhang, X.; Zheng, X.; et al. The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 2014, 6, 325–335. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Yang, S.; Song, N.; Zhou, X.; Gao, J.; Yu, N.; Shan, L.; Wang, Q.; Liang, J.; et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. USA 2014, 111, 7096–7101. [Google Scholar] [CrossRef]
- Gong, F.; Clouaire, T.; Aguirrebengoa, M.; Legube, G.; Miller, K.M. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell. Biol. 2017, 216, 1959–1974. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of Epigenetic Drugs in Disease: An Overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Højfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2014, 12, 917–930. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Trojer, P. Targeting histone lysine methylation in cancer. Pharmacol. Ther. 2015, 50, 1–22. [Google Scholar] [CrossRef]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [CrossRef]
- Spannhoff, A.; Hauser, A.T.; Heinke, R.; Sippl, W.; Jung, M. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. Chem. Med. Chem. 2009, 4, 1568–1582. [Google Scholar] [CrossRef]
- Hatch, S.B.; Yapp, C.; Montenegro, R.C.; Savitsky, P.; Gamble, V.; Tumber, A.; Ruda, G.F.; Bavetsias, V.; Fedorov, O.; Atrash, B.; et al. Assessing histone demethylase inhibitors in cells: Lessons learned. Epigenetics Chromatin 2017, 10, 9. [Google Scholar] [CrossRef]
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases—progress, challenges, and the future. Biochim. Biophys. Acta 2014, 1839, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- McAllister, T.E.; England, K.S.; Hopkinson, R.J.; Brennan, P.E.; Kawamura, A.; Schofield, C. Recent Progress in Histone Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1308–1329. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Sawada, H.; Suzuki, M.; Tojo, T.; Sasaki, R.; Hasegawa, M.; Mizukami, T.; Suzuki, T. Identification of Jumonji AT-Rich Interactive Domain 1A Inhibitors and Their Effect on Cancer Cells. ACS Med. Chem. Lett. 2015, 6, 665–670. [Google Scholar] [CrossRef]
- Gale, M.; Sayegh, J.; Cao, J.; Norcia, M.; Gareiss, P.; Hoyer, D.; Merkel, J.S.; Yan, Q. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget 2016, 7, 39931–39944. [Google Scholar] [CrossRef] [PubMed]
- Gehling, V.S.; Bellon, S.F.; Harmange, J.C.; LeBlanc, Y.; Poy, F.; Odate, S.; Buker, S.; Lan, F.; Arora, S.; Williamson, K.E.; et al. Identification of potent, selective KDM5 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4350–4354. [Google Scholar] [CrossRef]
- Mannironi, C.; Proietto, M.; Bufalieri, F.; Cundari, E.; Alagia, A.; Danovska, S.; Rinaldi, T.; Famiglini, V.; Coluccia, A.; La Regina, G.; et al. An high-throughput in vivo screening system to select H3K4-specific histone demethylase inhibitors. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Kristensen, L.H.; Nielsen, A.L.; Helgstrand, C.; Lees, M.; Cloos, P.; Kastrup, J.S.; Helin, K.; Olsen, L.; Gajhede, M. Studies of H3K4me3 demethylation by KDM5B/Jarid1B/PLU1 reveals strong substrate recognition in vitro and identifies 2,4-pyridine-dicarboxylic acid as an in vitro and in cell inhibitor. FEBS J. 2012, 279, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Sayegh, J.; Cao, J.; Zou, M.R.; Morales, A.; Blair, L.P.; Norcia, M.; Hoyer, D.; Tackett, A.J.; Merkel, J.S.; Yan, Q. Identification of small molecule inhibitors of Jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J. Biol. Chem. 2013, 288, 9408–9417. