Abstract
An uncomplicated, high-yielding synthetic route has been developed to constitute complicated heterocycles, applying domino, click and retro-Diels–Alder (RDA) reaction sequences. Starting from 2-aminocarboxamides, a new set of isoindolo[2,1-a]quinazolinones was synthesized with domino ring closure. A click reaction was performed to create the 1,2,3-triazole heterocyclic ring, followed by an RDA reaction resulting in dihydropyrimido[2,1-a]isoindole-2,6-diones. The absolute configuration, concluded by the norbornene structure that served as a chiral source, remained constant throughout the transformations. The structure of the synthesized compounds was examined by 1H and 13C Nuclear Magnetic Resonance (NMR) methods.
1. Introduction
Compounds containing quinazoline and triazole heterocyclic rings play a significant role in both organic and pharmaceutical chemistries. Quinazoline heterocycles are important subunits of a broad diversity of synthetic pharmaceuticals as well as natural compounds with antiviral [1,2], anti-inflammatory [3], antimalarial [4], and anticancer [5] activities. The synthesis of bioactive saturated quinazoline derivatives was also investigated [6,7,8]. In the past few years, researchers have exploited heterocycles containing the 1,2,3-triazole ring to generate many medicinal scaffolds exhibiting anti-HIV [9], antibacterial [10,11,12] and anticancer activities [13,14]. Building a triazole ring into a compound can change or even improve the pharmacokinetic properties of a drug [15,16,17,18,19,20,21,22,23].
There exist numerous methods, which are effective without using the classic and time-consuming protection–deprotection processes as well as purification methodology of intermediates [24,25,26,27] in the synthesis of diverse and complex chiral compounds from simple substrates in an economically suitable manner. At the same time, the use of multi-component domino reactions in asymmetric synthesis has gained a continuously increasing interest [28,29,30,31,32]. Asymmetric domino reactions are based on the application of removable chiral auxiliaries and chiral reagents [24].
The 1,4-disubstituted 1,2,3-triazole function still plays an important role in drug discovery, which justifies the continuous advancement of new strategies for their synthesis [33,34,35]. One of the most widely used click-chemistry methods in this field is the copper-catalyzed 1,3-dipolar cycloaddition between an alkyne and an azide (CuAAC), due to its simplicity and high selectivity [36,37,38,39].
For synthetic chemists, the RDA reaction has become a valuable tool for their research towards the design and synthesis of novel heterocyclic scaffolds, because of its efficiency in introducing a double bond into a heterocyclic skeleton [40] along with enantiocontrolled [41] and enantiodivergent [42] syntheses of heterocyclic compounds.
The skeletal transformations of heterocycles take its place in the construction of complex molecular frameworks from simple feedstock among the most powerful synthetic strategies [43,44,45]. Within this context, domino ring closure and RDA are the predominant approaches, since they lead to valuable nitrogenated heterocycles of high biological activity [46,47,48,49,50,51,52,53], such as isoindolo- and pyrroloquinazolinones. In our laboratory, their reactivity and skeletal deformation have been widely examined under mild conditions.
Continuing our work on the synthesis of novel N-heterocycles and focusing on the biological potential of fused quinazolinones [47,48,49,50,51,52,53], herein we report the synthesis of a new series of isoindolo[2,1-a]quinazolinones and pyrimido[2,1-a]isoindoles starting from 2-aminocarboxamides.
Our present aim was (i) to examine the diastereoselectivity of the domino ring-closure reaction of N-propargyl-substituted diendo- and diexo-2-aminonorbornenecarboxamides with 2-formylbenzoic acid, (ii) to develop Cu(I)-catalyzed azide/alkyne cycloaddition (CuAAC) in a regioselective manner, (iii) to investigate the RDA reaction of the created isoindolo[2,1-a]quinazolinones, and (iv) to extend this methodology to obtain different racemic and enantiomeric pyrimido[2,1-a]isoindole derivatives containing a triazole ring.
2. Results
First, all methods were performed and optimized with racemic starting materials followed by repeating the syntheses with the enantiomers. In Scheme 1 and Scheme 2 only a single enantiomer is represented for evidence. Please note that all spectroscopic data of the racemic compounds were identical to those of the enantiomeric samples.
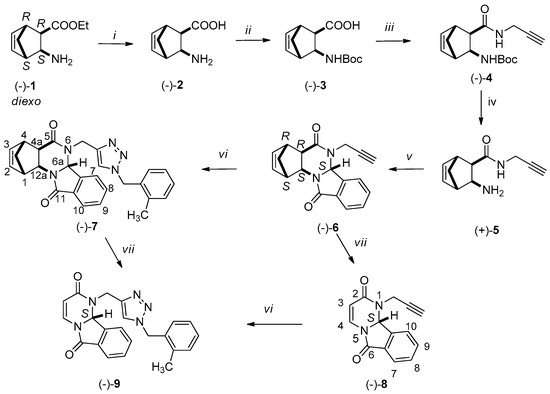
Scheme 1.
Reagents and conditions: (i) H2O, MW 100 °C, 30 min, 300 W, 90%; (ii) Boc2O, NaOH, dioxane/H2O, 0 °C, 30 min, r.t., 4 h, 87%; (iii) DIC, HOBt, propargylamine, THF, r.t., 24 h, 75%; (iv) 1. HCl/H2O 10%, r.t., 4 h 2. NaOH, H2O/CHCl3; (v) 2-formylbenzoic acid, pTSA, EtOH, MW, 100 °C, 30 min; 71%; (vi) 1. 2-methylbenzyl chloride, NaN3, Et3N, H2O/tBuOH, r.t., 1 h, 2. sodium ascorbate, (−)-6 or (−)-8, CuSO4, r.t. 8 h, 70–72%; (vii) MW, DCB, 220 °C, 60 min, 300 W, 70–72%.
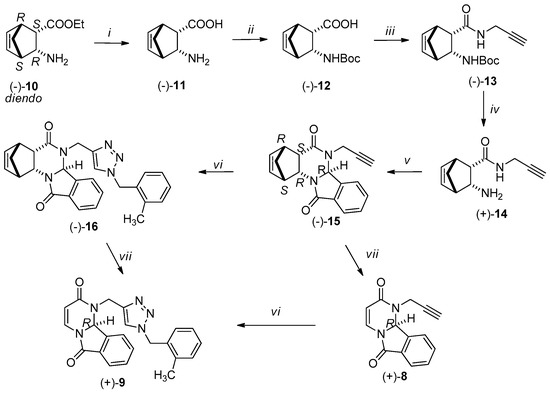
Scheme 2.
Reagents and conditions, see in Scheme 1, yields: (i) 85%; (ii) 85%; (iii) 72%; (iv-v) 70% (vi) 70–72%; (vii) 70–72%.
Racemic N-Boc-protected amino acids (±)-3 and (±)-12 were produced from the corresponding diendo- and diexo-3-aminobicyclo[2.2.1]hept-5-ene-2-carboxylic acids (±)-2 and (±)-11 according to an earlier procedure [54]. The synthesis of enantiomeric Boc-protected amino acids (−)-3 and (−)-12 started from enantiomeric 2-aminonorbornene esters (−)-1 and (−)-10, as depicted in Scheme 1 and Scheme 2. The starting enantiomeric 2-aminonorbornene esters (−)-1 and (−)-10 were prepared from racemic amino esters (±)-1 and (±)-4 by diastereomeric salt formation as previously described [45].
The free enantiomeric amino esters (−)-1 (ee > 95%) and (−)-10 (ee > 96%) were hydrolyzed with water under microwave irradiation to furnish amino acids (−)-2 and (–)-11, which were then reacted with di-tert-butyl dicarbonate affording N-Boc-protected amino acids (−)-3 and (−)-12. They were then transformed into propargylamides (−)-4 and (−)-13 in tetrahydrofuran using propargylamine in the presence of N,N′-diisopropylcarbodiimide (DIC) and hydroxybenzotriazole (HOBt). After acidic deprotection of amides (−)-4 and (−)-13, free amide bases (+)-5 and (+)-14 were used in the next step without purification.
The reaction of propargylamides 5 and 14 with 2-formylbenzoic acid is plausibly interpreted as a domino process, in which the first step is the Schiff base formation [47,50]. The Schiff base undergoes ring closure to give isoindolo[2,1-a]quinazolinones 6 and 15, respectively.
The reaction was implemented by dissolving (+)-5 and (+)-14 in ethanol using one equivalent of 2-formylbenzoic acid and stirring the solution at 100 °C for 30 min under microwave irradiation in the presence of p-toluenesulfonic acid. The 1H NMR spectra revealed the formation of the isoindolo[2,1-a]quinazoline (−)-6 and (−)-15 (Scheme 1 and Scheme 2).
The full NMR signal assignment was carried out for compounds (−)-6 and (−)-15. The relative configuration of the new hydrogen in the product from the diexo isomer, according to the characteristic NOE crosspeaks, is in trans arrangement with the annelated hydrogen atoms in (–)-6. The characteristic structure crosspeak was found for (−)-6 between protons C(6a)-H and C(13)-H. For the diendo isomer, in the product of the ring closure reaction, the annelated hydrogen atoms and the relative configuration of the new hydrogen are trans in (−)-15. For (−)-15 the characteristic structure crosspeaks were found between the C(6a)-H and C(3)-H protons.
In the next step the 1,2,3-triazole ring was formed by click reaction using Cu(I)-catalyzed azide/alkyne (CuAAc) cycloaddition. The azide was synthesized in situ by dissolving sodium azide in the mixture of 2-methylbenzyl chloride, trimethylamine, and tert-butyl alcohol and stirring at room temperature for 1 h [35]. Afterwards, (−)-6 or (−)-15 was added along with copper(II) sulfate and sodium ascorbate as a reducing agent. The nascent copper(I) acting as the catalyst is responsible for the regioselectivity [35]. The CuAAC reaction of the terminal alkyne moiety of (−)-6 and (−)-15 was completely regioselective affording 1,4-disubstituted triazoles (−)-7 and (−)-16.
In the last step, RDA reaction was performed with (−)-6, (−)-7 and (−)-15, (−)-16 resulting in (−)-8, (−)-9 and (+)-8, (+)-9, respectively. The reactions were carried out in 1,2-dichlorobenzene by stirring under microwave irradiation at 220 °C for 60 min.
3. Materials and Methods
3.1. General Methods
1H NMR spectra were recorded at 500.20 MHz, while the 13C NMR spectra were measured at 125.62 MHz in CDCl3 or in DMSO-d6 at ambient temperature, with a Bruker AV NEO Ascend 500 spectrometer (Bruker Biospin, Karlsruhe, Germany) with Double Resonance Broad Band Probe (BBO). Chemical shifts are given, relative to tetramethysilane (TMS) as internal standard, in δ (ppm). Elemental analyses were performed with a Perkin–Elmer CHNS-2400 Ser II Elemental Analyzer. Microwave-promoted reactions were carried out in sealed reaction vials (10 mL) in a microwave (CEM, Discover, SP) cavity (CEM Corporation, Matthwes, NC, USA). Optical rotations were measured with a Perkin–Elmer 341 polarimeter (Perkin–Elmer, Shelton, CT, USA). Melting points were determined with a Hinotex-X4 micro melting point apparatus (Hinotek, Ningbo, China) and are uncorrected. Racemic diendo- and diexo-3-aminobicyclo[2.2.1]hept-5-ene-2-carboxylic acids (±)-2 and (±)-11 were prepared according to a literature procedure [55,56].
The enantiomers of 2-aminonorbornene esters (+)-1, (−)-1, (+)-10 and (−)-10 were prepared from racemic 2-aminonorbornene esters via diastereomeric salt formation with O,O′-di-p-toluoyltartaric acid (DPTTA) and O,O′-dibenzoyltartaric acid (DBTA) as previously published [45]. The ee values of (+)-1 and (−)-1 were determined by a literature method [45]. The ee values for (+)-10 (92%) and (−)-10 (98%) were determined by HPLC using Phenomenex-IA column after derivatization with benzoyl chloride in the presence of TEA. Analytical conditions were as follows: eluent: a mixture of n-hexane and isopropyl alcohol (IPA) (70:30), flow rate: 0.3 mL·min−1, detection at 254 nm, retention times: (−)-10: 40.81 min (antipode: 25.33 min). The ee values for (−)-4 (98%), (+)-4 (95%), (−)-13 (95%) and (+)-13 (96%) were determined by GC on a Chirasil-L-Val column (25 m): 180 °C isotherm, 1 mL·min−1, retention times (−)-13: 4.91 min, (+)-13: 4.52 min; flow rate 1 mL·min−1, 160 °C for 5 min → 180 °C (rate of temperature rise 10 °C/min; retention times (−)-4: 11.84 (min), (+)-4: 12.64 (min). The ee values of the domino ring closure compounds (−)-6 and (+)-6 were identified by HPLC using Chiracel-OD-H column, eluent: a mixture of n-hexane and IPA (70:30), flow rate: 0,15 mL·min−1, detection at 254 nm, retention times (−)-6: 61.83 min, (+)-6: 66.29 min. The ee values of the domino ring closure products (−)-15 and (+)-15 were identified by HPLC using Phenomenex-IA column, eluent: a mixture of n-hexane and IPA (60:40), flow rate: 1 mL·min−1, detection at 254 nm, retention times (−)-15: 22.04 min, (+)-15: 42.99 min. The ee values of the RDA products (−)-8 and (+)-8 were determined by HPLC using Phenomenex-IA column, eluent: a mixture of n-hexane and IPA (70:30 containing 0.1% DEA), flow rate: 0.5 mL·min−1, detection at 254 nm, retention times (−)-8: 80.88 min, (+)-8: 76.58 min. The ee values of RDA products (−)-9 and (+)-9 were determined by HPLC using ChiralPak-IA column, eluent: a mixture of n-hexane and IPA (60:40 containing 0.1% DEA), flow rate: 0.5 mL·min−1, detection at 254 nm, retention times (−)-9: 26.76 min, (+)-9: 23.87 min.