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R.; Ng, S.; Mecinović, J.; Liénard, B.M.; Bello, S.H.; Sun, Z.; McDonough, M.A.; Oppermann, U.; Schofield, C.J. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 2008, 51, 7053–7056. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Liu, Y.; Kubicek, S.; Myllyharju, J.; Tumber, A.; Ng, S.; Che, K.H.; Podoll, J.; Heightman, T.D.; Oppermann, U.; et al. A Selective Inhibitor and Probe of the Cellular Functions of Jumonji C Domain-Containing Histone Demethylases. J. Am. Chem. Soc. 2011, 133, 9451–9456. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Shi, L.; Lai, C.; O’Connell, S.M.; Xu, J.; Stansfield, R.K.; Hosfield, D.J.; Veal, J.M.; Stafford, J.A. Structure-based design and discovery of potent and selective KDM5 inhibitors. Bioorg Med. Chem. Lett. 2018, 28, 1490–1494. [Google Scholar] [CrossRef]
- Yamamoto, S.; Wu, Z.; Russnes, H.G.; Takagi, S.; Peluffo, G.; Vaske, C.; Zhao, X.; Moen Vollan, H.K.; Maruyama, R.; Ekram, M.B.; et al. JARID1B is a luminal lineage-driving oncogene in breast cancer. Cancer Cell. 2014, 25, 762–777. [Google Scholar] [CrossRef]
- Kim, S.; Dere, E.; Burgoon, L.D.; Chang, C.C.; Zacharewski, T.R. Comparative analysis of AhR-mediated TCDD-elicited gene expression in human liver adult stem cells. Toxicol. Sci. 2009, 112, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Kim, W.T.; Kim, D.H.; Park, J.W.; Kang, T.H.; Chung, J.W.; Leem, S.H. Tristetraprolin suppresses AHRR expression through mRNA destabilization. FEBS Lett. 2013, 587, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, L.; Ahmed, S.; Tamblyn, L.; Krutmann, J.; Förster, I.; Weighardt, H.; Matthews, J. Aryl hydrocarbon receptor repressor and TiPARP (ARTD14) use similar, but also distinct mechanisms to repress aryl hydrocarbon receptor signaling. Int. J. Mol. Sci. 2014, 15, 7939–7957. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Takane, Y.; Izawa, T.; Nakahama, T.; Inouye, Y. The inhibitory effect of aryl hydrocarbon receptor repressor (AhRR) on the growth of human breast cancer MCF-7 cells. Biol. Pharm. Bull. 2006, 29, 1254–1257. [Google Scholar] [CrossRef][Green Version]
- Srikannathasan, V.; Wohlkonig, A.; Shillings, A.; Singh, O.; Chan, P.F.; Huang, J.; Gwynn, M.N.; Fosberry, A.P.; Homes, P.; Hibbs, M.; Theobald, A.J.; et al. Crystallization and initial crystallographic analysis of covalent DNA-cleavage complexes of Staphyloccocus aureus DNA gyrase with QPT-1, moxifloxacin and etoposide. Acta Crystallogr F Struct Biol Commun. 2015, 71, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Lanigan, R.M.; Ruda, G.F.; Atrash, B.; McLaughlin, M.G.; Tumber, A.; Mok, N.Y.; Le Bihan, Y.V.; Dempster, S.; Boxall, K.J.; et al. 1.8-Substituted Pyrido[3,4-D]Pyrimidin-4(3H)-One Derivatives as Potent, Cell Permeable, Kdm4 (Jmjd2) and Kdm5 (Jarid1) Histone Lysine Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1388–1409. [Google Scholar] [CrossRef]
- Tumber, A.; Nuzzi, A.; Hookway, E.S.; Hatch, S.B.; Velupillai, S.; Johansson, C.; Kawamura, A.; Savitsky, P.; Yapp, C.; Szykowska, A.; et al. Potent and Selective KDM5 Inhibitor Stops Cellular Demethylation of H3K4me3 at Transcription Start Sites and Proliferation of MM1S Myeloma Cells. Cell. Chem. Biol. 2017, 24, 371–380. [Google Scholar] [CrossRef]