3.2. Synthesis of New Compounds
3.2.1. Synthesis of Amino Acids [(+)-2, (−)-2, (+)-11, (−)-11]
2-Aminonorbornene ester hydrochlorides (+)-1, (−)-1 [45] or (+)-10, (−)-10 [45] (3.00 g, 13.82 mmol in 30 mL H2O) were treated with 10% aqueous NaOH solution (30 mL) to liberate the free acids. The aqueous layer was extracted with CHCl3 (3 × 30 mL). The combined organic phase was dried (Na2SO4) and the solvent was evaporated. The residue was diluted in water (2 mL) in a 10-mL pressurized reaction vial, and the solution was stirred and warmed to 100 °C for 30 min at max. 300 W microwave irradiation. The solvent was evaporated, and the crude product was filtered off from acetone and recrystallized from water/acetone. [(−)-2]: white crystals (90% yield), m.p. 258–262 °C. Lit m.p. >260 °C, = −12.1 (c = 0.5, H2O), lit [57] = = −12.3 [57] (c = 0.3, H2O). [(+)-2]: white crystals (91% yield), m.p. 257–260 °C = +12.0 (c = 0.5, H2O). Lit. [58] = +13.8 (c = 1, H2O). [(−)-11]: white crystals (85% yield) m.p. 236–238 °C, = −62 (c = 0.5, H2O). [(+)-11]: white crystals (91% yield) m.p. 233–237 °C, = +62 (c = 1, H2O). 1H and 13C NMR data of the enantiomeric amino acids were identical with those of the racemic compounds [55,56].
3.2.2. Synthesis of Boc-Protected Amino Acids [(+)-3, (−)-3, (+)-12, (−)-12]
To a solution of the appropriate 2-aminonorbornene carboxylic acid [(+)-2, (−)-2 (+)-11 or (−)-11, 3.1 g, 10 mmol] in 100 mL of a 2:1 dioxane/H2O mixture, 25 mL 1 M NaOH was added. The solution was cooled to 0 °C and Boc2O (2.4 g, 11 mmol) was added. The mixture was stirred at 0 °C for 1 h, warmed to r.t., stirred for 4 h and, finally, it was evaporated to 30 mL. The solvent was acidified with 10% H2SO4 (pH = 2.5) and the resulting mixture was extracted with EtOAc (3 × 50 mL). The solvent was evaporated, and the crude product was filtered off from Et2O and recrystallized from iPr2O. [(−)-3]: white crystals (87% yield), m.p. 133–135 °C, = −78 (c = 1, EtOH). [(+)-3]: white crystals (91% yield), m.p. 136–137 °C, = +76 (c = 1, EtOH). [(−)-12]: white crystals (85% yield), m.p. 132–135 °C, = −14 (c = 1, EtOH). [(+)-12]: white crystals (91% yield), m.p. 133–134 °C, = +13 (c = 1, EtOH). 1H and 13C NMR data of the enantiomeric amino acids were identical with those of the racemic compounds [54,55,56,57,58,59].
3.2.3. Synthesis of Boc-Protected Propargyl Amides [(+)-4, (−)-4, (+)-13, (−)-13]
A mixture of the appropriate amino acid [(+)-3, (−)-3 (+)-12 or (−)-12, 2.53 g, 10 mmol], hydroxybenzotriazole (1.83 g, 12 mmol), N,N′-diisopropylcarbodiimide (DIC) (1.51 g, 12 mmol), and propargylamine (0.55 g, 0.7 mL, 10 mmol) was stirred in THF (50 mL) overnight at r.t. After completion of the reaction (checked by thin layer chromatography), the solvent was evaporated. Purification of the residue by column chromatography over silica gel with EtOAc gave the desired products.
tert-Butyl ((1R,2R,3S,4S)-3-(prop-2-yn-1-ylcarbamoyl)bicyclo[2.2.1]hept-5-en-2-yl)carbamate [(−)-4]: White crystals (75% yield), m.p. 128–130 °C, = −38 (c = 1, EtOH), 1H-NMR (500 MHz, CDCl3, 30 °C): δ = 1.42 (s, 9H, CH3) 1.60 (m, 1H, H-7), 2.10 (m, 1H, H-7), 2.20 (t, J = 2.3 Hz, 1H, C≡CH) 2.35 (d, J = 8.2 Hz, 1H, H-2), 2.69 (m, 1H, H-1), 2.98 (m, 1H, H-4), 3.85–3.92 (m, H, H-4), 4.11–4.16 (m, 1H, NCH2), 5.09–5.21 (m, 1H, NCH2) 5,91 (s, 1H, NH) 6.14–6.23 (m, 2H, H-5, H-6) 13C NMR (125 MHz, CDCl3, 30 °C): δ = 28.4, 29.4, 44.9, 45.6, 48.0, 48.1, 53.0, 71.7, 79.3, 79.5, 137.5, 138.8, 155.9, 172.8 ppm. C16H22N2O3 (290.36): calcd. C, 66.18; H, 7.64; N, 9.65; found C 66.44; H 7.91; N 9.42.
tert-Butyl ((1S,2S,3R,4R)-3-(prop-2-yn-1-ylcarbamoyl)bicyclo[2.2.1]hept-5-en-2-yl)carbamate [(+)-4]: White crystals (74% yield). m.p. 129–131 °C, = +37 (c = 1, EtOH).
tert-Butyl ((1R,2S,3R,4S)-3-(prop-2-yn-1-ylcarbamoyl)bicyclo[2.2.1]hept-5-en-2-yl)carbamate [(−)-13]: White crystals (72% yield), m.p. 161–163 °C, = −3.7 (c = 0.5, EtOH), 1H-NMR (500 MHz, CDCl3, 30 °C): δ = 1.29–1.50 (m, 11H, CH3, H-7, H-7) 2.19 (t, J = 2.3 Hz, 1H, C≡CH) 2.98–3.10 (m, 3H, H-1, H-2, H-4), 3.75–3.83 (m, H, H-3), 4.03–4.13 (m, 1H, NCH2), 4.78–4.86 (m, 1H, NCH2) 6.06–6.11 (m, 1H, H-6) 6.20 (s, 1H, NH) 6.59–6.61 (m, 1H, H-5), 13C NMR (125 MHz, CDCl3, 30 °C): δ = 28.4, 29.4, 46.3, 47.1, 47.7, 53.0, 71.7, 79.3, 79.5, 137.5, 138.8, 155.9, 172.8 ppm. C16H22N2O3 (290.36): calcd. C, 66.18; H, 7.64; N, 9.65; found C 66.41; H 7.71; N 9.52.
tert-Butyl ((1S,2R,3S,4R)-3-(prop-2-yn-1-ylcarbamoyl)bicycle[2.2.1]hept-5-en-2-yl)carbamate [(+)-13]: White crystals (74% yield), m.p. 160–163 °C, = +4.1 (c = 0.5, EtOH).
3.2.4. Synthesis of Domino Ring Closure Products [(−)-6, (+)-6, (−)-15 and (+)-15]
The mixture of Boc-protected amides (+)-4, (−)-4 or (+)-13, (−)-13 (2.32 g, 8 mmol in 15 mL H2O) was stirred with 10% aqueous HCl solution (10 mL) at r.t. for 6 h. The aqueous layer was neutralized with 10% aqueous NaOH solution and extracted with CH2Cl2 (3 × 30 mL). The combined organic phase was dried (Na2SO4) and the solvent was evaporated. The resulting amides were used for the next steps without purification. Amide (+)-5, (−)-5, (+)-14 or (−)-14 (1.33 g, 7 mmol), 2-formylbenzoic acid (1.05 g, 7 mmol), and pTSA (20 mol%) were dissolved in EtOH (5 mL) in a 10-mL pressurized reaction vial, and the solution was warmed to 100 °C for 30 min at max. 300 W microwave irradiation. Upon completion of the reaction (monitored by TLC) the solvent was evaporated and the crude solid was filtered off from Et2O and recrystallized from EtOH.
(1S,4R,4aR,6aS,12aS)-6-(Prop-2-yn-1-yl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(−)-6]: White crystals (71% yield), m.p. 189–191 °C, = −19 (c = 0.5, EtOH), 1H-NMR (500 MHz, CDCl3, 30 °C): δ = 1.66–1.80 (m, 2H, H-13, H-13) 2.45 (t, J = 2.4 Hz, 1H, C≡CH), 2.63–2.69 (m, 1H, H-4a) 2.91–2.96 (m, 1H, H-4), 3.46–3.48 (m, 1H, H-1), 3.80–3.83 (m, 1H, H-12a), 4.53–4.59 (m, 1H, NCH2), 5.40–5.52 (m, 1H, NCH2) 6.18 (s, 1H, H-6a) 6.35–6.44 (m, 2H, H-2, H-3) 7.59–8.10 (m, 4H, Ar), 13C NMR (125 MHz, CDCl3, 30 °C): δ = 32.5, 42.4, 45.7, 49.2, 50.1, 50.4, 68.1, 74.5, 77.8, 125.1, 125.3, 130.8, 132.5, 138.5, 138.8, 139.5, 167.2, 171.3. ppm. C19H16N2O2 (304.34.36): calcd. C, 74.98; H, 5.30; N, 9.20; found C 74.91; H 5.42; N 9.35.
(1R,4S,4aS,6aR,12aR)-6-(Prop-2-yn-1-yl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(+)-6]: White crystals (69% yield), m.p. 189–192 °C, = +18 (c = 0.5, EtOH).
(1S,4R,4aS,6aR,12aR)-6-(Prop-2-yn-1-yl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(−)-15]: White crystals (70% yield), m.p. 189–191 °C, = −34.9 (c = 0.5, EtOH), 1H-NMR (500 MHz, CDCl3, 30 °C): δ = 1.61–1.62 (m, 2H, H-13, H-13) 2.40 (t, J = 2.1 Hz, 1H, C≡CH), 3.10–3.19 (m, 1H, H-4a) 3.25–3.35 (m, 1H, H-4), 3.47–3.54 (m, 1H, H-1), 3.60–3.73 (m, 1H, H-12a), 5.22–5.39 (m, 2H, NCH2), 5.97 (s, 1H, H-6a), 6.26–6.31 (m, 1H, H-3), 6.44–6.52 (m, 1H, H-2) 7.52–7.96 (m, 4H, Ar), 13C NMR (125 MHz, CDCl3, 30 °C): δ = 32.2, 32.5, 42.4, 45.3, 49.2, 50.1, 50.4, 68.1, 74.5, 78.2, 125.1, 155.3, 130.8, 132.5, 138.5, 138.8, 139.5, 167.2, 171.3. ppm. C19H16N2O2 (304.34.36): calcd. C, 74.98; H, 5.30; N, 9.20; found C 74.82; H 5.51; N 9.32.
(1R,4S,4aR,6aS,12aS)-6-(Prop-2-yn-1-yl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(+)-15]: White crystals (74% yield), m.p. 189–192 °C, = +36 (c = 0.5, EtOH).
3.2.5. General Procedure for the Synthesis of (−)-7, (+)-7, (−)-16 and (+)-16 by Click Reaction
A solution of 2-methylbenzyl chloride (1.1 mmol), NaN3 (1 mmol), and Et3N (1.3 mmol) in H2O/tBuOH (1:1, 10 mL) was stirred at r.t. for 1 h. Subsequently, a mixture of 1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione derivative ((−)-6, (+)-6, (−)-15 or (+)-15; 1 mmol), sodium ascorbate (0.15 mmol), and CuSO4 (10 mol%) was added to the freshly prepared azide derivative and the mixture was stirred at r.t. for 8 h. The mixture was diluted with H2O and extracted with EtOAc. The organic phase was dried (Na2SO4), the solvent was evaporated off. Purification of the residue by column chromatography over silica gel with EtOAc gave the desired products. (The same procedure was used for the synthesis of (−)-9 and (+)-9 by click reaction starting from (−)-8 and (+)-8).
(1S,4R,4aR,6aS,12aS)-6-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)-methyl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(−)-7]: White crystals (70% yield), m.p. 100–107 °C, = −20 (c = 0.5, EtOH), 1H-NMR (500 MHz, CDCl3, 30 °C): δ = 1.31–1.42 (m, 2H, H-13) 2.26 (s, 3H, CH3), 2.54–2.58 (m, 1H, H-4a) 2.81–2.85 (m, 1H, H-4), 3.17–3.21 (m, 1H, H-1), 4.50–4.54 (m, 1H, H-12a), 4.57–4.64 (d, J = 15.9 Hz, 1H, NCH2), 5.35–5.42 (d, J = 15.9 Hz, 1H, NCH2), 5.53 (s, 2H, NCH2Ar) 6.10 (s, 1H, H-6a), 6.26–6.41 (m, 2H, H-2, H-3), 7.03–7.33 (m, 4H, Ar), 7.37 (s, 1H, CHtriazol) 7.52–8.34 (m, 4H, Ar), 13C NMR (125 MHz, CDCl3, 30 °C): δ = 19.0, 37.5, 41.9, 44.5, 48.6, 49.4, 49.6, 52.4, 68.1, 122.6, 124.4, 126.5, 126.7, 129.2, 129.3, 130.3, 131.0, 131.9, 132.1, 132.4, 136.7, 138.2, 139.6, 144.0, 166.9, 171.5. ppm. C27H25N5O2 (451.52): calcd. C, 71.82; H, 5.58; N, 15.51; found C 71.93; H 5.47; N 15.35.