- Penterling, C.; Drexler, G.A.; Böhland, C.; Stamp, R.; Wilke, C.; Braselmann, H.; Caldwell, R.B.; Reindl, J.; Girst, S.; Greubel, C.; et al. Depletion of Histone Demethylase Jarid1A Resulting in Histone Hyperacetylation and Radiation Sensitivity Does Not Affect DNA Double-Strand Break Repair. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Rafehi, H.; Orlowski, C.; Georgiadis, G.T.; Ververis, K.; El-Osta, A.; Karagiannis, T.C. Clonogenic assay: Adherent cells. J. Vis. Exp. 2011, 49. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhang, Y.; Wang, X.; Zhang, X.; Gao, Y.; Yin, L.; Li, Q.; Li, J. Induction and inhibition of the pan-nuclear gamma-H2AX response in resting human peripheral blood lymphocytes after X-ray irradiation. Cell Death Discovery 2016, 2, 16011. [Google Scholar] [CrossRef]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, M.M.; Simon, J.A. Inner workings and regulatory inputs that control Polycomb repressive complex 2. Chromosoma 2012, 121, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Mocavini, I.; Pippa, S.; Licursi, V.; Paci, P.; Trisciuoglio, D.; Mannironi, C.; Presutti, C.; Negri, R. JARID1B expression and its function in DNA damage repair are tightly regulated by miRNAs in breast cancer. Cancer Sci. 2019, 110, 1232–1243. [Google Scholar] [CrossRef]
- Rath, B.H.; Waung, I.; Camphausen, K.; Tofilon, P.J. Inhibition of the histone H3K27 demethylase UTX enhances tumor cell radiosensitivity. Mol. Cancer Ther. 2018, 17, 1070–1078. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Ilari, A.; Fiorillo, A.; Poser, E.; Lalioti, V.S.; Sundell, G.N.; Ivarsson, Y.; Genovese, I.; Colotti, G. Structural basis of Sorcin-mediated calcium-dependent signal transduction. Sci Rep. 2015, 5, 16828. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput Aided Mol. Des. 2013, 3, 221–234. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banath, J.P.; Durand, R.E. Heterogeneity in radiation induced DNA damage and repair in tumor and normal cells using the “Comet” assay. Radiat. Res. 1990, 122, 86–94. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound RS5033 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Compd | R1 | Position | R2 |
| RS3195 | CH3O | 3 |  |
| RS3152 | CH3O | 3 |  |
| RS3183 | CH3O | 3 |  |
| RS5033 | CH3O | 3 |  |
| RS4995 | Br | 4 |  |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pippa, S.; Mannironi, C.; Licursi, V.; Bombardi, L.; Colotti, G.; Cundari, E.; Mollica, A.; Coluccia, A.; Naccarato, V.; La Regina, G.; et al. Small Molecule Inhibitors of KDM5 Histone Demethylases Increase the Radiosensitivity of Breast Cancer Cells Overexpressing JARID1B. Molecules 2019, 24, 1739. https://doi.org/10.3390/molecules24091739
Pippa S, Mannironi C, Licursi V, Bombardi L, Colotti G, Cundari E, Mollica A, Coluccia A, Naccarato V, La Regina G, et al. Small Molecule Inhibitors of KDM5 Histone Demethylases Increase the Radiosensitivity of Breast Cancer Cells Overexpressing JARID1B. Molecules. 2019; 24(9):1739. https://doi.org/10.3390/molecules24091739
Chicago/Turabian StylePippa, Simone, Cecilia Mannironi, Valerio Licursi, Luca Bombardi, Gianni Colotti, Enrico Cundari, Adriano Mollica, Antonio Coluccia, Valentina Naccarato, Giuseppe La Regina, and et al. 2019. "Small Molecule Inhibitors of KDM5 Histone Demethylases Increase the Radiosensitivity of Breast Cancer Cells Overexpressing JARID1B" Molecules 24, no. 9: 1739. https://doi.org/10.3390/molecules24091739
APA StylePippa, S., Mannironi, C., Licursi, V., Bombardi, L., Colotti, G., Cundari, E., Mollica, A., Coluccia, A., Naccarato, V., La Regina, G., Silvestri, R., & Negri, R. (2019). Small Molecule Inhibitors of KDM5 Histone Demethylases Increase the Radiosensitivity of Breast Cancer Cells Overexpressing JARID1B. Molecules, 24(9), 1739. https://doi.org/10.3390/molecules24091739