(1R,4S,4aS,6aR,12aR)-6-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)-methyl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(+)-7]: White crystals (74% yield), m.p. 100–102 °C, = +20 (c = 0.5, EtOH).
(1S,4R,4aS,6aR,12aR)-6-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)-methyl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(−)-16]: White crystals (72% yield), m.p. 128–130 °C, = −20 (c = 0.5, EtOH), 1H-NMR (500 MHz, CDCl3, 30 °C): δ = 1.53–1.65 (m, 2H, H-13) 2.28 (s, 3H, CH3), 3.08 (m, 1H, H-4a) 3.20–3.25 (m, 1H, H-4), 3.28–3.33 (m, 1H, H-1), 4.45–4.55 (m, 1H, H-12a), 5.20–5.29 (m, 2H, NCH2), 5.47 (m, 2H, NCH2Ar) 5.64 (m, 1H, H-3), 5.79 (s, 1H, H-6a), 5.98 (m, 1H, H-2) 7.03–7.32 (m, 4H, Ar), 7.40 (s, 1H, CHtriazol) 7.51–8.40 (m, 4H, Ar), 13C NMR (125 MHz, CDCl3, 30 °C): δ = 19.0, 37.2, 42.7, 47.8, 48.8, 48.9, 50.0, 52.4, 68.6, 122.9, 124.2, 126.8, 126.8, 129.3, 129.4, 130.0, 131.1, 131.9, 131.9, 132.4, 135.4, 136.7, 137.0, 139.7, 144.0, 167.5, 171.2 ppm. C27H25N5O2 (451.52): calcd. C, 71.82; H, 5.58; N, 15.51; found C 71.91; H 5.43; N 15.32.
(1R,4S,4aR,6aS,12aS)-6-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)-methyl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(+)-16]: White crystals (74% yield), m.p. 97–99 °C, = +19.3 (c = 0.5, EtOH).
3.2.6. RDA Protocols for the Synthesis of Pyrimido[2,1-a]isoindols [(−)-8 (+)-8, (−)-9, (+)-9]
Isoindoloquinazoline derivatives [(−)-6, (+)-6, (−)-7, (+)-7, (−)-15, (+)-15, (−)-16, (+)-16, 0.5 mmol] were dissolved in 5 mL 1,2-dichlorobenzene. The solution was stirred and heated to 220 °C for 60 min at max. 300 W microwave irradiation. Then the solvent was evaporated, the residue was dissolved in EtOAc and purified by column chromatography on silica gel eluting with EtOAc.
(S)-1-(Prop-2-yn-1-yl)-1,10b-dihydropyrimido[2,1-a]isoindole-2,6-dione [(−)-8]: White crystals (52% yield), m.p. 91–93 °C, = −376 (c = 0.2, EtOH), 1H-NMR (500 MHz, CDCl3, 30 °C): δ = 2.45 (t, J = 2.5 Hz, 1H, C≡CH), 3.73 (dd, J = 17.7, 2.1, 1H,CH2) 5.15 (d, J = 17.7 Hz, 1H, CH2) 5.54 (d, J = 7.6 Hz, 1H, H-3) 6.20 (s, 1H, H-10b) 7.66–8.10 (m, 5H, Ar, H-4), 13C NMR (125 MHz, CDCl3, 30 °C): δ = 31.3, 69.8, 73.7, 79.9, 106.8, 125.4, 125.4, 130.8, 131.7, 133.7, 133.7, 138.5, 164.3, 165.4. ppm. C14H10N2O2 (238.24): calcd. C, 70.58; H, 4.23; N, 11.76; found C 70.81; H 4.41; N 11.52.
(R)-1-(Prop-2-yn-1-yl)-1,10b-dihydropyrimido[2,1-a]isoindole-2,6-dione [(+)-8]: White crystals (51% yield), m.p. 93–95 °C, = +368 (c = 0.2, EtOH).
(S)-1-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)methyl)-1,10b-dihydropyrimido[2,1-a]isoindole-2,6-dione [(−)-9]: White crystals (72% yield), m.p. 199–200 °C, = −295 (c = 0.2, EtOH), 1H-NMR (500 MHz, CDCl3, 30 °C): δ = 2.31 (s, 3H, CH3), 4.45 (d, J = 15.7 Hz, 1H, NCH2CH) 5.15 (d, J = 15.8 Hz, 1H, NCH2CH) 5.43–5.64 (m, 3H, NCH2Ar, H-3) 6.18 (s, 1H, H-10b) 7.17–7.31 (m, 4H, Ar) 7.55 (1H, s, CHtriazol) 7.63–7.95 (m, 4H, Ar) 8.70 (d, J = 7.9 Hz 1H, H-4), 13C NMR (125 MHz, CDCl3, 30 °C): δ = 19.1, 37.2, 52.4, 70.3, 106.9, 123.5, 125.0, 126.7, 127.5, 129.2, 129.6, 130.6, 131.1, 131.6, 132.3, 133.6, 133.6, 136.9, 138.9, 143.8, 164.4, 165.8. ppm. C22H19N5O2 (385.42): calcd. C, 68.56; H, 4.97; N, 18.17; found C 68.71; H 4.81; N 18.39.
(R)-1-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)methyl)-1,10b-dihydropyrimido[2,1-a]isoindole-2,6-dione [(+)-9]: White crystals (72% yield), m.p. 199–200 °C, = +287 (c = 0.2, EtOH).
3.2.7. Representative Data for the Racemates (±)-4–(±)-16
tert-Butyl ((1R*,2R*,3S*,4S*)-3-(prop-2-yn-1-ylcarbamoyl)bicyclo[2.2.1]hept-5-en-2-yl)carbamate [(±)-4]: Yield 75%, white crystals, m.p. 133–135 °C.
(1S*,4R*,4aR*,6aS*,12aS*)-6-(Prop-2-yn-1-yl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(±)-6]: Yield 68%, white crystals, m.p. 235–240 °C.
(1S*,4R*,4aR*,6aS*,12aS*)-6-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)-methyl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(±)-7]: Yield 79%, pale-yellow crystals, m.p. 163–165 °C.
1-(Prop-2-yn-1-yl)-1,10b-dihydropyrimido[2,1-a]isoindole-2,6-dione [(±)-8]: Yield 68%, white crystals, m.p. 166–168 °C.
1-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)methyl)-1,10b-dihydropyrimido[2,1-a]isoindole-2,6-dione [(±)-9]: Yield 61%, white crystals, m.p. 192–195 °C.
tert-Butyl ((1R*,2S*,3R*,4S*)-3-(prop-2-yn-1-ylcarbamoyl)bicyclo[2.2.1]hept-5-en-2-yl)carbamate [(±)-13]: Yield 71%, white crystals, m.p. 160–161 °C.
(1S*,4R*,4aS*,6aR*,12aR*)-6-(Prop-2-yn-1-yl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(±)-15]: Yield 71%, white crystals, m.p. 215–220 °C.
(1S*,4R*,4aS*,6aR*,12aR*)-6-((1-(2-Methylbenzyl)-1H-1,2,3-triazol-4-yl)-methyl)-1,4,4a,6,6a,12a-hexahydro-1,4-methanoisoindolo[2,1-a]quinazoline-5,11-dione [(±)-16]: Yield 75%, white crystals, m.p. 163–165 °C.
Supplementary materials contain obtained 1H and 13C NMR spectra of compounds 4–16, HPLC chromatograms of domino products (+)-6, (−)-6, (+)-15 and (−)-15 and RDA products (+)-8, (−)-8, (+)-9 and (−)-9 presented in the manuscript.
4. Conclusions
In summary, we have developed an efficient procedure for the synthesis of novel racemic and enantiomeric 1,2,3-triazole pharmacophore-based pyrimido[2,1-a]isoindoles in the reaction of N-propargyl-substituted norbornenecarboxamides through a traceless chirality transfer strategy. It involves a three-step reaction using a domino and a copper-catalyzed click reaction as well as an RDA reaction step. The stereochemical information to the newly formed stereogenic centers of the isoindoloquinazolinones was facilitated by the high rigidity of the norbornene skeleton. The absolute configuration of the final products was characterized, because the configuration did not change during subsequent steps. The main advantages of this protocol are simplicity, high yield, short time, mild reaction conditions and easy work-up.
Supplementary Materials
The following are available online. The 1H and 13C NMR spectra of compounds 4–16 and HPLC chromatograms of domino products (+)-6, (−)-6, (+)-15 and (−)-15 and RDA products (+)-8, (−)-8, (+)-9 and (−)-9 are available as supporting data.
Author Contributions
F.F. and M.P. planned and designed the project. M.E.H., M.P. and Z.K. performed the syntheses and characterized the synthesized compounds. M.P and M.E.H. prepared the manuscript for publication, all authors discussed the results and commented on the manuscript.
Funding
We are grateful to the Hungarian Research Foundation (OTKA No. K 115731). The financial support of the GINOP-2.3.2-15-2016-00038 project is acknowledged. Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT is acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Held, F.E.; Guryev, A.A.; Fröhlich, T.; Hampel, F.; Kahnt, A.; Hutterer, C.; Steingruber, M.; Bahsi, H.; von Bojničić-Kninski, C.; Mattes, D.S. Facile Access to Potent Antiviral Quinazoline Heterocycles with Fluorescence Properties via Merging Metal-Free Domino Reactions. Nat. Commun. 2017, 8, 15071–15080. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, C.; Hamilton, S.; Steingruber, M.; Zeitträger, I.; Bahsi, H.; Thuma, N.; Naing, Z.; Örfi, Z.; Örfi, L.; Socher, E. The Chemical Class of Quinazoline CoMpounds Provides a Core Structure for The Design of Anticytomegaloviral Kinase Inhibitors. Antiviral. Res. 2016, 134, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Alafeefy, A.M.; Kadi, A.A.; Al-Deeb, O.A.; El-Tahir, K.E.; Al-jaber, N.A. Synthesis, Analgesic and Anti-inflammatory Evaluation of Some Novel Quinazoline Derivatives. Eur. J. Med. Chem. 2010, 45, 4947–4952. [Google Scholar] [CrossRef] [PubMed]
- Madapa, S.; Tusi, Z.; Mishra, A.; Srivastava, K.; Pandey, S.; Tripathi, R.; Puri, S.; Batra, S. Search for New Pharmacophores for Antimalarial Activity. Part II: Synthesis and Antimalarial Activity of New 6-ureido-4-anilinoquinazolines. Bioorg. Med. Chem. 2009, 17, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jin, L.; Xiang, H.; Wu, J.; Wang, P.; Hu, D.; Xue, W.; Yang, S. Synthesis and Anticancer Activities of 5,6,7-trimethoxy-N-phenyl (ethyl)-4-aminoquinazoline Derivatives. Eur. J. Med. Chem. 2013, 66, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.; Zhang, Y.; Heinrich, J.-C.; Haupt, J.; Donakonda, S.; Lennig, P. Thymine derivatives and quinazoline-dione derivatives for the inhibition of HSP27. WO 2016016268 A1, 4 February 2016. [Google Scholar]
- Primeau, J.L.; Garrick, L.M.; Ocain, T.D.; Soll, R.M.; Dollings, P.J. Preparation of pyrimido-cycloalkanes as angiotensin II antagonists and antihyperlipidemics. WO 9308171 A1, 29 April 1993. [Google Scholar]
- Hasegawa, T.; Nakajima, H.; Kubota, D.; Okuma, K. Preparation of pyrimidine derivatives as poly(ADP-ribose) polymerase inhibitors. WO 2000042025 A1, 20 July 2000. [Google Scholar]
- Da Silva, F.d.C.; de Souza, M.C.B.; Frugulhetti, I.I.; Castro, H.C.; Silmara, L.d.O.; de Souza, T.M.L.; Rodrigues, D.Q.; Souza, A.M.; Abreu, P.A.; Passamani, F. Synthesis, HIV-RT Inhibitory Activity and SAR of 1-benzyl-1H-1,2,3-triazole Derivatives of Carbohydrates. Eur. J. Med. Chem. 2009, 44, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Katzung, B.G.; Trevor, A.J. Beta-Lactam and Other Cell Wall- and Membrane-Active Antibiotics. In Basic and Clinical Pharmacology 13E; McGraw-Hill Education: New York, NY, USA, 2014; p. 1145. [Google Scholar]
- Lauria, A.; Delisi, R.; Mingoia, F.; Terenzi, A.; Martorana, A.; Barone, G.; Almerico, A.M. 1,2,3-Triazole in Heterocyclic Compounds, Endowed with Biological Activity, Through 1,3-Dipolar Cycloadditions. Eur. J. Org. Chem. 2014, 2014, 3289–3306. [Google Scholar] [CrossRef]
- Xia, Y.; Qu, F.; Peng, L. Triazole Nucleoside Derivatives Bearing Aryl Functionalities on the Nucleobases Show Antiviral and Anticancer Activity. Mini-Rev. Med. Chem. 2010, 10, 806–821. [Google Scholar] [CrossRef]
- Peterson, L.B.; Blagg, B.S. Click chemistry to probe Hsp90: Synthesis and Evaluation of a Series of Triazole-Containing Novobiocin Analogues. Bioorg. Med. Chem. Lett. 2010, 20, 3957–3960. [Google Scholar] [CrossRef]
- Doiron, J.; Richard, R.; Touré, M.M.; Picot, N.; Richard, R.; Čuperlović-Culf, M.; Robichaud, G.A.; Touaibia, M. Synthesis and Structure–Activity Relationship of 1-and 2-Substituted-1,2,3-Triazole Letrozole-Based Analogues as Aromatase Inhibitors. Eur. J. Med. Chem. 2011, 46, 4010–4024. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal Attributes of 1,2,3-Triazoles: Current Developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Keivanloo, A.; Bakherad, M.; Lotfi, M. Use of Ligand-Assisted Click Reactions for the Rapid Synthesis of Novel 1,2,3-Triazole Pharmacophore-Based 1,2,4-Triazines and their Benzo-Fused Analogues. Tetrahedron 2017, 73, 5872–5882. [Google Scholar] [CrossRef]
- Maračić, S.; Kraljević, T.G.; Paljetak, H.Č.; Perić, M.; Matijašić, M.; Verbanac, D.; Cetina, M.; Raić-Malić, S. 1,2,3-Triazole Pharmacophore-Based Benzofused Nitrogen/Sulfur Heterocycles with Potential Anti-Moraxella Catarrhalis Activity. Bioorg. Med. Chem. 2015, 23, 7448–7463. [Google Scholar] [CrossRef] [PubMed]
- Ouahrouch, A.; Ighachane, H.; Taourirte, M.; Engels, J.W.; Sedra, M.H.; Lazrek, H.B. Benzimidazole-1,2,3-Triazole Hybrid Molecules: Synthesis and Evaluation for Antibacterial/Antifungal Activity. Arch. Pharm. Chem. Life Sci. 2014, 347, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Massarotti, A.; Aprile, S.; Mercalli, V.; Del Grosso, E.; Grosa, G.; Sorba, G.; Tron, G.C. Are 1,4- and 1,5-Disubstituted 1,2,3-Triazoles Good Pharmacophoric Groups? Chem. Med. Chem. 2014, 9, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Kume, M. Synthesis and Structure-Activity Relation-ships of new 7β-[(Z)-2-(2-aminothiazol-4-yl)-2-hydroxyiminoacetamido]-cephalosporins with 1,2,3-Triazole in C-3 Side Chain. J. Antibiot. 1993, 46, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Masui, Y.; Goto, Y.; Kitaura, Y.; Mizutani, T.; Matsumura, I.; Sugata, Y.; Ide, Y.; Takayama, M.; Takahashi, H. Practical Large-Scale Synthesis of Cefmatilen, a New Cephalosporin Antibiotic. Org. Process. Res. Dev. 2004, 8, 744–753. [Google Scholar] [CrossRef]
- Chang, K.Y.; Kwon, S.H.; Nam, G.; Seo, J.H.; Kim, S.H.; Choi, K.I.; Kim, J.H.; Ha, D.C. New Cephalosporin Antibiotics with 3-Triazolylpyridiniummethyl Substituents. J. Antibiot. 2001, 54, 460–462. [Google Scholar] [CrossRef]
- Chitasombat, M.N.; Kontoyiannis, D.P. The ‘Cephalosporin Era’of Triazole Therapy: Isavuconazole, a Welcomed Newcomer for the Treatment of Invasive Fungal Infections. Expert Opin. Pharmaco. 2015, 16, 1543–1558. [Google Scholar] [CrossRef]
- Pellissier, H. Stereocontrolled Domino Reactions. Chem. Rev. 2012, 113, 442–524. [Google Scholar] [CrossRef]
- Bharate, J.B.; Vishwakarma, R.A.; Bharate, S.B. Metal-free Domino One-pot Protocols for Quinoline Synthesis. RSC Adv. 2015, 5, 42020–42053. [Google Scholar] [CrossRef]
- Tu, S.J.; Jiang, B. Microwave-Assisted Domino Reaction in Organic Synthesis. In Advances in Induction and Microwave Heating of Mineral and Organic Materials; Grundas, S.A., Ed.; InTech: Xuzhou, China, 2011; pp. 673–696. Available online: https://www.intechopen.com/books/advances-in-induction-and-microwave-heating-of-mineral-and-organic-materials/microwave-assisted-domino-reaction-in-organic-synthesis (accessed on 6 December 2018).
- Zhao, Y.H.; Li, Y.; Guo, T.; Tang, Z.; Deng, K.; Zhao, G. CuI-Catalyzed Domino Reactions for the Synthesis of Benzoxazine-Fused Isoquinolines under Microwave Irradiation. Synth. Commun. 2016, 46, 355–360. [Google Scholar] [CrossRef]
- Tietze, L.F.; Brasche, G.; Gericke, K. Domino Reactions in Organic Synthesis; Weinheim Wiley-VCH: Gottingen, Germany, 2006. [Google Scholar]
- Padwa, A.; Bur, S.K. The Domino Way to Heterocycles. Tetrahedron 2007, 63, 5341–5378. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Ye, P.; Sprague, K.; Sargent, K.; Yohannes, D.; Baldino, C.M.; Wilson, C.J.; Ng, S.C. Novel One-pot Total Syntheses of Deoxyvasicinone, Mackinazolinone, Isaindigotone, and Their Derivatives Promoted by Microwave Irradiation. Org. Lett. 2005, 7, 3363–3366. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Yamada, K. Amination/Cyclization Cascade by Acid-Catalyzed Activation of Indolenine for the One-Pot Synthesis of Phaitanthrin E. Org. Lett. 2016, 18, 6504–6507. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Terasaki, M. Synthesis of Phaitanthrin E and Tryptanthrin through Amination/Cyclization Cascade. Helv. Chim. Acta. 2018, 101, e1700284. [Google Scholar] [CrossRef]
- Rolfe, A.; Painter, T.O.; Asad, N.; Hur, M.Y.; Jeon, K.O.; Brzozowski, M.; Klimberg, S.V.; Porubsky, P.; Neuenswander, B.; Lushington, G.H. Triazole-Containing Isothiazolidine 1,1-dioxide Library Synthesis: One-pot, Multi-Component Protocols for Small Molecular Probe Discovery. Am. Chem. Soc. Comb. Sci. 2011, 13, 511–517. [Google Scholar] [CrossRef]
- Lundberg, P.; Hawker, C.J.; Hult, A.; Malkoch, M. Click Assisted One-pot Multi-step Reactions in Polymer Science: Accelerated Synthetic Protocols. Macromol. Rapid Commun. 2008, 29, 998–1015. [Google Scholar] [CrossRef]
- Esmaeili-Marandi, F.; Saeedi, M.; Yavari, I.; Mahdavi, M.; Shafiee, A. Synthesis of Novel Isoindolo [2,1-a] quinazolinedione Derivatives Containing a 1,2,3-Triazole Ring System. Helv. Chim. Acta. 2016, 99, 37–40. [Google Scholar] [CrossRef]
- Totobenazara, J.; Burke, A.J. New Click-chemistry Methods for 1,2,3-Triazoles Synthesis: Recent Advances and Applications. Tetrahedron Lett. 2015, 56, 2853–2859. [Google Scholar] [CrossRef]
- Wang, C.; Ikhlef, D.; Kahlal, S.; Saillard, J.Y.; Astruc, D. Metal-Catalyzed Azide-Alkyne “Click” Reactions: Mechanistic Overview and Recent Trends. Coord. Chem. Rev. 2016, 316, 1–20. [Google Scholar] [CrossRef]
- Singh, M.S.; Chowdhury, S.; Koley, S. Advances of Azide-Alkyne Cycloaddition-Click Chemistry over the Recent Decade. Tetrahedron 2016, 72, 5257–5283. [Google Scholar] [CrossRef]
- Tăbăcaru, A.; Furdui, B.; Ghinea, I.O.; Carac, G.; Dinică, R.M. Recent Advances in Click Chemistry Reactions Mediated by Transition Metal Based Systems. Inorg. Chim. Acta. 2017, 455, 329–349. [Google Scholar] [CrossRef]
- Stájer, G.; Csende, F.; Fülöp, F. The Retro Diels-Alder Reaction as a Valuable Tool for the Synthesis of Heterocycles. Curr. Org. Chem. 2003, 7, 1423–1432. [Google Scholar] [CrossRef]
- Suzuki, K.; Inomata, K.; Endo, Y. Enantiocontrolled Synthesis of Jasmonates via Tandem Retro-Diels− Alder− Ene Reaction Activated by a Silyl Substituent. Org. Lett. 2004, 6, 409–411. [Google Scholar] [CrossRef] [PubMed]
- González-Temprano, I.; Osante, I.; Lete, E.; Sotomayor, N. Enantiodivergent Synthesis of Pyrrolo [2,1-a] Isoquinolines Based on Diastereoselective Parham Cyclization and α-Amidoalkylation Reactions. J. Org. Chem. 2004, 69, 3875–3885. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.A.; Kerr, M.A. Heterocycles from Cyclopropanes: Applications in Natural Product Synthesis. Chem. Soc. Rev. 2009, 38, 3051–3060. [Google Scholar] [CrossRef]
- Yoder, R.A.; Johnston, J.N. A Case Study in Biomimetic Total Synthesis: Polyolefin Carbocyclizations to Terpenes and Steroids. Chem. Rev. 2005, 105, 4730–4756. [Google Scholar] [CrossRef]
- Cheng, Y.; Huang, Z.T.; Wang, M.X. Heterocyclic Enamines: The Versatile Intermediates in the Synthesis of Heterocyclic Compounds and Natural Products. Curr. Org. Chem. 2004, 8, 325–351. [Google Scholar] [CrossRef]
- Palkó, M.; Sohár, P.; Fülöp, F. Synthesis and Transformations of diendo-3-Aminobicyclo[2.2.2]oct-5-ene-2-carboxylic Acid Derivatives. Molecules 2011, 16, 7691–7705. [Google Scholar] [CrossRef]
- Fekete, B.; Palkó, M.; Haukka, M.; Fülöp, F. Synthesis of Pyrrolo[1,2-a]pyrimidine Enantiomers via Domino Ring-Closure followed by Retro Diels-Alder Protocol. Molecules 2017, 22, 613. [Google Scholar] [CrossRef] [PubMed]
- Fekete, B.; Palkó, M.; Mándity, I.; Haukka, M.; Fülöp, F. A Domino Ring-Closure Followed by Retro-Diels–Alder Reaction for the Preparation of Pyrimido[2,1-a]isoindole Enantiomers. Eur. J. Org. Chem. 2016, 3519–3527. [Google Scholar] [CrossRef]
- Fülöp, F.; Miklós, F.; Forró, E. Diexo-3-aminonorbornane-2-carboxylic Acid as Highly Applicable Chiral Source for the Enantioselective Synthesis of Heterocycles. Synlett 2008, 1687–1689. [Google Scholar] [CrossRef]
- Miklós, F.; Tóth, Z.; Hänninen, M.M.; Sillanpää, R.; Forró, E.; Fülöp, F. Retro-Diels–Alder Protocol for the Synthesis of Pyrrolo[1,2-a]pyrimidine and Pyrimido[2,1-a]isoindole Enantiomers. Eur. J. Org. Chem. 2013, 4887–4894. [Google Scholar] [CrossRef]
- Miklós, F.; Bozó, K.; Galla, Z.; Haukka, M.; Fülöp, F. Traceless Chirality Transfer from a Norbornene β-Amino Acid to Pyrimido[2,1-a]isoindole Enantiomers. Tetrahedron: Asymmetry 2017, 28, 1401–1406. [Google Scholar] [CrossRef]
- Nekkaa, I.; Palko, M.; Mandity, I.; Miklos, F.; Fülöp, F. Continuous-Flow retro-Diels–Alder Reaction: A Process Window for Designing Heterocyclic Scaffolds. Eur. J. Org. Chem. 2018, 2018, 4456–4464. [Google Scholar] [CrossRef]
- Miklós, F.; Fülöp, F. “Dry” and “Wet” Green Synthesis of 2,2′-Disubstituted Quinazolinones. Eur. J. Org. Chem. 2010, 2010, 959–965. [Google Scholar] [CrossRef]
- Palkó, M.; Sándor, E.; Sohár, P.; Fülöp, F. Synthesis and Stereostructure of 3-amino-5- and -6-hydroxybicyclo[2.2.1]heptane-2-carboxylic Acid Diastereomers. Monats. Chem. 2005, 136, 2051–2058. [Google Scholar] [CrossRef]
- Stájer, G.; Szabó, E.A.; Fülöp, F.; Bernáth, G.; Sohár, P. Stereochemical Studies. 58. Saturated Heterocycles. 39. Preparation and Steric Structures of dihydro-1,3-oxazines, 1,3-oxazin-2-ones and 1,3-oxazine-2-thiones Fused with Norbornane and Norbornene. J. Heterocycl. Chem. 1983, 20, 1181–1185. [Google Scholar] [CrossRef]
- Stájer, G.; Mód, L.; Szabó, A.E.; Fülöp, F.; Bernáth, G.; Sohár, P. Stereochemical Studies—79 Synthesis and Kinetic Study on the Retrodiene Decomposition of Norbornene-condensed 1,3-oxazin-4-ones. Tetrahedron 1984, 40, 2385–2393. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Vapour-assisted Enzymatic Hydrolysis of β-Lactams in a Solvent-free System. Tetrahedron: Asymmetry 2008, 19, 1005–1009. [Google Scholar] [CrossRef]
- Lloyd, M.; Lloyd, R.; Keene, P.; Osborne, A. A Concise Synthesis of Single-Enantiomer β-Lactams and β-Amino acids using Rhodococcus Globerulus. J. Chem. Technol. Biotechnol 2007, 82, 1099–1106. [Google Scholar] [CrossRef]
- Canonne, P.; Akssira, M.; Dahdouh, A.; Kasmi, H.; Boumzebra, M. A Convenient Synthesis of Bridged Azatricyclic Anhydrides. Tetrahedron 1993, 49, 1985–1992. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–16 are not available from the authors. |


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).